

Abstract
Background
To better understand the pathogenesis of cervical cancer (CC), we systematically analysed the genomic variation and human papillomavirus (HPV) integration profiles of cervical intraepithelial neoplasia (CIN) and CC.
Methods
We performed whole-genome sequencing or whole-exome sequencing of 102 tumour-normal pairs and human papillomavirus probe capture sequencing of 45 CCs, 44 CIN samples and 25 normal cervical samples, and constructed strict integrated workflow of genomic analysis.
Results
Mutational analysis identified eight significantly mutated genes in CC including four genes (FAT1, MLL3, MLL2 and FADD), which have not previously been reported in CC. Targetable alterations were identified in 55.9% of patients. In addition, HPV integration breakpoints occurred in 97.8% of the CC samples, 70.5% of the CIN samples and 42.8% of the normal cervical samples with HPV infection. Integrations of high-risk HPV strains in CCs, including HPV16, 18, 33 and 58, also occurred in the CIN samples. Moreover, gene mutations were detected in 52% of the CIN specimens, and 54.8% of these mutations occurred in genes that also mutated in CCs.
Conclusion
Our results lay the foundation for a deep understanding of the molecular mechanisms and finding new diagnostic and therapeutic targets of CC.
Keywords: cervical cancer, pathogenesis, genomic variation, human papillomavirus integration
Introduction
Human papillomavirus (HPV) infection can lead to a variety of human cancers,1 2 including cervical cancer (CC), which is the fourth most common cause of cancer and a leading cause of mortality in women worldwide.3 It has been estimated that approximately 989 000 Chinese women were diagnosed with CC and 305 000 died from it in 2015.4 Despite improved multidisciplinary treatments, the prognosis of advanced CC is still poor, with a 5-year survival rate of approximately 15% for late-stage patients.5
CC screening as a secondary prevention is important in CC control. HPV DNA and liquid-based cytology have been widely used in CC screenings; however, there are limitations to both approaches.6 Many studies have reported the use of new approaches, such as assessing E6/E7 mRNA and protein expression or p16ink4a overexpression, in CC screening.7 The identification and validation of new molecular biomarkers play an important role in CC prevention. Previous studies have identified recurrent genetic mutations in PIK3CA, FBXW7, EP300, MAPK1, HLA-B, NFE2L2, TP53, ERBB2, ELF3 and CBFB in CC.8 However, the patterns of cellular alterations and virus integrations in cervical lesions, especially in cervical intraepithelial neoplasia (CIN), are largely unknown.
Treatments for CC are mainly based on surgery and chemoradiotherapy. Recent advances in targeted therapies against specific somatic alterations have transformed the management of cancers in general.9 The discovery of new therapeutic targets could improve the current strategies to combat CC, such as agents targeting the PI(3)K pathway.10 Thus, it is necessary to evaluate genetic alterations in cancer and to subclassify cancers based on genetic alterations.
Here, we systematically analysed the genomic variation profiles of 102 tumour–normal pairs and 25 CIN specimens with matched germline DNA. Somatic mutations, copy number alterations (CNAs), genomic rearrangements and HPV integration events were identified and used to define the genomic landscape in CC and to find potential biomarkers for the screening and treatment of CC.
Methods
Samples
This project and protocols were conducted according to the Declaration of Helsinki. Tumour tissues and matched adjacent normal tissues or peripheral blood were collected. Tissue cells of normal or CIN were collected from female patients attending a sexually transmitted disease clinic at Shanghai Skin Disease Hospital. Sample collection, HPV typing, liquid-based cytology and biopsy were performed under the guidelines or demands of the patients with appropriate informed consent.
Next-generation sequencing and data analysis
Whole-genome sequencing (WGS) or whole-exome sequencing (WES), HPV probe-capture sequencing (HPCS) and transcriptome using RNAseq were performed as described in the online supplementary materials. Read alignment and processing were performed using the BWA,11 Picard (http://broadinstitute.github.io/picard/) and GATK tools.12 We detected somatic single nucleotide variants (SNVs), insertions and deletions (InDels), CNAs and structural variations (SVs) using MuTect,13 Strelka,14 Varscan2,15 CLImAT16 and Clipping REveals STructure (CREST).17 MutSigCV and GISTIC V.2.0 were employed to analyse the genes significantly affected by mutations and CNAs.
Statistical analysis
P values were calculated using Fisher’s exact test, t-test, Mann-Whitney U test and Delong’s test as indicated. Receiver operating characteristic (ROC) curves with the area under the curve (AUC) were performed to evaluate the specificity and sensitivity of HPV testing for detecting CC using pROC package.18
Data availability
All the sequence data have been deposited in the database NCBI SRA: SRA315538 and the project number PRJNA305342.
See the online supplementary materials and methods for more detailed methods.
Results
Profiles of somatic mutations in CCs
To explore the profiles of molecular variants in CC samples, we selected 102 paired samples, including tumours (95 HPV-positive cases and 7 HPV-negative cases) and matched-normal tissues (n=8) or peripheral blood (n=94) (see online supplementary table S1). We then performed WES analysis of 76 tumour-normal sample pairs using the Illumina HiSeq 2000 platform and the Ion Proton platform, WGS analysis of 27 tumour-normal samples using the HiSeq X-Ten platform and the complete genomics (CG) platform (one sample sequenced by WES and WGS), and transcriptome analysis of 6 tumour-normal samples (all six samples overlapped with WGS/WES) using the HiSeq 2000 platform (see online supplementary figure S1 and supplementary table S1). The average depth of coverage was ~100× for WES data and ~30× (HiSeq X-Ten) or 100× (CG) for WGS (see online supplementary table S2), respectively.
jmedgenet-2018-105745supp001.pdf (8.6MB, pdf)
jmedgenet-2018-105745supp002.pdf (389.5KB, pdf)
Using multiple tools, we identified 17 034 somatic mutations in the exons and splice regions, including 8479 missense, 3755 synonymous, 691 nonsense, 166 splice, 40 in-frame InDel, 112 frameshift, 11 initiation-site loss, 10 read-through and 3770 UTR mutations (see online supplementary table S3). There was an average of 167 exonic and splicing mutations (ranging from 2 to 1361) per sample, suggesting high heterogeneity in the CC samples. The mean and median exonic mutation rates were 3.53/Mb and 2.05/Mb, respectively (figure 1A). These rates are in the same range as the rates observed in the majority of adult solid tumours.19 In total, 91.9% (226/246) of the randomly selected somatic mutations were completely validated by mass spectrometry (MS) genotyping or Sanger sequencing (see online supplementary table S3).
Figure 1.

Genomic alteration profiles of cervical carcinomas. (A) Total mutational rates per Mb, including the non-silent and silent mutations in each sample. The HPV infection status, pathology type and International Federation of Gynecology and Obstetrics (FIGO) stage for all the cases are displayed below. (B) The alteration spectrum of the significantly mutated genes in 102 cervical cancers (CCs). The grey bar on the right indicates the q value calculated using MutsigCV. The frequency of non-silent mutations in each gene is shown on the left. (C) A summary of the significantly amplified or deleted genes in 102 CCs, with the frequency of each gene shown on the left.
Through mutation spectrum analysis, we observed a high C>T transition rate and a high C>G transversion rate in the CC samples, accounting for 55.0% and 18.9% of the total mutations, respectively (see online supplementary figure S2A-S2B). Context analysis revealed that Tp*CpN>(T/G) was the predominant mutation type, which is consistent with previous report.8 (Tp*CpW(W=A or T)>(T/G)) mutations constituted 42.3% of all the mutations. It has been reported that the overexpression of the cytidine deaminases in the APOBEC family,20–22 may cause this mutation signature. Indeed, transcriptomic analysis demonstrated that the APOBEC family member APOBEC3H was expressed at higher levels in CC samples than those in the adjacent normal tissues (p=0.02, online supplementary table S4), while other members of APOBEC family including APOBEC3A,APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G showed no significant dysregulation in the same samples (see online supplementary table S4, all p>0.05). These data suggest that the mechanisms underlying the major APOBEC3H-characterised mutation types (Tp*CpW >(T/G)) play an important role in cervical carcinogenesis.
Driver mutations in CCs
To identify recurrently mutated genes, we analysed all somatic SNVs and InDels in 102 CC samples using MutsigCV.19 Five genes showed a statistically significant level of recurrent mutations (with false-discovery rates (FDRs)<0.1): PIK3CA (16.7%, 17/102), FBXW7 (12.8%, 13/102), MLL3 (7.8%, 8/102), CASP8 (3.9%, 4/102) and FADD (3.9%, 4/102) (figure 1B and online supplementary table S5). Although the results were not statistically significant, FAT1 (8.8%, 9/102), MLL2 (5.9%, 6/102) and EP300 (5.9%, 6/102), which had high mutation rates and were enriched in truncation mutations (see online supplementary table S6), were predicted to have roles in CC. Mutations in these genes were validated by MS genotyping or Sanger sequencing (see online supplementary table S3). Recurrent mutations in PIK3CA, FBXW7 and EP300, which have been previously reported in CC,8 were also identified in our cohort. The majority of PIK3CA mutations clustered in the helical domain (73.7%, including four E542K, one E545G and nine E545K) but not in the kinase domain (15.8%, 3/19). Moreover, several hotspot mutations were identified in FBXW7 (three R465C, two R465H, five R505G and one R505L), and nonsense mutations were frequently observed in EP300 (50%, 3/6; online supplementary table S3). We found recurrent mutations in CASP8, FADD, MLL2, MLL3 and FAT1, which are, to our knowledge, reported here for the first time in CC (figure 1B; online supplementary table S5-S6). Mutations in these genes were more frequent than those previously reported in the COSMIC database (see online supplementary table S6).
In our cohort, CASP8 and FADD each harboured four mutations, most of which were inactivating mutations (75%, 6/8; figure 1B and online supplementary table S6). Interestingly, although the results were not statistically significant (p=0.15), FADD mutations were found only in cases without CASP8 mutations (figure 1B). CASP8 is normally recruited to death receptors by binding to the apoptotic adaptor FADD, which subsequently initiates caspase-mediated apoptosis. Loss of function of these two genes may promote carcinogenesis by inducing resistance to cell death.
The MLL2 and MLL3 genes, which each encodes a histone 3-lysine 4 methyltransferase, contained truncation mutations in 85.7% (6/7) and 77.8% (7/9) of the samples, respectively (figure 1B and online supplementary table S6). Both genes are frequently mutated in various tumours, including bladder cancer23 and breast cancer.24 The enrichment in loss-of-function mutations in these genes suggested that the MLL2 and MLL3 genes play roles in tumour suppression in multiple cancer types, including CC.
Another frequently mutated gene was FAT1, which was mutated in 8.8% (9/102) of tumours (figure 1B and online supplementary table S6). Reportedly, recurrent somatic mutations in FAT1 lead to aberrant Wnt activation in multiple human cancers.25 Mutations in related family members, such as FAT2, FAT3 and FAT4, were also found in our cohort (see online supplementary table S3). FAT2 gene-silencing by small interfering RNAs promoted the growth of CC cells, suggesting that FAT2 may be a new tumour-suppressor gene in CC (see online supplementary figure S2C).
Somatic CNAs and structure variations in CCs
In this study, we first compared differences in CNAs between WGS at a low depth and WES in eight samples. The results showed a high correlation across platforms (average correlation coefficient=0.93, 0.87–0.97; online supplementary figure S3A), suggesting that the WES data were also suitable for CNA analysis.
We performed CNA analysis on the WES or WGS data from 102 sample pairs, using a modified CLImAT algorithm.16 The results revealed that 17.6% (18/102) of the samples presented polyploid genome features (the average ploidy was >3.5), of these, 52.2% (14.8%–76.5%) of the chromosomal regions contained CNAs (online supplementary figures S3B-S3C); 82.4% (84/102) of the samples presented diploid genome features, and of these, 23.8% (0.0%–59.7%) of the chromosomal regions contained CNAs. This value was significantly lower than the proportion observed in the polyploidy group (p<0.05, Fisher’s exact test; online supplementary figure S3B).
Among the 84 diploid samples, genome-wide segmented copy number analysis showed that many chromosome arms had undergone large-scale gains or losses in copy number, with frequent gains observed on chromosomes 1q, 3q, 5p, 8q, 9q and 20q and frequent losses observed on chromosomes 3p, 4p, 4q, 6q, 8p, 11p, 11q, 17p, 18q and 19p (see online supplementary table S7). The overall CNA pattern was broadly consistent with the results of other published studies of CC.8 26 27
We identified 23 focal events using the GISTIC2.0 algorithm,28 including 10 amplification peaks and 13 deletion peaks that involved 2541 genes (see online supplementary figure S3D and table S8). These genes included well-known oncogenes and tumour-suppressor genes, such as BIRC3 (11q22.1), PIK3CA/SOX2 (3q29), EGFR (7P11.2), CCND1 (11q13.3), BAP1 (3P11.2) and ATM (11q23.3) (figures 1C and 2A).
Figure 2.

Structural alterations in cervical carcinomas. (A) A genome-wide view of copy number alterations (CNAs). The upper regions represent amplification (red), the lower regions represent deletion (blue) and the peak regions represent the potential driver genes. The three inserts show the common CNA regions (regions between two vertical lines) of the three potential drivers, including TERT, BIRC2/BIRC3 and E2F1. (B and C) Chromothripsis in two samples: CC-X009 (B) and CC-X019 (C). The upper image shows the SVs that are coloured red (insertion), blue (deletion), orange (inversion), green (intrachromosome translocation) and grey (interchromosome translocation). The lower images show the copy number (CN), logR ratio and B allele frequency (BAF).
Considering the limitations of the GISTIC tool, we used an in-house-designed algorithm to identify more potential CNA peaks. We identified seven peaks with a high ratio that involved 1461 genes (see online supplementary table S8). For example, MCL1, which is one of the most frequently amplified genes in human cancer,29 was amplified in 39.3% of the CC samples. Another frequently amplified gene, E2F1 (20q11.21), is a key transcription factor controlling the cell cycle and was amplified in 25.0% of the CC samples.
CREST software17 was used to identify SVs in CCs. A total of 770 SVs were identified, including 109 interchromosomal translocations, 161 intrachromosomal translocations, 115 insertions and 385 deletions (see online supplementary figure S3E and table S9). Chromothripsis occurred in two samples: CC-X009 and CC-X019 (figure 2B,C). We therefore deemed it useful to further examine the relationship between SVs and cervical carcinogenesis.
Driver pathway analysis in CCs
Using the list of driver genes that we found to be altered by mutations or copy number changes, we searched for over-represented pathways with known roles.30
The results showed that well-defined cancer-related pathways, including the RTK/RAS/PI(3)K, cell cycle and apoptosis pathways, were altered in 88%, 74% and 73% of cases, respectively (figure 3A). The activation of EGFR, ERBB2, ERBB3, ERBB4 (mutations and amplifications) and NRAS (amplification) affected the RTK/RAS pathway, and the activation of PIK3CA (mutations and amplifications) predominately resulted in the dysregulation of the PI(3)K/AKT/mTOR pathway. Altered genes in the cell-cycle pathway were mainly related to the G1/S transition, including activated CCND1, CDK4, E2F1 and E2F3 (amplifications) and inactivated CDKN1C, CDKN2D and RB1 (deletions). We also identified mutations that frequently affect the apoptosis pathway, including deletions and mutations of FAS, TNF, FADD and caspases 3, 6, 7, 8, 9 and 10. In addition, the amplification of the BIRC2/3 genes on 11q22 played a role in the deregulation of the cancer cell-death pathway.
Figure 3.

Frequently altered pathways and potential therapeutic targets and drugs in cervical carcinomas. Somatic mutations and copy number alterations (CNAs) in components of the RTK/RAS/PI(3)K, apoptosis, chromatin remodelling, cell cycle, oxidative stress response and squamous differentiation pathways. Red, activating genetic alterations; blue, inactivating genetic alterations. The percentages shown indicate the activation or inactivation of at least one allele. (B) The left panel represents the alteration frequency for each potential drug-targeted gene in the different pathways. The middle panel represents the recurrent somatic mutations (which reoccurred in this cohort or were previously reported in the COSMIC database) and CNAs (≥5 copies). The right panel represents the potential drugs that can be used against the alterations shown in the middle panel.
Genes involved in the chromatin remodelling pathway were recurrently altered by mutations or CNAs. Potential driver gene analysis identified several histone modifiers (MLL2, MLL3 and EP300), and many other genes in this pathway were also mutated or deleted. In addition, 87% of cases had mutations or deletions in components of the SWI/SNF nucleosome remodelling complex, including ARID1A, ARID1B, SMARCAD1, SMARCA4 and PBRM1. We also found alterations in genes involved in oxidative stress response in 59% of cases, including mutations and deletions in CUL3 and KEAP1, as well as amplifications and mutations in NFE2L2. Specifically, genes with known roles in squamous cell differentiation were also frequently altered. Amplifications in SOX2 and TP63 were widely identified in CC samples as were mutations and deletions in NOTCH family members (NOTCH1/2/3).
To identify genes that could potentially be targeted by drugs in the treatment of CC, we used the TARGET (tumour alterations relevant for genomics-driven therapy) database31 for the integrated analysis of mutations and CNAs. The results showed that 34 genes with targetable oncogenic mutations were revealed in 55.9% (57/102) of the samples. Of these 57 samples, each contained at least one mutation or one CNA (≥five copies) in a gene that predicts sensitivity or resistance to anticancer agents (ranging from one to five) (figure 3B). These results suggested that patients with CC may potentially benefit from next-generation sequencing (NGS)-based molecular genotyping and that newly identified genetic alterations may serve as potential candidate drug targets for CC.
Somatic mutations in both CINs and CCs
To study the relationship between somatic mutations and the initiation and progression of CC, we performed WES in 35 additional sample pairs, including 10 CIN1, 9 CIN2, 6 CIN3 and paired blood cell DNA, as well as 10 CC tumour-normal specimens. The average depth of coverage was 95×, and the average percentage of region covered at least 1× was 99.42% (see online supplementary table S2). We found 1747 SNVs and 27 InDels in all 35 samples, 1743 of which occurred in CCs and involved 1552 genes (see online supplementary table S10). Interestingly, 31 gene mutations were detected in 52% (13/25) of the CIN specimens, and 54.8% (17/31) of these mutations (in 32% (8/25) of the CIN specimens) occurred in genes that also mutated in CCs (figure 4). All mutations observed in CINs were validated manually by IGV or by Sanger sequencing (see online supplementary figure S4). Our current findings suggested that mutations could be detected in CIN samples. These results provide a solid foundation for further study of the CC pathogenesis and provide new molecular biomarkers for the early warning and screening of CC.
Figure 4.

Somatic mutations in cervical intraepithelial neoplasia (CINs) and cervical carcinomas. All somatic mutations that occurred in the 25 CINs and 112 cervical cancers (CCs) are shown in the left panel, with each row indicating a gene and each column representing a sample. Genes mutated in the CIN specimens are zoomed in and shown in the right panel.
HPV DNA integrations in both CINs and CCs
To study the relationship between HPV integration in the human genome and the occurrence and development of CC, we performed an HPCS technology. We analysed HPV integration breakpoints in 25 normal cervical samples (including 18 HPV-negative and 7 HPV-positive samples), 44 CIN samples (including 24 CIN1 and 20 CIN2-3 samples) and 45 CC samples (see online supplementary table S1).
A total of 2466 HPV integration breakpoints were identified in 84.3% (75/89) of the abnormal samples, including 97.8% (44/45) of the CC samples and 70.5% (31/44) of the CIN samples (figure 5A, online supplementary table S2 and S11). To confirm the HPV integration events, we randomly chose 29 integration junctions for validation using Sanger sequencing. The results showed that 82.8% (24/29) of the integration junctions were validated (see online supplementary table S11), indicating that the results were reliable for further analysis.
Figure 5.
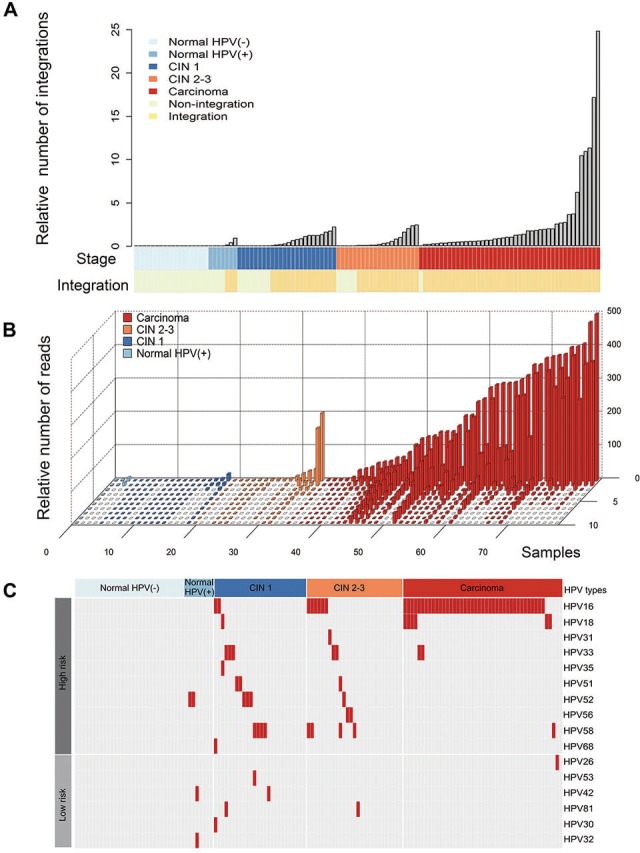
Human papillomavirus (HPV) integration events in the normal cervical samples, cervical intraepithelial neoplasia (CIN) samples and cervical carcinomas. (A) The relative number of HPV integration events in the normal cervix samples that were HPV positive or HPV negative and in the CIN1, CIN2-3 and cervical cancer (CC) samples. All panels are aligned with the vertical tracks representing 114 individuals. (B) The relative number of the supported reads from the normal cervix, CIN1, CIN2-3 and CC samples that were integration-positive. The X-axis represents 78 individuals, the Y-axis represents the relative number of supported reads with integrations and the Z-axis represents the top 10 integration events in each sample. (C) HPV types that were integrated into different samples are shown. The right panel indicates the HPV types (high-risk strains in the upper panel and low-risk strains in the lower panel); the upper panel indicates the clinical stage, including the normal cervical samples that were HPV negative (light blue) and HPV positive (blue) as well as the CIN1 (dark blue), CIN2-3 (orange) and cervical carcinoma (red) samples.
As expected, no HPV integration breakpoint was detected in the normal samples with HPV negative. HPV integration events were found in 42.8% (3/7) of the HPV-positive normal cervical samples (figure 5A). Moreover, we found that HPV integration breakpoints had more supported reads in CC samples than those in the normal samples (p=2.88E-05, Mann-Whitney U test), CIN1 samples (p<2.2E-16, Mann-Whitney U test) and CIN2-3 samples (p=3.79E-09, Mann-Whitney U test), whereas the HPV integration breakpoints had more supported reads in the CIN samples than those in the normal samples (p=8.78E-05 for CIN2-3 vs normal and p=1.94E-02 for CIN1 vs normal, Mann-Whitney U test; figure 5B). Interestingly, we found that the integrations of certain HPV strains in the CCs, including HPV16, 18, 33 and 58, had also occurred in the CIN1 and CIN2-3 samples (figure 5C). The integration occurrence of high-risk HPV was higher than that of low-risk HPV in CINs and CC as compared with normal tissues with HPV positive (p=0.004 for CIN1, p=0.0015 for CIN2-3 and p=1.34e-06 for CC, respectively; Fisher’s exact test; online supplementary figure 5B). These data suggest that oncogenic HPV integration events may occur early in preneoplastic lesions.
To validate the diagnostic performance using HPV integration, ROC curve analysis was conducted in CC and CIN compared with normal individuals (see online supplementary table S12 and figure S5). We found that HPV integration alone or combined with HPV DNA testing could distinguish CC and normal. We used multiple HPV integration as biomarkers to estimate its diagnostic value, the AUC values were 76.5% (95% CI 64.17% to 88.83%), 81.2% (95% CI 69% to 93.4%) and 95.5% (95% CI 88.93% to 100%) for CIN1, CIN2-3 and CC, respectively. Specifically, HPV16/18 integration exclusively showed an AUC value of 96.7% (95% CI 92.98% to 100%; DeLong’s test) for CC versus normal, suggesting an ideal biomarker for detecting CC. The combination of HPV integration and DNA testing had a trend towards higher AUC value than HPV DNA testing (DeLong’s test; online supplementary table S12) in CIN and CC, suggesting a better biomarker for cervical cancer screening.
Our results showed that HPV integration breakpoints are distributed over the whole HPV genome (see online supplementary figure S6A). Statistical analyses indicated that integration junctions were more likely to be located in the E1 (p=7.2E-4) and E2 genes (p=4.5E-4), whereas integration junctions were less likely to be located in the E6 gene (p=4.7E-2), the long control region (LCR, p=2.8E-3), the L1 gene (p=2.8E-2) or the L2 gene (p=1.2E-3; online supplementary figure S6B). Our data suggested that intact forms of the E6 gene and the LCR were more likely to be observed in the human genome.
Local algorithms predicted that the integrated HPV fragment sizes ranged from 43 to 7885 bp (see online supplementary figure S6C). To validate the HPV fragment sizes predicted by NGS, we randomly selected 20 HPV integration events and performed long PCR to amplify the full-length fragments. Junctions residing on both sides of the six successful amplicons were validated by Sanger sequencing (see online supplementary figure S6D). The results revealed a 6649 bp HPV integrant in the CC-H011 genome (see online supplementary figure S7A), and the other five HPV fragments, ranging from 1897 to 4648 bp, were inserted into the human genome of CC samples (see online supplementary figures S7B-7F). These results indicated that HPV fragments of varied fragment sizes randomly insert into the human genome. Transcriptomic data showed that integration of the E4, E5, E6 and E7 genes led to the expression of full length of viral genes (see online supplementary figure S7G), whereas integration of the E1, E2, L1 and L2 genes resulted in only partial transcription or no transcription (see online supplementary figure S7G). These data indicate that although all HPV genes can be integrated into the host genome, only the oncogenes are completely expressed.
Furthermore, HPV integration sites were found within the exons or introns of 886 human genes, including MYC, RAD51B, FHIT, KIF1B, ALK and PDE4D (see online supplementary figure S8A and table S11). We identified 53 integration breakpoints in the MYC gene and its flanking regions (the upstream or downstream 500K) in 24.4% (11/45) of the samples (see online supplementary table S11). Reportedly, integration breakpoints are closely associated with genomic instability.32 Indeed, we found (1) focal amplification of the MYC gene in CC-H011 and CC-H012, in which the HPV integration breakpoints were located in regions both upstream and downstream of the MYC gene; (2) a 17 Mb gain in 8q that included integration into MYC in CC-H024, in which the HPV integration breakpoints were located ~240 kb upstream of MYC and (3) focal amplification of POU5F1B (317 kb upstream of MYC) in CC-H044, in which HPV integration breakpoints were located both upstream and downstream of POU5F1B (see online supplementary figure S8B). Focal amplification may be identified using a ‘looping’ model, as previously reported.32 Gene mutations were also enriched in the host cell genome regions adjacent to the integration breakpoints (p=9.05E-9) in addition to the CNAs and SVs (p<2.2E-16 and p=2.17E-7, respectively; online supplementary figure S8C). Therefore, the host genome instability caused by HPV integration may drive cervical carcinogenesis.
Discussion
Here, we report a comprehensive analysis of genomic alterations and HPV integrations in normal cervical samples, CINs and CCs. To our knowledge, this study is the first to combine cellular genomic alterations and virus integration profiles in both CCs and CIN, to document the mechanisms that underline cervical carcinogenesis and provide potential biomarkers for the screening and therapy.
We report comprehensive mutation profile of CINs and CC samples and a comparison analysis of mutation profiling was conducted. It is worth noting that frequently mutant genes, such as PIK3CA in CC, were not identified in CINs. Moreover, TP53 mutation, a highly prevalent event in various cancers, was rarely identified in both CC and CINs. These observations were consistent with previous study that low mutation frequency of the TP53 and PIK3CA genes were found in CIN3 samples.33 Interestingly, certain other genes that were mutated in CCs were also mutated in CINs, some of which were reported to be related with cancer development, including PTBP3,34 ESX1,35 PER3 36 37 and CIP2A.38
Consistent with previous reports,39 40 we identified HPV integration in CIN samples and normal cervical samples with HPV infection. However, Liu et al did not find HPV integration in CIN1 using HIVID.40 Particularly, HPV integrations were identified especially in CIN1 in this study and the study by Hu et al.39 The reason might be ascribed to the discrepancies in sample size and sequencing depth (~10× for samples by Liu et al,41 ~6427× for specimens by Hu et al and ~61 217× in our cohort, respectively). And the integration events were validated by Sanger with a ratio of 82.8%, indicating that high-depth HIVID could be a sensitive method to detect integrated HPV. The different types of virus integration spectra were observed in different stages of cervical samples. HPV16 and HPV18 are the dominant HPV types integrated into CC samples. Integration of multiple HPV types, including HPV16, HPV18, HPV33 and HPV58, has been identified in CIN samples. Big data of CC screening showed that HPV 16, 33 and 58 were the most common HPV types in high-grade squamous intraepithelial lesions in Chinese women.42 It is possible that different integrated HPV types indicate different outcomes of CIN samples, which may require further follow-up and validation for these patients. Novel disease-specific biomarkers, such as gene mutations and HPV integrations, may serve as secondary markers after positive HPV DNA tests to identify women with prevalent precancers who require immediate colposcopy or treatment.
In clinical practice, advanced patients with CC often choose platinum-containing chemotherapies, but their prognosis remains poor. Therefore, it is necessary to search for new therapeutic drugs, especially targeted drugs, via NGS-based molecular genotyping. Our results showed that 55.9% of the CC samples harboured at least one actionable alteration. PI(3)K/AKT/mTOR inhibitors,43 tyrosine-kinase inhibitors,44 receptor tyrosine kinase antibodies45 and cyclin-dependent kinase inhibitors46 have been approved by the Food and Drug Administration or are in clinical trials for several cancers but not for CCs. Patients with alterations in the targets of these drugs might benefit from targeted therapies. In addition, 50.9% (29/57) of these patients harboured more than one actionable alteration, indicating that combinational therapies may allow patients with CC to receive greater treatment benefits.
In summary, we systematically analysed the genomic variations and HPV integration profiles of CIN and CC, our findings provide the foundation for detecting precancerous lesions early and developing new biotechnology for the screening and therapy of CC.
Footnotes
JH, ZQ, YG and YW contributed equally.
Contributors: JH, PZ and ZL conceived the project and JH and XY designed the experiments. YG, XX, YG, YH, SC, XG, YZ, SZ, JX, RW, HZ, LY and XY performed sequencing and ZQ, YG, LJ, XC, MZ, CB and JH analysed the sequencing data. WL and BZ performed the pathology experiments. QY performed the RNA interference experiments. MS performed the pathway analysis. JH, PZ and XY contributed reagents, materials and analysis tools. ZL, PZ, YW and HX contributed the samples. JH, ZQ and YG integrated, analysed and interpreted all data. JH contributed to the supervision of the work. JH, YHG and ZYQ wrote the manuscript with the assistance and final approval of all authors.
Funding: This work was supported by grants from the National Natural Science Foundation of China (81872274), the China National Key Projects for Infectious Disease (2017ZX10203207), the project of Precision Medicine of Southwestern Hospital (SWH2016ZDCX1013), the Clinical Research Plan of Shanghai Hospital Development Center (16CR1029B and 16CR3111B), the Chinese National Key Program on Basic Research (2014CB965002) and the Shanghai Commission for Science and Technology (15431902900).
Competing interests: None declared.
Ethics approval: This study was approved by the Research Ethics Committee of Southwestern Hospital (No. 2014 – 016) and EthicsCommittee of the Shanghai Skin Disease Hospital (No. SKIN2015-010).
Provenance and peer review: Not commissioned; externally peer reviewed.
Correction notice: This article has been corrected since it was published Online First. Dr Pingyu Zhou’s corresponding address has been corrected.
Patient consent for publication: Not required.
References
- 1. Zandberg DP, Bhargava R, Badin S, Cullen KJ. The role of human papillomavirus in nongenital cancers. CA Cancer J Clin 2013;63:57–81. 10.3322/caac.21167 [DOI] [PubMed] [Google Scholar]
- 2. Zaravinos A. An updated overview of HPV-associated head and neck carcinomas. Oncotarget 2014;5:3956–69. 10.18632/oncotarget.1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–E386. 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 4. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin 2016;66:115–32. 10.3322/caac.21338 [DOI] [PubMed] [Google Scholar]
- 5. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010;17:1471–4. 10.1245/s10434-010-0985-4 [DOI] [PubMed] [Google Scholar]
- 6. Szarewski A, Ambroisine L, Cadman L, Austin J, Ho L, Terry G, Liddle S, Dina R, McCarthy J, Buckley H, Bergeron C, Soutter P, Lyons D, Cuzick J. Comparison of predictors for high-grade cervical intraepithelial neoplasia in women with abnormal smears. Cancer Epidemiol Biomarkers Prev 2008;17:3033–42. 10.1158/1055-9965.EPI-08-0508 [DOI] [PubMed] [Google Scholar]
- 7. Cuschieri K, Wentzensen N. Human papillomavirus mRNA and p16 detection as biomarkers for the improved diagnosis of cervical neoplasia. Cancer Epidemiol Biomarkers Prev 2008;17:2536–45. 10.1158/1055-9965.EPI-08-0306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Treviño V, Vazquez-Santillan K, Guadarrama AS, Wright AA, Rosenberg MW, Duke F, Kaplan B, Wang R, Nickerson E, Walline HM, Lawrence MS, Stewart C, Carter SL, McKenna A, Rodriguez-Sanchez IP, Espinosa-Castilla M, Woie K, Bjorge L, Wik E, Halle MK, Hoivik EA, Krakstad C, Gabiño NB, Gómez-Macías GS, Valdez-Chapa LD, Garza-Rodríguez ML, Maytorena G, Vazquez J, Rodea C, Cravioto A, Cortes ML, Greulich H, Crum CP, Neuberg DS, Hidalgo-Miranda A, Escareno CR, Akslen LA, Carey TE, Vintermyr OK, Gabriel SB, Barrera-Saldaña HA, Melendez-Zajgla J, Getz G, Salvesen HB, Meyerson M. Landscape of genomic alterations in cervical carcinomas. Nature 2014;506:371–5. 10.1038/nature12881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arteaga CL, Baselga J. Impact of genomics on personalized cancer medicine. Clin Cancer Res 2012;18:612–8. 10.1158/1078-0432.CCR-11-2019 [DOI] [PubMed] [Google Scholar]
- 10. Lou H, Villagran G, Boland JF, Im KM, Polo S, Zhou W, Odey U, Juárez-Torres E, Medina-Martínez I, Roman-Basaure E, Mitchell J, Roberson D, Sawitzke J, Garland L, Rodríguez-Herrera M, Wells D, Troyer J, Pinto FC, Bass S, Zhang X, Castillo M, Gold B, Morales H, Yeager M, Berumen J, Alvirez E, Gharzouzi E, Dean M. Genome analysis of latin american cervical cancer: Frequent activation of the pik3ca pathway. Clin Cancer Res 2015;21:5360–70. 10.1158/1078-0432.CCR-14-1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997, 2013. [Google Scholar]
- 12. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. 10.1093/bioinformatics/bts271 [DOI] [PubMed] [Google Scholar]
- 15. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu Z, Liu Y, Shen Y, Wang M, Li A. CLImAT: accurate detection of copy number alteration and loss of heterozygosity in impure and aneuploid tumor samples using whole-genome sequencing data. Bioinformatics 2014;30:2576–83. 10.1093/bioinformatics/btu346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, Holmfeldt L, Payne-Turner D, Fan X, Wei L, Zhao D, Obenauer JC, Naeve C, Mardis ER, Wilson RK, Downing JR, Zhang J. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 2011;8:652–4. 10.1038/nmeth.1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77 10.1186/1471-2105-12-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van ’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Australian Pancreatic Cancer Genome Initiative ICGC Breast Cancer Consortium ICGC MMML-Seq Consortium ICGC PedBrain. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jönsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerød A, Tutt A, Martens JW, Aparicio SA, Borg Å, Salomon AV, Thomas G, Børresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR. Breast Cancer Working Group of the International Cancer Genome Consortium. Mutational processes molding the genomes of 21 breast cancers. Cell 2012;149:979–93. 10.1016/j.cell.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olson ME, Harris RS, Harki DA. APOBEC Enzymes as targets for virus and cancer therapy. Cell Chem Biol 2018;25 10.1016/j.chembiol.2017.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315–22. 10.1038/nature12965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012;486:353–60. 10.1038/nature11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan Ş, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, Vakiani E, Solit D, Singh B, Ganly I, Liau L, Cloughesy TC, Mischel PS, Mellinghoff IK, Chan TA. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet 2013;45:253–61. 10.1038/ng.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirchhoff M, Rose H, Petersen BL, Maahr J, Gerdes T, Lundsteen C, Bryndorf T, Kryger-Baggesen N, Christensen L, Engelholm SA, Philip J. Comparative genomic hybridization reveals a recurrent pattern of chromosomal aberrations in severe dysplasia/carcinoma in situ of the cervix and in advanced-stage cervical carcinoma. Genes Chromosomes Cancer 1999;24:144–50. [DOI] [PubMed] [Google Scholar]
- 27. van den Tillaart SA, Corver WE, Ruano Neto D, ter Haar NT, Goeman JJ, Trimbos JB, Fleuren GJ, Oosting J. Loss of heterozygosity and copy number alterations in flow-sorted bulky cervical cancer. PLoS One 2013;8:e67414 10.1371/journal.pone.0067414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41 10.1186/gb-2011-12-4-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899–905. 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013;153:17–37. 10.1016/j.cell.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 31. Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, McKenna A, Sivachenko A, Rosenberg M, Kiezun A, Voet D, Lawrence M, Lichtenstein LT, Gentry JG, Huang FW, Fostel J, Farlow D, Barbie D, Gandhi L, Lander ES, Gray SW, Joffe S, Janne P, Garber J, MacConaill L, Lindeman N, Rollins B, Kantoff P, Fisher SA, Gabriel S, Getz G, Garraway LA. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med 2014;20:682–8. 10.1038/nm.3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, He D, Ried T, Symer DE, Gillison ML. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res 2014;24:185–99. 10.1101/gr.164806.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tornesello ML, Annunziata C, Buonaguro L, Losito S, Greggi S, Buonaguro FM. TP53 and PIK3CA gene mutations in adenocarcinoma, squamous cell carcinoma and high-grade intraepithelial neoplasia of the cervix. J Transl Med 2014;12:255 10.1186/s12967-014-0255-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen B, Zhao AG, Shao J, Mu XY, Jiang L, Liu JW. The effects of PTBP3 silencing on the proliferation and differentiation of MKN45 human gastric cancer cells. Life Sci 2014;114:29–35. 10.1016/j.lfs.2014.07.038 [DOI] [PubMed] [Google Scholar]
- 35. Figueiredo AL, Salles MG, Albano RM, Porto LC. Molecular and morphologic analyses of expression of ESX1L in different stages of human placental development. J Cell Mol Med 2004;8:545–50. 10.1111/j.1582-4934.2004.tb00479.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X, Yan D, Teng M, Fan J, Zhou C, Li D, Qiu G, Sun X, Li T, Xing T, Tang H, Peng X, Peng Z. Reduced expression of PER3 is associated with incidence and development of colon cancer. Ann Surg Oncol 2012;19:3081–8. 10.1245/s10434-012-2279-5 [DOI] [PubMed] [Google Scholar]
- 37. Climent J, Perez-Losada J, Quigley DA, Kim IJ, Delrosario R, Jen KY, Bosch A, Lluch A, Mao JH, Balmain A. Deletion of the PER3 gene on chromosome 1p36 in recurrent ER-positive breast cancer. J Clin Oncol 2010;28:3770–8. 10.1200/JCO.2009.27.0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu Y, Gu TT, Zheng PS. CIP2A cooperates with H-Ras to promote epithelial-mesenchymal transition in cervical-cancer progression. Cancer Lett 2015;356:646–55. 10.1016/j.canlet.2014.10.013 [DOI] [PubMed] [Google Scholar]
- 39. Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, Liu H, Liu X, Zhao Y, Fang X, Li S, Chen W, Tang T, Fu A, Wang Z, Chen G, Gao Q, Li S, Xi L, Wang C, Liao S, Ma X, Wu P, Li K, Wang S, Zhou J, Wang J, Xu X, Wang H, Ma D. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet 2015;47:158–63. 10.1038/ng.3178 [DOI] [PubMed] [Google Scholar]
- 40. Liu Y, Zhang C, Gao W, Wang L, Pan Y, Gao Y, Lu Z, Ke Y. Genome-wide profiling of the human papillomavirus DNA integration in cervical intraepithelial neoplasia and normal cervical epithelium by HPV capture technology. Sci Rep 2016;6:35427 10.1038/srep35427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y, Lu Z, Xu R, Ke Y. Comprehensive mapping of the human papillomavirus (HPV) DNA integration sites in cervical carcinomas by HPV capture technology. Oncotarget 2016;7:5852–64. 10.18632/oncotarget.6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jing L, Zhong X, Huang W, Liu Y, Wang M, Miao Z, Zhang X, Zou J, Zheng B, Chen C, Liang X, Yang G, Jing C, Wei X. HPV genotypes and associated cervical cytological abnormalities in women from the pearl river delta region of guangdong province, China: a cross-sectional study. BMC Infect Dis 2014;14:388 10.1186/1471-2334-14-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010;363:1801–11. 10.1056/NEJMoa1001671 [DOI] [PubMed] [Google Scholar]
- 44. Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R. FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist 2005;10:461–6. 10.1634/theoncologist.10-7-461 [DOI] [PubMed] [Google Scholar]
- 45. Blumenthal GM, Scher NS, Cortazar P, Chattopadhyay S, Tang S, Song P, Liu Q, Ringgold K, Pilaro AM, Tilley A, King KE, Graham L, Rellahan BL, Weinberg WC, Chi B, Thomas C, Hughes P, Ibrahim A, Justice R, Pazdur R. First FDA approval of dual anti-HER2 regimen: pertuzumab in combination with trastuzumab and docetaxel for HER2-positive metastatic breast cancer. Clin Cancer Res 2013;19:4911–6. 10.1158/1078-0432.CCR-13-1212 [DOI] [PubMed] [Google Scholar]
- 46. Dhillon S. Palbociclib: first global approval. Drugs 2015;75:543–51. 10.1007/s40265-015-0379-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2018-105745supp001.pdf (8.6MB, pdf)
jmedgenet-2018-105745supp002.pdf (389.5KB, pdf)
Data Availability Statement
All the sequence data have been deposited in the database NCBI SRA: SRA315538 and the project number PRJNA305342.
See the online supplementary materials and methods for more detailed methods.