

Because almost all amino acids in nature are l-amino acids, the functioning of d-amino acids has received little attention. Thus, there is little information available on the activity of enzymes toward d-amino acids or synthetic methods for d-amino acid-containing dipeptides. Recently, d-amino acids and d-amino acid-containing peptides have attracted attention as novel functional compounds, and d-amino acid-activating enzymes and synthetic methods are required for the development of the d-amino acid-containing-peptide industry. This study provides additional knowledge regarding d-amino acid-activating enzymes and proposes a unique synthetic method for d-amino acid-containing peptides, including ld-, dl-, and dd-dipeptides.
KEYWORDS: adenylation domain, d-amino acid, dipeptide synthesis, nonribosomal peptide synthetase
ABSTRACT
Recent papers have reported dipeptides containing d-amino acids to have novel effects that cannot be observed with ll-dipeptides, and such dipeptides are expected to be novel functional compounds for pharmaceuticals and food additives. Although the functions of d-amino acid-containing dipeptides are gaining more attention, there are few reports on the synthetic enzymes that can accept d-amino acids as substrates, and synthetic methods for d-amino acid-containing dipeptides have not yet been constructed. Previously, we developed a chemoenzymatic system for amide synthesis that comprised enzymatic activation and a subsequent nucleophilic substitution reaction. In this study, we demonstrated the application of the system for d-amino acid-containing-dipeptide synthesis. We chose six adenylation domains as targets according to our newly constructed hypothesis, i.e., an adenylation domain located upstream from the epimerization domain may activate d-amino acid as well as l-amino acid. We successfully synthesized over 40 kinds of d-amino acid-containing dipeptides, including ld-, dl-, and dd-dipeptides, using only two adenylation domains, TycA-A from tyrocidine synthetase and BacB2-A from bacitracin synthetase. Furthermore, this study offered the possibility that the epimerization domain could be a clue to the activity of the adenylation domains toward d-amino acid. This paper provides additional information regarding d-amino acid-containing-dipeptide synthesis through the combination of enzymatic adenylation and chemical nucleophilic reaction, and this system will be a useful tool for dipeptide synthesis.
IMPORTANCE Because almost all amino acids in nature are l-amino acids, the functioning of d-amino acids has received little attention. Thus, there is little information available on the activity of enzymes toward d-amino acids or synthetic methods for d-amino acid-containing dipeptides. Recently, d-amino acids and d-amino acid-containing peptides have attracted attention as novel functional compounds, and d-amino acid-activating enzymes and synthetic methods are required for the development of the d-amino acid-containing-peptide industry. This study provides additional knowledge regarding d-amino acid-activating enzymes and proposes a unique synthetic method for d-amino acid-containing peptides, including ld-, dl-, and dd-dipeptides.
INTRODUCTION
The chemical and physical properties of l-amino acids and d-amino acids are nearly identical, but they have different bioactivities. Also, dipeptides that contain a d-amino acid moiety in their structures (called d-amino acid-containing dipeptides in this paper) have different biological properties from peptides comprising only l-amino acids. For instance, d-Glu-d-Trp, d-γ-Glu-d-Trp, d-Glu-l-Trp, and d-γ-Glu-l-Trp inhibit the proliferation of CFU-spleen cells in intact bone marrow, while the dipeptides containing only l-amino acids, such as l-Glu-l-Trp and l-γ-Glu-l-Trp, have no effect on their proliferation (1). Cyclo(d-Tyr-d-Phe), a cyclic dipeptide isolated from the fermented broth of Bacillus sp., had greater antibacterial and anticancer activities than cyclo(l-Tyr-l-Phe) (2). Some kinds of dd-diketopiperazines, such as cyclo(d-Pro-d-Phe), inhibit the growth of Vibrio anguillarum more effectively than other stereoisomers (3). An interesting report revealed that cyclo(l-Trp-d-Pro) increased the heart rate, whereas cyclo(d-Trp-l-Pro) reduced the heart rate (4). These reports are great examples that show the potential of d-amino acid-containing dipeptides as new bioactive molecules.
Dipeptides can be synthesized chemically or enzymatically. Almost all dipeptides can be synthesized by chemical reactions, but multistep reactions for protection, condensation, and deprotection are required. Enzymatic methods provide one-pot procedures for dipeptide synthesis. Many enzymatic methods for synthesizing ll-dipeptides have been reported, but insufficient methods for d-amino acid-containing-dipeptide synthesis have been proposed. Using the reverse reaction of amidohydrolase from Streptomyces sp. strain 82F2, various d-amino acid-containing dipeptides were successfully synthesized (5). However, the enzyme was not suitable for dd-dipeptide synthesis, and also, the substrates should be methyl or benzyl ester because of the substrate specificity of the amidohydrolase (5). Aminoacyltransferase from Saccharothrix sp. strain AS-2 is a unique enzyme for d-amino acid-containing-dipeptide synthesis, but some of the substrates should be methyl esters (6). d-Alanine-d-alanine ligase is also a useful enzyme, but only d-Ala can be used as the N terminus of the dipeptide (7). Still, a process for synthesis of d-amino acid-containing dipeptides from free amino acids using these conventional enzymatic methods has not been constructed due to the restricted substrate specificities of the enzymes involved.
We have focused on the mechanism of nonribosomal peptide synthetase (NRPS). NRPS is a multimodule synthetase that produces secondary metabolites like tyrocidine, bacitracin, and paenibacterin. NRPS catalyzes three consecutive reactions: (i) the l-amino acid is activated by the adenylation domain and (ii) is then thioesterized with the phosphopantetheine group that is connected to the thiolation domain, before (iii) being finally coupled to the amino acyl or peptidyl intermediate by the condensation domain (8). When a d-amino acid is used as the member of the secondary metabolite, the l-form substrate is converted into the d-form by the epimerization domain before condensation (9). Previously, we focused on the function of the adenylation domain and constructed a chemoenzymatic method for amide synthesis. In this mechanism, a free amino acid was activated by the adenylation domain to be aminoacyl-AMP, and the amide bond was formed with the subsequent nucleophilic attack by the amine (10). Using this system, various amines, such as linear amines, cyclic amines, and amino acids, can be used as nucleophiles and various amides can be synthesized (11).
We assumed that various d-amino acid-containing dipeptides, such as ld-, dl-, and dd-dipeptides, could be synthesized using an adenylation domain that can activate d-amino acids. Because few studies describing such adenylation domains have been reported, the goal of this study was to identify adenylation domains that activate d-amino acids and to extend the application of these systems to d-amino acid-containing-dipeptide synthesis.
RESULTS
Substrate specificity of TycA-A.
Previously, we constructed a novel colorimetric assay for evaluation of the substrate specificities of adenylation domains, in which we found that TycA-A could activate a number of l-amino acids (10). Here, we tested the activity of TycA-A toward d-amino acids. The results revealed that TycA-A could activate d-Ala, d-Leu, d-Met, d-Phe, d-Tyr, and d-Trp (Fig. 1), confirming that TycA-A had broad substrate specificity. This result indicated that TycA-A had the potential to synthesize d-amino acid-containing dipeptides. Our interest moved to the screening of d-amino acid-activating adenylation domains from a pool of adenylation domains in nature.
FIG 1.

Substrate specificity of TycA-A. Results shown are those of hydroxamate-based colorimetric assays. Filled bars indicate the absorbances at 490 nm (ABS490) of reaction mixtures with TycA-A, and open bars indicate those of reaction mixtures without TycA-A. Error bars indicate the standard deviations based on the results of three independent experiments.
Identification of d-amino acid-activating adenylation domains.
The adenylation domain chosen as the target should activate d-amino acids. Enough information about adenylation domains that activate l-amino acids has been reported, such as the amino acid residues around the catalytic site and crystallization (12–15). However, there are few reports about the d-amino acid-activating adenylation domains, and we needed to screen adenylation domains. We hypothesized that an adenylation domain located upstream from the epimerization domain, which is the key domain incorporating d-amino acid into the secondary metabolite, may activate d-amino acids in addition to l-amino acids. The process of derivation of this hypothesis is described in Discussion. According to this hypothesis, we additionally chose the following five adenylation domains: BacB2-A and BacC4-A in bacitracin synthetase from Bacillus licheniformis strain NBRC 12199 (16) and PbtA1-A, PbtB2-A, and PbtB3-A in the paenibacterin synthetase homologs from Paenibacillus alvei strain NBRC 3343. PbtA1-A, PbtB2-A, and PbtB3-A in this study were paenibacterin synthetase homologs of previously reported paenibacterin synthetase from Paenibacillus thiaminolyticus strain OSY-SE (17). All the targets, including TycA-A, were located upstream from the epimerization domain (Fig. 2). BacB2-A, BacC4-A, PbtA1-A, PbtB2-A, and PbtB3-A are the adenylation domains in the modules that incorporate d-Orn, d-Asp, d-Orn, d-Lys, and d-Ser, respectively, into the secondary metabolite (16–18).
FIG 2.

Locations of the six adenylation domains in the NRPS modules. TycA-A in tyrocidine synthetase (a), BacB2-A and BacC4-A in bacitracin synthetase (b), and PbtA1-A, PbtB2-A, and PbtB3-A in paenibacterin synthetase (c). All six adenylation domains are located upstream from the epimerization domains. Domain labeling is as follows: T, thiolation domain; C, condensation domain; E, epimerization domain; and Te, thioesterase domain. The three-letter code represents the adenylation domain and amino acid as its substrate (e.g., Phe represents the adenylation domain whose amino acid substrate is phenylalanine).
Substrate specificities of other adenylation domains.
The substrate specificities of BacB2-A, BacC4-A, PbtA1-A, PbtB2-A, and PbtB3-A toward d-amino acids and l-amino acids were evaluated with a previously reported colorimetric assay for the adenylation domains (10). Three adenylation domains activated d-Lys and no other d-amino acids: BacB2-A, PbtA1-A, and PbtB2-A (Fig. 3). This result indicated that these adenylation domains have the potential to synthesize d-amino acid-containing dipeptides. Interestingly, our assay indicated that BacB2-A and PbtA1-A have no activity toward l-Orn and d-Orn (Fig. 3; Fig. S1 in the supplemental material), even though the two are adenylation domains in the modules that incorporate d-Orn into the secondary metabolites (17). The other two adenylation domains activated only l-amino acids: BacC4-A activated l-Asp, and PbtB3-A activated l-Ser (Fig. S1).
FIG 3.

Substrate specificities of BacB2-A (a), BacC4-A (b), PbtA1-A (c), PbtB2-A (d), and PbtB3-A (e) toward d-amino acids. Filled bars indicate the absorbances of reaction mixtures with the enzymes, and open bars indicate those of reaction mixtures without enzymes. Error bars indicate the standard deviations based on the results of three independent experiments.
Synthesis of ld-, dl-, and dd-dipeptides by TycA-A and BacB2-A.
We demonstrated ld-, dl-, and dd-dipeptide synthesis using these adenylation domains. BacB2-A, PbtA1-A, and PbtB2-A have the same substrate specificity, so BacB2-A was chosen as a representative for dipeptide synthesis. As a result, under all conditions, the assumed dipeptide was detected through high-performance liquid chromatography (HPLC), mass spectrometry (MS), and tandem MS (MS/MS) analysis (Table 1), which indicated that various kinds of d-amino acid-containing dipeptides could be synthesized through this chemoenzymatic method. Because homodipeptides, such as d-Trp-d-Trp, were detected as by-products, we further performed the dipeptide synthesis using different concentrations of nucleophiles. When 0, 10, 20, 50, 100, 200, or 300 mM d-Pro was added, d-Trp-d-Pro was more efficiently produced as the d-Pro concentration increased, and the by-product, d-Trp-d-Trp, was more successfully suppressed (Fig. 4).
TABLE 1.
ld-, dl-, and dd-dipeptide synthesis
| Adenylation domain | Substrate | Nucleophile | Retention time (min) | Parent ion (m/z) |
|---|---|---|---|---|
| TycA-A | d-Ala | d-Pro | 7.70 | 187.1085 |
| d-Ala | l-Pro | 8.52 | 187.1082 | |
| l-Ala | d-Pro | 8.47 | 187.1084 | |
| d-Leu | d-Pro | 3.45 | 229.1555 | |
| d-Leu | l-Pro | 4.60 | 229.1535 | |
| l-Leu | d-Pro | 4.57 | 229.1529 | |
| d-Met | d-Pro | 1.59 | 247.1118 | |
| d-Met | l-Pro | 3.27 | 247.1116 | |
| l-Met | d-Pro | 3.19 | 247.1118 | |
| d-Phe | d-Pro | 4.89 | 263.1396 | |
| d-Phe | l-Pro | 6.28 | 263.1396 | |
| l-Phe | d-Pro | 6.29 | 263.1396 | |
| d-Tyr | d-Pro | 2.73 | 279.1348 | |
| d-Tyr | l-Pro | 3.61 | 279.1347 | |
| l-Tyr | d-Pro | 3.65 | 279.1396 | |
| d-Trp | d-Pro | 6.35 | 302.1507 | |
| d-Trp | l-Pro | 6.97 | 302.1503 | |
| l-Trp | d-Pro | 6.96 | 302.1501 | |
| BacB2-A | d-Lys | d-Pro | 14.06 | 244.1661 |
| d-Lys | l-Pro | 11.94 | 244.1661 | |
| l-Lys | d-Pro | 12.00 | 244.1663 |
FIG 4.
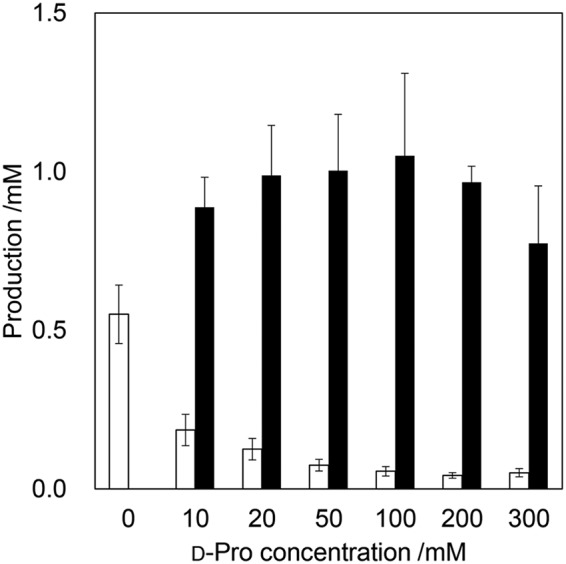
Relations between concentrations of d-Pro and production levels of dipeptides d-Trp-d-Trp (open bars) and d-Trp-d-Pro (filled bars). Error bars indicate the standard deviations based on the results of three independent experiments.
Synthesis of d-Trp-d-Xaa and d-Lys-d-Xaa by TycA-A and BacB2-A.
A previous study showed that various kinds of amines, such as linear amines, cyclic amines, cyclic amino acids, and derivatives, could be used as nucleophiles for amide synthesis (11). Here, our results suggested that all the d-amino acids could be used for nucleophiles in this system; thus, we demonstrated dipeptide synthesis using general d-amino acids as nucleophiles. As a result, under all conditions tested, the expected dipeptides were detected with HPLC, MS, and MS/MS analyses (Tables 2 and 3). MS/MS fragment patterns are shown in Fig. 5, which are similar to those in the previous study (11). When amino acids that could be activated by TycA-A, such as d-Phe, were used for the C terminus of the product, the expected d-Trp-d-Phe, d-Phe-d-Trp, and d-Phe-d-Phe dipeptides were synthesized (Fig. S2). Also, homodipeptides, such as d-Trp-d-Trp and d-Lys-d-Lys, were detected in almost all samples. Considering all these results, although there are still some points to improve, over 20 kinds of d-amino acid-containing dipeptides could be synthesized through this hybrid reaction mechanism using only a single adenylation domain.
TABLE 2.
Products synthesized by TycA-A
| Product | Parent ion (m/z) |
MS/MS ion fragment (m/z)a
: |
||||
|---|---|---|---|---|---|---|
| Calculated | Detected | 1 | 2 | 3 | 4 | |
| d-Trp-Gly | 262.1192 | 262.1198 | 130.0657 | 131.0455 | 159.0923 | 102.0208 |
| d-Trp-d-Ala | 276.1348 | 276.1350 | 130.0654 | 145.0613 | 159.0922 | 116.0366 |
| d-Trp-d-Val | 304.1661 | 304.1670 | 130.0659 | 173.0920 | 159.0925 | 144.0568 |
| d-Trp-d-Leu | 318.1818 | 318.1820 | 130.0662 | 187.1096 | 159.0926 | 158.0842 |
| d-Trp-d-Ile | 318.1818 | 318.1822 | 130.0662 | 187.1113 | 159.0925 | 158.0848 |
| d-Trp-d-Ser | 292.1297 | 292.1301 | 130.0658 | ND | 159.0923 | 132.0275 |
| d-Trp-d-Thr | 306.1454 | 306.1461 | 130.0649 | 175.0734 | 159.0924 | 146.0477 |
| d-Trp-d-Cys | 308.1069 | 308.1074 | 130.0657 | 177.0330 | 159.0923 | 148.0041 |
| d-Trp-d-Met | 336.1382 | 336.1389 | 130.0659 | 205.0646 | 159.0925 | 176.0442 |
| d-Trp-d-Pro | 302.1505 | 302.1511 | 130.0658 | 171.0704 | 159.0924 | 142.0486 |
| d-Trp-d-Asn | 319.1406 | 319.1412 | 130.0659 | 188.0706 | 159.0925 | 159.0454 |
| d-Trp-d-Gln | 333.1563 | 333.1566 | 130.0640 | 202.0828 | 159.0924 | 173.0520 |
| d-Trp-d-Asp | 320.1246 | 320.1253 | 130.0658 | 189.0513 | 159.0924 | 160.0189 |
| d-Trp-d-Glu | 334.1403 | 334.1406 | 130.0656 | 203.0622 | 159.0923 | 174.0465 |
| d-Trp-d-Lys | 333.1927 | 333.1932 | 130.0659 | 202.1197 | 159.0924 | 173.0971 |
| d-Trp-d-Arg | 361.1988 | 361.1990 | 130.0658 | 230.1257 | 159.0924 | 201.0988 |
| d-Trp-d-His | 342.1566 | 342.1568 | 130.0659 | 211.0900 | 159.0921 | 182.0624 |
| d-Trp-d-Phe | 352.1661 | 352.1667 | 130.0659 | 221.0918 | 159.0925 | 192.0658 |
| d-Trp-d-Tyr | 368.1610 | 368.1612 | 130.0657 | 237.0898 | 159.0923 | 208.0615 |
MS/MS fragments are depicted in Fig. 5. ND, not detected.
TABLE 3.
Products synthesized by BacB2-A
| Product | Parent ion (m/z) |
MS/MS ion fragment (m/z)a
: |
||||
|---|---|---|---|---|---|---|
| Calculated | Detected | 1 | 2 | 3 | 4 | |
| d-Lys-Gly | 204.1348 | 204.1352 | 84.0814 | ND | 129.1030 | 74.0268 |
| d-Lys-d-Ala | 218.1505 | 218.1507 | 84.0815 | 116.0360 | 129.1027 | 88.0403 |
| d-Lys-d-Val | 246.1818 | 246.1817 | 84.0811 | 144.0621 | 129.1025 | 116.0713 |
| d-Lys-d-Leu | 260.1974 | 260.1974 | 84.0815 | 158.0828 | 129.1025 | 130.0858 |
| d-Lys-d-Ile | 260.1974 | 260.1974 | 84.0812 | ND | 129.1031 | 130.0863 |
| d-Lys-d-Ser | 234.1454 | 234.1444 | 84.0814 | 132.0218 | 129.1023 | 104.0391 |
| d-Lys-d-Thr | 248.161 | 248.1612 | 84.0815 | 146.0462 | 129.1028 | 118.0549 |
| d-Lys-d-Cys | 250.1225 | 250.1228 | 84.0824 | 147.9908 | 129.1015 | 120.0120 |
| d-Lys-d-Met | 278.1538 | 278.1542 | 84.0814 | 176.0419 | 129.1028 | 148.0455 |
| d-Lys-d-Pro | 244.1661 | 244.1660 | 84.0812 | 142.0518 | 129.1027 | 114.0555 |
| d-Lys-d-Asn | 261.1563 | 261.1563 | 84.0816 | 159.0387 | 129.1031 | 131.0510 |
| d-Lys-d-Gln | 275.1719 | 275.1719 | 84.0810 | 173.0522 | 129.1024 | ND |
| d-Lys-d-Asp | 262.1403 | 262.1405 | 84.0811 | ND | 129.1029 | 132.0318 |
| d-Lys-d-Glu | 276.1559 | 276.1559 | 84.0814 | ND | 129.1028 | 146.0456 |
| d-Lys-d-Arg | 303.2145 | 152.0876b | 84.0815 | 230.1219 | 129.1030 | 173.1073 |
| d-Lys-d-His | 284.1723 | 284.1728 | 84.0805 | 182.0543 | 129.1041 | 154.0724 |
| d-Lys-d-Phe | 294.1818 | 294.1820 | 84.0813 | 192.0709 | 129.1027 | 164.0713 |
| d-Lys-d-Tyr | 310.1767 | 310.1769 | 84.0818 | 208.0622 | 129.1024 | 180.0598 |
| d-Lys-d-Trp | 333.1927 | 333.1928 | 84.0814 | 231.0796 | 129.1025 | 203.0820 |
MS/MS fragments are depicted in Fig. 5. ND, not detected.
[M + 2H]2+ was detected.
FIG 5.

Fragmentation patterns of the synthesized dipeptides. d-Trp-d-Xaa (a) and d-Lys-d-Xaa (c) could be detected as the four fragments shown in panels b and d, respectively.
Activity of BacB2-A toward d-Orn.
When d-Orn is incorporated into bacitracin, l-Orn is activated by BacB2-A and is converted into d-Orn by the downstream epimerization domain (9, 16). Therefore, BacB2-A should activate l-Orn at least, but the result of the hydroxamate-based colorimetric assay indicated that BacB2-A activated l-Lys and d-Lys instead of l-Orn and d-Orn. Thus, in an additional study, 10 mM d-Orn, 100 mM d-Pro (acyl acceptor), 20 mM ATP, 20 mM MgSO4, 50 mM Tris-HCl (pH 8.0), and 1.0 mg/ml BacB2-A were mixed and incubated at 37°C for 24 h. As a result, as shown by the results in Fig. 6a and b, the cyclized amide was detected instead of the dipeptide in both cases of the reaction, with and without nucleophiles. Therefore, it was assumed that d-Orn could be activated by BacB2-A and that an intramolecular nucleophilic reaction proceeded more promptly than the intermolecular nucleophilic attack by hydroxylamine or d-Pro (Fig. 6c). When d-Lys was used as the substrate, the expected dipeptides were successfully synthesized, but the cyclized d-Lys was detected as well (Fig. S3). Thus, the cyclization of d-Lys would be less prompt than the cyclization of d-Orn. We estimated this difference is derived from the stability of the transition state; the seven-membered cyclic amide from d-Lys may be less stable than the six-membered cyclic amide from d-Orn.
FIG 6.

Activity evaluation of BacB2-A toward d-Orn. (a and b) LC-MS chromatograms of the reaction mixture containing d-Orn without nucleophiles (a) and with d-Pro (b). (c) The results in panels a and b indicate that the reaction mechanism could produce the cyclic amide as shown.
DISCUSSION
In this paper, the combined reaction of the enzymatic adenylation of amino acids and the subsequent chemical reaction with nucleophiles was applied to provide a broad range of d-amino acid-containing dipeptides. The previous studies using this system undertook the synthesis of amide compounds (10, 11) or ll-dipeptides (19–21). Here, we extended and verified this application to d-amino acid-containing-dipeptide synthesis.
Many articles and reviews about NRPS engineering have been published to date (12, 22–28). In most of the previous studies about NRPS, substrate specificity evaluation was performed using an ATP-PPi exchange assay toward the whole module. However, the module accepts restricted substrates because of the strict substrate specificity of both the adenylation domain and the condensation domain. Recent research has tended to analyze the adenylation domain alone (10, 13, 17) but has not tested substrate specificity toward d-amino acids. Thus, there is still little information about the adenylation domain itself.
The key point in this research was to identify adenylation domains that activate d-amino acids in addition to l-amino acids. As a starting point, we proposed a hypothesis: adenylation domains located upstream from the epimerization domain activate not only l-amino acids but also d-amino acids. Generally, most adenylation domains activate only l-amino acids, not d-amino acids, because l-amino acids are natural sources and exist abundantly in bacteria. Even if d-amino acids were required as a component of a secondary metabolite, l-amino acids were activated by the adenylation domain, were converted into the d form by the epimerization domain, and were then incorporated into the structure by the condensation domain (9). Taking this into consideration, activation of d-amino acids is a misrecognition, and this is a problem for the biosynthesis of secondary metabolites. If general adenylation domains accidentally activate d-amino acids, the biosynthesis of secondary metabolites will stop because of the strict substrate specificity of the condensation domain. On the other hand, in the case of an adenylation domain in the d-amino acid-incorporating module, d-amino acid may be acceptable because the downstream condensation domain will accept the d-amino acid, and biosynthesis will proceed successfully. Thus, we assumed that the epimerization domain could be the mark of the chiral substrate specificity of the adenylation domain. Previous reports followed this hypothesis as well. For example, another adenylation domain in tyrocidine synthetase, TycB3-A from Bacillus brevis strain ATCC 8185, accepted d-Phe as well as l-Phe (18), and NpnC3 in nostophycin synthetase from cyanobacterium Nostoc sp. strain 152 activated d-allo-Ile as well as l-Ile and l-allo-Ile (29).
The results of the colorimetric assay revealed that four adenylation domains, TycA-A, BacB2-A, PbtA1-A, and PbtB2-A, obey our hypothesis and could activate d-amino acid (Fig. 1 and 3). We compared the amino acid sequences of these adenylation domains with those of other general adenylation domains, but a unique sequence for d-amino acid activation was not found. The biological meaning of this unique characteristic is still unknown, and there is room to revise this hypothesis. We believe the hypothesis could be a significant hint toward understanding the NRPS system.
Still, this system has a few problems to be solved. TycA-A has a very broad substrate specificity, a characteristic that sometimes results in by-production. Although d-Trp-d-Phe is the desired product, d-Phe-d-Trp will be produced as well, because both d-Trp and d-Phe can be accepted by TycA-A (Fig. S2). Thus, we should pay attention and choose the appropriate adenylation domain to avoid by-production. Also, as described in Results, BacB2-A was assumed to be able to activate d-Orn, and it was indicated that the intramolecular nucleophilic reaction was dominant rather than the intermolecular reaction (Fig. 6). Thus, this hybrid system cannot be applied to the synthesis of the dipeptide that has d-Orn at its N terminus. Another problem is the low yield. A previous study indicated that the reason for the low efficiency was the accumulation of PPi released by substrate activation (21). The improved hybrid system using the adenylation domain and pyrophosphatase was more effective in the synthesis of dipeptides (21). Because Escherichia coli has its own pyrophosphatase, we are sure that the whole-cell reaction was preferred to the purified enzymatic reaction. Moreover, proteases and peptidases in bacteria cannot degrade the d-amino acid-containing dipeptides; thus, the d-amino acid-containing dipeptide is expected to be produced by the whole-cell reaction as well.
This study proposed a unique system for the synthesis of various kinds of dipeptides containing d-amino acids with a single adenylation domain. Now, further improvement and application of this system is proceeding in our laboratory.
MATERIALS AND METHODS
Reagents.
All chemical reagents were purchased from Fujifilm Wako Pure Chemical (Osaka, Japan), Kanto Chemical (Tokyo, Japan), and Tokyo Chemical Industry (Tokyo, Japan). The host strains used in this study were Escherichia coli JM109 for the cloning process and Escherichia coli BL21(DE3) for protein expression. Bacillus licheniformis NBRC 12199 and Paenibacillus alvei NBRC 3343 were purchased from the Biological Resource Center, NITE (Tokyo, Japan).
Preparation of the adenylation domains.
The targeted genes coding for adenylation domains were amplified from genomic DNA by PCR using KOD Plus Neo polymerase (Toyobo, Osaka, Japan). The amplicons were digested using restriction enzymes (Nippon Gene, Tokyo, Japan) and were ligated into the digested pET-21d(+) (Novagen, Madison, WI, USA). The plasmid with the TycA-A gene inserted was constructed previously (10), and other genes were cloned in this study. Primer sequences and restriction enzymes are listed in Table 4.
TABLE 4.
Primer sequences used in this study
| Adenylation domain | Direction | Primera | Restriction site |
|---|---|---|---|
| BacB2-A | Fw | CCCCCATGGCCCTTTCAGAAGAAGAAAGACACACAG | NcoI |
| Rv | AAAGCGGCCGCGACGTCTCCGGCAGGTTC | NotI | |
| BacC4-A | Fw | CATGCCATGGCGCACATGCCGTTGAGCGAC | NcoI |
| Rv | AATCTCGAGCGGCGGTTCGGTTCGTATTCCGTCTC | XhoI | |
| PbtA1-A | Fw | CCCCCATGGCCAGCGATAAAGAGGCCCAAACG | NcoI |
| Rv | AAAGCGGCCGCTGCTTGTCCGGATGGAGC | NotI | |
| PbtB2-A | Fw | CCCCCATGGCCACACAGGAAGAACAAGAACAGATTC | NcoI |
| Rv | AAAGCGGCCGCACCGCCTTCCGGAGCCG | NotI | |
| PbtB3-A | Fw | CCCCCATGGCCACTGAAGCAGAGAAAGCGGAG | NcoI |
| Rv | AAAGCGGCCGCAAGGCTTCTCTCTGGAGCCG | NotI |
Underlined regions indicate the restriction site.
E. coli BL21(DE3) harboring each plasmid was cultivated in 300 ml Luria-Bertani (LB) medium (10 g/liter Bacto tryptone, 5 g/liter Bacto yeast extract, and 10 g/liter NaCl) containing 50 μg/ml ampicillin and 100 μM isopropyl-β-d-thiogalactopyranoside at 25°C for 18 h. The culture medium was centrifuged (4°C, 3,000 × g, 10 min), and then bacteria were resuspended in 5 ml binding buffer (the composition is described below) and sonicated with a UD-200 ultrasonic disruptor equipped with a microtip TP-040 (Tomy Seiko, Tokyo, Japan). After the sonication, the crude solution was centrifuged (4°C, 20,000 × g, 30 min) and the recombinant protein was purified using a HisTrap HP 5-ml column (GE Healthcare, Tokyo, Japan) on an ÄKTA prime plus instrument (GE Healthcare). The composition of the binding buffer was 50 mM imidazole, 20 mM Na2HPO4, and 500 mM NaCl (pH 7.0, adjusted using HCl); the composition of the elution buffer was 500 mM imidazole, 20 mM Na2HPO4, and 500 mM NaCl (pH 7.0, adjusted using HCl). After that, the eluent was desalted using a PD-10 column (GE Healthcare). The expression and purification of the adenylation domains were confirmed by SDS-PAGE analysis (Fig. S4 in the supplemental material).
Substrate specificity evaluation of the adenylation domains.
Substrate specificity evaluation was carried out according to the method of the previous study using a hydroxamate-based colorimetric assay (10). Concentrations of 5 mM d-amino acid (acyl donor), 200 mM hydroxylamine (acyl acceptor), 10 mM ATP, 10 mM MgSO4, 50 mM Tris-HCl (pH 8.0), and 1.0 mg/ml enzyme (16.5 μM TycA-A, 16.5 μM BacB2-A, 16.3 μM BacC4-A, 16.7 μM PbtA1-A, 16.4 μM PbtB2-A, or 16.7 μM PbtB3-A) were mixed and incubated at 37°C for 24 h. The volume of the reaction mixture in all experiments, including dipeptide synthesis, was 200 μl. After incubation, the reaction was quenched by 100 μl 8% trichloroacetic acid, and 100 μl 3.4% FeCl3 in 2 M HCl was added as a color reagent. The mixture was centrifuged (4°C, 20,000 × g, 10 min), and the absorbance at 490 nm of 100 μl supernatant was measured with a model 550 96-well microplate reader (Bio-Rad, Hercules, CA, USA).
Dipeptide synthesis by TycA-A and BacB2-A.
For the dl-, ld-, and dd-dipeptide synthesis evaluation, 10 mM substrates, 100 mM d-Pro or l-Pro (acyl acceptor), 20 mM ATP, 20 mM MgSO4, 50 mM Tris-HCl (pH 8.0), and 1.0 mg/ml enzyme (16.5 μM TycA-A or 16.5 μM BacB2-A) were mixed and incubated at 37°C for 24 h. The substrates were d-Ala, d-Trp, d-Tyr, d-Phe, d-Leu, d-Met, l-Ala, l-Trp, l-Tyr, l-Phe, l-Leu, or l-Met for TycA-A and d-Lys or l-Lys for BacB2-A. The mixture volume was 200 μl as well. After incubation, the reaction was quenched by heating at 80°C for 10 min, and the mixture was centrifuged (4°C, 20,000 × g, 10 min). The supernatant was stored at −20°C until HPLC and MS analysis.
For the d-Trp-d-Xaa and d-Lys-d-Xaa dipeptide synthesis, 10 mM d-Trp for TycA-A or d-Lys for BacB2-A, 10 mM d-amino acid (acyl acceptor), 20 mM ATP, 20 mM MgSO4, 50 mM Tris-HCl (pH 8.0), and 1.0 mg/ml enzyme (16.5 μM TycA-A or 16.5 μM BacB2-A) were mixed and incubated at 37°C for 24 h. The process after incubation was identical to that described for the dl-, ld-, and dd-dipeptide synthesis evaluation above.
HPLC and LC-MS analyses.
HPLC analysis was performed using a Chromaster HPLC system (Hitachi High-Technologies, Tokyo, Japan) to detect and purify the dipeptides synthesized by TycA-A. Solvent A was 0.1% (wt/vol) formic acid in water, and solvent B was 0.1% (wt/vol) formic acid in acetonitrile. The column was an XTerra MS C18 IS column (Waters, Milford, MA, USA). The pump program was the following: solvent A-solvent B were used at a gradient of 100:0 to 80:20 for 15 min, at 0:100 for 2 min, and at 100:0 for 3 min. The flow rate was 1.0 ml/min, and the absorbance at 214 nm was detected. Because l-Ala-d-Pro, d-Ala-l-Pro, and d-Ala-d-Pro produced by TycA-A could not be detected with this HPLC method, these samples were analyzed with LC-MS program 2 described below.
LC-MS analysis was performed using an Acquity ultraperformance liquid chromatography (UPLC) system (Waters) and Xevo G2-XS QTof quadrupole-time of flight MS system (Waters). Two programs were used in this study. Program 1 was used to detect the dipeptide purified using the HPLC method described above, and program 2 was used to detect the dipeptide synthesized by BacB2-A. In program 1, the column was an Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 100 mm; Waters), solvent A was 0.1% (wt/vol) formic acid in water, and solvent B was 0.1% (wt/vol) formic acid in acetonitrile. The pump program was the following: solvent A-solvent B at 95:5 for 1 min, at a gradient of 95:5 to 80:20 for 15 min, at 5:95 for 3 min, and at 95:5 for 3 min. The flow rate was 0.25 ml/min. The conditions for MS analysis were as follows: capillary, 0.5 kV; sampling cone, 20; source temperature, 120°C; desolvation temperature, 450°C; desolvation gas flow, 800 liters/h; and collision energy, 6. In program 2, the column was an Acquity BEH amide column (1.7 μm, 2.1 × 100 mm; Waters), solvent C was 10 mM ammonium acetate solution (pH 10.0, adjusted with ammonia water) in organic solution (95:5 acetonitrile/water), and solvent D was 10 mM ammonium acetate solution (pH 10.0, adjusted with ammonia water) in organic solution (40:60 acetonitrile/water). The pump program was the following: solvent C-solvent D at 80:20 for 1 min, at a gradient of 80:20 to 28:72 for 15.0 min; at 28:72 for 6 min, and at 80:20 for 6 min. The flow rate was 0.2 ml/min. The conditions of the MS analysis were as follows: capillary, 0.5 kV; sampling cone, 20; source temperature, 120°C; desolvation temperature, 300°C; desolvation gas flow, 1,000 liters/h; collision energy, 6. The data were analyzed using MassLynx (Waters).
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI grant no. 16K14495 (to K.K.), by the Institute for Fermentation, Osaka (IFO), Japan (to K.K.), and by Waseda University grants for special research projects no. 2017S-111 and 2018K-251 (to S.S.).
S.K. conceived the experiments, analyzed the data, and wrote the manuscript. S.S. supported the experiment and improved the manuscript. R.H. supervised the project. K.K. supervised the project and improved the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00120-19.
REFERENCES
- 1.Deigin VI, Semenets TN, Zamulaeva IA, Maliutina YV, Selivanova EI, Saenko AS, Semina OV. 2007. The effects of the EW dipeptide optical and chemical isomers on the CFU-S population in intact and irradiated mice. Int Immunopharmacol 7:375–382. doi: 10.1016/j.intimp.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Nishanth Kumar S, Dileep C, Mohandas C, Nambisan B, Ca J. 2014. Cyclo (D-Tyr-D-Phe): a new antibacterial, anticancer, and antioxidant cyclic dipeptide from Bacillus sp. N strain associated with a rhabditid entomopathogenic nematode. J Pept Sci 20:173–185. doi: 10.1002/psc.2594. [DOI] [PubMed] [Google Scholar]
- 3.Fdhila F, Vazquez V, Sánchez JL, Riguera R. 2003. dd-Diketopiperazines: antibiotics active against Vibrio anguillarum isolated from marine bacteria associated with cultures of Pecten maximus. J Nat Prod 66:1299–1301. doi: 10.1021/np030233e. [DOI] [PubMed] [Google Scholar]
- 4.Jamie H, Kilian G, Dyason K, Milne PJ. 2002. The effect of the isomers of cyclo (Trp-Pro) on heart and ion-channel activity. J Pharm Pharmacol 54:1659–1665. doi: 10.1211/002235702252. [DOI] [PubMed] [Google Scholar]
- 5.Arima J, Usuki H, Hatanaka T, Mori N. 2011. One-pot synthesis of diverse DL-configuration dipeptides by a Streptomyces D-stereospecific amidohydrolase. Appl Environ Microbiol 77:8209–8218. doi: 10.1128/AEM.05543-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugihara A, Shimada Y, Sugihara S, Nakai T, Kakuno T, Nagao T, Watanabe Y, Tominaga Y. 2002. A new type of aminoacyltransferase from Saccharothrix sp. AS-2 favorable for the synthesis of D-amino acid-containing peptides. J Biochem 131:247–254. doi: 10.1093/oxfordjournals.jbchem.a003095. [DOI] [PubMed] [Google Scholar]
- 7.Sato M, Masuda Y, Kirimura K, Kino K. 2007. Thermostable ATP regeneration system using polyphosphate kinase from Thermosynechococcus elongatus BP-1 for D-amino acid dipeptide synthesis. J Biosci Bioeng 103:179–184. doi: 10.1263/jbb.103.179. [DOI] [PubMed] [Google Scholar]
- 8.Sieber SA, Marahiel MA. 2005. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev 105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 9.Luo L, Kohli RM, Onishi M, Linne U, Marahiel MA, Walsh CT. 2002. Timing of epimerization and condensation reactions in nonribosomal peptide assembly lines: kinetic analysis of phenylalanine activating elongation modules of tyrocidine synthetase B. Biochemistry 41:9184–9196. doi: 10.1021/bi026047+. [DOI] [PubMed] [Google Scholar]
- 10.Hara R, Suzuki R, Kino K. 2015. Hydroxamate-based colorimetric assay to assess amide bond formation by adenylation domain of nonribosomal peptide synthetases. Anal Biochem 477:89–91. doi: 10.1016/j.ab.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Hara R, Hirai K, Suzuki S, Kino K. 2018. A chemoenzymatic process for amide bond formation by an adenylating enzyme-mediated mechanism. Sci Rep 8:2950. doi: 10.1038/s41598-018-21408-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stachelhaus T, Mootz H, Marahiel M. 1999. The specificity-conferring code of adenylation nonribosomal peptide synthetases. Chem Biol 6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 13.Kaljunen H, Schiefelbein SHH, Stummer D, Kozak S, Meijers R, Christiansen G, Rentmeister A. 2015. Structural elucidation of the bispecificity of A domains as a basis for activating non-natural amino acids. Angew Chem Int Ed 54:8833–8836. doi: 10.1002/anie.201503275. [DOI] [PubMed] [Google Scholar]
- 14.Conti E, Stachelhaus T, Marahiel MA, Brick P. 1997. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J 16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyanaga A, Cieślak J, Shinohara Y, Kudo F, Eguchi T. 2014. The crystal structure of the adenylation enzyme VinN reveals a unique β-amino acid recognition mechanism. J Biol Chem 289:31448–31457. doi: 10.1074/jbc.M114.602326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konz D, Klens A, Schörgendorfer K, Marahiel MA. 1997. The bacitracin biosynthesis operon of Bacillus licheniformis ATCC 10716: molecular characterization of three multi-modular peptide synthetases. Chem Biol 4:927–937. doi: 10.1016/S1074-5521(97)90301-X. [DOI] [PubMed] [Google Scholar]
- 17.Huang E, Guo Y, Yousef AE. 2014. Biosynthesis of the new broad-spectrum lipopeptide antibiotic paenibacterin in Paenibacillus thiaminolyticus OSY-SE. Res Microbiol 165:243–251. doi: 10.1016/j.resmic.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Mootz HD, Màrahiel MA. 1997. The tyrocidine biosynthesis operon of Bacillus brevis: Complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J Bacteriol 179:6843–6850. doi: 10.1128/jb.179.21.6843-6850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dieckmann R, Neuhof T, Pavela-Vrancic M, von Döhren H. 2001. Dipeptide synthesis by an isolated adenylate-forming domain of non-ribosomal peptide synthetases (NRPS). FEBS Lett 498:42–45. doi: 10.1016/S0014-5793(01)02471-1. [DOI] [PubMed] [Google Scholar]
- 20.Abe T, Hashimoto Y, Sugimoto S, Kobayashi K, Kumano T, Kobayashi M. 2017. Amide compound synthesis by adenylation domain of bacillibactin synthetase. J Antibiot (Tokyo) 70:435–442. doi: 10.1038/ja.2016.117. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki S, Hara R, Kino K. 2018. Production of aminoacyl prolines using the adenylation domain of nonribosomal peptide synthetase with class III polyphosphate kinase 2-mediated ATP regeneration. J Biosci Bioeng 125:644–648. doi: 10.1016/j.jbiosc.2017.12.023. [DOI] [PubMed] [Google Scholar]
- 22.Eppelmann K, Stachelhaus T, Marahiel MA. 2002. Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry 41:9718–9726. doi: 10.1021/bi0259406. [DOI] [PubMed] [Google Scholar]
- 23.Kries H, Niquille DL, Hilvert D. 2015. A subdomain swap strategy for reengineering nonribosomal peptides. Chem Biol 22:640–648. doi: 10.1016/j.chembiol.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Kries H, Wachtel R, Pabst A, Wanner B, Niquille D, Hilvert D. 2014. Reprogramming nonribosomal peptide synthetases for “clickable” amino acids. Angew Chem Int Ed 53:10105–10108. doi: 10.1002/anie.201405281. [DOI] [PubMed] [Google Scholar]
- 25.Villiers B, Hollfelder F. 2011. Directed evolution of a gatekeeper domain in nonribosomal peptide synthesis. Chem Biol 18:1290–1299. doi: 10.1016/j.chembiol.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 26.Kries H. 2016. Biosynthetic engineering of nonribosomal. J Pept Sci 22:564–570. doi: 10.1002/psc.2907. [DOI] [PubMed] [Google Scholar]
- 27.Winn M, Fyans JK, Zhuo Y, Micklefield J. 2016. Recent advances in engineering nonribosomal peptide assembly lines. Nat Prod Rep 33:317–347. doi: 10.1039/c5np00099h. [DOI] [PubMed] [Google Scholar]
- 28.Roderich DS, Mainz A. 2017. Nonribosomal peptide synthesis—principles and prospects. Angew Chem Int Ed 56:3770–3821. doi: 10.1002/anie.201609079. [DOI] [PubMed] [Google Scholar]
- 29.Fewer DP, Julia O, Rouhiainen L, Jokela J, Wahlsten M, Sivonen K. 2011. Nostophycin biosynthesis is directed by a hybrid polyketide synthase-nonribosomal peptide synthetase in the toxic cyanobacterium Nostoc sp. strain 152. Appl Environ Microbiol 77:8034–8040. doi: 10.1128/AEM.05993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.