

Curli are proteins produced by many bacteria as a structural component of biofilms, and they have recently emerged as a platform for fabrication of biological materials. Curli fibers are very robust and resistant to degradation, and the curli subunits can tolerate many protein fusions, facilitating the biosynthesis of novel functional materials. A serious bottleneck in the development of more sophisticated engineered curli systems is the rapid quantification of curli production by the bacteria. In this work we address this issue by developing a technique to monitor curli production directly in bacterial cultures, allowing for rapid curli quantification in a manner compatible with many powerful high-throughput techniques that can be used to engineer complex biological material systems.
KEYWORDS: synthetic biology, biofilms, biotechnology, curli, quantitative methods
ABSTRACT
Curli are amyloid proteins that are assembled into extracellular polymeric fibers by bacteria during biofilm formation. The beta-sheet-rich protein CsgA, the primary structural component of the fibers, is secreted through dedicated machinery and self-assembles into cell-anchored fibers many times longer than the cell. Here, we have developed an in situ fluorescence assay for curli production that exploits the fluorescent properties of Congo red (CR) dye when bound to amyloid, allowing for rapid and robust curli quantification. We initially evaluated three amyloid-binding dyes for the fluorescent detection of curli in bacterial culture and found only Congo red compatible with in situ quantification. We further characterized the fluorescent properties of the dye directly in bacterial culture and calibrated the fluorescence using purified CsgA protein. We then used the Congo red assay to rapidly develop and characterize inducible curli-producing constructs in both an MC4100-derived lab strain of Escherichia coli and a derivative of the probiotic strain E. coli Nissle. This technique can be used to evaluate curli production in a minimally invasive manner using a range of equipment, simplifying curli quantification and the development of novel engineered curli systems.
IMPORTANCE Curli are proteins produced by many bacteria as a structural component of biofilms, and they have recently emerged as a platform for fabrication of biological materials. Curli fibers are very robust and resistant to degradation, and the curli subunits can tolerate many protein fusions, facilitating the biosynthesis of novel functional materials. A serious bottleneck in the development of more sophisticated engineered curli systems is the rapid quantification of curli production by the bacteria. In this work we address this issue by developing a technique to monitor curli production directly in bacterial cultures, allowing for rapid curli quantification in a manner compatible with many powerful high-throughput techniques that can be used to engineer complex biological material systems.
INTRODUCTION
Curli are amyloid proteins that are secreted from bacterial cells to form protein fibers that make up the major proteinaceous component of many biofilms (1). Escherichia coli produces these beta-sheet-rich protein fibers made up of CsgA and CsgB proteins during surface colonization, cell aggregation, and biofilm growth (2, 3). These proteins fold after secretion by dedicated machinery (4, 5) and self-assemble into micron-length fibers in the extracellular space, with the minor CsgB subunit nucleating fibers made of the major CsgA subunit (6). Curli synthesis and function are relatively well understood in enteric bacteria, such as E. coli, and homologous genes are present in a diverse range of bacterial species (7). Curli proteins provide mechanical stability to bacterial biofilms (8, 9) and are resistant to many forms of degradation, such as proteases, detergents, or boiling (10). Furthermore, the proteins in curli fibers can be functionalized through mutation (11) or fusion to functional protein domains (12, 13), providing a platform for the engineering of living functional materials. Regulating and exploiting the properties of such living materials is an active field of research (14) with a variety of applications, such as bioremediation (15) and medical therapeutics (16, 17).
Quantitatively monitoring curli fiber production is a critical aspect of curli research, both in the context of microbiology and for the engineering of synthetic curli systems. However, elucidating protein concentration is complicated by the same factors that make curli fibers unique, with their extreme robustness leading to insoluble, cell-anchored fibers that mediate cell aggregation. Furthermore, since the curli are secreted into the extracellular medium, single-cell assays of curli production are inherently not viable. Although assays exist for quantitatively detecting curli, they rely on burdensome sample processing steps that are not compatible with in situ monitoring.
A simplified assay would facilitate the development of more complex engineered curli systems, allowing for the rapid evaluation of designed curli systems and the selection of high-performing constructs. Such an assay would ideally be performed in the solution phase and tolerant of the heterogenous and inherently complex environment of cell culture. An ideal assay would be optical in nature, relying on in situ measurements that are minimally invasive and are compatible with many quantitative experimental techniques. Fluorescence assays are commonly employed for quantification in a wide variety of situations, ranging from genetic component quantification (18) to the quantification of desired metabolites in engineered systems with biosensors (19) or fluorescent probes (20). Fluorescence assays also have the benefit of allowing real-time monitoring (21) and high-throughput screening (22), both of which are incompatible with current curli assays.
Amyloid fiber concentrations have been measured in various ways in the many fields studying such proteins (23). The first amyloid stain, described in 1922, used Congo red (CR) dye (24) to detect amyloidosis in tissue samples. More staining techniques for tissues have been developed subsequently, including thioflavin T (ThT) (25) and immunostaining (26). In the context of bacterial amyloids such as curli, amyloid stains are also used to detect the presence of curli in biofilms, as are immunostaining techniques (27) and luminescent probes (28). In vitro, protocols with purified curli protein commonly measure ThT dye fluorescence to monitor curli polymerization (29). Curcumin dye has also been shown to be fluorescent in the presence of purified assembled CsgA (30).
Curli quantification in bacterial culture has generally been performed with colorimetric CR assays (12, 29). The CR dye binds with some specificity to amyloid, and the binding induces shifts in the absorbance spectrum (31) and birefringence (32), as well as producing a fluorescent product (33). Quantitative assays are typically performed by centrifugation of a bacterial culture, pelleting the culture, and resuspending it in phosphate-buffered saline (PBS) with CR before a further centrifugation to measure the supernatant absorbance. This assay reveals how much CR dye bound to curli fibers and was subsequently removed from solution into the pellet with the bacteria. The centrifugation steps here are a bottleneck in sample processing and, as such, provide a significant hurdle to many higher-throughput assays. Another CR protocol is also used to detect curli in bacteria growing on plates (29); however, colonies are only assessed qualitatively for red coloration.
The use of dyes for fluorescent in situ curli assays with growing bacterial cultures has not yet been systematically studied. Culturing bacteria together with the dye would allow curli measurement in real time, as well as with high-throughput assays, due to the minimally invasive nature of the optical measurement. Past studies have shown that cells can grow and produce curli in the presence of both CR (34) and curcumin (30); however, the fluorescent properties of such dyes in bacterial culture have not been addressed. Furthermore, many amyloid dyes are inexpensive and readily available, with a history of use making them simple to obtain and employ in a range of experiments.
In this work, we assess the use of amyloid dyes as in situ reporters for curli concentration, evaluating the performance of Congo red, thioflavin T and curcumin. We found that CR was the only viable dye, and developed a method based on CR fluorescence to measure the concentration of curli fibers in a bacterial culture. We then applied the method to characterize the performance of several synthetic inducible curli constructs in two E. coli strains, PQN4 (derived from MC4100 lab strain) and PBP5 (derived from the probiotic Nissle 1917 strain). Our results demonstrate the efficacy of in situ CR dye fluorescence to quantitatively assess curli production, allowing for rapid curli measurement compatible with many high-throughput experimental techniques.
RESULTS
Dye evaluation.
We tested CR, ThT, and curcumin for their efficacy in quantitatively measuring curli concentrations in bacterial culture, as these dyes have a long history of use for amyloid detection, exhibit amyloid-specific fluorescence, and are readily available. To test these dyes, we produced curli material in bacterial culture and assessed whether there was a fluorescent signal specific to the presence of curli. We used E. coli PQN4 cells, which are an E. coli MC4100 (LSR10)-derived strain lacking the entire curli operon (ΔcsgBACDEFG) (35). Curli was produced with plasmid pBbB8k_csgBACEFG (36), which contained a synthetic curli operon csgBACEFG regulated by an arabinose-inducible promoter (PBAD). The csgBACEFG operon contained genes for the curli fiber subunits (CsgBA), coexpressed with the machinery to secrete monomers from the periplasm (CsgEG) (2), as well as an inhibitor of intracellular amyloid folding (CsgC) (37).
We grew PQN4 bacteria with or without plasmid pBbB8k_csgBACEFG overnight (24 h) in LB medium in the presence of arabinose inducer (100 μM) and the three dyes, each at 4 different concentrations. We then measured the fluorescence and absorbance spectra of the overnight cultures (Fig. S1a to c), reading the emission and excitation fluorescence spectra at two distinct imaging settings. Emission spectra were found at a fixed excitation wavelength of either 435 nm or 525 nm, and the excitation spectra at a fixed emission wavelength of either 510 nm or 625 nm. These optical settings were chosen to coincide with known fluorescence peaks of CR (33), ThT (29), and curcumin (30). Only CR provided a curli-specific fluorescent signal (Fig. 1a to c), with ThT dye and curcumin fluorescence being mostly correlated with cell density rather than curli. A CR concentration of 25 μg/ml led to a greater fluorescent signal at 525 to 625 nm (Fig. 1a), with higher concentrations reducing the fluorescence emission signal. We found reductions in cell density of the overnight culture only at high concentrations of ThT dye (Fig. 1d to f), suggesting little toxicity from CR or curcumin at the concentrations tested.
FIG 1.

Fluorescence and absorbance measurements of overnight PQN4 cultures with or without curli-producing plasmid pBbB8k_csgBACEFG at a range of amyloid dye concentrations. In each case, samples were incubated for 24 h in LB medium with 100 μM arabinose inducer to induce transcription of the csgBACEFG operon. (a) CR fluorescence at 625 nm with excitation at 525 nm, (b) ThT fluorescence at 510 nm with excitation at 435 nm, (c) curcumin fluorescence at 510 nm with excitation at 435 nm. Absorbance readings at 600 nm are shown for samples incubated in the presence of (d) CR, (e) ThT, or (f) curcumin. Error bars show standard deviation for readings within a 10-nm wavelength band (n = 3).
The fluorescence spectra of overnight bacterial cultures grown in the presence of 25 μg/ml CR are shown in Fig. 2a. Excitation at 525 nm and detection at 625 nm led to a clearly observable signal from induced cultures with the plasmid present, after background subtraction of signal originating from the culture itself (Fig. 2b). Cellular autofluorescence in this channel was negligible comparable to LB medium autofluorescence, indicating that CR fluorescence was not enhanced by the presence of the cells alone.
FIG 2.
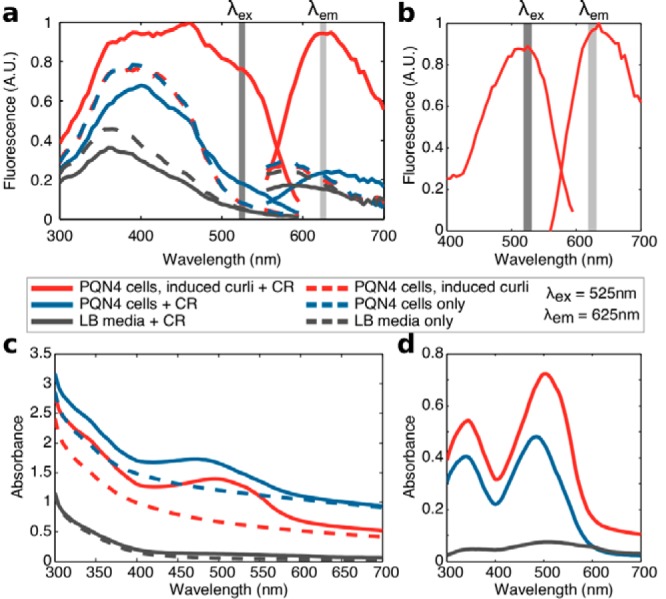
Spectra of overnight bacterial cultures with CR dye. Curli-producing cultures were PQN4 cells harboring the pBbB8k_csgBACEFG plasmid induced with 100 μM arabinose. Cultures without curli were PQN4 cells without plasmid, and the medium control contained LB only, which was used for all samples. (a) Fluorescence spectra of bacterial cultures grown overnight with 25 μg/ml CR dye (solid lines) or without dye (dashed lines). Excitation spectra (left curves) were found at a fixed emission wavelength (λem) of 625 nm, and emission spectra (right) at a fixed excitation wavelength (λex) of 525 nm. (b) Fluorescence spectrum of the CR in the presence of curli-producing cells, normalized to remove the background signal from cell culture control without dye. (c) Absorbance spectra of the overnight cultures with and without CR. (d) Normalized absorbance spectra for CR-containing cultures, displaying the characteristic shift in CR absorbance with curli binding.
The absorbance spectra of the CR dye with curli-producing cultures (Fig. 2d) reproduced the characteristic shift found in previous in vitro research (31), suggesting that the bacterial culture did not significantly disrupt the typical CR-amyloid interaction. A further notable result was that in curli-producing cultures, the CR dye led to an increase in background absorbance at all of the wavelengths tested (300 nm to 700 nm). As such, the dye interferes with typical measurements of cell density in a curli-dependent manner and could thus complicate calculations of curli production per cell. This suggests that curli production should be measured for the culture as a whole. Additionally, we noted that CR dye aggregated and precipitated in LB over time, reducing dye absorbance during incubation. However, this phenomenon was limited to wells without bacteria, suggesting that the presence of bacteria disrupted dye aggregation, facilitating more robust quantification of the cell cultures.
We further assessed and validated curli production in PQN4 with 25 μg/ml CR in greater detail. Alongside plasmid pBbB8k_csgBACEFG, we used pBbB8k_csgBACEFG-A6xHis, which is the same as the former but encodes CsgA fused to a 36-amino-acid (aa) flexible linker and a polyhistidine sequence. PQN4 cells harboring one of these plasmids, or no plasmid, were grown for 24 h overnight in LB with 25 μg/ml CR at a range of arabinose concentrations (Fig. 3a). The resulting cultures showed a CR fluorescent signal at arabinose concentrations greater than ∼1 μM. To confirm the presence of curli in these cultures, we performed immunostaining for CsgA. For each plasmid type, we grew 7 overnight culture samples at a range of arabinose concentrations in the presence of CR, and first quantified their CR fluorescence (shown as crosses in Fig. 3a). These same cultures were then used for Western blotting (Fig. 3b) with either anti-CsgA antibodies for samples with pBbB8k_csgBACEFG (red crosses) or anti-6×His antibodies for the pBbB8k_csgBACEFG-A6xHis plasmid (blue crosses). The immunostaining revealed appropriately sized CsgA bands in samples that had a high CR fluorescent signal, demonstrating that curli protein abundance corresponded to the CR fluorescent signal.
FIG 3.

Validation of curli production. (a) PQN4 cells with plasmid pBbB8k_csgBACEFG, pBbB8k_csgBACEFG-A6xHis, or no plasmid were grown overnight at a range of arabinose concentrations with 25 μg/ml CR dye. The CR fluorescence of overnight cultures is shown with shaded areas showing averages and standard deviations of 3 replicates for each condition. For each plasmid, 7 samples were taken for Western blotting, each at a particular arabinose concentration, with their specific CR fluorescence reading shown as crosses on the curve. (b) Western blot of an SDS-PAGE gel, using (red ×) anti-CsgA for samples from plasmid pBbB8k_csgBACEFG-bearing cells, or (blue +) anti-6×His for the His-tagged variant. Curli-producing cultures were also imaged with CR dye in a confocal microscope, showing fluorescent aggregates (c and d) with 100 μM arabinose (ara) that were not present without induction (e and f). The CR fluorescence channel is shown in red, overlaid onto the grayscale brightfield channel. Bar, 10 μm.
To further visualize curli production and in situ CR-curli fluorescence, we imaged PQN4 liquid cultures with pBbB8k_csgBACEFG and CR dye in a confocal microscope (Fig. 3c to f), finding fluorescent cell aggregates in samples with arabinose induction. The aggregates were brightly fluorescent at the CR-curli wavelengths and highlighted that curli fibers mediated the aggregation of the bacterial cells.
To assess the viability of this technique to detect curli production in wild-type bacterial samples, we grew E. coli MC4100 and PQN4 strains in several medium conditions in the presence of CR dye. In LB medium at 37°C (Fig. S2a), neither strain should produce any curli, so their levels of CR fluorescence should be similar. However, MC4100 cells, which harbor the curli operons in their genome, should produce curli in yeast extract-Casamino Acids (YESCA) medium at 26°C for 2 days (34) (Fig. S2), unlike PQN4 cells, which lack the curli operons. The assay could not detect a significant difference between PQN4 and MC4100 cells in YESCA medium, suggesting that the curli concentration was too low to be detected in the bacterial strains alone. When curli was expressed from a plasmid, however, the CR-curli signal was also present in YESCA medium (Fig. S2b), although it was lower than that in LB at 37°C.
Congo red calibration.
In order to characterize the CR-curli fluorescent signal, we quantified this relationship with purified proteins. Unassembled CsgA-6×His monomers were purified from E. coli BL21 cultures transformed with pET21d-csgAhis (35). The purified monomers were allowed to polymerize in 2-(N-morpholino)ethanesulfonic acid (MES) buffer, and the subsequent dynamics were observed in a plate reader with both ThT and CR dyes (Fig. 4a). A clear CR fluorescence signal was detected in response to curli polymerization; however, we found the CR signal to be noisy, more so than the characteristic sigmoidal ThT fluorescence curve. This was due to the large insoluble aggregates containing both dye and curli fibers that formed in these wells. However, the fluorescence signal for CR was clearly visible above background, and we obtained a calibration curve using a range of CsgA monomer concentrations allowed to polymerize for 24 h at 37°C. In order to more accurately measure the CR fluorescence signal from the insoluble aggregates, we performed fluorescence readings of a 25-point grid at the bottom of each well, taking the mean of the spatially distinct points (Fig. 4b).
FIG 4.
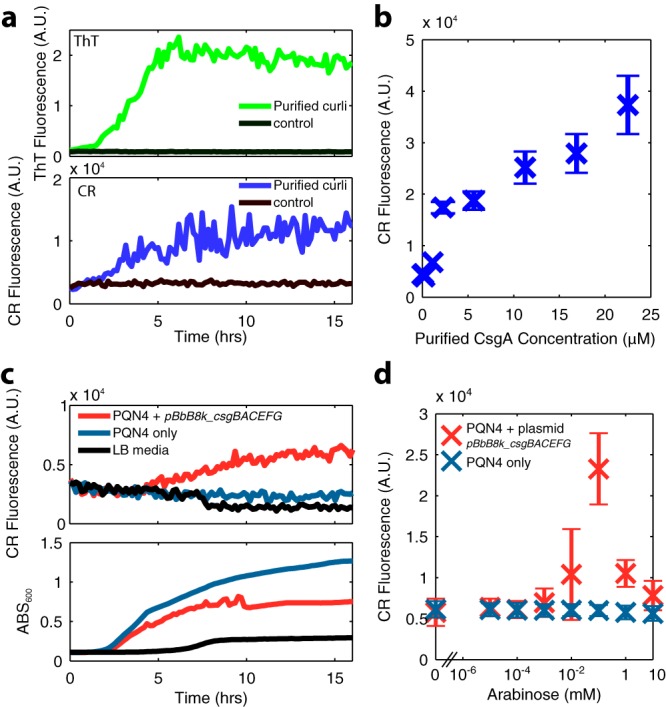
(a) Polymerization dynamics of 23 μM purified CsgA monomers visualized with ThT dye (emission, 438 nm; excitation, 495 nm; top) or CR dye (emission, 525 nm; excitation, 625 nm; bottom) measured in a plate reader every 10 min for 16 h. (b) CR dye fluorescence of purified CsgA monomers left to polymerize with 25 μg/ml CR dye for 24 h, as measured by area scanning in a plate reader, with error bars showing the standard deviation of 25 spatially distinct data points within a well. (c) CR fluorescence dynamics (top) and 600 nm absorbance (bottom) of bacterial cultures incubated at 37°C in LB medium, assayed every 10 min for 16 h. (d) CR dye fluorescence of curli-producing bacterial cultures incubated for 24 h in a range of arabinose concentrations, measured by scanning the area of the well in a plate reader, with error bars showing standard deviation of an area scan as in panel b.
We used CR fluorescence to observe the production of curli in a bacterial culture over time with arabinose induction (Fig. 4c). These kinetic curves show a clear CR fluorescent signal with plasmid pBbB8k_csgBACEFG, appearing after around 4 h of growth, when the culture was in the mid-exponential phase. Curli production also noticeably slowed growth (Fig. 4c), suggesting that curli expression was burdensome to the cells. We also varied the concentration of arabinose, finding a peak CR fluorescence signal from overnight cultures harboring pBbB8k_csgBACEFG at 100 μM (Fig. 4d). At high inducer concentrations, overall curli production was limited due to the poor growth of cells. The results, when properly normalized and compared to those in Fig. 4b, suggested that PQN4 cells with curli induction from the pBbB8k_csgBACEFG plasmid produced around 10 μM curli subunits in overnight culture (Fig. 3d), corresponding to around 0.1 g/liter curli.
Growing cell cultures can alter the pH of their environment, and pH is known to alter the optical properties of CR (38). To assess the pH effects on the CR-curli signal, we grew PQN4 cells with plasmid pBbB8k_csgBACEFG overnight in LB with 100 μM arabinose without CR. The cells were pelleted by centrifugation and resuspended in PBS buffer with 25 μg/ml CR that had been pH readjusted to between pH 3 and 11. After an hour of shaking incubation with CR to ensure binding, we measured the CR fluorescent signal, and longer incubation times did not affect the results. We found that acidic conditions decreased the fluorescent signal (Fig. 5). However, with the LB medium growth conditions used throughout this work, the pH of the overnight cultures was not found to drop below 6.5, suggesting only a limited effect from pH in our results.
FIG 5.
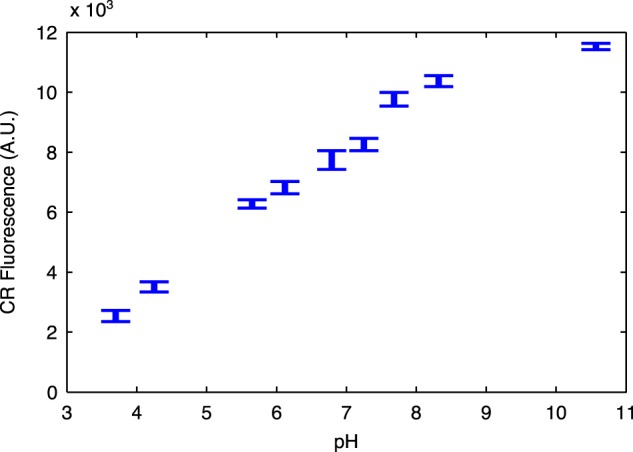
Influence of pH on the fluorescence of curli-bound CR. A curli-producing overnight culture was resuspended in PBS buffer containing 25 μg/ml CR at a range of pH values. Error bars show standard deviation between 4 replicates.
Synthetic curli system design.
We then applied this method to develop and characterize two new inducible plasmid systems for curli production. LacI and TetR derepression systems were used to express the curli-producing synthetic operon csgBACEFG (Fig. 6b and c) containing the wild-type ribosome-binding sites (RBSs) for each gene, as well as a 5′-untranslated region (UTR) RiboJ ribozyme (39) to insulate the operon expression from promoter sequence effects. To build a construct with high curli expression levels, we screened a library of RBS variants driving the repressor proteins for curli production with CR dye in the plate reader. The plasmids that expressed high levels of curli upon induction but were tightly repressed otherwise were selected, resulting in plasmids pL6FO and pT1FO, containing the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible and anhydrotetracycline (ATC)-inducible constructs, respectively.
FIG 6.

CR dye fluorescence characterization of synthetic curli systems in the PQN4 lab strain (red) and the probiotic Nissle-derived PBP5 strain (green). We assessed 3 inducible curli fiber-producing plasmids, inducing transcription of the csgBACEFG operon by (a) arabinose, (b) IPTG, or (c) ATC. Cells harboring these curli plasmid systems, and controls without plasmids, were assessed after overnight incubation in LB medium with CR dye at a range of inducer concentrations. In each case, each data point shows the mean of 3 separate experiments.
The constructs pL6FO and pT1FO were characterized alongside plasmid pBbB8k-csgBACEFG in both the PQN4 strain and PBP5, an E. coli Nissle 1917 derivative containing a deletion of the genomic csg operon. Nissle is a probiotic E. coli strain and thus a prime target for therapeutic engineering (16, 40). All constructs produced detectable curli expression in both strains (Fig. 6), with the IPTG and ATC constructs performing similarly to pBbB8k-csgBACEFG in PQN4. In PBP5, we found arabinose to be a poor inducer of curli, unlike IPTG and ATC (Fig. 6a). Nissle has a 17% larger genome than those of most lab strains of E. coli (41) and can digest arabinose, unlike most lab strains, suggesting that arabinose metabolism reduced the efficacy of the PBAD induction system.
Throughout our results, the PBP5 strain produced a higher CR background signal than that of the PQN4 strain. Since E. coli Nissle is known to be a much better biofilm former than lab strains, we investigated whether PBP5 produced cellulose, as cellulose also binds to CR (42). To assess this, we performed a knockout of the bacterial cellulose operons bcsEFG and bcsRQABZC (43) in strain PBP5, making strain AIK2 (EcN ΔcsgBACDEFG ΔbcsEFGRQABZC). We transformed this strain with pL6FO and assessed the CR signal at a range of IPTG concentrations (Fig. 7). We observed a similarly high background signal with AIK2 as that with PBP5 and slightly reduced peak curli production in the cellulose knockout. The results suggested that little cellulose was produced under our assay conditions, and that the background signal was caused by CR interacting with some other molecules present in the Nissle-derived strains. Despite these high background fluorescence levels in the PBP5 strain, detectable curli production could be induced with the synthetic systems as with the lab strains, and in the cases of pL6FO and pT1FO, the PBP5 strain gave significantly higher CR-curli fluorescent signals than those of the PQN4 lab strain, even when the background signal was removed.
FIG 7.
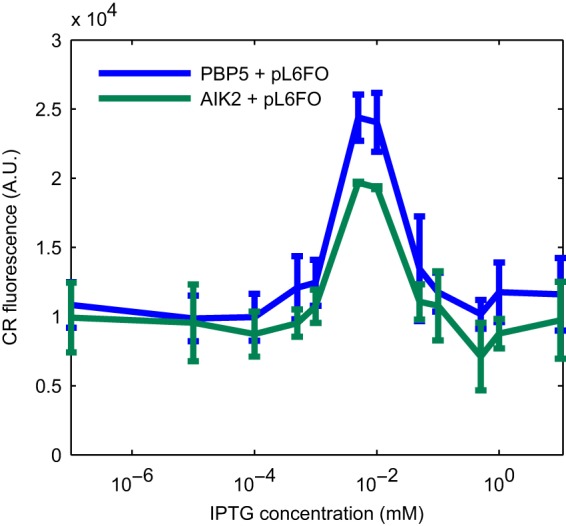
CR fluorescent signal in a cellulose knockout Nissle strain. Plasmid pL6FO, harboring an IPTG-inducible csgBACEFG operon, was used to induce curli production in 2 Nissle-derived strains, PBP5 and AIK2. PBP5 contains a knockout of the native curli operon csgBACDEFG, and AIK2 is PBP5 with a further knockout of the cellulose-producing operon bcsEFGRQABZC. Aside from a minor reduction in the peak curli production, removal of the cellulose operon did not influence the background CR signal. Error bars show standard deviation from experiments performed in duplicate.
DISCUSSION
In this work, we have developed a technique using CR fluorescence to characterize synthetic curli systems in E. coli. The technique involves adding CR dye to bacterial cultures before growth and monitoring the fluorescence emission at 625 nm with excitation at 525 nm. Growing bacteria in the presence of CR significantly simplified assays, doing away with any sample processing steps and allowing direct curli quantification from the culture. We then used this method to rapidly develop and characterize inducible curli-producing constructs in both a lab strain and a probiotic strain of E. coli.
Curli has recently emerged as an important protein material (44, 45) due to its ability to gain an impressive array of function through protein fusion and its mechanical robustness. Furthermore, curli is produced by bacteria and secreted into the extracellular space by design, allowing for manufacture directly in bacterial culture. The bacteria within such synthetic biofilms can also be modified to exhibit desirable biological or chemical functions, acting as engineered living materials with the potential to grow and react to their environment (14). The design and construction of synthetic biological systems require the assessment of the system in question to characterize and test features of interest for further design refinement (46). As such, rapid and convenient assays are critical to accelerating bioengineering. Our work has shown how CR fluorescence can be used for rapid curli quantification to design and build synthetic curli material systems.
Fluorescence detection is a popular quantification method, making this technique compatible with a large range of laboratory equipment in assays such as plate fluorometry, colony imaging, or confocal microscopy. Furthermore, this method is compatible with high-throughput techniques, such as flow cytometry or microfluidic sorting, facilitating the rapid screening of large numbers of samples. In recent decades, high-throughput techniques have become powerful tools for engineering biological systems (47), facilitating both improved biological systems and a deeper understanding of the underlying biological principles. Looking forward, such techniques are required for the development of synthetic curli systems, allowing the fabrication of more sophisticated engineered living material systems.
Throughout this work, we cultured bacteria in LB medium due to the high levels of curli expression with synthetic curli systems. However, LB medium has several drawbacks, such as autofluorescence and variability between batches. We repeated the construct characterization for pL6FO and pT1FO in several formulations of defined M9 minimal medium (Fig. S3), finding curli production similar to that in LB medium when Casamino Acids alone were used as the carbon source. M9 medium also gave a significantly lower background fluorescence signal than that of LB medium, suggesting that it is preferable for more precise curli construct characterization in the future.
The aggregation of the CR posed a challenge for accurate quantification for the case of purified protein due to the spatial inhomogeneity of the large aggregates of dye and curli. ThT dye, by comparison, produced a less noisy signal in vitro (Fig. 4a), despite the polymerization of CsgA into insoluble fibers throughout all the in vitro assays. Given the propensity of CR, unlike ThT, to aggregate in bacterial medium, PBS, and MES buffer, this additional aggregation pathway likely sped the formation of larger insoluble aggregates. Crucially to our assays, when bacteria were present with the dye, CR did not aggregate significantly over the course of overnight shaking incubation. While aggregates were formed when cells were producing curli, rapid shaking homogenized the cultures enough to allow for simple and reproducible curli detection with CR fluorescence.
From our results, it was not clear whether the CR altered the dynamics of CsgA polymerization or if it influenced the curli polymer morphology in any way. We microscopically observed aggregates containing cells and curli (Fig. 3c to f), and these fluorescent aggregates appeared similar in cultures that had been grown with CR and in cultures for which CR was “spiked in” after an incubation without CR. While this suggested that CR did not significantly influence the aggregative function of curli fibers, it is possible that CR could alter the material properties of a curli polymer matrix.
Our method can simply and robustly characterize engineered curli production from E. coli in a noninvasive manner; however, there are several points to keep in mind for the analysis of such data. When CR dye is bound to curli, it absorbs light at 600 nm (Fig. 2d), which would introduce errors into optical density measurements in a curli-dependent fashion. If accurate measurements are desired of curli production on a per cell level, this error could be corrected for with appropriate fluorescence measurements in a more complex normalization procedure.
The background CR fluorescence can vary significantly between E. coli strains. Although under our culture conditions we did not detect any cellulose production, for other bacteria and in different growth conditions this may not be the case. CR binds to cellulose to form a fluorescent complex and, in fact, it binds more tightly to cellulose than to curli (42). If cellulose were present, it would likely obscure the curli quantification, as would silk, which also binds to CR. However, this also suggests that a similar technique could be extended to investigate the biosynthesis of these materials in bacterial cultures. Furthermore, in samples with both curli and cellulose, novel CR dye derivatives like 1-fluoro-2,5-bis[(E)-3-carboxy-4-hydroxystyryl]benzene (FSB) (48) could be used to obtain a curli-specific signal.
Conclusion.
In this work, we have developed a rapid and reliable assay for curli production in bacterial culture. Given the wide range of functionality that can be displayed on curli fibers, the scope for curli-based engineered living materials is vast. Our work presents a new tool for the rapid testing of engineered curli systems and opens up a range of powerful tools to characterize and develop complex high-throughput curli material systems. Furthermore, the propensity of CR to bind to other biological polymers, such as silk and cellulose, could allow for an extension of this technique to other biologically fabricated materials.
MATERIALS AND METHODS
Bacterial culture.
All E. coli cells were grown in Miller’s LB medium (Sigma-Aldrich) unless indicated otherwise. Cells containing plasmids were grown with 50 mg/ml kanamycin, and cells without plasmids were grown without antibiotics. Where appropriate, the inducer l-arabinose (Sigma-Aldrich), isopropyl-β-d-thiogalactopyranoside (RPI), or anhydrotetracycline (Cayman Chem) was added to the culture. Also, Congo red (≥85% purity, C6277 BioXrta; Sigma-Aldrich), thioflavin T (Sigma-Aldrich), and curcumin (Sigma-Aldrich) dyes were used with bacterial culture or with purified proteins where indicated.
Western blotting.
Immunostaining was performed per the protocol in reference 29. Briefly, bacterial cultures were grown in LB overnight with shaking at 37°C. The cultures were then normalized to the same 600-nm absorbance, and 150 μl of each culture was pelleted by centrifugation at 16,000 × g for 3 min. The supernatant was carefully decanted, and the pellet was resuspended in 100% hexafluoroisopropanol (HFIP; Oakwood Chemical). The HFIP was subsequently removed through vacuum concentration (45°C for 30 min), and the pellet was resuspended in SDS-PAGE sample buffer and heated to 98°C for 10 min. Samples were run on a NuPAGE Novex 4% to 12% bis-Tris gel and transferred onto an iBlot polyvinylidene difluoride (PVDF) membrane (Invitrogen). After blocking with 5% Blotto nonfat milk (Santa Cruz Biotechnology) in Tris-buffered saline and 0.1% Tween 20 buffer (TBST), the membrane was washed in TBST and treated with antibodies. In the case of the anti-CsgA antibodies, the membrane was first treated with rabbit anti-CsgA antibodies (4), washed five times in TBST, and then treated with goat anti-rabbit antibody horseradish peroxidase (HRP) conjugate (Bethyl). For anti-His treatment, a monoclonal mouse anti-His antibody HRP conjugate (Thermo Fisher) was used. The membranes were then washed five times in TBST, and chemiluminescence was induced using the Clarity Western enhanced chemiluminescence (ECL) substrate (Bio-Rad) and imaged in a FluorChem M system (Protein Simple).
Plasmid construction and selection.
All plasmids were constructed with Gibson assembly (49). Plasmids pL6FO and pT1FO were selected from pools of plasmids initially designed to express curli in a tightly regulated, inducible fashion. These plasmids contained constitutively induced LacI or TetR repressor proteins, a terminator, and then promoter pLac (BBa_R0011; Biobricks Registry) or pTet (BBa_R0040; Biobricks Registry) to express the synthetic csgBACEFG operon. Curli production optimization was performed by designing an RBS library for the repressor proteins using the Salis Lab RBS calculator (50), synthesizing these libraries on DNA oligonucleotides with degenerate nucleotides, and using the oligonucleotides for plasmid assembly. Assembled plasmids were transformed into PQN4 cells, which were plated onto LB agar plates with 25 μg/ml CR. Red colonies were selected and further assessed in liquid culture with CR in the plate reader.
Strain development.
Deletion mutants of E. coli strain Nissle 1917 (EcN) were created by the lambda red recombineering method described previously by Datsenko and Wanner (51). The chloramphenicol acetyltransferase (CAT) cassette with 500-bp homology arms upstream and downstream of the curli operon was transformed into EcN with preinduced lambda red genes from the pKD46 plasmid using 0.5% arabinose during the early log phase. The transformants were recovered in super optimal broth with catabolite repression (SOC) medium at 30°C for 3 h to enable the genomic replacement of the target with the CAT cassette and plated onto LB plates containing 25 μg/ml chloramphenicol (RPI) to select resistant colonies. Genomic knockout was verified by colony PCR and Sanger DNA sequencing to confirm the replacement of the target DNA with the CAT cassette. Then, a verified mutant was transformed with the pE-FLP plasmid (Addgene plasmid 45978; a gift from Drew Endy and Keith Shearwin [52]) to remove the CAT cassette. The successful colonies were then selected from the loss of chloramphenicol resistance. Then, pE-FLP was cured using a blank transformation technique and selection for colonies that had lost the ampicillin resistance in pE-FLP. Successful knockouts were confirmed again by Sanger sequencing of an amplicon of the region surrounding the knockout.
Protein purification.
CsgA was purified per a protocol previously described by Sleutel et al. (53). CsgA was cloned into pET21d without its N-terminal 19-residue secretion signal sequence and with a C-terminal 6×His tag. Expression was induced in BL21(DE3) cells in LB medium by addition of 1 mM IPTG at an optical density at 600 nm (OD600) of 0.9. After 1 h of induction, cells were harvested by centrifugation at 5,000 × g for 10 min at 25°C. Pellets were lysed overnight in lysis buffer (50 mM KPi and 8 M guanidinium chloride [GdnHCl] [pH 7.2]) under constant stirring at 4°C. Cell lysate was centrifuged at 30,000 × g for 20 min at 20°C to remove cell debris. Supernatant was sonicated and passed through a 0.22-μm filter. SuperFlow Ni-nitrilotriacetic acid (NTA) agarose beads (Thermo Fisher Scientific) were added to filtered supernatant, and the mixture was incubated on a rocking platform at room temperature for 1 h. Supernatant and beads were then loaded into a PD-10 disposable column with frit (GE Healthcare). After washing in 10 column volumes of wash buffer (50 mM KPi, 8 M GdnHCl, and 12.5 mM imidazole [pH 7.2]), the purified CsgA protein was eluted in 1-ml fractions of elution buffer (50 mM KPi, 8 M GdnHCl and 125 mM imidazole [pH 7.2]).
Proteins were eluted in 8 M GdnHCl to prevent aggregation and maintain the monomeric form of CsgA. Elution fraction samples were desalted using Zeba Spin desalting columns (7K molecular weight cutoff) (Thermo Fisher Scientific) and analyzed by SDS-PAGE for purity. Pure, highly concentrated samples were combined, split into aliquots, and frozen at −80°C. Assays were subsequently performed by thawing frozen protein in a cold-water bath and triggering polymerization after desalting with a Zeba Spin desalting column.
Plate reader assays.
For all plate fluorometry assays with bacteria, bacterial starter cultures were grown overnight at 37°C in an Infors high-throughput (HT) Multitron shaking incubator (225 rpm) in the medium that would be used for the assay. Overnight cultures were then diluted 1:1,000 into fresh medium and placed into flat clear-bottomed black 96-well plates (Greiner). The medium was supplemented with the appropriate antibiotics, inducers, and dyes at the start of the experiment. For kinetic runs, plates were placed into a BioTek Neo HTS plate reader at 37°C, where CR fluorescence (excitation [ex], 525 nm; emission [em], 625 nm) and absorbance (600 nm) were read every 10 min, with shaking between reads. For endpoint curli quantification, the 96-well plates were incubated for 24 h in a 37°C shaking incubator (900 rpm), and fluorescence and absorbance were read in a BioTek Synergy H1 plate reader, reading the CR fluorescence (ex, 525 nm; em, 625 nm) and absorbance (600 nm). Spectral scans were also performed in the BioTek Synergy H1 plate reader.
Area-scanning plate fluorometry was employed with purified CsgA protein, and for cell cultures in Fig. 3d, using the BioTek Synergy H1 plate reader machine to scan a 5 by 5 grid at the bottom of each well to capture the spatial variation of the samples. Grid spacing was set automatically by the Gen5 plate reader software to cover the entire area of the well.
CsgA polymerization assays were performed using freshly desalted CsgA buffer switched to 15 mM MES (pH 6.0) using a Zeba Spin desalting column. Protein concentration was measured using absorbance at 280 nm. Protein samples were pipetted into a black flat-bottomed 96-well microplate (Greiner Bio One) in the presence of 50 μM thioflavin T (ThT) or 25 μg/ml Congo red (CR) dye. Fluorescence measurements were performed using the Biotek Neo HTS plate reader at 25°C with excitation at 438 nm and emission at 495 nm for ThT and excitation at 525 nm and emission at 625 nm for CR. Fluorescence readings were taken every 10 min, and the plates were shaken for 5 min before each reading.
Microscopy.
Bacteria harboring plasmids were grown overnight in LB medium with appropriate antibiotics, and this culture was used to inoculate 1:100 fresh LB medium containing specified dyes, inducers, and antibiotics. After 5 h of growth, 1-μl drops of culture were placed onto a glass slide, covered with a coverslip, and imaged in a Leica SP5 confocal microscope reading a bright-field (BF) channel as well as the CR fluorescence channel by exciting at 525 nm and measuring emission at 625 nm.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NSF Division of Materials Research (grant 1410751), the National Institutes of Health (grant 1R01DK11077001A1), and the Wyss Institute for Biologically Inspired Engineering.
We also thank the reviewers for their insightful comments, which led to an improvement of the manuscript.
We declare no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00434-19.
REFERENCES
- 1.Beloin C, Roux A, Ghigo J-M. 2008. Escherichia coli biofilms. Curr Top Microbiol Immunol 322:249–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnhart MM, Chapman MR. 2006. Curli biogenesis and function. Annu Rev Microbiol 60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans ML, Chapman MR. 2014. Curli biogenesis: order out of disorder. Biochim Biophys Acta 1843:1551–1558. doi: 10.1016/j.bbamcr.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. 2002. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Gerven N, Klein RD, Hultgren SJ, Remaut H. 2015. Bacterial amyloid formation: structural insights into curli biogensis. Trends Microbiol 23:693–706. doi: 10.1016/j.tim.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shu Q, Crick SL, Pinkner JS, Ford B, Hultgren SJ, Frieden C. 2012. The E. coli CsgB nucleator of curli assembles to β-sheet oligomers that alter the CsgA fibrillization mechanism. Proc Natl Acad Sci U S A 109:6502–6507. doi: 10.1073/pnas.1204161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dueholm MS, Albertsen M, Otzen D, Nielsen PH. 2012. Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS One 7:e51274. doi: 10.1371/journal.pone.0051274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galy O, Latour-Lambert P, Zrelli K, Ghigo J-M, Beloin C, Henry N. 2012. Mapping of bacterial biofilm local mechanics by magnetic microparticle actuation. Biophys J 103:1400–1408. doi: 10.1016/j.bpj.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng G, Vad BS, Dueholm MS, Christiansen G, Nilsson M, Tolker-Nielsen T, Nielsen PH, Meyer RL, Otzen DE. 2015. Functional bacterial amyloid increases Pseudomonas biofilm hydrophobicity and stiffness. Front Microbiol 6:1099. doi: 10.3389/fmicb.2015.01099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collinson SK, Emödy L, Müller KH, Trust TJ, Kay WW. 1991. Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J Bacteriol 173:4773–4781. doi: 10.1128/jb.173.15.4773-4781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Courchesne N-M, DeBenedictis EP, Tresback J, Kim JJ, Duraj-Thatte A, Zanuy D, Keten S, Joshi NS. 2018. Biomimetic engineering of conductive curli protein films. Nanotechnology 29:454002. doi: 10.1088/1361-6528/aadd3a. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen PQ, Botyanszki Z, Tay PKR, Joshi NS. 2014. Programmable biofilm-based materials from engineered curli nanofibres. Nat Commun 5:4945. doi: 10.1038/ncomms5945. [DOI] [PubMed] [Google Scholar]
- 13.Gerven NV, Goyal P, Vandenbussche G, Kerpel MD, Jonckheere W, Greve HD, Remaut H. 2014. Secretion and functional display of fusion proteins through the curli biogenesis pathway. Mol Microbiol 91:1022–1035. doi: 10.1111/mmi.12515. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen PQ, Courchesne N-M, Duraj-Thatte A, Praveschotinunt P, Joshi NS. 2018. Engineered living materials: prospects and challenges for using biological systems to direct the assembly of smart materials. Adv Mater 30:1704847. doi: 10.1002/adma.201704847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tay PKR, Nguyen PQ, Joshi NS. 2017. A synthetic circuit for mercury bioremediation using self-assembling functional amyloids. ACS Synth Biol 6:1841–1850. doi: 10.1021/acssynbio.7b00137. [DOI] [PubMed] [Google Scholar]
- 16.Duraj-Thatte AM, Praveschotinunt P, Nash TR, Ward FR, Joshi NS. 2018. Modulating bacterial and gut mucosal interactions with engineered biofilm matrix proteins. Sci Rep 8:3475. doi: 10.1038/s41598-018-21834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Li Z, Xiao H, Wang N, Li Y, Xu X, Chen Z, Tan H, Li J. 2018. A universal and ultrastable mineralization coating bioinspired from biofilms. Adv Funct Mater 28:1802730. doi: 10.1002/adfm.201802730. [DOI] [Google Scholar]
- 18.Rudge TJ, Brown JR, Federici F, Dalchau N, Phillips A, Ajioka JW, Haseloff J. 2016. Characterization of intrinsic properties of promoters. ACS Synth Biol 5:89–98. doi: 10.1021/acssynbio.5b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan S-A, Nadler DC, Yokoo R, Savage DF. 2016. Biofuel metabolic engineering with biosensors. Curr Opin Chem Biol 35:150–158. doi: 10.1016/j.cbpa.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hounslow E, Noirel J, Gilmour DJ, Wright PC. 2017. Lipid quantification techniques for screening oleaginous species of microalgae for biofuel production. Eur J Lipid Sci Technol 119:1500469. doi: 10.1002/ejlt.201500469. [DOI] [Google Scholar]
- 21.Young JW, Locke JCW, Altinok A, Rosenfeld N, Bacarian T, Swain PS, Mjolsness E, Elowitz MB. 2012. Measuring single-cell gene expression dynamics in bacteria using fluorescence time-lapse microscopy. Nat Protoc 7:80–88. doi: 10.1038/nprot.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agresti JJ, Antipov E, Abate AR, Ahn K, Rowat AC, Baret J-C, Marquez M, Klibanov AM, Griffiths AD, Weitz DA. 2010. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc Natl Acad Sci U S A 107:4004–4009. doi: 10.1073/pnas.0910781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elghetany MT, Saleem A. 1988. Methods for staining amyloid in tissues: a review. Stain Technol 63:201–212. doi: 10.3109/10520298809107185. [DOI] [PubMed] [Google Scholar]
- 24.Bennhold H. 1922. Eine specifische amyloidfärbung mit kongorot. Münch Med Wochenschr 44:1537–1538. [Google Scholar]
- 25.Levine H. 2008. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci 2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallo GR, Feiner HD, Chuba JV, Beneck D, Marion P, Cohen DH. 1986. Characterization of tissue amyloid by immunofluorescence microscopy. Clin Immunol Immunopathol 39:479–490. doi: 10.1016/0090-1229(86)90175-3. [DOI] [PubMed] [Google Scholar]
- 27.Macarisin D, Patel J, Bauchan G, Giron JA, Sharma VK. 2012. Role of curli and cellulose expression in adherence of Escherichia coli O157:H7 to spinach leaves. Foodborne Pathog Dis 9:160–167. doi: 10.1089/fpd.2011.1020. [DOI] [PubMed] [Google Scholar]
- 28.Choong FX, Bäck M, Fahlén S, Johansson LB, Melican K, Rhen M, Nilsson KPR, Richter-Dahlfors A. 2016. Real-time optotracing of curli and cellulose in live Salmonella biofilms using luminescent oligothiophenes. NPJ Biofilms Microbiomes 2:16024. doi: 10.1038/npjbiofilms.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y, Smith D, Hufnagel D, Chapman M. 2013. Experimental manipulation of the microbial functional amyloid called curli, p 53–75. In Delcour AH. (ed), Bacterial cell surfaces. Humana Press, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCrate OA, Zhou X, Cegelski L. 2013. Curcumin as an amyloid-indicator dye in E. coli. Chem Commun (Camb) 49:4193–4195. doi: 10.1039/c2cc37792f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klunk WE, Pettegrew JW, Abraham DJ. 1989. Quantitative evaluation of Congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. J Histochem Cytochem 37:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 32.Howie AJ, Brewer DB. 2009. Optical properties of amyloid stained by Congo red: history and mechanisms. Micron 40:285–301. doi: 10.1016/j.micron.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Clement CG, Truong LD. 2014. An evaluation of Congo red fluorescence for the diagnosis of amyloidosis. Hum Pathol 45:1766–1772. doi: 10.1016/j.humpath.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Reichhardt C, Jacobson AN, Maher MC, Uang J, McCrate OA, Eckart M, Cegelski L. 2015. Congo red interactions with curli-producing E. coli and native curli amyloid fibers. PLoS One 10:e0140388. doi: 10.1371/journal.pone.0140388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dorval Courchesne N-M, Duraj-Thatte A, Tay PKR, Nguyen PQ, Joshi NS. 2017. Scalable production of genetically engineered nanofibrous macroscopic materials via filtration. ACS Biomater Sci Eng 3:733–741. doi: 10.1021/acsbiomaterials.6b00437. [DOI] [PubMed] [Google Scholar]
- 36.Praveschotinunt P, Dorval Courchesne N-M, den Hartog I, Lu C, Kim JJ, Nguyen PQ, Joshi NS. 2018. Tracking of engineered bacteria in vivo using nonstandard amino acid incorporation. ACS Synth Biol 7:1640–1650. doi: 10.1021/acssynbio.8b00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans ML, Chorell E, Taylor JD, Åden J, Götheson A, Li F, Koch M, Sefer L, Matthews SJ, Wittung-Stafshede P, Almqvist F, Chapman MR. 2015. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell 57:445–455. doi: 10.1016/j.molcel.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mera SL, Davies JD. 1984. Differential Congo red staining: the effects of pH, non-aqueous solvents and the substrate. Histochem J 16:195–210. doi: 10.1007/BF01003549. [DOI] [PubMed] [Google Scholar]
- 39.Lou C, Stanton B, Chen Y-J, Munsky B, Voigt CA. 2012. Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat Biotechnol 30:1137–1142. doi: 10.1038/nbt.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danino T, Prindle A, Kwong GA, Skalak M, Li H, Allen K, Hasty J, Bhatia SN. 2015. Programmable probiotics for detection of cancer in urine. Sci Transl Med 7:289ra84. doi: 10.1126/scitranslmed.aaa3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grozdanov L, Raasch C, Schulze J, Sonnenborn U, Gottschalk G, Hacker J, Dobrindt U. 2004. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J Bacteriol 186:5432–5441. doi: 10.1128/JB.186.16.5432-5441.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reichhardt C, McCrate OA, Zhou X, Lee J, Thongsomboon W, Cegelski L. 2016. Influence of the amyloid dye Congo red on curli, cellulose, and the extracellular matrix in E. coli during growth and matrix purification. Anal Bioanal Chem 408:7709–7717. doi: 10.1007/s00216-016-9868-2. [DOI] [PubMed] [Google Scholar]
- 43.Römling U, Galperin MY. 2015. Bacterial cellulose biosynthesis: diversity of operons, subunits, products and functions. Trends Microbiol 23:545–557. doi: 10.1016/j.tim.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knowles TPJ, Mezzenga R. 2016. Amyloid fibrils as building blocks for natural and artificial functional materials. Adv Mater 28:6546–6561. doi: 10.1002/adma.201505961. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen PQ. 2017. Synthetic biology engineering of biofilms as nanomaterials factories. Biochem Soc Trans 45:585–597. doi: 10.1042/BST20160348. [DOI] [PubMed] [Google Scholar]
- 46.Agapakis CM. 2014. Designing synthetic biology. ACS Synth Biol 3:121–128. doi: 10.1021/sb4001068. [DOI] [PubMed] [Google Scholar]
- 47.Cheng AA, Lu TK. 2012. Synthetic biology: an emerging engineering discipline. Annu Rev Biomed Eng 14:155–178. doi: 10.1146/annurev-bioeng-071811-150118. [DOI] [PubMed] [Google Scholar]
- 48.Reichhardt C, Cegelski L. 2018. The Congo red derivative FSB binds to curli amyloid fibers and specifically stains curliated E. coli. PLoS One 13:e0203226. doi: 10.1371/journal.pone.0203226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 50.Tian T, Salis HM. 2015. A predictive biophysical model of translational coupling to coordinate and control protein expression in bacterial operons. Nucleic Acids Res 43:7137–7151. doi: 10.1093/nar/gkv635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.St-Pierre F, Cui L, Priest DG, Endy D, Dodd IB, Shearwin KE. 2013. One-step cloning and chromosomal integration of DNA. ACS Synth Biol 2:537–541. doi: 10.1021/sb400021j. [DOI] [PubMed] [Google Scholar]
- 53.Sleutel M, Van den Broeck I, Van Gerven N, Feuillie C, Jonckheere W, Valotteau C, Dufrêne YF, Remaut H. 2017. Nucleation and growth of a bacterial functional amyloid at single fiber resolution. Nat Chem Biol 13:902–908. doi: 10.1038/nchembio.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.