

Multidrug-resistant (MDR) Escherichia coli isolates pose global threats to public health due to the decreasing availability of treatment options. To better understand the characteristics of MDR E. coli isolated from food-producing animals with no antibiotic exposure, we employed genomic comparison, high-resolution phylogenetics, and functional characterization. Our findings highlight the potential capacity of MDR E. coli to cause severe disease and suggest that these strains are widespread intercontinentally. This study underlines the occurrence of MDR E. coli in food-producing animals raised without antibiotic use, which has alarming, critical ramifications within animal and human medical practice.
KEYWORDS: antimicrobial resistance, ESBLs, multidrug resistance
ABSTRACT
The effectiveness of antibiotics has been challenged by the increasing frequency of antimicrobial resistance (AMR), which has emerged as a major threat to global health. Despite its negative impact on the development of AMR, there are few effective strategies for reducing AMR in food-producing animals. Using whole-genome sequencing and comparative genomics of 36 multidrug-resistant (MDR) Escherichia coli strains isolated from beef cattle with no previous exposure to antibiotics, we obtained results suggesting that the occurrence of MDR E. coli also arises in animals with no antibiotic selective pressure. Extended-spectrum-β-lactamase-producing E. coli strains with enhanced virulence capacities for toxin production and adherence have evolved, which implies important ramifications for animal and human health. Gene exchanges by conjugative plasmids and insertion elements have driven widespread antibiotic resistance in clinically relevant pathogens. Phylogenetic relatedness of E. coli strains from various geographic locations and hosts, such as animals, environmental sources, and humans, suggests that transmission of MDR E. coli strains occurs intercontinentally without host barriers.
IMPORTANCE Multidrug-resistant (MDR) Escherichia coli isolates pose global threats to public health due to the decreasing availability of treatment options. To better understand the characteristics of MDR E. coli isolated from food-producing animals with no antibiotic exposure, we employed genomic comparison, high-resolution phylogenetics, and functional characterization. Our findings highlight the potential capacity of MDR E. coli to cause severe disease and suggest that these strains are widespread intercontinentally. This study underlines the occurrence of MDR E. coli in food-producing animals raised without antibiotic use, which has alarming, critical ramifications within animal and human medical practice.
INTRODUCTION
Antimicrobial-resistant microorganisms (ARMs) pose severe clinical challenges for human and animal health. Of the ARMs, extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae are resistant to most third- and some fourth-generation cephalosporins that are important for the treatment of human bacterial diseases (1, 2). The prevalence of ESBL-producing Enterobacteriaceae is increasing not only in human medicine but also in the various environmental and agricultural settings (3–7). Escherichia coli are major producers of ESBLs, with increasing detection of ESBL-producing E. coli strains in livestock (8), making it of particular concern due to the potential for transfer of resistance to human isolates through food. Although the use of certain cephalosporins in food-producing animals was banned by the Food and Drug Administration’s Center for Veterinary Medicine in 2012 (9), high levels of ESBL-producing E. coli strains in food-producing animals continue to occur (10–12).
Cefotaxime, a third-generation cephalosporin, is banned for prophylactic use and treatment in food-producing animals, but the prevalence of cefotaxime-resistant bacteria (CRB) has continued to rise in beef cattle (10, 11, 13). Due to its strong, selective antimicrobial activity, cefotaxime has been widely used to select ESBL-producing bacteria from animal and environmental samples. Resistance to cefotaxime has been attributed to the acquisition of plasmid-mediated CTX-M genes (14). CTX-M genes are found on plasmids within the major human pathogens, such as pathogenic E. coli and Klebsiella pneumoniae, and have been found to originate from environmental Kluyvera species (15, 16). Another well-known plasmid-mediated β-lactamase gene, CMY-2 type, has also been reported to confer resistance to cefotaxime (17). In previous studies, we reported that the presence of CRB in beef cattle arose without antibiotics on pasture (10, 11), indicating that the emergence of ARMs in food-producing animals is caused by factors other than antibiotic use. However, the underlying mechanisms by which commensal bacteria in the gastrointestinal tract acquire cefotaxime resistance in animals grazing on pasture without antibiotics remain unclear.
In this study, we employed two research beef cattle farms to understand the occurrence of CRB on farms that not only have limited exposure to human activities but also have beef cattle raised without antibiotics, in particular, third-generation cephalosporins, including cefotaxime. By using whole-genome sequencing and comparative genomics, we explore drivers for environmental transmission of clinically relevant multidrug-resistant Escherichia coli strains in food-producing animals.
RESULTS
Multidrug-resistant E. coli strains in beef cattle raised without antibiotics.
A total of 2,769 cefotaxime-resistant bacteria (CRB) were isolated from 1,535 cattle raised on pasture without antibiotics during their entire life span at two different research facilities (Fig. 1A). The prevalence of CRB in beef cattle was 42.6% on both farms. Of the CRB, 293 isolates from 200 cattle (prevalence = 13.0%) carried either CTX-M or CMY-2 genes confirmed by PCR typing, and 176 isolates were identified as E. coli by using selective media, ChromAgar E. coli, and 16S rRNA genotyping (Fig. 1B). Most of the E. coli isolates carried either a CTX-M (33.5%) or CMY-2 (64.2%) gene, while 4 isolates carried both genes. Of the E. coli isolates, we selected 36 strains (9 CMY-2 positive and 27 CTX-M positive), based on farm location and animal sources, to conduct an antibiotic susceptibility test (AST) to evaluate whether these CRB were multidrug resistant (MDR). All isolates were resistant to cefotaxime (>4 μg/ml), and 83.3% of them were resistant to a clinically important level (>64 μg/ml), as shown by the MICs (Fig. 1C) (18). Thirteen antibiotics belonging to 8 classes, including sulfonamides, aminoglycosides, tetracyclines, fluoroquinolones, chloramphenicol, penicillins, cephalosporins, and polymyxins, were tested. All isolates were resistant to ampicillin, ceftiofur, and cephalothin, but relatively low or no resistance was observed against gentamicin, amikacin, nalidixic acid, or colistin. None of the isolates, including those with intermediate colistin resistance, carried the MCR-1 gene that confers resistance to colistin by modifying lipopolysaccharide (LPS) (19). All isolates were MDR, being resistant against three or more different antibiotic classes, with 10 (27.8%), 6 (16.7%), 7 (19.4%), and 13 (36.1%) isolates resistant to 3, 4, 5, or 6 different antibiotic classes, respectively. In particular, these isolates showed either resistance (10 isolates) or intermediate susceptibility (20 isolates) against amoxicillin-clavulanic acid, a combination of a penicillin class antibiotic and an ESBL inhibitor, indicating that these strains also produce traditional β-lactamase.
FIG 1.

Occurrence of multidrug-resistant E. coli strains in beef cattle raised without antibiotics. (A) Fecal samples were collected from beef cattle on two research farms, the North Florida Research and Education Center (NFREC, blue dot) and the Beef Research Unit (BRU, orange dot). (B) Prevalences of cefotaxime-resistant bacteria (CRB) and cefotaxime-resistant E. coli strains in beef cattle. Isolates carrying the CMY-2 or/and CTX-M gene(s) were identified by PCR genotyping. (C) Antimicrobial susceptibility testing (AST) against 13 different antibiotics belonging to 8 classes (S, sulfonamides; A, aminoglycosides; T, tetracyclines; F, fluoroquinolones; C, chloramphenicol; PEN, penicillisn; CEP, cephalosporins; P, polymyxins) was conducted following the CLSI guidelines. Thirty-six isolates, 22 from the BRU (orange) and 14 from the NFREC (blue), were selected based on origin and animal source. Colored blocks represent resistance, intermediate susceptibility, and susceptibility. MICs of cefotaxime were determined by the broth microdilution method. The presence (+) and absence (−) of CMY-2 and CTX-M genes in the isolates are indicated to the right of the AST results.
Phylogeny of ESBL- and CMY-2-producing MDR E. coli isolates.
To investigate the genetic relationship of these ESBL- and CMY-2-producing MDR E. coli isolates in food-producing animals raised in two different research facilities about 350 kilometers away from each other, whole genomes of the MDR E. coli isolates were sequenced and subjected to phylogenetic analysis. Of these 36 isolates, 3 (JEONG5446, JEONG5776, and JEONG9567) belonged to a single isolate clade according to core-genome-based single-nucleotide polymorphism (SNP) analysis, while the other 33 isolates clustered into 7 multi-isolate clades. Strains clustered into the same clades had limited numbers of SNPs (0 to 30 SNPs), indicating they are clonal variants. Strains were not interspersed between the two farms but clustered together based on farm location, suggesting no transmission occurred between the farms (Fig. 2). Mauve alignment of whole genomes within the same clade were conducted in order to seek genome rearrangement and genome architecture. The alignments revealed that isolates within the same clade had extensive similarity in the chromosomal contents, with minimal genome rearrangement (Fig. S1 in the supplemental material). Based on these data, we concluded that strains in the same clade are clonal variants and selected 11 genetically distinctive ESBL- or CMY-2-producing MDR E. coli strains, belonging to 10 sequence types (STs), from each clade for comparative and functional genomic analyses to understand MDR occurrence.
FIG 2.
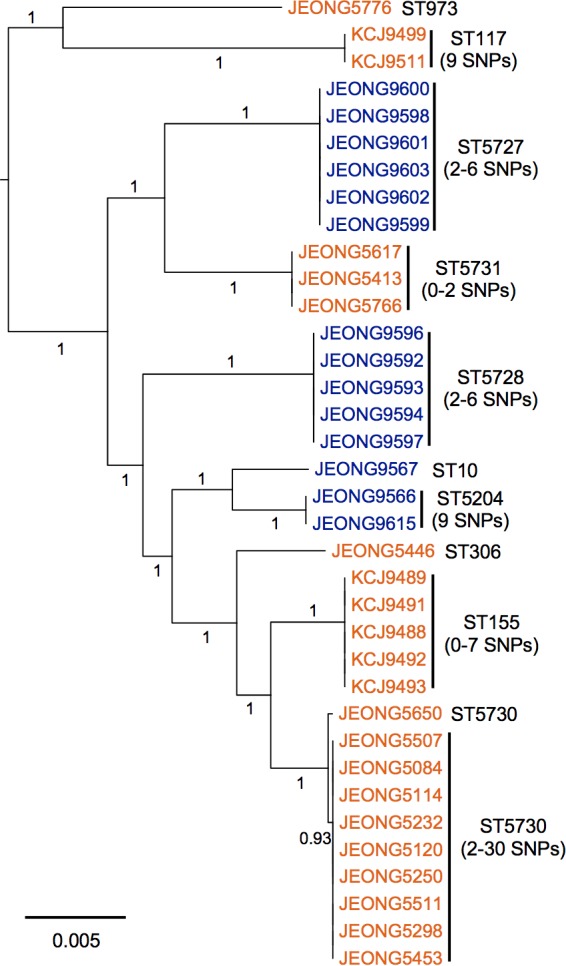
Multidrug-resistant E. coli isolates clustered based on farm location. The maximum-likelihood phylogenetic tree was constructed based on the single-nucleotide polymorphisms (SNPs) identified in the core genomes of the 36 multidrug-resistant E. coli isolates by Parsnp. Of the 36 isolates, 14 were from the NFREC farm (blue) and 22 were from the BRU farm (orange). Sequencing types (ST) of isolates were identified by using the MLST 1.8 software of the Center for Genomic Epidemiology (CGE). The numbers of SNPs were analyzed by using NCBI Pathogen Detection. The numbers listed on the horizontal branches indicate bootstrap values. The scale bar indicates the mean number of nucleotide substitutions per site.
Antibiotic resistance genes of MDR E. coli strains.
The 11 representative ESBL- and CMY-2-producing MDR E. coli were identified as 10 different serotypes, with 3 O-antigen nontypeable and 10 unique multilocus sequence types (MLSTs). Except for one strain, all isolates carried Inc group plasmids (Table 1). All genome-sequencing contigs were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) and submitted to the Comprehensive Antibiotic Resistance Database (CARD) to further identify antibiotic resistance genes (ARGs), resulting in a total of 74 antibiotic resistance-related genes belonging to 19 categories and 9 classes (Fig. 3). All isolates encoded efflux pumps that confer multidrug resistance potential. In addition, we found genes conferring resistance to aminoglycoside, β-lactam, fluoroquinolone, sulfonamide, polymyxin, and peptide antibiotics. This ARG profile supports the results of AST analysis, which described all of the isolates as MDR (Fig. 1C).
TABLE 1.
Characterization of E. coli isolates
| Strain | MLST | Serotype | Plasmid replicon(s) |
|---|---|---|---|
| JEONG5232 | 5730 | O128:H31 | IncN, IncX2, CoI, CoIRNAI |
| JEONG5446 | 306 | O84:H2 | IncFII, IncA/C2, CoI |
| JEONG5650 | 5730 | :H10 | IncN, CoI, CoIRNAI |
| JEONG5766 | 5731 | O169:H41 | IncN, CoIRNAI |
| JEONG5776 | 973 | O25:H15 | IncFIA, IncFIB, IncFII |
| JEONG9566 | 5204 | :H20 | IncX1 |
| JEONG9567 | 10 | :H32 | IncR |
| JEONG9592 | 5728 | O142:H29 | Not detected |
| JEONG9598 | 5727 | O27:H9 | IncY, IncR, CoIRNAI |
| KCJ9489 | 155 | O28:H12 | IncY |
| KCJ9499 | 117 | O119:H4 | IncFIB, IncFII |
FIG 3.

Antibiotic resistance gene profiles of multidrug-resistant E. coli strains. The antibiotic resistance genes (ARGs) of 11 representative multidrug-resistant E. coli strains were identified by comparing their whole-genome sequences against the Comprehensive Antibiotic Resistance Database (CARD). ARGs with ≥70% identity are indicated by dark gray blocks, and those with <70% identity are shown in light gray blocks. ARGs were classified into different categories (colored blocks with numbers) based on their functions, and the categories are listed next to the gene names. Genes with mutations conferring antibiotic resistance are marked by asterisks.
To determine whether ESBL (CTX-M-1 and CTX-M-32) and CMY-2 genes were borne by plasmids or chromosomes, we performed PLACNET analysis to assign contigs to either a chromosomal or plasmid network. Contigs carrying the CTX-M or CMY-2 genes were identified (Fig. 4A to C). Contigs with plasmid-encoded replication initiator protein (RIP) and relaxase protein are shown with green and red dots, respectively (Fig. 4A to C). Both CTX-M and CMY-2 genes were found on the plasmid or chromosomal network highlighted with purple dots (Fig. 4A to C). Three strains, JEONG9566, JEONG9567, and JEONG9592, carried CTX-M-32 gene in the chromosomal DNA, while the other 6 isolates carried CTX-M-1/CTX-M-32 or CMY-2 genes on their plasmids. Interestingly, CMY-2 genes were located only within plasmids, whereas CTX-M genes were found within plasmids and chromosomes. Additionally, plasmid replication types were identified using PlasmidFinder. Various types of Inc plasmids, including IncA/C2, IncFIA, IncFIB, IncFII, IncN, IncR, and IncY, were identified (Table 1). The genetic environments surrounding the CTX-M and CMY-2 genes were found to be associated with mobile elements, as insertion sequence (IS) elements were identified near the CTX-M and CMY-2 gene sequences (Fig. 4D). In JEONG5446 and JEONG5776, insertion sites of the plasmid-carried CMY-2 genes were distinct, mediated by different IS elements, indicating that the transposition events of the CMY-2 genes originated independently. Although insertion sites of CTX-M-32 genes in JEONG9566 and JEONG9567 were in the chromosomes at different loci, transposition events were associated with the same mobile elements, IS1380 and IS5.
FIG 4.

Loci and genetic environments of CTX-M and CMY-2 genes. (A to D) The loci of CTX-M/CMY-2 genes in JEONG9598 (A), JEONG9566 (B), and JEONG5446 (C). The contigs (blue, green, red, and purple nodes) of the isolates were assigned to either the bacterial chromosome (gray background) or a plasmid (white background) based on their homology to reference genomes/plasmids (orange nodes) using PLACNETw and manual pruning. The sizes of the blue, green, red, or purple nodes are in proportion to the lengths of the contigs, while the size of the orange nodes (reference genomes/plasmids) is the same. Purple nodes represent contigs containing a CTX-M or CMY-2 gene. (D) Comparison of genetic environments surrounding the CTX-M or CMY-2 gene in representative isolates. The genes surrounding each CTX-M or CMY-2 (red arrows) gene contain insertion sequences (yellow arrows) and other genes as indicated (green arrows). Similarities (gray shading) of the genetic environments between isolates were analyzed using BLASTn.
Versatile virulence genes carried in MDR E. coli.
To investigate potential pathogenicity of these 11 representative MDR E. coli isolates, we analyzed the virulence gene profiles of the isolates by comparing them to clinically relevant pathogenic E. coli strains, one isolated from pig, ExPEC PCN033 (20), and one from a human, KCJ1409 (10). The virulence factors were identified by using the Virulence Factor Database (VFDB). Numerous virulence factors necessary for adherence, chemotaxis, invasion, and iron uptake were identified (Fig. 5). Genes encoding curli (csgDFG), enterobactin (entABCEFS), and the iron uptake system (fepABCDG) were conserved in all isolates, whereas fimACDEFG (type 1 fimbriae), yagZYXWV (ecpABCDE; E. coli common pilus), and gspDEFGHIJK (general secretion pathway) were missing in two isolates, JEONG9566 and JEONG9592. Genes (iucABCD) encoding proteins for aerobactin synthesis, which is related to iron uptake, were only present in 3 isolates, JEONG5446, JEONG5776, and JEONG9499. The afa-VIII gene cluster encoding the AfaE-VIII adhesin was found in JEONG5776 and KCJ9499, and yersiniabactin biosynthetic proteins (ybtAEPQSTUX, fyuA, irp1, and irp2) were found in JEONG5253 and KCJ9499. K88 fimbriae (faeCDEFHIJ) and P fimbriae (papBCDFG) genes were found in JEONG5776 and KCJ9499, respectively. Among 11 representative MDR E. coli isolates, KCJ9499 showed a virulence gene profile that was more similar to that of the human clinical isolate KCJ1409 than the other 10 isolates. These two strains shared unique adhesins and iron uptake proteins, including AfaE-VIII adhesin, P-fimbriae, and yersiniabactin biosynthetic proteins, consistent with KCJ9499 having the capacity to colonize in the gastrointestinal (GI) tract of humans. Additionally, JEONG5446, identified as serotype O84:H2, carried the type 3 secretion system (T3SS) and its secreted effector proteins, including translocated intimin receptor (Tir), several E. coli secreted proteins (Esp), and non-LEE effectors (Nle) (Fig. 5). Furthermore, JEONG5446 carried a Shiga toxin-producing gene, stx1AB, the product of which causes hemolytic uremic syndrome (HUS) in humans (21). Taken together, ESBL- or CMY-2-producing MDR E. coli strains carried virulence factor profiles similar to those of the clinically relevant isolates from human and swine origins.
FIG 5.

Versatile virulence genes in multidrug-resistant E. coli isolates. The virulence factors of 11 representative isolates from this study and reference hypervirulent isolates in swine (PCN033) and human (KCJ1409) were identified by aligning protein sequences of the representative isolates (query sequences) against the reference sequences (subject sequences) in the Virulence Factor Database (VFDB) using BLASTp. The similarity of each virulence factor (query sequence) to the subject sequence in VFDB was calculated using the following formula: subject coverage (%) × query coverage (%) × identity of query and subject sequences (%). The high-to-low similarities are presented using different colors, ranging from dark blue to light blue.
Functional pathogenicity determinants.
As adhesion to epithelial cells is the first step in bacterial colonization of the intestinal tract, the adhesion capabilities of the 11 ESBL- or CMY-2-producing MDR E. coli isolates were evaluated in Caco-2 human epithelial colorectal adenocarcinoma cells. E. coli O157:H7 EDL933, a strain encoding T3SS and Shiga toxins, and E. coli DH5α were compared as positive and negative controls, respectively. All strains except JEONG5650 showed significantly greater adherence capability (P < 0.05) than the negative-control DH5α (Fig. 6A), consistent with a role as a human pathogen.
FIG 6.

Functional pathogenicity determinants of JEONG5446. (A) Adherence of 11 representative isolates to Caco-2 cells. Assays of the adherence of isolates were conducted in three independent experiments at a multiplicity of infection (MOI) of 10 (10 bacterial cells to 1 Caco-2 cell). Bars indicate the mean values ± standard errors of the means (SEM), and asterisks (*) indicate statistical differences (P < 0.05) compared with the result for the negative control, DH5α. (B) Survival curves of JEONG5446, EDL933, and DH5α with (MMC) or without (not treated [NT]) mitomycin C treatment. Bacterial cell cultures were treated with mitomycin C when the OD600 reached 0.5. (C) Dot blot analysis for detecting Shiga toxin type 1 (Stx1) and Stx2 in JEONG5446 treated with (+) or without (−) mitomycin C. EDL933 (Stx1 and Stx2 positive) and DH5α (Stx1 and Stx2 negative) were used as the positive and negative control, respectively.
The stxAB genes are generally present in lambdoid prophage, and the expression of Shiga toxin is controlled by the phage regulatory system mediated by the bacterial SOS response (22). Due to mutations in the phage transcription system, the expression of Shiga toxin is frequently impaired (23). To determine whether JEONG5446 produces Shiga toxin by phage induction, the expression of Shiga toxin was measured by an immuno-dot blot assay after mitomycin C (MMC) treatment, which causes DNA damage that triggers the SOS response, thereby resulting in prophage induction and cell lysis. Bacterial cell lysis was evaluated by measuring the optical density at 600 nm (OD600) after treatment with MMC. JEONG5446 was lysed with MMC, like EDL933, encoding Stx1 and Stx2 in prophage BP-933V and BP-933W, respectively, indicating phage induction with MMC treatment, whereas DH5α (negative control without an stx gene) maintained a normal OD600 during the experiment in the presence of MMC (Fig. 6B). Immuno-dot blot analysis showed that the positive-control strain, EDL933, expressed both Shiga toxin 1 and 2 with MMC, whereas the negative-control strain, DH5α, did not express them regardless of the presence of MMC. JEONG5446 produced Stx1 in the manner of MMC dependency (Fig. 6C), which is consistent with the cell lysis data, indicating that JEON5446 carries the stx1 gene, which is expressed by phage induction triggered by lytic factors, including antibiotic treatments.
Phylogenetic analysis of MDR E. coli strains with other global lineages.
Since the beef cattle employed in this study were raised without antibiotics and with limited human activities within the nearby vicinity, we hypothesized that the occurrence of these MDR E. coli strains did not likely originate on the farms, but rather, the origins of these pathogens are plausibly from environmental sources. To identify the potential sources from which these MDR E. coli strains were introduced into the farms, we conducted a phylogenetic tree analysis with other relevant global lineages using genome sequences. Recently, efforts have been made for outbreak investigation of infectious diseases by targeting a variety of pathogens through the use of whole-genome-sequence data (24). First, genomes of MDR E. coli isolates were submitted to the NCBI GenomeTrakr pathogen detection system to generate phylogenetic trees with 55,062 E. coli/Shigella genomes (25). Although none of the MDR E. coli strains were identified as clonal variants of known outbreak-associated pathogens, isolates were closely clustered with clinically relevant E. coli strains (Fig. S2A to J). For example, JEONG5776 was clustered with strain PCN033, an ESBL-producing E. coli strain that caused outbreaks in swine in China (20). Furthermore, as shown by the results in Fig. 7, a phylogenetic analysis of sequence type 306 (ST306), which has been reported to cause hemolytic uremic syndrome in humans, using 88 available genomes in EnteroBase, showed three major clades. In the phylogeny, Shiga toxin-producing isolate JEONG5446 clustered with a variety of isolates from humans, animals, and environmental sources (Fig. 7). The two closest strains, PNUSAE004879 and 2015 C-3863, isolated from humans, had relatively small numbers of SNPs in their core genomes compared to the number in JEONG5446, 1,325 SNPs in PNUSAE004879 and 1,836 SNPs in 2015 C-3863, indicating that these isolates have genetically close relatedness. These data suggest that MDR E. coli strains isolated in the GI tract of animals with no antibiotic treatment history were likely transmitted from the environment through unknown vehicles.
FIG 7.

Phylogenetic relatedness between JEONG5446 and other ST306 E. coli isolates. The maximum-likelihood phylogenetic tree based on core-genome single-nucleotide polymorphisms was constructed using Parsnp. Genome sequences of the 87 ST306 E. coli strains other than JEONG5446 (ST306) were downloaded from Enterobase. Based on the sources, different colors are used (blue, cattle; red, human; pink, food; green, environment; light blue, wild animals, including reptiles and deer; black, unknown). JEONG5446 is marked by an asterisk (*). The bootstrap values are listed on the horizontal branches. The scale bar indicates the mean number of nucleotide substitutions per site.
DISCUSSION
In this study, we identified and characterized ESBL- or CMY-2-producing MDR E. coli strains isolated from beef cattle grazing on pasture without a history of antibiotic treatment. MDR E. coli isolates carried genes encoding well-recognized virulence factors, in combination with a variety of antibiotic resistance genes (ARGs). Functional virulence determination revealed that these isolates have the capability to cause severe diseases in humans. Gene exchanges have driven acquisition of ARGs, and the ecological success of MDR E. coli poses a potential threat to human and animal health globally.
Natural occurrence of antibiotic resistance has recently been recognized by identifying natural resistomes of ARGs (26) and the dissemination of bacteria that carry resistance genes (27). In a previous study, we reported high prevalence of cefotaxime-resistant bacteria (CRB) in cattle with no known exposure to antibiotics through prophylactic or therapeutic antibiotics during their entire life span, suggesting that the occurrence of antibiotic resistance in cattle might originate in the environment or from commensal bacteria in the gastrointestinal tract of animals (11). In this study, we characterized 36 MDR E. coli isolates with high MICs for cefotaxime and with antibiotic resistance against various classes of drugs, including sulfonamides, aminoglycosides, tetracyclines, fluoroquinolones, chloramphenicol, penicillins, cephalosporins, and polymyxins (Fig. 1). These isolates were resistant to most classes of antibiotics available in veterinary medicine, carrying a high level of animal health significance. By genome analysis, we revealed that these isolates carried various ARGs with high similarity to ARGs found in human clinical isolates in their chromosomal or plasmid DNA. Horizontal gene transfer by conjugative plasmids and IS elements may have driven the spread of antibiotic resistance (Fig. 4D and Table 1), indicating potent and wide spread of ARGs among animals and humans. This phenomenon is supported by multiple lines of evidence that have identified plasmid-mediated CMY-type and CTX-M-type β-lactamases in Klebsiella pneumoniae, E. coli, Proteus mirabilis, Enterobacter aerogenes, and Salmonella in food animals and human clinics around the world (28–31).
Virulence gene profiles and functional analyses of MDR E. coli strains showed that these strains have evolved in a manner which supports having severe pathogenic capacities (Fig. 5). Notably, JEONG5446, serotype O84, carried genes encoding T3SS, effectors, and Shiga toxin 1 with versatile adherence. Although there is no evidence that JEONG5446 is associated with any human outbreaks, it may have the potential to cause disease outbreaks which could lead to hemolytic uremic syndrome (HUS) once infected within the human GI tract, since this strain encodes robust virulence factors necessary for adherence, chemotaxis, invasion, and iron uptake. In fact, serotype O84 has been associated with isolates causing outbreaks in New Zealand among humans, cattle, and sheep (32). Previously, it has been shown that an enteroaggregative E. coli O104:H4 strain acquired Shiga toxin-encoding genes and ARGs (CTX-M-15 and TEM-1) that led to a large number of HUS cases in Germany during 2011 (33). Moreover, MDR E. coli isolates showed virulence gene profiles similar to those of clinically relevant human and swine isolates, suggesting that these strains may cause outbreaks in humans and animals. Since these isolates are resistant to medically important antibiotics, treatment options would be limited.
Antibiotic-resistant bacteria are widespread through multiple routes, including human travel, precipitation, and migratory birds (34). Phylogenetic analyses have been widely used to determine the relatedness of isolates and trace the sources of potential reservoirs of pathogens (24, 25, 35). In this study, having less than 10 SNPs in the core genome, we could not identify clonal variants among the strains isolated from two farms (Fig. 2), suggesting that MDR E. coli isolates in these farms were independently introduced or divergently evolved. Interestingly, JEONG5776 had very close phylogenetic relatedness with swine pathogen PCN033 (isolated from China), indicating that continental transmissions of MDR E. coli might have occurred via unknown routes or vehicles. In addition, phylogenetic analysis of STs (973, 117, 5727, 5731, 5728, 10, 5204, 306, 155, and 5730) in GenomeTrakr showed genetic relatedness of MDR E. coli strains with other strains isolated from different regions and hosts (Fig. 7 and Fig. S2 in the supplemental material). Previous studies have shown that migratory birds were responsible for the dissemination of antibiotic-resistant E. coli found in the Arctic, an environment where no selective pressures for antibiotic resistance exist (36). Furthermore, some studies have shown that up to 30% of migratory Franklin’s gulls in Chile and up to 17% of wild gulls in Canada were carriers of ESBL-producing E. coli strains (37, 38). Wild gull species have also been found to acquire antibiotic-resistant organisms in their countries of origin and to spread the bacteria throughout their migration (39). Moreover, potential spread to farms is possible, as migratory birds often interact with cattle during migration periods. There are several species of migratory birds which have large breeding distributions and often frequent animal farms in order to feed on animal waste (40), suggesting migratory birds could be one of the routes that have transmitted MDR E. coli into beef cattle farms. However, many questions regarding transmission routes remain unanswered.
The findings of our study have critical ramifications for animal and human medical practice. Not only do potentially clinically relevant pathogens acquire multidrug resistance mechanisms, but these mechanisms appear to disseminate globally. There is also a strong possibility that pathogens with MDR profiles have evolved to adapt within new hosts by acquisition of genes conferring traits like virulence factors that are critically necessary for survival. More detailed analyses of MDR E. coli strains in wildlife, livestock, and the environment could provide information regarding pathogen transmission. In addition, we were unable to generate complete genome sequences due to the constraints inherent in using short-read Illumina sequencing data and the repetitive sequences like phage genomes and mobile elements that might have limited specificity and sensitivity for comparative bioinformatic analyses. For example, we used the PlasmidFinder database to identify plasmid replicons based on sequence homologies, which might have provided false-positive data. Therefore, further analyses using long-read sequencing technology, such as PacBio sequencing, along with functional analyses, may be helpful to validate the conclusions drawn in this study.
MATERIALS AND METHODS
Statement of ethics.
Standard practices of animal husbandry were applied to all animals in this study. Research protocols were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC number 2015-68003-22971).
Fecal sample collection.
A total of 1,535 fecal samples were collected from beef calves belonging to two different herds housed at two different farms in North Central Florida. The herds were located at the Beef Research Unit (BRU) in Waldo, FL, and the North Florida Research and Education Center (NFREC) in Marianna, FL. The animals were grazing on pasture. None of the animals in this study had been previously exposed to antibiotics for treatment purpose. Sterile cotton swabs were used to collect fecal samples directly from the recto-anal junction (RAJ) of each animal. Following sample collection, fecal swabs were placed in sterile 15-ml centrifuge tubes, transported on ice to the Emerging Pathogens Institute at the University of Florida, and immediately processed.
Isolation and identification of ESBL-producing E. coli strains.
Samples were serially diluted (up to 10−4) with Luria Bertani (LB) broth and then plated on MacConkey agar (BD, USA) containing cefotaxime (4 μg/ml). Plates were incubated at 37°C and examined after 24 h to enumerate bacterial colonies. Resistance to cefotaxime due to the production of extended-spectrum β-lactamase was identified by streaking cefotaxime-resistant isolates on ChromAgar ESBL (CHROMagar, France) as previously described (41, 42). Up to four colonies from each fecal sample with the presence of cefotaxime-resistant bacteria were purified and stored at −80°C in 15% glycerol for future use. Frozen cefotaxime-resistant isolates (n = 2,769) were revived on MacConkey agar plates containing 4 μg/ml cefotaxime and screened by PCR for the presence of the CTX-M gene using primers KCP 685 (5′-TTTGCGATGTGCAGTACCAGTAA-3′) and KCP 686 (5′-CGATATCGTTGGTGGTGCCATA-3′) (544 bp) as described previously (43). In addition, we confirmed the presence of the CMY-2 and MCR-1 genes using primers KCP556 (5′-ATGATGAAAAAATCGTTATGC-3′) and KCP557 (5′-TTGCAGCTTTTCAAGAATGCGC-3′) (1,200 bp) for the CMY-2 gene (44) and primers KCP830 (5′-CGGTCAGTCCGTTTGTTC-3′) and KCP831 (5′-CTTGGTCGGTCTGTAGGG-3′) (305 bp) for the MCR-1 gene (45). Taxonomic identification was conducted at the species level to identify cefotaxime-resistant E. coli. Genomic DNA was extracted with a Qiagen DNA minikit (Qiagen, Valencia, CA) and used as a template for PCR to amplify the 16S rRNA gene using primers KCP 812 (5′-CAGGCCTAACACATGCAAGTC-3′) and KCP 813 (5′-GGGCGGWGTGTACAAGGC-3′) (∼1,300 bp) (46). The PCR products were purified using the QIAquick PCR purification kit and sequenced by the Sanger sequencing method at the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. The resulting sequences were analyzed using the NCBI nucleotide BLAST program to compare the homology of the sequences to the 16S rRNA gene sequences of other organisms.
Characterization of cefotaxime-resistant E. coli strains.
A total of 36 cefotaxime-resistant E. coli isolates, as identified by the 16S rRNA gene sequencing, were further subjected to MIC testing against cefotaxime using the broth microdilution method (47). Isolates were further tested for susceptibility to 13 different antimicrobial compounds according to the Clinical and Laboratory Standards Institute guidelines (18). Briefly, the isolates were tested using the standard Kirby-Bauer disk diffusion method on Mueller-Hinton agar to generate antibiograms of the cefotaxime-resistant isolates. The control strains used for the antibiotic susceptibility testing were Escherichia coli (ATCC 35401), Staphylococcus aureus (ATCC 25923), and Pseudomonas aeruginosa (ATCC 27853). The following antimicrobial-disk concentrations were used: amikacin (30 μg), ampicillin (10 μg), amoxicillin-clavulanic acid (30 μg), ceftiofur (30 μg), cephalothin (30 μg), chloramphenicol (30 μg), colistin (10 μg), gentamicin (10 μg), nalidixic acid (30 μg), streptomycin (10 μg), sulfamethoxazole-trimethoprim (23.75 μg/1.25 μg), sulfisoxazole (250 μg), and tetracycline (30 μg) (BD, USA).
Whole-genome sequencing and phylogenetic-tree analysis.
For whole-genome sequencing of 36 isolates, DNA was extracted from each isolate using the DNeasy blood and tissue kit (Qiagen, Valencia, CA) following the protocol for Gram-negative bacteria. DNA libraries were constructed using the Nextera XT sample preparation kit (Illumina, San Diego, CA) according to the manufacturer's protocol. Sequencing was performed using an Illumina MiSeq with cartridges providing 2 × 250-bp paired-end read coverage. The resulting sequence reads were trimmed for quality and length using Sickle (48) and then assembled using SPAdes (version 3.0) (49). The assembled genome sequences of 36 isolates were deposited in NCBI (Table S1 in the supplemental material). Next, a multiple alignment of the de novo assemblies was performed using progressiveMauve (version 2.4.0) (http://darlinglab.org/mauve/user-guide/progressivemauve.html) to understand genome rearrangement and compare whole-genome architecture (50). The phylogenetic trees of 36 sequenced genomes and 88 ST306 strains were generated using Parsnp (https://harvest.readthedocs.io/en/latest/) (51) based on the core-genome single-nucleotide polymorphisms (SNPs). The assembled genomes were used as input files of Parsnp that used PhiPack (52) to detect recombination and generated reliable core-genome SNPs. The set of core-genome SNPs was used to generate maximum-likelihood phylogenetic trees using 1,000 bootstrap replicates with FastTree2 (53) embedded in the Parsnp program. The number of SNPs in each phylogenetic clade was calculated by using NCBI Pathogen Detection (https://www.ncbi.nlm.nih.gov/pathogens/) (54). The phylogenetic trees of each representative strain were generated using Genome Workbench (https://www.ncbi.nlm.nih.gov/tools/gbench/) after uploading the raw reads to GenomeTrakr (25).
Identification of virulence genes and antibiotic resistance genes.
Virulence genes were identified through PATRIC (https://patricbrc.org/) (55) by aligning whole-genome sequences against the Virulence Factor Database (VFDB) (56) using BLAST. The identity of each virulence gene was defined by multiplying gene coverage times query identity with subject identity. The Comprehensive Antibiotic Resistance Database (CARD, version 2.0.1, https://card.mcmaster.ca/analyze) (57) was also employed for discovery of additional antibiotic resistance genes. Briefly, the whole-genome sequence of each isolate was submitted to Resistance Gene Identifier (RGI, version 4.0.3) in CARD to predict resistance genes using homology and SNP models.
Identification of MLST, plasmid replicon type, and serotype.
The multilocus sequence type (MLST), serotype, plasmid replicons, and plasmid MLST (pMLST) of each isolate were determined using MLST 1.8 (58), SerotypeFinder (59), PlasmidFinder and pMLST 1.4 (60) of the Center for Genomic Epidemiology (CGE) (http://www.genomicepidemiology.org/).
Identification of CTX-M and CMY-2 gene loci and their genetic environments.
To identify whether the CTX-M and CMY-2 genes were in the chromosome or plasmid, we employed PLACNETw (https://castillo.dicom.unican.es/upload/) as previously described (61). PLACNETw assembled sequence reads to generate contigs, and then the contigs were aligned to reference sequences of complete chromosomes and plasmids from NCBI. An original network was generated to illustrate the relatedness among contigs and reference sequences. Following the manual pruning of the original network, the contigs were assigned to either chromosome or plasmid. To find out the location of a CTX-M or CMY-2 gene, alignment was conducted using the sequence of the CTX-M or CMY-2 gene against the chromosomal and plasmid sequences by BLASTn. The genetic environments of CTX-M and CMY-2 genes, i.e., the genes surrounding a CTX-M or CMY-2 gene, were acquired from GenBank files of sequenced strains. Similarity of genetic environments of CTX-M or CMY-2 genes was determined by BLASTn embedded in EasyFig (http://mjsull.github.io/Easyfig/) (62).
Adherence assay.
An adherence assay was conducted to evaluate the adherence of the representative isolates to Caco-2 cells (human epithelial colon cancer cell). EDL933 and DH5α were used as a positive and a negative control, respectively. Caco-2 cells were maintained in Dulbecco modified Eagle medium (DMEM) (product number 10-017-CV; Corning, USA) supplemented with 20% (vol/vol) heat-inactivated fetal bovine serum at 37°C and 5% CO2. A total of 105 Caco-2 cells were seeded into each well of a 24-well polystyrene plate, followed by incubation until 90% confluence. Overnight bacterial cultures in LB broth were seeded into new tubes of LB broth (1:250) to produce the main cultures, followed by incubation at 37°C with shaking for 8 h. A total of 106 bacterial cells were washed with sterile phosphate-buffered saline (PBS) three times, resuspended in 500 μl DMEM, and added to each well with 105 Caco-2 cells (multiplicity of infection [MOI] of 10). After a 3-h incubation at 37°C with 5% CO2, the medium in each well was replaced by 500 μl of new DMEM, followed by another 3-h incubation. After the medium was removed, each well was washed with sterile PBS three times to remove the unattached bacteria. Then, 1 ml of 1% Triton X-100 was added to each well to lyse the Caco-2 cells. Finally, 100 μl of the diluted suspension was spread on LB agar and incubated at 37°C overnight, followed by the enumeration of colonies on the plates. This experiment was conducted in duplicate three times.
Mitomycin C treatment and phage induction.
Mitomycin C (MMC) treatment was previously described (63). Briefly, the optical densities of bacteria were measured to determine whether the bacterial cells were lysed by phage induction. Overnight bacterial cultures were seeded to LB medium. When the OD600 of the bacterial culture reached between 0.5 and 0.7, MMC was added to a final concentration of 0.5 μg/ml. The negative-control and positive-control strains were DH5α and EDL933, respectively. Phage induction was conducted as previously described with minor changes (23). To collect phage particles in the bacterial cultures, 25-ml amounts of bacterial cell cultures were treated without or with MMC (final concentration of 1 μg/ml) when the OD600 reached 0.7. After 24 h of incubation, bacterial cultures were centrifuged at 3,700 × g for 30 min at 4°C, followed by filtration of the supernatants through 0.22-μm-pore-size membrane filters (catalog number 09-719A; Fisher Scientific, USA). To precipitate phage particles, 25% (vol/vol) polyethylene glycol 8000 (PEG 8000) (product number 81268; Sigma-Aldrich, USA)-and-NaCl solution (20% PEG 8000 and 10% NaCl) was added to the supernatant. The mixture was incubated at 4°C overnight, followed by centrifugation at 12,000 × g for 1 h. The pellets acquired were resuspended using STE buffer (1 M Tris [pH 8], 0.5 M EDTA [pH 8], and 5 M NaCl), and used for immuno-dot blotting.
Immuno-dot blot assay.
To detect the expression of Shiga toxin type 1 (Stx1) and Shiga toxin type 2 (Stx2), we used an immuno-dot blot assay. Briefly, 3 μl of each phage particle sample (bacterial cell cultures of strains EDL933, JEONG5446, and DH5α treated with or without MMC) was loaded to a nitrocellulose membrane (0.2-μm pores; Bio-Rad, USA). The membranes were blocked with 5% skim milk (BD, USA) at room temperature for 1 h, followed by incubation with Stx1 antibody (sc-52726; Santa Cruz, USA) diluted 1:100 in 5% skim milk or Stx2 antibody (sc-52727; Santa Cruz, USA) diluted 1:100 in 5% skim milk for 1 h at room temperature. After washing with PBS with 0.2% Tween 20 (TPBS), the membranes were incubated at room temperature for 1 h with goat anti-mouse IgG1 secondary antibody (IRDye 800CW, product number 926-32350; LI-COR, USA) diluted 1:10,000 in 5% skim milk. After washing with TPBS, the blots were imaged with an Odyssey CLs (LI-COR, USA) to identify Stx1 and Stx2.
Statistical analysis.
The data from the adherence assay were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test.
Accession number(s).
Whole-genome sequences of 36 strains have been deposited in the NCBI database. The accession numbers of these sequenced genomes are listed in Table S1 in the supplemental material, including JEONG5084 (NSED00000000), JEONG5114 (NSEE00000000), JEONG5120 (NSEF00000000), JEONG5232 (NSEG00000000), JEONG5250 (NSEH00000000), JEONG5298 (NSEI00000000), JEONG5413 (NSEJ00000000), JEONG5446 (NSEK00000000), JEONG5453 (NSEL00000000), JEONG5507 (NSEM00000000), JEONG5511 (NSEN00000000), JEONG5617 (NSEO00000000), JEONG5650 (NSEP00000000), JEONG5766 (NSEQ00000000), JEONG5776 (NSER00000000), JEONG9566 (PKKV00000000), JEONG9567 (PKKU00000000), JEONG9592 (PKKW00000000), JEONG9593 (QDJW00000000), JEONG9594 (QDJX00000000), JEONG9596 (QDJY00000000), JEONG9597 (QDKF00000000), JEONG9598 (PKKX00000000), JEONG9599 (QDJZ00000000), JEONG9600 (QDKA00000000), JEONG9601 (QDKB00000000), JEONG9602 (QDKC00000000), JEONG9603 (QDKD00000000), JEONG9615 (QDKE00000000), KCJ9488 (QGNB00000000), KCJ9489 (QGNC00000000), KCJ9491 (QGND00000000), KCJ9492 (QGNE00000000), KCJ9493 (QGNF00000000), KCJ9499 (QGNS00000000), and KCJ9511 (QGNG00000000).
Supplementary Material
ACKNOWLEDGMENTS
This material is based upon work that is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, under award number 2015-68003-22971 to K.C.J. L.T. was partially supported by the Chinese Scholarship Council (grant number 201608030002).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.03030-18.
REFERENCES
- 1.Livermore DM, Canton R, Gniadkowski M, Nordmann P, Rossolini GM, Arlet G, Ayala J, Coque TM, Kern-Zdanowicz I, Luzzaro F, Poirel L, Woodford N. 2007. CTX-M: changing the face of ESBLs in Europe. J Antimicrob Chemother 59:165–174. doi: 10.1093/jac/dkl483. [DOI] [PubMed] [Google Scholar]
- 2.Brolund A. 2014. Overview of ESBL-producing Enterobacteriaceae from a Nordic perspective. Infect Ecol Epidemiol 4:24555. doi: 10.3402/iee.v4.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. 2014. Antimicrobial resistance: global report on surveillance. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 4.Reuland EA, Al Naiemi N, Kaiser AM, Heck M, Kluytmans JA, Savelkoul PH, Elders PJ, Vandenbroucke-Grauls CM. 2016. Prevalence and risk factors for carriage of ESBL-producing Enterobacteriaceae in Amsterdam. J Antimicrob Chemother 71:1076–1082. doi: 10.1093/jac/dkv441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Hoek AH, Veenman C, van Overbeek WM, Lynch G, de Roda Husman AM, Blaak H. 2015. Prevalence and characterization of ESBL- and AmpC-producing Enterobacteriaceae on retail vegetables. Int J Food Microbiol 204:1–8. doi: 10.1016/j.ijfoodmicro.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 6.Ben Said L, Jouini A, Klibi N, Dziri R, Alonso CA, Boudabous A, Ben Slama K, Torres C. 2015. Detection of extended-spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae in vegetables, soil and water of the farm environment in Tunisia. Int J Food Microbiol 203:86–92. doi: 10.1016/j.ijfoodmicro.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 7.Haque A, Yoshizumi A, Saga T, Ishii Y, Tateda K. 2014. ESBL-producing Enterobacteriaceae in environmental water in Dhaka, Bangladesh. J Infect Chemother 20:735–737. doi: 10.1016/j.jiac.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Smet A, Martel A, Persoons D, Dewulf J, Heyndrickx M, Herman L, Haesebrouck F, Butaye P. 2010. Broad-spectrum beta-lactamases among Enterobacteriaceae of animal origin: molecular aspects, mobility and impact on public health. FEMS Microbiol Rev 34:295–316. doi: 10.1111/j.1574-6976.2009.00198.x. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt CW. 2012. FDA proposes to ban cephalosporins from livestock feed. Environ Health Perspect 120:A106. doi: 10.1289/ehp.120-a106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mir RA, Weppelmann TA, Johnson JA, Archer D, Morris JG Jr, Jeong KC. 2016. Identification and characterization of cefotaxime resistant bacteria in beef cattle. PLoS One 11:e0163279. doi: 10.1371/journal.pone.0163279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mir RA, Weppelmann TA, Teng L, Kirpich A, Elzo MA, Driver JD, Jeong KC. 2018. Colonization dynamics of cefotaxime resistant bacteria in beef cattle raised without cephalosporin antibiotics. Front Microbiol 9:500. doi: 10.3389/fmicb.2018.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonggrijp MA, Santman-Berends I, Heuvelink AE, Buter GJ, van Schaik G, Hage JJ, Lam T. 2016. Prevalence and risk factors for extended-spectrum beta-lactamase- and AmpC-producing Escherichia coli in dairy farms. J Dairy Sci 99:9001–9013. doi: 10.3168/jds.2016-11134. [DOI] [PubMed] [Google Scholar]
- 13.Hansen KH, Damborg P, Andreasen M, Nielsen SS, Guardabassi L. 2013. Carriage and fecal counts of cefotaxime M-producing Escherichia coli in pigs: a longitudinal study. Appl Environ Microbiol 79:794–798. doi: 10.1128/AEM.02399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry JA, Wright GD. 2013. The antibiotic resistance “mobilome”: searching for the link between environment and clinic. Front Microbiol 4:138. doi: 10.3389/fmicb.2013.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humeniuk C, Arlet G, Gautier V, Grimont P, Labia R, Philippon A. 2002. beta-lactamases of Kluyvera ascorbata, probable progenitors of some plasmid-encoded CTX-M types. Antimicrob Agents Chemother 46:3045–3049. doi: 10.1128/AAC.46.9.3045-3049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livermore DM, Hawkey PM. 2005. CTX-M: changing the face of ESBLs in the UK. J Antimicrob Chemother 56:451–454. doi: 10.1093/jac/dki239. [DOI] [PubMed] [Google Scholar]
- 17.Phan MD, Peters KM, Sarkar S, Forde BM, Lo AW, Stanton-Cook M, Roberts LW, Upton M, Beatson SA, Schembri MA. 2015. Third-generation cephalosporin resistance conferred by a chromosomally encoded blaCMY-23 gene in the Escherichia coli ST131 reference strain EC958. J Antimicrob Chemother 70:1969–1972. doi: 10.1093/jac/dkv066. [DOI] [PubMed] [Google Scholar]
- 18.CLSI. 2015. Performance standards for antimicrobial susceptibility testing; twenty-fifth informational supplement (M100-S25). Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 19.Gao R, Hu Y, Li Z, Sun J, Wang Q, Lin J, Ye H, Liu F, Srinivas S, Li D, Zhu B, Liu YH, Tian GB, Feng Y. 2016. Dissemination and mechanism for the MCR-1 colistin resistance. PLoS Pathog 12:e1005957. doi: 10.1371/journal.ppat.1005957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Zheng H, Yang M, Xu Z, Wang X, Wei L, Tang B, Liu F, Zhang Y, Ding Y, Tang X, Wu B, Johnson TJ, Chen H, Tan C. 2015. Genome analysis and in vivo virulence of porcine extraintestinal pathogenic Escherichia coli strain PCN033. BMC Genomics 16:717. doi: 10.1186/s12864-015-1890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. 2012. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins (Basel) 4:1261–1287. doi: 10.3390/toxins4111261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimmitt PT, Harwood CR, Barer MR. 2000. Toxin gene expression by Shiga toxin-producing Escherichia coli: the role of antibiotics and the bacterial SOS response. Emerg Infect Dis 6:458–465. doi: 10.3201/eid0605.000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park D, Stanton E, Ciezki K, Parrell D, Bozile M, Pike D, Forst SA, Jeong KC, Ivanek R, Dopfer D, Kaspar CW. 2013. Evolution of the Stx2-encoding prophage in persistent bovine Escherichia coli O157:H7 strains. Appl Environ Microbiol 79:1563–1572. doi: 10.1128/AEM.03158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang SY, Weller D, Falardeau J, Strawn LK, Mardones FO, Adell AD, Switt A. 2016. Food safety trends: from globalization of whole genome sequencing to application of new tools to prevent foodborne diseases. Trends Food Sci Technol 57:188–198. doi: 10.1016/j.tifs.2016.09.016. [DOI] [Google Scholar]
- 25.Timme RE, Rand H, Sanchez Leon M, Hoffmann M, Strain E, Allard M, Roberson D, Baugher JD. 2018. GenomeTrakr proficiency testing for foodborne pathogen surveillance: an exercise from 2015. Microb Genom 4:185. doi: 10.1099/mgen.0.000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. 2010. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 8:251–259. doi: 10.1038/nrmicro2312. [DOI] [PubMed] [Google Scholar]
- 27.Petty NK, Ben Zakour NL, Stanton-Cook M, Skippington E, Totsika M, Forde BM, Phan MD, Gomes Moriel D, Peters KM, Davies M, Rogers BA, Dougan G, Rodriguez-Bano J, Pascual A, Pitout JD, Upton M, Paterson DL, Walsh TR, Schembri MA, Beatson SA. 2014. Global dissemination of a multidrug resistant Escherichia coli clone. Proc Natl Acad Sci U S A 111:5694–5699. doi: 10.1073/pnas.1322678111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winokur PL, Vonstein DL, Hoffman LJ, Uhlenhopp EK, Doern GV. 2001. Evidence for transfer of CMY-2 AmpC beta-lactamase plasmids between Escherichia coli and Salmonella isolates from food animals and humans. Antimicrob Agents Chemother 45:2716–2722. doi: 10.1128/AAC.45.10.2716-2722.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mata C, Navarro F, Miro E, Walsh TR, Mirelis B, Toleman M. 2011. Prevalence of SXT/R391-like integrative and conjugative elements carrying blaCMY-2 in Proteus mirabilis. J Antimicrob Chemother 66:2266–2270. doi: 10.1093/jac/dkr286. [DOI] [PubMed] [Google Scholar]
- 30.Pournaras S, Poulou A, Voulgari E, Vrioni G, Kristo I, Tsakris A. 2010. Detection of the new metallo-beta-lactamase VIM-19 along with KPC-2, CMY-2 and CTX-M-15 in Klebsiella pneumoniae. J Antimicrob Chemother 65:1604–1607. doi: 10.1093/jac/dkq190. [DOI] [PubMed] [Google Scholar]
- 31.Lee SH, Jeong SH, Park YM. 2003. Characterization of blaCMY-10 a novel, plasmid-encoded AmpC-type beta-lactamase gene in a clinical isolate of Enterobacter aerogenes. J Appl Microbiol 95:744–752. doi: 10.1046/j.1365-2672.2003.02040.x. [DOI] [PubMed] [Google Scholar]
- 32.Cookson AL, Croucher D, Pope C, Bennett J, Thomson-Carter F, Attwood GT. 2006. Isolation, characterization, and epidemiological assessment of Shiga toxin-producing Escherichia coli O84 isolates from New Zealand. J Clin Microbiol 44:1863–1866. doi: 10.1128/JCM.44.5.1863-1866.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H. 2011. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perron GG, Inglis RF, Pennings PS, Cobey S. 2015. Fighting microbial drug resistance: a primer on the role of evolutionary biology in public health. Evol Appl 8:211–222. doi: 10.1111/eva.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grad YH, Lipsitch M, Feldgarden M, Arachchi HM, Cerqueira GC, Fitzgerald M, Godfrey P, Haas BJ, Murphy CI, Russ C, Sykes S, Walker BJ, Wortman JR, Young S, Zeng Q, Abouelleil A, Bochicchio J, Chauvin S, Desmet T, Gujja S, McCowan C, Montmayeur A, Steelman S, Frimodt-Moller J, Petersen AM, Struve C, Krogfelt KA, Bingen E, Weill FX, Lander ES, Nusbaum C, Birren BW, Hung DT, Hanage WP. 2012. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc Natl Acad Sci U S A 109:3065–3070. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sjolund M, Bonnedahl J, Hernandez J, Bengtsson S, Cederbrant G, Pinhassi J, Kahlmeter G, Olsen B. 2008. Dissemination of multildrug-resistant bacteria into the Arctic. Emerg Infect Dis 14:70–72. doi: 10.3201/eid1401.070704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez J, Johansson A, Stedt J, Bengtsson S, Porczak A, Granholm S, Gonzalez-Acuna D, Olsen B, Bonnedahl J, Drobni M. 2013. Characterization and comparison of extended-spectrum beta-lactamase (ESBL) resistance genotypes and population structure of Escherichia coli isolated from Franklin’s gulls (Leucophaeus pipixcan) and humans in Chile. PLoS One 8:e76150. doi: 10.1371/journal.pone.0076150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonnedahl J, Stedt J, Waldenstrom J, Svensson L, Drobni M, Olsen B. 2015. Comparison of Extended-spectrum beta-lactamase (ESBL) CTX-M genotypes in Franklin gulls from Canada and Chile. PLoS One 10:e0141315. doi: 10.1371/journal.pone.0141315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baez J, Hernandez-Garcia M, Guamparito C, Diaz S, Olave A, Guerrero K, Canton R, Baquero F, Gahona J, Valenzuela N, Del Campo R, Silva J. 2015. Molecular characterization and genetic diversity of ESBL-producing Escherichia coli colonizing the migratory Franklin’s gulls (Leucophaeus pipixcan) in Antofagasta, north of Chile. Microb Drug Resist 21:111–116. doi: 10.1089/mdr.2014.0158. [DOI] [PubMed] [Google Scholar]
- 40.Stedt J, Bonnedahl J, Hernandez J, Waldenstrom J, McMahon BJ, Tolf C, Olsen B, Drobni M. 2015. Carriage of CTX-M type extended spectrum beta-lactamases (ESBLs) in gulls across Europe. Acta Vet Scand 57:74. doi: 10.1186/s13028-015-0166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glupczynski Y, Berhin C, Bauraing C, Bogaerts P. 2007. Evaluation of a new selective chromogenic agar medium for detection of extended-spectrum beta-lactamase-producing Enterobacteriaceae. J Clin Microbiol 45:501–505. doi: 10.1128/JCM.02221-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reglier-Poupet H, Naas T, Carrer A, Cady A, Adam JM, Fortineau N, Poyart C, Nordmann P. 2008. Performance of chromID ESBL, a chromogenic medium for detection of Enterobacteriaceae producing extended-spectrum beta-lactamases. J Med Microbiol 57:310–315. doi: 10.1099/jmm.0.47625-0. [DOI] [PubMed] [Google Scholar]
- 43.Edelstein M, Pimkin M, Palagin I, Edelstein I, Stratchounski L. 2003. Prevalence and molecular epidemiology of CTX-M extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae in Russian hospitals. Antimicrob Agents Chemother 47:3724–3732. doi: 10.1128/AAC.47.12.3724-3732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiiru J, Kariuki S, Goddeeris BM, Butaye P. 2012. Analysis of beta-lactamase phenotypes and carriage of selected beta-lactamase genes among Escherichia coli strains obtained from Kenyan patients during an 18-year period. BMC Microbiol 12:155. doi: 10.1186/1471-2180-12-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newton-Foot M, Snyman Y, Maloba MRB, Whitelaw AC. 2017. Plasmid-mediated mcr-1 colistin resistance in Escherichia coli and Klebsiella spp. clinical isolates from the Western Cape region of South Africa. Antimicrob Resist Infect Control 6:78. doi: 10.1186/s13756-017-0234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ, Dymock D, Wade WG. 1998. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol 64:795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.CLSI. 2012. Methods for dilution antimicrobial susceptibility testes for bacteria that grow aerobically; approved standard, 9th ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 48.Joshi NA, Fass JN. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33). https://github.com/najoshi/sickle. Accessed 6 June 2017.
- 49.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Treangen TJ, Ondov BD, Koren S, Phillippy AM. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.NCBI Resource Coordinators. 2017. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 45:D12–D17. doi: 10.1093/nar/gkw1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, Conrad N, Dietrich EM, Disz T, Gabbard JL, Gerdes S, Henry CS, Kenyon RW, Machi D, Mao C, Nordberg EK, Olsen GJ, Murphy-Olson DE, Olson R, Overbeek R, Parrello B, Pusch GD, Shukla M, Vonstein V, Warren A, Xia F, Yoo H, Stevens RL. 2017. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res 45:D535–D542. doi: 10.1093/nar/gkw1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L, Zheng D, Liu B, Yang J, Jin Q. 2016. VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res 44:D694–D697. doi: 10.1093/nar/gkv1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, Doshi S, Courtot M, Lo R, Williams LE, Frye JG, Elsayegh T, Sardar D, Westman EL, Pawlowski AC, Johnson TA, Brinkman FS, Wright GD, McArthur AG. 2017. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Ponten T, Ussery DW, Aarestrup FM, Lund O. 2012. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joensen KG, Tetzschner AMM, Iguchi A, Aarestrup FM, Scheutz F. 2015. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J Clin Microbiol 53:2410–2426. doi: 10.1128/JCM.00008-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carattoli A, Zankari E, Garcia-Fernandez A, Larsen MV, Lund O, Villa L, Aarestrup FM, Hasman H. 2014. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vielva L, de Toro M, Lanza VF, de la Cruz F. 2017. PLACNETw: a web-based tool for plasmid reconstruction from bacterial genomes. Bioinformatics 33:3796–3798. doi: 10.1093/bioinformatics/btx462. [DOI] [PubMed] [Google Scholar]
- 62.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jeon SJ, Oh M, Yeo WS, Galvao KN, Jeong KC. 2014. Underlying mechanism of antimicrobial activity of chitosan microparticles and implications for the treatment of infectious diseases. PLoS One 9:e92723. doi: 10.1371/journal.pone.0092723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.