

Abstract
Fatigue is one of the most common symptoms in multiple sclerosis (MS), with a major impact on patients’ quality of life. Currently, treatment proceeds by trial and error with limited success, probably due to the presence of multiple different underlying mechanisms. Recent neuroscientific advances offer the potential to develop tools for differentiating these mechanisms in individual patients and ultimately provide a principled basis for treatment selection. However, development of these tools for differential diagnosis will require guidance by pathophysiological and cognitive theories that propose mechanisms which can be assessed in individual patients. This article provides an overview of contemporary pathophysiological theories of fatigue in MS and discusses how the mechanisms they propose may become measurable with emerging technologies and thus lay a foundation for future personalised treatments.
Keywords: network, lesion, inflammation, dopamine, dyshomeostasis, interoception, metacognition
Introduction
Multiple sclerosis (MS) is the most common neurological disease that causes disability in young adults (for recent reviews, see1–3). With an estimated prevalence of up to 83%, fatigue is one of the most common symptoms in MS4–6 and exerts the greatest impact on patients’ quality of life.4 7 Fatigue therefore represents one of the most pressing clinical problems in the management of MS. Beyond MS, fatigue represents a relevant symptom in numerous disorders from internal medicine, neurology and psychiatry.7–9 In general practice, 20% of patients complain of fatigue; this increases up to 50% in diseases involving the dysregulation of the immune system such as chronic infections, cancer or autoimmune diseases.10 It is also a frequently reported feature of psychiatric diseases and represents a core diagnostic criterion of depression in DSM-5.
As for other neuropsychiatric symptoms, fatigue likely results from different underlying causes.7 10 At present, diagnostic tools are missing which differentiate, in individual patients, between alternative potential causes of fatigue. As a consequence, we lack a principled basis for treatment selection: while several pharmacological and physical therapies are used in practice—for example, drugs like amantadine11 or modafinil12 that affect glutamatergic and dopaminergic transmission and reuptake of monoaminergic transmitters, respectively—treatment selection proceeds by trial and error and under consideration of side effects, rather than by assessment of individual pathophysiological mechanisms. The current absence of objective clinical tests for differentiating alternative disease mechanisms constitutes a critical barrier to improving individual treatment decisions.
Developing tools for differential diagnosis of fatigue in MS requires pathophysiological theories that propose mechanisms which can, in principle, be assessed in individual patients. Previous review articles (eg,10) have mostly treated fatigue as a clinical symptom across disorders. In this paper, we concentrate on MS and discuss possible pathophysiological mechanisms. Our review article provides an overview of contemporary theories of fatigue in MS and discusses how the mechanisms they propose may become measurable with emerging technologies, potentially leading to differential diagnosis and treatment predictions in the future.
Initially, we briefly revisit the definition of fatigue and standard diagnostic procedures. This serves as a reminder that ‘fatigue’ is not a well-defined concept. This is not a trivial issue: one reason for the heterogeneity of the literature on fatigue is that past studies have assessed different constructs of fatigue, for example, not always distinguishing between the subjective perception of fatigue and externally observable fatigability of cognitive or motor functions.
Heterogeneous concepts of ‘fatigue’
Concepts of fatigue vary remarkably in the literature. For example, fatigue has been described as ‘a feeling arising from difficulty in initiation of or sustaining voluntary effort’,8 ‘an overwhelming sense of tiredness that is out of proportion (in relation to the performed activity)’13 or as a ‘feeling that relates to the lack of motivation to deploy resources and engage in high effort performance to cope with their situation’.10
In an attempt towards standardisation, a recent taxonomy distinguishes two major dimensions of fatigue: perception of fatigue and performance fatigability.5 The latter refers to objectively measurable aspects of fatigue, for example, the observable decrease in performance during a cognitive or motor task. By contrast, the perceptual dimension is inherently subjective and cannot be assessed directly by an external observer. From a pathomechanistic perspective, these two dimensions are distinct: explanations of fatigability can, in principle, be derived from physiological and biochemical principles. By contrast, understanding the subjective perception of fatigue requires a cognitive perspective, in particular, concepts of interoception and metacognition.14–16
For clinical practice, operationalised definitions of fatigue are provided by various fatigue questionnaires; see7 17 for overview. Some questionnaires were specifically designed for MS.18 Again, these questionnaires show notable differences in how they operationalise fatigue, which represents an important source of heterogeneity in pathophysiological studies of fatigue.17
Pathophysiological concepts of fatigue
This article focuses on four main classes of potential pathophysiological mechanisms of fatigue in MS (figure 1):
Figure 1.

Pathophysiological mechanisms of fatigue discussed in this article. White and grey boxes represent classes of mechanisms and specific mechanisms, respectively; directed arrows and circle-ended arrows represent direct and mediating effects, respectively. Due to space limitations, only one mechanism per arrow is shown; see main text for other mechanisms. CNS, central nervous system; DA, dopamine; GM, grey matter; NAWM, normally appearing white matter; WM, white matter.
Structural damage of white matter (WM) and grey matter (GM),
Inflammatory processes (within or outside the central nervous system, CNS),
Maladaptive network recruitment due to distributed lesions or inflammation,
Metacognition (self-monitoring) of interoception of dyshomeostatic states.
Structural brain damage
MS is characterised by lesions in the CNS that are disseminated in space (eg, in the cerebrum, brainstem, spinal cord) and time (dynamic changes in lesion load). A key pathological feature of MS are WM lesions with demyelination and inflammation that lead to axonal damage and progressive degeneration.1 Additionally, GM lesions are present, resulting from cortical demyelination due to subpial inflammation, retrograde neuronal degeneration following axonal transection and degenerative mechanisms due to oxidative injury.19 Beyond demarcated lesions, the above processes induce diffuse damage of WM and GM that may lead to regional and whole-brain atrophy. Morphometric studies reported mixed results on the potential link between global measures of brain atrophy and fatigue20–22 and between local measures of WM/GM atrophy and fatigue,23–27 respectively. In the following, we discuss how different forms of WM and GM damage may play a role for the experience of fatigue.
WM lesions
A general idea is that fatigue may result from global impairment of brain function due to distributed WM lesions.28 29 Several studies have used structural MRI and morphometric analyses of WM lesion load to test this idea. Across studies, the overall results are mixed: while some studies found a correlation between fatigue and global structural damage of WM,20 21 23 30 other studies failed to do so.31–34 This inconsistency may partially result from differences in methodology, for example, quantifying structural brain damage with different morphometric methods.35 An alternative explanation for variability across studies is that individual degrees of fatigue may be determined more by the locations of WM lesions than by global lesion load.24 36 Individual differences in normal-appearing white matter might additionally contribute to this variability.24 29 As a final factor of variability, there is an ongoing debate whether individual disease trajectories of MS may be driven differentially by inflammation and neurodegeneration37 38; such differences in disease mechanisms could also relate to differences in fatigue.
Assuming a correlation between WM damage and fatigue can be ascertained, what exactly mediates this link? One explanation concerns fatigability (cognitive or motor): this might originate from reduced activation of the central or peripheral targets of synaptic connexions due to diminished speed and reliability of axonal transmission. In particular, demyelination is known to result in the slowing of conduction speed.39 40 Additionally, activity-dependent conduction block may contribute to fatigability.41 Therapeutically, fampridine (4-aminopyridine) is thought to improve the conduction of action potentials in demyelinated nerves by blocking voltage-gated potassium channels.42 Clinical studies on the efficacy of fampridine for treating fatigue in MS have obtained mixed results so far.43 44
A second explanation refers to structural disconnection, specifically disruption of communication between brain regions with functions of relevance for fatigue, such as motor planning and execution.45 46 Diffusion-weighted imaging studies demonstrated that WM changes are correlated with fatigue, particularly in the anterior internal capsule and anterior thalamic tract.23 24 28 29 47 48 Alternatively, WM damage could disconnect areas of importance for arousal and motivation.46 This is less well investigated, but a tractography study reported altered connectivity between posterior hypothalamus and mesencephalon,49 a tract thought to contain wakefulness-promoting ascending monoaminergic connexions.
GM lesions
GM lesions were long hypothesised to be part of the disease.50 51 However, it was not until the beginning of the 21st century that improved immunohistochemical techniques, high-resolution structural MRI and quantitative morphometry techniques provided clear evidence for GM lesions in MS. GM lesions in MS are discussed by several recent reviews.52–54
The spatial distribution of GM lesions in MS is diffuse.55 Recent neuropathological probability maps of lesions in both WM and GM19 emphasised the occurrence of GM lesions in regions with deep invaginations, such as insular and anterior cingulate cortex (ACC); notably, these are areas of central relevance for interoception, a topic discussed below. Furthermore, this study suggested two different degenerative mechanisms in cortex, that is, inflammation-induced oxidative injury of neurons and retrograde neurodegeneration due to axonal transection.19 In subcortical regions, frequent sites of GM lesions include the thalamus, basal ganglia, amygdala, substantia nigra and hypothalamus.56
How might GM lesions cause fatigue? First, similarly to WM lesions, GM lesions could disturb coordinated activity and connectivity in large-scale networks that mediate motor and cognitive processes, leading to compensatory activity in additional nodes and reducing the scope of adaptive changes.57 This change in network function might be detected by metacognitive mechanisms (see below and figure 1). Empirical investigations of cerebral networks in fatigue patients by neuroimaging demonstrated altered functional connectivity of the basal ganglia,58 among sensorimotor regions,59 or of the default mode network.60
Second, some of the commonly found deep GM lesions affect structures that are directly involved in vigilance, arousal and motivation. One prominent example is the hypothalamus, which neuropathological studies identified as a frequent lesion site in MS61–64 and which is critical for homeostatic regulation (see below). Moreover, the lateral hypothalamus hosts neurons producing orexin, a neuropeptide of fundamental importance for arousal and vigilance. In narcolepsy, a disease with dramatically reduced vigilance, autoimmunological reactions against orexin-producing neurons strongly decrease orexin levels.65 This led to the hypothesis that less pronounced reductions of orexin might produce fatigue in MS. For example, hypothalamic lesions in MS could lead to partial depletion of orexin; alternatively, immunological processes could affect the synthesis and/or postsynaptic efficacy of orexin. So far, studies correlating orexin levels in the cerebrospinal fluid (CSF) and fatigue scores have provided conflicting results.66 67
A third possibility of how GM lesions may cause fatigue refers to the frequent involvement of the brainstem in MS. Specifically, lesions of dopaminergic, serotonergic or noradrenergic nuclei in the brainstem and the consequent reduction of monoaminergic transmitter supply to cortex and basal ganglia could explain the reduction in motivation, mood and arousal that characterise fatigue.
Finally, GM lesions in hypothalamus or brainstem nuclei could disturb the hypothalamus–pituitary–adrenal axis and descending neural control of the autonomic nervous system leading to persistent endocrine and autonomic disturbances, respectively.62 68 This could cause fatigue directly, for example, due to diminished energy supply or hypotension; additionally, perception of prolonged dyshomeostasis has been postulated to underlie subjective experience of fatigue by metacognitive theories that are described below.14 16
Immunological and inflammatory processes
The immune system plays a key role in aetiology and progression of MS.2 3 69 In general, immunological processes in the CNS and the body can interact through multiple pathways.70 71 In MS, the relative contributions of central and peripheral immunological events during the induction and early inflammatory phase of MS are not fully understood. In particular, it remains to be clarified whether a primary immunological process takes place in the brain and spreads to the periphery or whether immune activation begins peripherally before being transferred to the initially unaffected CNS (for review, see69). The latter possibility is supported by the fact that highly effective immunomodulatory treatments for MS (eg, fingolimod, rituximab) have peripheral targets. Regardless of where the initial immune response occurred, myelin damage in the CNS is thought to lead to the release of antigens to the periphery.2 This, in turn, primes immune responses in lymphoid tissue and triggers the invasion of lymphocytes into the CNS.2 While peripheral immune responses may be the driving force at the early stage of MS, evidence suggests that later in the disease, the immune response is shifted and compartmentalised to the CNS in lymphoid-like follicles in the meninges that maintain chronic inflammation.72
Peripheral immunological and inflammatory processes are likely to play a central role for fatigue, in general,10 and in the specific context of MS.73 74 This is illustrated by ‘sickness behaviour’, a syndrome of fatigue, social withdrawal and lowered mood during common infections that trigger the production of proinflammatory cytokines.75 Furthermore, fatigue can be induced by immunomodulatory drugs like interferon-α76 77 or vaccinations that trigger production of proinflammatory cytokines.78 These findings raise the question of how peripheral immunological and inflammatory processes could affect the CNS in MS and impact the experience of fatigue. Several direct and indirect immune-to-brain pathways have been unearthed in the past few decades, including humoral, cellular and neuronal interfaces.10 70 71
Humoral links are established via circumventricular organs where inflammatory cytokines can cross the blood–brain barrier and bind to neurons with specific receptors; furthermore, cytokines such as IL-6 can exert direct actions on brain endothelial cells to produce inflammatory factors such as prostaglandin E2. These processes can trigger both central (eg, microglia activation, projections to the hypothalamus and nucleus of the solitary tract, NTS) and peripheral (eg, fever) processes. Peripheral immune states can also be signalled to the brain by trafficking of immune cells, such as monocytes; this constitutes a cellular pathway. Finally, neural immune-to-brain pathways consist of visceral (especially vagal) afferents that are activated by proinflammatory mediators such as interleukin-1.79 Anatomically, afferent vagal projections are relayed via the NTS and ventromedial posterior thalamus to posterior and mid-insula.80 81 Insular responses to peripheral inflammatory processes have been documented in humans, for example, using functional neuroimaging after induction of acute inflammation by typhoid vaccination80 82 or after injection of endotoxins.83 This interoceptive pathway is an important immune-to-brain link in sickness behaviour and plays a central role in metacognitive hypotheses of fatigue (see below). It also represents the afferent part of a reflex arc that regulates, through NTS projections to the vagal dorsal motor nucleus and nucleus ambiguus, peripheral immunological processes via hypothalamus–pituitary axis activation and anti-inflammatory cholinergic pathways.84
Additionally, several indirect pathways exist how peripheral inflammation can affect the CNS. One notable upstream consequence of peripheral inflammation is a reduction in synthesis of monoaminergic neurotransmitters such as dopamine, norepinephrine and serotonin.10 71 In brief, peripheral inflammation leads to a deficit of tetrahydrobiopterin, an essential cofactor of aromatic amino acid hydroxylase enzymes which are critical for the synthesis of monoamines. This is a potentially highly relevant mechanism for fatigue: given the well-documented involvement of these neuromodulatory transmitters in motivation (dopamine), arousal (norepinephrine) and mood (serotonin), it is conceivable that a general reduction in their synthesis could produce the clinical picture of fatigue. This idea has been investigated experimentally in relation to dopamine. For example, decreased activity of the dopaminergic midbrain was recently demonstrated using functional MRI (fMRI) following typhoid vaccination as a model of systemic inflammation.80 85 Similarly, 18F-dopa PET in patients undergoing therapy with interferon-α showed significant changes in presynaptic striatal dopamine function, consistent with a decrease in synthesis of dopamine.77
A further indirect mechanism of how peripheral inflammation affects the CNS is provided by the kynurenine pathway. This pathway is involved in the metabolism of tryptophan, an essential precursor for monoamine synthesis. In brief, inflammatory mediators activate indoleamine-2,3-dioxygenase which degrades tryptophan along the kynurenine pathway and thus limits monoamine synthesis. Inside the CNS, kynurenine is further metabolised to neurotoxic metabolites such as quinolinic acid, an NMDA receptor agonist which may trigger excitotoxicity and cerebral inflammation.86 Kynurenine has been studied in animal models of relapsing–remitting MS, such as experimental autoimmune encephalomyelitis, as well as in MS in humans (see87 for a review of empirical evidence for altered kynurenine pathway activity in MS). While the exact contribution of kynurenines to fatigue remains to be established, a recent study in mice demonstrated that physical exercise activates molecular pathways in muscle that accelerate conversion of kynurenine (which can pass the blood–brain barrier) into kynurenic acid (which cannot), and that this mechanism protects against depressinogenic effects of both direct kynurenine administration and chronic stress.88 In humans, the volume of the striatum, which is of relevance for reward and motivational processes, is inversely associated with activation of the kynurenine pathway.89
Following this brief overview of how peripheral inflammation may relate to the experience of fatigue, the question remains how inflammation inside the CNS might lead to fatigue. One possibility relates to dopamine: CNS inflammation triggers activation of microglia, and the ensuing production of cytokines in situ negatively affects dopaminergic transmission, for example, through direct effects on dopamine transporters and receptor function (for review, see10). This has motivated a view of fatigue as resulting from altered connectivity between striatum and prefrontal cortex.8 90 The efficacy of this connexion depends on dopaminergic meso-prefrontal afferents and has been implicated in reward-oriented learning and motivation.91 In brief, from this view, fatigue is conceptualised as a variation of altered response to reward and the ensuing decrease in motivation.
An alternative mechanism for how inflammation within the CNS could produce fatigue rests on orexin, a neuropeptide produced by neurons in the lateral hypothalamus.65 In addition to its critical role for vigilance and arousal (as visible in narcolepsy, see above), orexin is involved in the sleep–wake cycle, reward processing and food intake. Animal studies found that inflammation-induced lethargy is mediated by suppression of orexin neuron activity by interleukin-1β and TNF-α92 and that orexin levels correlate with diminished vigilance and exploratory behaviour.93 Concerning MS, the role of orexin for fatigue has not yet been firmly established: studies on the correlation between fatigue and CSF orexin levels have provided contradictory results.66 67
Maladaptive network recruitment during task performance
Functional imaging studies have demonstrated that patients with MS affected by fatigue, compared with patients with MS without fatigue and healthy controls, frequently show an increase of distributed brain activity during the performance of tasks.45 47 94 95 In the spinal cord, patients with MS with fatigue, despite smaller WM lesion load, show higher functional recruitment of the cervical cord than patients with MS without fatigue96 (for discussion, see97). Moreover, patients with MS with fatigue often fail to show physiological adaptation of brain activity during tasks57 (but see98). This may differ across disease stages and brain regions.99 100
One possible explanation for altered cortical activity in MS is that networks mediating specific cognitive operations are perturbed by one of the mechanisms described above, that is, WM/GM lesions or functional impairments due to inflammatory processes. In order to maintain cognitive performance despite lesion-induced or inflammation-induced loss of network function, compensatory recruitment of neuronal tissue may be needed, either in terms of additional regions not usually contributing to a particular task and/or in terms of unusually high levels of activation. Analogous findings have been obtained in other diseases with discrete lesions, such as stroke.101 Whatever the cause of altered cortical activity, the question remains how aberrant activity levels are linked to subjective experience of fatigue. One possibility is that the activation of ‘atypical’ and the compensatory (non-adapting) activation of ‘typical’ regions might be detected by self-monitoring mechanisms, a metacognitive perspective we discuss below.
An alternative possibility is that impairment of neuromodulatory projections from the brainstem—by lesions or by inflammation-induced decrease of transmitter synthesis102—could lead to a functional reorganisation of cortical networks. This is because neuromodulatory transmitters profoundly influence activity and connectivity in cortex, by two major mechanisms. First, they regulate neuronal gain and excitability via slow afterhyperpolarisation currents mediated by calcium-dependent potassium channels103; second, they alter both short-term and long-term synaptic plasticity by modulating NMDA receptors.104 Rapid functional reorganisation of cortical networks in response to manipulations of neuromodulatory transmitters was demonstrated in human and animal studies,105 106 and it is conceivable that similar effects could arise from brainstem lesions in MS or through effects of inflammation on monoamine synthesis.102
Metacognitive perspective on fatigue
The disease theories discussed so far offer potential physiological mechanisms but do not explain how the subjective experience of fatigue might arise. A metacognitive perspective may provide a crucial bridge here. Metacognition corresponds to cognition about cognition, such as judging the accuracy of a perceptual decision.107 In cybernetic theories of brain function, metacognition is understood as ‘self-monitoring of one’s level of mastery in acting on the world… and can be seen as a high-level form of inference about one’s capacity for control’16 (figure 2). Three metacognitive mechanisms of fatigue have been proposed—with emphasis on (1) interoception, that is, the perception of bodily states, (2) network-level function and (3) perceived effort of movements, respectively.
Figure 2.
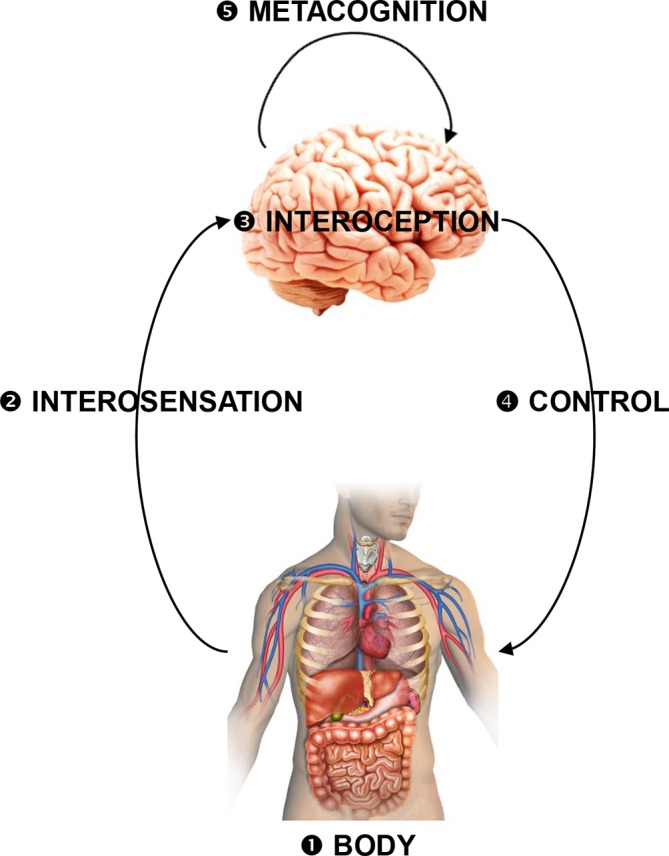
A coarse schematic overview of the inference–control–metacognition loop for bodily regulation (for details, see16). Interoceptive surprise as a possible computational substrate of fatigue can arise from perturbations of any components of this loop: (1) actual perturbations of bodily state that evade cerebral attempts of correction (eg, chronic inflammation, cancer); (2) altered interosensations (due to pathologies of interoceptors or afferent pathways); (3) disturbances of interoception (eg, inflammatory lesions of insula); (4) disturbances of interoactions (neuronally or endocrinologically mediated cerebral influences on bodily functions), for example, inflammatory lesions of ACC, brainstem, hypothalamus or their projections; (5) altered metacognitive processes (eg, changes in expected performance levels). The multiple failure loci offer a potential explanation for the clinical heterogeneity of fatigue and speaks to the necessity of developing tools for differential diagnostics at the circuit level.
First, interoception—the perception of the physiological state of the body, including blood oxygenation, acidity and osmolality; heart rate; plasma concentration of glucose, hormones, cytokines and so on—is disturbed in MS108 and is increasingly recognised as an important factor for the experience of fatigue.70 109 The interoception-related metacognitive theory views fatigue as resulting from the brain’s inference about its capacity for control.14 Specifically, it postulates that fatigue reflects the metacognitive diagnosis that the brain is failing to exert control over bodily states and does not have any action at its disposal to overcome a state of dyshomeostasis. Given this inferred helplessness or low ‘allostatic self-efficacy’, fatigue would correspond to a feeling state that signals the futility of any further actions. A central notion of this theory is that the brain’s capacity to regulate bodily states is represented by a compact information-theoretic quantity (ie, interoceptive surprise) that can be accessed by metacognitive areas. Neuronally, interoceptive surprise can be computed from prediction error signals that index the mismatch between expected and actual bodily states (figure 3). These prediction errors are thought to be signalled by pyramidal cells in supragranular layers of interoceptive areas (eg, insula, ACC) and to require ionotropic glutamatergic receptors, particularly NMDA receptors.14 110 111 While this exact mechanism remains to be demonstrated experimentally, the general theory is supported by empirical evidence from different investigations, including the correlation of activity in insula and ACC with subjectively perceived fatigue during experimentally controlled states of dyshomeostasis, such as induced inflammation.80 82 Moreover, the theory explains why fatigue is a frequent symptom of any disease that involves chronic dyshomeostatic states, including immunological, metabolic, endocrine, cardiovascular, hepatic and renal diseases.9 10 Notably, recent neuroimaging investigations of patients with MS demonstrated that electrophysiological markers of interoception are altered; at the same time, insula and ACC were found to show structural atrophy and abnormal functional connectivity.108
Figure 3.

Neuroanatomically specific circuit model of interoception that plays a central role in theories of fatigue from computational psychiatry.14 The regions are based on anatomical investigations of interoeptive circuitry81; the network they form is thought to instantiate a predictive model of bodily states.111 in this hierarchical network, predictions are sent from higher to lower areas, while prediction errors (PEs; the difference between actual and predicted states) are signalled in the opposite direction and used to update predictions (‘predictive coding’110). Specifically, hierarchically higher visceromotor areas, such as the anterior insula (AI) and anterior cingulate cortex (ACC), are thought to tune homeostatic reflex arcs by means of allostatic predictions computed from bodily and environmental information. In turn, AI/ACC inform hierarchically lower areas, such as posterior and mid-insula, about the expected interosensory consequences (corollary discharge). The latter areas compare these predictions against actual interosensory input and return PE that serve to update the predictions by AI/ACC. At the top of the hierarchy, metacognitive areas (possibly medial prefrontal cortex, mPFC) monitor the level of PE and compute interoceptive surprise. The better the predicted bodily states can be achieved through regulatory action, the smaller PE and interoceptive surprise. Importantly, because of the closed-loop nature of brain–body interactions (figure 2), impairments of any part of the network can lead to chronic interoceptive surprise. This may lead to the metacognitive diagnosis of helplessness or low allostatic self-efficacy (lack of control over bodily states) and has been posited as the substrate for fatigue as a feeling state.14
A clinically important corollary of this theory is that fatigue necessarily has very different causes. This is because interoceptive surprise can arise from disturbances of any component of the closed-loop relation between interoception, bodily regulation and metacognition16 (see figure 2). Notably, interoceptive surprise can result from merely perceived dyshomeostasis. This could occur when the cortical areas that infer bodily states are perturbed, for example, due to inflammatory lesions of the insula which are frequent in MS.19 The ensuing ‘illusion’ of dyshomeostasis would elicit misinformed interoactions, creating dyshomeostatic bodily states that reify the initially spurious interoceptive surprise and render it chronic. Similarly, primary disturbances of interoactions—for example, due to inflammatory lesions of visceromotor areas like ACC,19 hypothalamus62 or brainstem nuclei—would produce lasting perturbations of bodily states.
Second, fatigue might also arise from other forms of prediction error or surprise that the brain finds itself unable to reduce.14 109 The brain’s self-monitoring of performance is not restricted to interoception and its associated circuitry; instead, expectations are held about any domain of cognition, and domain-independent mechanisms of metacognition exist112 which might ‘read out’ error signals from domain-specific functional networks.16 In MS, lesions outside interoceptive pathways impair performance levels of many cognitive and motor acts, as reflected by progressive changes in functional networks.113 Similar to fatigue resulting from bodily dyshomeostasis, this might lead to fatigue as a metacognitive diagnosis of network function: the brain’s interpretation of its own state as a chronic mismatch between actual and expected performance levels that is not amenable to actions.14
Finally, a third proposed metacognitive mechanism of fatigue focuses exclusively on the sensorimotor system.15 This concept assumes that diminished sensory attenuation during the execution of movements would lead to proprioceptive prediction errors, requiring the brain to conclude that movements require more effort than predicted. Thus, here the emphasis is on fatigue as a direct consequence of unexpectedly high perceived effort during movements (proprioceptive surprise) as a cognitive cause of fatigue.
Towards differential diagnosis and targeted treatment of fatigue
Treatment of fatigue in MS involves the initial exclusion of MS-unrelated causes, for example, anaemia, hypothyroidism or sleep disturbances such as obstructive sleep apnoea. Subsequently, the choice of disease-modifying drugs could be oriented towards drugs with potentially beneficial effects on fatigue. For example, glatiramer acetate might reduce fatigue more effectively than β-interferon.114 Some studies demonstrated that natalizumab may decrease fatigue in patients with MS,115 116 although this might result from a primary effect of natalizumab on depression.117
A central question for this review is how fatigue-specific treatments could be personalised, given that fatigue in MS likely arises through multiple mechanisms (figure 1). In clinical practice, we are presently unable to differentiate between alternative causes of fatigue in individual patients. As a consequence, current treatment of fatigue is driven by trial and error and consideration of side effects. Treatment strategies for fatigue in MS include both pharmacological approaches (eg, the NMDA receptor antagonist amantadine, stimulants such as modafinil, or antidepressants) and non-pharmacological strategies (eg, mindfulness-based cognitive therapy (MBCT), cognitive-behavioural therapy or exercise) (for recent reviews, see118 119). Additionally, social factors, cognitive profiles and psychological traits modulate fatigue120 121; these may offer additional entry points for therapeutic interventions.
In order to select treatments in a rational and predictive manner, novel clinical tests are needed. Importantly, these need to go beyond detecting fatigue; instead, they should inform the choice of patient-specific treatment—by enabling differential diagnosis of alternative mechanisms and/or predicting individual therapeutic response. Some of the candidate mechanisms discussed above map onto existing treatment approaches; others suggest novel treatment strategies. How could these be identified, in individual patients, with existing or emerging techniques?
As all of the pathophysiological mechanisms discussed above eventually lead to changes in network function, measures of brain connectivity are of central importance. In the domain of fMRI, methodological developments have enabled a transition from simple estimates of undirected coupling (functional connectivity) to model-based measures of directed influences between neuronal populations (effective connectivity). The latter have become increasingly sophisticated—with an ability to resolve neuronal versus haemodynamic contributions to layer-wise fMRI signals and characterising transient modulatory influences122—and are increasingly used in translational neuroimaging.123 Furthermore, recent developments allow for computationally efficient whole-brain estimates of effective connectivity,124 which is important for diseases with distributed pathology as MS.
In conjunction with high-resolution fMRI, models of effective connectivity could probe specific candidate mechanisms of fatigue in individual patients, with implications for treatment choice. For example, occult dysfunction of specific nuclei—say through inflammation-induced reduction of monoaminergic transmitter synthesis in brainstem nuclei or of orexin in lateral hypothalamus—could be inferred by assessing efferent connexion strengths of the corresponding nuclei to known target regions (or across the whole brain). For assays of monoaminergic nuclei, this approach could be further enhanced by computational models of transmitter release, respectively.125 If abnormal connectivity of specific nuclei can be established (eg, in comparison with reference distributions), this may predict beneficial therapeutic effects of stimulants (modafinil) or selective serotonin reuptake inhibitors/selective norepinephrine reuptake inhibitors.
Similarly, estimates of effective connectivity between areas involved in interoception, homeostatic control and metacognition could help identify patients in whom fatigue results from a metacognitive diagnosis of helplessness with regard to overcoming dyshomeostasis (compare figure 3). This could be combined with inflammation-sensitive MRI sequences82 and designed perturbations of interoceptive and homeostatic processes16 to detect abnormalities in key circuit components frequently affected by inflammation and lesions in MS.19 62 If investigations of this sort point to a metacognitive/interoceptive origin of fatigue, this may predict therapeutic efficacy of MBCT126 or related forms of contemplative training with a focus on interoception.
Moving from MRI to electrophysiological techniques, several approaches could contribute to differential diagnosis and prognosis of fatigue in MS.127 128 For example, multimodal (visual, auditory, somatosensory, motor) evoked potentials (EPs) measured by electroencephalography (EEG) and transcranial magnetic stimulation (TMS), respectively, have predictive value with regard to future disability status in general.129 130 By contrast, there are hardly any prospective EP studies with a specific focus on predicting fatigue. One such study highlighting the potential use of EPs suggested that the auditory P300 potential may predict treatment response to modafinil, although with moderate sensitivity and specificity.131
Recently developed computational methods might enhance the use of EEG data for differential diagnosis of fatigue and treatment prediction. For example, biophysical models of EEG data have shown promising potential for detecting pathologies at the synaptic level, including dysfunction of the NMDA receptor.132 Models of this kind might help inform individual application of amantadine, one of the few agents featuring in official guidelines (National Institute for Health and Care Excellence, UK), but with only weak efficacy in non-selected patients.118 Similar models of magnetoencephalography data from patients with monogenetic channelopathies have demonstrated that it may be possible to identify selective alterations of specific ion channels, such as voltage-gated potassium channels.133 This approach could help in identifying those fatigue patients who might benefit from treatment with fampridine. This question might also be addressed by TMS: a prospective study using motor EPs in patients with MS fatigue prior to therapy with fampridine showed that increased central motor conduction time might predict individual treatment response.134
Conclusions and outlook
Despite its frequency and pronounced impact on the lives of patients with MS, techniques for differential diagnosis of fatigue and mechanism-guided treatment selection in individual patients do not exist. A critical basis for developing such methods are pathophysiological theories about the mechanisms of fatigue. This paper has reviewed contemporary theories about four major classes of disease mechanisms leading to fatigue: structural damage of WM/GM, inflammatory processes, maladaptive network recruitment during task performance and metacognitive interpretations of brain states that suggest ‘helplessness’. We anticipate that these theories together with recent advances in high-resolution functional neuroimaging and computational modelling will guide the development of tools for differential diagnosis. Similar to ongoing efforts in psychiatry,123 computational neuroimaging tools may provide indices of different causes of fatigue that support treatment decisions in individual patients. This represents an exciting and promising endeavour, but will require prospective patient studies for validating the clinical use of candidate tools.
Acknowledgments
We gratefully acknowledge support by the Wilhelm-Schulthess Foundation, René and Susanne Braginsky Foundation, the University of Zurich and the Clinical Research Priority Program Multiple Sclerosis from the University of Zurich and the University Hospital of Zurich.
Footnotes
Contributors: Z-MM and KES performed the literature search, selected the relevant articles, wrote the paper and led the discussion. NAH, HDC, CTD, GS, NW, AL and AM added further references, contributed to the discussion and edited the paper.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Brownlee WJ, Hardy TA, Fazekas F, et al. Diagnosis of multiple sclerosis: progress and challenges. Lancet 2017;389:1336–46. 10.1016/S0140-6736(16)30959-X [DOI] [PubMed] [Google Scholar]
- 2. Thompson AJ, Baranzini SE, Geurts J, et al. Multiple sclerosis. Lancet 2018;391:1622–36. 10.1016/S0140-6736(18)30481-1 [DOI] [PubMed] [Google Scholar]
- 3. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med 2018;378:169–80. 10.1056/NEJMra1401483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stuke K, Flachenecker P, Zettl UK, et al. Symptomatology of MS: results from the German MS registry. J Neurol 2009;256:1932–5. 10.1007/s00415-009-5257-5 [DOI] [PubMed] [Google Scholar]
- 5. Kluger BM, Krupp LB, Enoka RM. Fatigue and fatigability in neurologic illnesses: proposal for a unified taxonomy. Neurology 2013;80:409–16. 10.1212/WNL.0b013e31827f07be [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giovannoni G. Multiple sclerosis related fatigue. J Neurol Neurosurg Psychiatry 2006;77:2–3. 10.1136/jnnp.2005.074948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Penner IK, Paul F. Fatigue as a symptom or comorbidity of neurological diseases. Nat Rev Neurol 2017;13:662–75. 10.1038/nrneurol.2017.117 [DOI] [PubMed] [Google Scholar]
- 8. Chaudhuri A, Behan PO. Fatigue in neurological disorders. Lancet 2004;363:978–88. 10.1016/S0140-6736(04)15794-2 [DOI] [PubMed] [Google Scholar]
- 9. Wessely S. Chronic fatigue: symptom and syndrome. Ann Intern Med 2001;134:838–43. 10.7326/0003-4819-134-9_Part_2-200105011-00007 [DOI] [PubMed] [Google Scholar]
- 10. Dantzer R, Heijnen CJ, Kavelaars A, et al. The neuroimmune basis of fatigue. Trends Neurosci 2014;37:39–46. 10.1016/j.tins.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tur C. Fatigue management in multiple sclerosis. Curr Treat Options Neurol 2016;18:26 10.1007/s11940-016-0411-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Asano M, Finlayson ML. Meta-analysis of three different types of fatigue management interventions for people with multiple sclerosis: exercise, education, and medication. Mult Scler Int 2014;2014:1–12. 10.1155/2014/798285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Induruwa I, Constantinescu CS, Gran B. Fatigue in multiple sclerosis—a brief review. J Neurol Sci 2012;323:9–15. 10.1016/j.jns.2012.08.007 [DOI] [PubMed] [Google Scholar]
- 14. Stephan KE, Manjaly ZM, Mathys CD, et al. Allostatic self-efficacy: a metacognitive theory of dyshomeostasis-induced fatigue and depression. Front Hum Neurosci 2016;10:550 10.3389/fnhum.2016.00550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuppuswamy A. The fatigue conundrum. Brain 2017;140:2240–5. 10.1093/brain/awx153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petzschner FH, Weber LAE, Gard T, et al. Computational psychosomatics and computational psychiatry: toward a joint framework for differential diagnosis. Biol Psychiatry 2017;82:421–30. 10.1016/j.biopsych.2017.05.012 [DOI] [PubMed] [Google Scholar]
- 17. Flachenecker P, Kümpfel T, Kallmann B, et al. Fatigue in multiple sclerosis: a comparison of different rating scales and correlation to clinical parameters. Mult Scler 2002;8:523–6. 10.1191/1352458502ms839oa [DOI] [PubMed] [Google Scholar]
- 18. Penner IK, Raselli C, Stöcklin M, et al. The Fatigue Scale for Motor and Cognitive Functions (FSMC): validation of a new instrument to assess multiple sclerosis-related fatigue. Mult Scler 2009;15:1509–17. 10.1177/1352458509348519 [DOI] [PubMed] [Google Scholar]
- 19. Haider L, Zrzavy T, Hametner S, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016;139:807–15. 10.1093/brain/awv398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tedeschi G, Dinacci D, Lavorgna L, et al. Correlation between fatigue and brain atrophy and lesion load in multiple sclerosis patients independent of disability. J Neurol Sci 2007;263:15–19. 10.1016/j.jns.2007.07.004 [DOI] [PubMed] [Google Scholar]
- 21. Marrie RA, Fisher E, Miller DM, et al. Association of fatigue and brain atrophy in multiple sclerosis. J Neurol Sci 2005;228:161–6. 10.1016/j.jns.2004.11.046 [DOI] [PubMed] [Google Scholar]
- 22. Nourbakhsh B, Azevedo C, Nunan-Saah J, et al. Longitudinal associations between brain structural changes and fatigue in early MS. Mult Scler Relat Disord 2016;5:29–33. 10.1016/j.msard.2015.10.006 [DOI] [PubMed] [Google Scholar]
- 23. Sepulcre J, Masdeu JC, Goñi J, et al. Fatigue in multiple sclerosis is associated with the disruption of frontal and parietal pathways. Mult Scler 2009;15:337–44. 10.1177/1352458508098373 [DOI] [PubMed] [Google Scholar]
- 24. Rocca MA, Parisi L, Pagani E, et al. Regional but not global brain damage contributes to fatigue in multiple sclerosis. Radiology 2014;273:511–20. 10.1148/radiol.14140417 [DOI] [PubMed] [Google Scholar]
- 25. Calabrese M, Rinaldi F, Grossi P, et al. Basal ganglia and frontal/parietal cortical atrophy is associated with fatigue in relapsing–remitting multiple sclerosis. Mult Scler 2010;16:1220–8. 10.1177/1352458510376405 [DOI] [PubMed] [Google Scholar]
- 26. Derache N, Grassiot B, Mézenge F, et al. Fatigue is associated with metabolic and density alterations of cortical and deep gray matter in relapsing–remitting-multiple sclerosis patients at the earlier stage of the disease: a PET/MR study. Mult Scler Relat Disord 2013;2:362–9. 10.1016/j.msard.2013.03.005 [DOI] [PubMed] [Google Scholar]
- 27. Patejdl R, Penner IK, Noack TK, et al. Multiple sclerosis and fatigue: a review on the contribution of inflammation and immune-mediated neurodegeneration. Autoimmun Rev 2016;15:210–20. 10.1016/j.autrev.2015.11.005 [DOI] [PubMed] [Google Scholar]
- 28. Bester M, Lazar M, Petracca M, et al. Tract-specific white matter correlates of fatigue and cognitive impairment in benign multiple sclerosis. J Neurol Sci 2013;330:61–6. 10.1016/j.jns.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bisecco A, Caiazzo G, d'Ambrosio A, et al. Fatigue in multiple sclerosis: the contribution of occult white matter damage. Mult Scler 2016;22:1676–84. 10.1177/1352458516628331 [DOI] [PubMed] [Google Scholar]
- 30. Chalah MA, Riachi N, Ahdab R, et al. Fatigue in multiple sclerosis: neural correlates and the role of non-invasive brain stimulation. Front Cell Neurosci 2015;9:460 10.3389/fncel.2015.00460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Werf SP, Jongen PJ, Lycklama à Nijeholt GJ, et al. Fatigue in multiple sclerosis: interrelations between fatigue complaints, cerebral MRI abnormalities and neurological disability. J Neurol Sci 1998;160:164–70. 10.1016/S0022-510X(98)00251-2 [DOI] [PubMed] [Google Scholar]
- 32. Bakshi R, Miletich RS, Henschel K, et al. Fatigue in multiple sclerosis: cross-sectional correlation with brain MRI findings in 71 patients. Neurology 1999;53:1151–3. 10.1212/WNL.53.5.1151 [DOI] [PubMed] [Google Scholar]
- 33. Gobbi C, Rocca MA, Riccitelli G, et al. Influence of the topography of brain damage on depression and fatigue in patients with multiple sclerosis. Mult Scler 2014;20:192–201. 10.1177/1352458513493684 [DOI] [PubMed] [Google Scholar]
- 34. Papadopoulou A, Müller-Lenke N, Naegelin Y, et al. Contribution of cortical and white matter lesions to cognitive impairment in multiple sclerosis. Mult Scler 2013;19:1290–6. 10.1177/1352458513475490 [DOI] [PubMed] [Google Scholar]
- 35. Popescu V, Schoonheim MM, Versteeg A, et al. Grey matter atrophy in multiple sclerosis: clinical interpretation depends on choice of analysis method. PLoS One 2016;11:e0143942 10.1371/journal.pone.0143942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Altermatt A, Gaetano L, Magon S, et al. Clinical correlations of brain lesion location in multiple sclerosis: voxel-based analysis of a large clinical trial dataset. Brain Topogr 2018;31:886–94. 10.1007/s10548-018-0652-9 [DOI] [PubMed] [Google Scholar]
- 37. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009;132:1175–89. 10.1093/brain/awp070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 2008;31:247–69. 10.1146/annurev.neuro.30.051606.094313 [DOI] [PubMed] [Google Scholar]
- 39. McDonald WI, Sears TA. The effects of experimental demyelination on conduction in the central nervous system. Brain 1970;93:583–98. 10.1093/brain/93.3.583 [DOI] [PubMed] [Google Scholar]
- 40. Snooks SJ, Swash M. Motor conduction velocity in the human spinal cord: slowed conduction in multiple sclerosis and radiation myelopathy. J Neurol Neurosurg Psychiatry 1985;48:1135–9. 10.1136/jnnp.48.11.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vucic S, Burke D, Kiernan MC. Fatigue in multiple sclerosis: mechanisms and management. Clin Neurophysiol 2010;121:809–17. 10.1016/j.clinph.2009.12.013 [DOI] [PubMed] [Google Scholar]
- 42. Hayes KC. The use of 4-aminopyridine (fampridine) in demyelinating disorders. CNS Drug Rev 2004;10:295–316. 10.1111/j.1527-3458.2004.tb00029.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossini PM, Pasqualetti P, Pozzilli C, et al. Fatigue in progressive multiple sclerosis: results of a randomized, double-blind, placebo-controlled, crossover trial of oral 4-aminopyridine. Mult Scler 2001;7:354–8. 10.1177/135245850100700602 [DOI] [PubMed] [Google Scholar]
- 44. Morrow SA, Rosehart H, Johnson AM. The effect of Fampridine-SR on cognitive fatigue in a randomized double-blind crossover trial in patients with MS. Mult Scler Relat Disord 2017;11:4–9. 10.1016/j.msard.2016.10.011 [DOI] [PubMed] [Google Scholar]
- 45. Filippi M, Rocca MA, Colombo B, et al. Functional magnetic resonance imaging correlates of fatigue in multiple sclerosis. Neuroimage 2002;15:559–67. 10.1006/nimg.2001.1011 [DOI] [PubMed] [Google Scholar]
- 46. Filippi M, Preziosa P, Rocca MA. Brain mapping in multiple sclerosis: lessons learned about the human brain. Neuroimage 2017. 10.1016/j.neuroimage.2017.09.021 [DOI] [PubMed] [Google Scholar]
- 47. Genova HM, Rajagopalan V, Deluca J, et al. Examination of cognitive fatigue in multiple sclerosis using functional magnetic resonance imaging and diffusion tensor imaging. PLoS One 2013;8:e78811 10.1371/journal.pone.0078811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gobbi C, Rocca MA, Pagani E, et al. Forceps minor damage and co-occurrence of depression and fatigue in multiple sclerosis. Mult Scler 2014;20:1633–40. 10.1177/1352458514530022 [DOI] [PubMed] [Google Scholar]
- 49. Hanken K, Eling P, Kastrup A, et al. Integrity of hypothalamic fibers and cognitive fatigue in multiple sclerosis. Mult Scler Relat Disord 2015;4:39–46. 10.1016/j.msard.2014.11.006 [DOI] [PubMed] [Google Scholar]
- 50. Sander, Dr. M. Hirnrindenbefunde bei multipler Sklerose. Eur Neurol 1898;4:427–36. 10.1159/000228765 [DOI] [Google Scholar]
- 51. Schob F. Ein Beitrag Zur pathologischen Anatomie Der multiplen Sklerose. Eur Neurol 1907;22:62–87. 10.1159/000211848 [DOI] [Google Scholar]
- 52. Calabrese M, Magliozzi R, Ciccarelli O, et al. Exploring the origins of grey matter damage in multiple sclerosis. Nat Rev Neurosci 2015;16:147–58. 10.1038/nrn3900 [DOI] [PubMed] [Google Scholar]
- 53. Honce JM. Gray matter pathology in MS: neuroimaging and clinical correlations. Mult Scler Int 2013;2013:1–16. 10.1155/2013/627870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Calabrese M, Castellaro M. Cortical gray matter MR imaging in multiple sclerosis. Neuroimaging Clin N Am 2017;27:301–12. 10.1016/j.nic.2016.12.009 [DOI] [PubMed] [Google Scholar]
- 55. Bø L, Vedeler CA, Nyland HI, et al. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol 2003;62:723–32. 10.1093/jnen/62.7.723 [DOI] [PubMed] [Google Scholar]
- 56. Vercellino M, Masera S, Lorenzatti M, et al. Demyelination, inflammation, and neurodegeneration in multiple sclerosis deep gray matter. J Neuropathol Exp Neurol 2009;68:489–502. 10.1097/NEN.0b013e3181a19a5a [DOI] [PubMed] [Google Scholar]
- 57. Morgen K, Kadom N, Sawaki L, et al. Training-dependent plasticity in patients with multiple sclerosis. Brain 2004;127:2506–17. 10.1093/brain/awh266 [DOI] [PubMed] [Google Scholar]
- 58. Finke C, Schlichting J, Papazoglou S, et al. Altered basal ganglia functional connectivity in multiple sclerosis patients with fatigue. Mult Scler 2015;21:925–34. 10.1177/1352458514555784 [DOI] [PubMed] [Google Scholar]
- 59. Cruz Gómez ÁJ, Ventura Campos N, Belenguer A, et al. Regional brain atrophy and functional connectivity changes related to fatigue in multiple sclerosis. PLoS One 2013;8:e77914 10.1371/journal.pone.0077914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bisecco A, Nardo FD, Docimo R, et al. Fatigue in multiple sclerosis: the contribution of resting-state functional connectivity reorganization. Mult Scler 2018;24:1696–705. 10.1177/1352458517730932 [DOI] [PubMed] [Google Scholar]
- 61. Huitinga I, De Groot CJA, Van der Valk P, et al. Hypothalamic lesions in multiple sclerosis. J Neuropathol Exp Neurol 2001;60:1208–18. 10.1093/jnen/60.12.1208 [DOI] [PubMed] [Google Scholar]
- 62. Huitinga I, Erkut ZA, van Beurden D, et al. Impaired hypothalamus–pituitary–adrenal axis activity and more severe multiple sclerosis with hypothalamic lesions. Ann Neurol 2004;55:37–45. 10.1002/ana.10766 [DOI] [PubMed] [Google Scholar]
- 63. Zellini F, Niepel G, Tench CR, et al. Hypothalamic involvement assessed by T1 relaxation time in patients with relapsing–remitting multiple sclerosis. Mult Scler 2009;15:1442–9. 10.1177/1352458509350306 [DOI] [PubMed] [Google Scholar]
- 64. Qiu W, Raven S, Wu J-S, et al. Hypothalamic lesions in multiple sclerosis. J Neuropathol Exp Neurol 2011;82:819–22. 10.1136/jnnp.2009.198192 [DOI] [PubMed] [Google Scholar]
- 65. Bonvalet M, Ollila HM, Ambati A, et al. Autoimmunity in narcolepsy. Curr Opin Pulm Med 2017;23:522–9. 10.1097/MCP.0000000000000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Papuć E, Stelmasiak Z, Zbigniew S, et al. CSF hypocretin-1 concentrations correlate with the level of fatigue in multiple sclerosis patients. Neurosci Lett 2010;474:9–12. 10.1016/j.neulet.2010.02.062 [DOI] [PubMed] [Google Scholar]
- 67. Constantinescu CS, Niepel G, Patterson M, et al. Orexin A (hypocretin-1) levels are not reduced while cocaine/amphetamine regulated transcript levels are increased in the cerebrospinal fluid of patients with multiple sclerosis: no correlation with fatigue and sleepiness. J Neurol Sci 2011;307:127–31. 10.1016/j.jns.2011.04.024 [DOI] [PubMed] [Google Scholar]
- 68. Flachenecker P, Rufer A, Bihler I, et al. Fatigue in MS is related to sympathetic vasomotor dysfunction. Neurology 2003;61:851–3. 10.1212/01.WNL.0000080365.95436.B8 [DOI] [PubMed] [Google Scholar]
- 69. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 2015;14:406–19. 10.1016/S1474-4422(14)70305-9 [DOI] [PubMed] [Google Scholar]
- 70. Savitz J, Harrison NA. Interoception and inflammation in psychiatric disorders. Biol Psychiatry Cogn Neurosci Neuroimaging 2018;3:514–24. 10.1016/j.bpsc.2017.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dantzer R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev 2018;98:477–504. 10.1152/physrev.00039.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011;134:2755–71. 10.1093/brain/awr182 [DOI] [PubMed] [Google Scholar]
- 73. Gold SM, Krüger S, Ziegler KJ, et al. Endocrine and immune substrates of depressive symptoms and fatigue in multiple sclerosis patients with comorbid major depression. J Neurol Neurosurg Psychiatry 2011;82:814–8. 10.1136/jnnp.2010.230029 [DOI] [PubMed] [Google Scholar]
- 74. Heesen C, Nawrath L, Reich C, et al. Fatigue in multiple sclerosis: an example of cytokine mediated sickness behaviour? J Neurol Neurosurg Psychiatry 2006;77:34–9. 10.1136/jnnp.2005.065805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 2007;21:153–60. 10.1016/j.bbi.2006.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Capuron L, Gumnick JF, Musselman DL. Neurobehavioral effects of interferon-α in cancer patients phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002;26:643–52. 10.1016/S0893-133X(01)00407-9 [DOI] [PubMed] [Google Scholar]
- 77. Capuron L, Pagnoni G, Drake DF, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry 2012;69:1044–53. 10.1001/archgenpsychiatry.2011.2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harrison NA, Voon V, Cercignani M, et al. A Neurocomputational account of how inflammation enhances sensitivity to punishments versus rewards. Biol Psychiatry 2016;80:73–81. 10.1016/j.biopsych.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goehler LE, Gaykema RPA, Hansen MK, et al. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci 2000;85:49–59. 10.1016/S1566-0702(00)00219-8 [DOI] [PubMed] [Google Scholar]
- 80. Harrison NA, Brydon L, Walker C, et al. Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry 2009;66:415–22. 10.1016/j.biopsych.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron 2013;77:624–38. 10.1016/j.neuron.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 82. Harrison NA, Cooper E, Dowell NG, et al. Quantitative magnetization transfer imaging as a biomarker for effects of systemic inflammation on the brain. Biol Psychiatry 2015;78:49–57. 10.1016/j.biopsych.2014.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hannestad J, Subramanyam K, Dellagioia N, et al. Glucose metabolism in the insula and cingulate is affected by systemic inflammation in humans. J Nucl Med 2012;53:601–7. 10.2967/jnumed.111.097014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tracey KJ. Reflex control of immunity. Nat Rev Immunol 2009;9:418–28. 10.1038/nri2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Harrison NA, Cercignani M, Voon V, et al. Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology 2015;40:831–8. 10.1038/npp.2014.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schwarcz R, Bruno JP, Muchowski PJ, et al. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 2012;13:465–77. 10.1038/nrn3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rajda C, Majláth Z, Pukoli D, et al. Kynurenines and multiple sclerosis: the dialogue between the immune system and the central nervous system. Int J Mol Sci 2015;16:18270–82. 10.3390/ijms160818270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Agudelo LZ, Femenía T, Orhan F, et al. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014;159:33–45. 10.1016/j.cell.2014.07.051 [DOI] [PubMed] [Google Scholar]
- 89. Savitz J, Dantzer R, Meier TB, et al. Activation of the kynurenine pathway is associated with striatal volume in major depressive disorder. Psychoneuroendocrinology 2015;62:54–8. 10.1016/j.psyneuen.2015.07.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dobryakova E, Genova HM, DeLuca J, et al. The dopamine imbalance hypothesis of fatigue in multiple sclerosis and other neurological disorders. Front Neurol 2015;6:52 10.3389/fneur.2015.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Haber SN. The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 2014;282:248–57. 10.1016/j.neuroscience.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Grossberg AJ, Zhu X, Leinninger GM, et al. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci 2011;31:11376–86. 10.1523/JNEUROSCI.2311-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gaykema RP, Park SM, McKibbin CR, et al. Lipopolysaccharide suppresses activation of the tuberomammillary histaminergic system concomitant with behavior: a novel target of immune-sensory pathways. Neuroscience 2008;152:273–87. 10.1016/j.neuroscience.2007.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tartaglia MC, Narayanan S, Arnold DL. Mental fatigue alters the pattern and increases the volume of cerebral activation required for a motor task in multiple sclerosis patients with fatigue. Eur J Neurol 2008;15:413–9. 10.1111/j.1468-1331.2008.02090.x [DOI] [PubMed] [Google Scholar]
- 95. White AT, Lee JN, Light AR, et al. Brain activation in multiple sclerosis: a BOLD fMRI study of the effects of fatiguing hand exercise. Mult Scler 2009;15:580–6. 10.1177/1352458508100034 [DOI] [PubMed] [Google Scholar]
- 96. Rocca MA, Absinta M, Valsasina P, et al. Abnormal cervical cord function contributes to fatigue in multiple sclerosis. Mult Scler 2012;18:1552–9. 10.1177/1352458512440516 [DOI] [PubMed] [Google Scholar]
- 97. Kearney H, Miller DH, Ciccarelli O. Spinal cord MRI in multiple sclerosis--diagnostic, prognostic and clinical value. Nat Rev Neurol 2015;11:327–38. 10.1038/nrneurol.2015.80 [DOI] [PubMed] [Google Scholar]
- 98. Mancini L, Ciccarelli O, Manfredonia F, et al. Short-term adaptation to a simple motor task: a physiological process preserved in multiple sclerosis. Neuroimage 2009;45:500–11. 10.1016/j.neuroimage.2008.12.006 [DOI] [PubMed] [Google Scholar]
- 99. Rocca MA, Colombo B, Falini A, et al. Cortical adaptation in patients with MS: a cross-sectional functional MRI study of disease phenotypes. Lancet Neurol 2005;4:618–26. 10.1016/S1474-4422(05)70171-X [DOI] [PubMed] [Google Scholar]
- 100. Rocca MA, Meani A, Riccitelli GC, et al. Abnormal adaptation over time of motor network recruitment in multiple sclerosis patients with fatigue. Mult Scler 2016;22:1144–53. 10.1177/1352458515614407 [DOI] [PubMed] [Google Scholar]
- 101. Grefkes C, Ward NS. Cortical reorganization after stroke: how much and how functional? Neuroscientist 2014;20:56–70. 10.1177/1073858413491147 [DOI] [PubMed] [Google Scholar]
- 102. Dipasquale O, Cooper EA, Tibble J, et al. Interferon-α acutely impairs whole-brain functional connectivity network architecture—a preliminary study. Brain Behav Immun 2016;58:31–9. 10.1016/j.bbi.2015.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. McCormick DA, Wang Z, Huguenard J. Neurotransmitter control of neocortical neuronal activity and excitability. Cereb Cortex 1993;3:387–98. 10.1093/cercor/3.5.387 [DOI] [PubMed] [Google Scholar]
- 104. Gu Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience 2002;111:815–35. 10.1016/S0306-4522(02)00026-X [DOI] [PubMed] [Google Scholar]
- 105. Bouret S, Sara SJ. Network reset: a simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci 2005;28:574–82. 10.1016/j.tins.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 106. van den Brink RL, Pfeffer T, Warren CM, et al. Catecholaminergic neuromodulation shapes intrinsic MRI functional connectivity in the human brain. J. Neurosci. 2016;36:7865–76. 10.1523/JNEUROSCI.0744-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fleming SM, Dolan RJ, Frith CD. Metacognition: computation, biology and function. Philos Trans R Soc Lond B Biol Sci 2012;367:1280–6. 10.1098/rstb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Salamone PC, Esteves S, Sinay VJ, et al. Altered neural signatures of interoception in multiple sclerosis. Hum Brain Mapp 2018;39:4743–54. 10.1002/hbm.24319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. McMorris T, Barwood M, Corbett J. Central fatigue theory and endurance exercise: toward an interoceptive model. Neurosci Biobehav Rev 2018;93:93–107. 10.1016/j.neubiorev.2018.03.024 [DOI] [PubMed] [Google Scholar]
- 110. Seth AK, Suzuki K, Critchley HD. An interoceptive predictive coding model of conscious presence. Front Psychol 2011;2:395 10.3389/fpsyg.2011.00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Seth AK. Interoceptive inference, emotion, and the embodied self. Trends Cogn Sci 2013;17:565–73. 10.1016/j.tics.2013.09.007 [DOI] [PubMed] [Google Scholar]
- 112. Morales J, Lau H, Fleming SM. Domain-general and domain-specific patterns of activity supporting metacognition in human prefrontal cortex. J Neurosci 2018;38:3534–46. 10.1523/JNEUROSCI.2360-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Loitfelder M, Fazekas F, Koschutnig K, et al. Brain activity changes in cognitive networks in relapsing–remitting multiple sclerosis—insights from a longitudinal FMRI study. PLoS One 2014;9:e93715 10.1371/journal.pone.0093715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Metz LM, Patten SB, Archibald CJ, et al. The effect of immunomodulatory treatment on multiple sclerosis fatigue. J Neurol Neurosurg Psychiatry 2004;75:1045–7. 10.1136/jnnp.2002.007724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Svenningsson A, Falk E, Celius EG, et al. Natalizumab treatment reduces fatigue in multiple sclerosis. Results from the TYNERGY trial; a study in the real life setting. PLoS One 2013;8:e58643 10.1371/journal.pone.0058643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Planche V, Moisset X, Morello R, et al. Improvement of quality of life and its relationship with neuropsychiatric outcomes in patients with multiple sclerosis starting treatment with natalizumab: a 3-year follow-up multicentric study. J Neurol Sci 2017;382:148–54. 10.1016/j.jns.2017.10.008 [DOI] [PubMed] [Google Scholar]
- 117. Penner IK, Sivertsdotter EC, Celius EG, et al. Improvement in fatigue during natalizumab treatment is linked to improvement in depression and day-time sleepiness. Front Neurol 2015;6:18 10.3389/fneur.2015.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang TT, Wang L, Deng XY, et al. Pharmacological treatments for fatigue in patients with multiple sclerosis: a systematic review and meta-analysis. J Neurol Sci 2017;380:256–61. 10.1016/j.jns.2017.07.042 [DOI] [PubMed] [Google Scholar]
- 119. Miller P, Soundy A. The pharmacological and non-pharmacological interventions for the management of fatigue related multiple sclerosis. J Neurol Sci 2017;381:41–54. 10.1016/j.jns.2017.08.012 [DOI] [PubMed] [Google Scholar]
- 120. Johansson S, Ytterberg C, Hillert J, et al. A longitudinal study of variations in and predictors of fatigue in multiple sclerosis. J Neurol Neurosurg Psychiatry 2008;79:454–7. 10.1136/jnnp.2007.121129 [DOI] [PubMed] [Google Scholar]
- 121. Hanken K, Eling P, Hildebrandt H. Is there a cognitive signature for MS-related fatigue? Mult Scler 2015;21:376–81. 10.1177/1352458514549567 [DOI] [PubMed] [Google Scholar]
- 122. Friston KJ, Preller KH, Mathys C, et al. Dynamic causal modelling revisited. Neuroimage 2017. 10.1016/j.neuroimage.2017.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Stephan KE, Schlagenhauf F, Huys QJM, et al. Computational neuroimaging strategies for single patient predictions. Neuroimage 2017;145:180–99. [DOI] [PubMed] [Google Scholar]
- 124. Frässle S, Lomakina EI, Kasper L, et al. A generative model of whole-brain effective connectivity. Neuroimage 2018;179:505–29. 10.1016/j.neuroimage.2018.05.058 [DOI] [PubMed] [Google Scholar]
- 125. Iglesias S, Tomiello S, Schneebeli M, et al. Models of neuromodulation for computational psychiatry. Wiley Interdiscip Rev Cogn Sci 2017;8:e1420 10.1002/wcs.1420 [DOI] [PubMed] [Google Scholar]
- 126. Hoogerwerf AEW, Bol Y, Lobbestael J, et al. Mindfulness-based cognitive therapy for severely fatigued multiple sclerosis patients: a waiting list controlled study. J Rehabil Med 2017;49:497–504. 10.2340/16501977-2237 [DOI] [PubMed] [Google Scholar]
- 127. Leocani L, Comi G. Neurophysiological investigations in multiple sclerosis. Curr Opin Neurol 2000;13:255–61. 10.1097/00019052-200006000-00004 [DOI] [PubMed] [Google Scholar]
- 128. Hardmeier M, Leocani L, Fuhr P. A new role for evoked potentials in MS? Repurposing evoked potentials as biomarkers for clinical trials in MS. Mult Scler 2017;23:1309–19. 10.1177/1352458517707265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Giffroy X, Maes N, Albert A, et al. Multimodal evoked potentials for functional quantification and prognosis in multiple sclerosis. BMC Neurol 2016;16:83 10.1186/s12883-016-0608-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Leocani L, Rovaris M, Boneschi FM, et al. Multimodal evoked potentials to assess the evolution of multiple sclerosis: a longitudinal study. J Neurol Neurosurg Psychiatry 2006;77:1030–5. 10.1136/jnnp.2005.086280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nagels G, D'hooghe MB, Vleugels L, et al. P300 and treatment effect of modafinil on fatigue in multiple sclerosis. J Clin Neurosci 2007;14:33–40. 10.1016/j.jocn.2005.10.008 [DOI] [PubMed] [Google Scholar]
- 132. Symmonds M, Moran CH, Leite MI, et al. Ion channels in EEG: isolating channel dysfunction in NMDA receptor antibody encephalitis. Brain 2018;141:1691–702. 10.1093/brain/awy107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gilbert JR, Symmonds M, Hanna MG, et al. Profiling neuronal ion channelopathies with non-invasive brain imaging and dynamic causal models: case studies of single gene mutations. Neuroimage 2016;124:43–53. 10.1016/j.neuroimage.2015.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zeller D, Reiners K, Bräuninger S, et al. Central motor conduction time may predict response to fampridine in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry 2014;85:707–9. 10.1136/jnnp-2013-306860 [DOI] [PMC free article] [PubMed] [Google Scholar]