

Summary
Alternative 3′untranslated regions (3′UTRs) are widespread but their functional roles are largely unknown. We investigated the function of the long BIRC3 3′UTR which is upregulated in leukemia. The 3′UTR does not regulate BIRC3 protein localization or abundance, but is required for CXCR4-mediated B cell migration. We established an experimental pipeline to study the mechanism of regulation and used mass spectrometry to identify BIRC3 protein interactors. In addition to 3′UTR-independent interactors involved in known BIRC3 functions, we detected interactors that bind only to BIRC3 protein encoded from the mRNA with the long 3′UTR. They regulate several functions, including CXCR4 trafficking. We further identified RNA-binding proteins differentially bound to the alternative 3′UTRs and found that cooperative binding of Staufen and HuR mediates 3′UTR-dependent complex formation. We show that the long 3′UTR is required for the formation of specific protein complexes that enable additional functions of BIRC3 protein beyond its 3′UTR-independent functions.
Graphical Abstract
eTOC blurb:
Lee & Mayr show that the E3 ligase BIRC3 has several different functions that are exclusively accomplished by BIRC3 protein encoded from the mRNA transcript containing the long but not the short 3′UTR. 3′UTR-dependent protein functions are caused by BIRC3 protein complexes that require the long 3′UTR for their formation.
Introduction
Over half of human genes use alternative cleavage and polyadenylation (APA) to generate mRNA transcripts with alternative 3′ untranslated regions (3′UTRs) (Lianoglou et al., 2013). APA is a regulated process as the expression ratios of alternative 3′UTR isoforms change in a coordinated manner during many biological processes, including differentiation, immune cell activation or cancer (Sandberg et al., 2008; Mayr and Bartel, 2009; Gruber et al., 2014; Brumbaugh et al., 2018). Initially, when alternative 3′UTRs were discovered to be widespread, it was thought that their major role is the regulation of protein abundance (Sandberg et al., 2008; Mayr and Bartel, 2009). Indeed, many genes that encode short-lived mRNAs, including cytokines, cell cycle regulators or oncogenes use their 3′UTRs to regulate protein abundance (Mayr, 2018). However, several genome-wide studies have observed that generally less than 20% of significant 3′UTR isoform changes are associated with changes in their corresponding mRNA or protein expression levels (Lianoglou et al., 2013; Spies et al., 2013; Gruber et al., 2014; Brumbaugh et al., 2018). Instead, when changes in APA were assessed during diverse biological processes, it was repeatedly observed that genes that changed their mRNA abundance levels largely did not change their 3′UTR isoform expression and vice versa (Lianoglou et al., 2013; Zhang et al., 2016; Jia et al., 2017). Therefore, it is still mostly unclear, how the changes in alternative 3′UTR isoform ratios contribute to biology (Mayr, 2017).
One way to use alternative 3′UTRs for the regulation of biological processes is through 3′UTR-mediated protein-protein interactions (Berkovits and Mayr, 2015; Ma and Mayr, 2018). It was shown that the long 3′UTR of CD47 increases CD47 plasma membrane localization, thus protecting cells better from phagocytosis by macrophages (Berkovits and Mayr, 2015). The increase in plasma membrane trafficking was caused by the binding of the adaptor SET to CD47. SET transfer from the 3′UTR to the newly synthesized protein occurs in a membraneless organelle that is associated with the endoplasmic reticulum (Ma and Mayr, 2018).
Here, we set out to establish an experimental pipeline to identify 3′UTR-dependent protein interactors and to study the functions of long 3′UTRs. We used BIRC3 as candidate as its long 3′UTR isoform was significantly upregulated in malignant B cells derived from chronic lymphocytic leukemia (CLL). BIRC3 encodes an E3 protein ubiquitin ligase that does not contain a transmembrane domain. It is known to regulate cell death and immune functions through negative regulation of the NF-κB pathway (Beug et al., 2012).
Despite upregulation of the long BIRC3 3′UTR in CLL, we observed that overall BIRC3 mRNA and BIRC3 protein levels were similar between normal and malignant B cells. To study the function of the long 3′UTR, we identified 3′UTR-dependent protein interactors of BIRC3. We call BIRC3 protein encoded from the long BIRC3 3′UTR isoform, BIRC3-LU, and BIRC3 protein encoded from the short 3′UTR isoform, BIRC3-SU. Both BIRC3-SU and BIRC3-LU can accomplish 3′UTR-independent functions, such as the cell-intrinsic control of cell death. However, only BIRC3-LU has the ability to regulate CXCR4-mediated B cell migration; an event that is crucial for CLL cell survival by enabling the homing of B cells to protective bone marrow niches (Burger et al., 2005). BIRC3-LU accomplishes this function through 3′UTR-mediated protein complex assembly involving IQGAP1 and RALA. In addition to regulating protein trafficking, BIRC3-LU binds to numerous other 3′UTR-dependent interactors allowing it to perform additional roles. Cooperative binding of RNA-binding proteins throughout the long 3′UTR enables the unique functions of the long 3′UTR by recruiting specific factors that participate in the formation of 3′UTR-dependent protein complexes. This allows BIRC3 protein to achieve several 3′UTR-dependent functions in addition to its 3′UTR-independent functions.
Results
Similar BIRC3 mRNA level in normal and malignant B cells, but upregulation of the long 3′UTR isoform in CLL
Our goal was to study regulatory functions of 3′UTRs that are independent of protein abundance regulation. We analyzed 3′UTR isoform expression between normal CD5+ B cells from healthy individuals (NB, N = 4) and CLL patients (N = 13) (Lee et al., 2018). We observed shortening and lengthening of 3′UTRs, but 66% of mRNA isoforms (412/627) expressed higher levels of longer 3′UTRs in leukemia (Figure S1A, Table S1). BIRC3 was our top candidate for follow-up experiments as expression of the long 3′UTR was significantly upregulated in all tested CLL samples (Figures 1A, 1B, S1B), but overall BIRC3 mRNA and protein levels were similar between normal and malignant B cells (Figures 1C, 1D, S1C, S1D).
Figure 1. The alternative BIRC3 3′UTRs do not regulate BIRC3 protein abundance or localization.

(A) The short and long BIRC3 3′UTRs are shown as 3′-seq data in transcripts per million (TPM) for normal CD5+ B cells (NB) and CLL B cells. The terminal exons are shown in blue. SU, short 3′UTR, LU, long 3′UTR.
(B) Fraction of long BIRC3 3′UTR isoform in normal (N = 4) and CLL B (N = 10) cells as mean ± SD, Mann Whitney test, **, p = 0.003.
(C) Overall BIRC3 mRNA abundance of samples from (B). Mann Whitney test, p = NS (not significant).
(D) BIRC3 protein level quantification from Figure S1D. Mann Whitney test, p = NS.
(E) Constructs: GFP fusion with BIRC3 coding region, followed by either the short (SU) or long (LU) BIRC3 3′UTR.
(F) Single molecule RNA-FISH against GFP after transfection of constructs from (E) in HeLa cells. The dotted line demarcates the nucleus. Representative cells are presented.
(G) Live cell fluorescence microscopy shows GFP-BIRC3 protein as in (F).
Subcellular mRNA and protein localization of GFP-BIRC3-SU and GFP-BIRC3-LU is similar
To test if the alternative 3′UTR isoforms may influence mRNA or protein localization, we generated GFP-BIRC3 fusion constructs either containing the short (631 nucleotides) or long (2,299 nucleotides) BIRC3 3′UTRs (Figure 1E). Single molecule RNA-FISH on the transfected constructs did not reveal a difference in mRNA localization caused by the alternative 3′UTRs (Figure 1F). Also, the alternative BIRC3 3′UTRs did not influence BIRC3 protein localization (Figure 1G).
Knock-down of the long BIRC3 3′UTR isoform to study the function of BIRC3-LU in B cells
We next set out to find a phenotype that is caused by BIRC3-LU, but not by BIRC3-SU, and worked with the malignant B cell line Raji as CLL cells cannot be cultivated. Using CRISPR-Cas9, we generated Raji cells that lack BIRC3 protein (BIRC3 knock-out (KO); Figures 2A, S2A). Parental Raji (wild-type, WT) and control (ctrl) KO cells that underwent the KO procedure, but still express BIRC3 protein served as controls (Figure S2B). To study the functions of BIRC3-LU which is BIRC3 protein encoded by the long BIRC3 3′UTR isoform, we stably expressed shRNAs that target exclusively the long 3′UTR (LU knock-down (KD), Figure 2A). Northern blots were used to validate the extent of KD (Figure S2C). LU KD cells are expected to only express BIRC3-SU, whereas Raji cells stably expressing a control shRNA (ctrl KD) are expected to express both BIRC3-SU and BIRC3-LU proteins (Figure 2A).
Figure 2. 3′UTR-independent and 3′UTR-dependent functions of BIRC3.

(A) Experimental systems for phenotypic analysis. Raji cells (N = 2) with BIRC3 protein knock-out (KO). Controls: parental Raji cells (WT) and Raji clone after CRISPR procedure that retains BIRC3 protein (ctrl KO). Raji cells with stable expression of shRNAs against the long BIRC3 3′UTR isoform (LU KD, N = 2) are compared to cells expressing a control shRNA (ctrl KD). Arrow indicates transcriptional start and pA indicates polyadenylation signal.
(B) Viability at day 2 after Fludarabine treatment for biological replicates of ctrl Raji (N = 4), LU KD (N = 4), and BIRC3 KO cells (N = 2). T-test for independent samples, *, p < 0.039.
(C) B cell migration after 3.5 hours in media with 50 ng/ml CXCL12 as mean ± SD from biological replicates. Mann Whitney test, *, p = 0.02.
(D) FACS analysis of endogenous surface CXCR4 expression in ctrl and KO cells as mean fluorescence intensity (MFI) ± SD of biological replicates. Mann Whitney test, *, p = 0.03. Representative example in Figure S2J.
(E) As in (D), but for ctrl and LU KD cells. Mann Whitney test, **, p = E-5. Representative example in Figure S2K.
(F) FACS analysis of endogenous surface CXCR4 levels in KO3 cells after expression of constructs from Figure 1E. GFP expression in left panel and mean MFI of surface CXCR4 ± SD from biological replicates in right panel. Mann Whitney test, **, p = 0.003. Representative example in Figure S2L.
(G) FACS analysis of endogenous surface CXCR4 levels from Figure S2M. Shown is mean MFI ± SD from biological replicates. Mann Whitney test, *, p = 0.03.
(H) CXCR4 mRNA level of NB (N = 4) and CLL B (N = 10) as mean 3′-seq TPM ± SD. Mann Whitney test, p = NS.
(I) FACS analysis of endogenous surface CXCR4 shown as MFI ± SD for normal (N = 2) and CLL B (N = 8) cells. Mann Whitney test, *, p = 0.037.
(J) 3′UTR-dependent trafficking regulation by BIRC3 is important for CLL biology. Similar mRNA level of BIRC3 and CXCR4, but increased expression of the long BIRC3 3′UTR in CLL which correlates with increased surface CXCR4 protein, allowing CLL cell migration towards the CXCL12 cytokine expressed in bone marrow niches.
BIRC3 protein regulates cell death in a 3′UTR-independent manner, but it regulates B cell migration in a 3′UTR-dependent manner
BIRC3 is known as anti-apoptotic protein (Beug et al., 2012). We tested B cell survival in our experimental system after treatment with Fludarabine. As expected, BIRC3 KO cells had fewer surviving cells than ctrl Raji cells (Figure 2B), but loss of BIRC3-LU did not significantly influence B cell survival (Figure 2B). This confirms that BIRC3 has anti-apoptotic activity and this function of BIRC3 protein is 3′UTR-independent.
To identify BIRC3 functions that are specific for BIRC3-LU, we performed additional phenotypic assays. BIRC3 belongs to the family of IAP (inhibitor of apoptosis) proteins and homologues from flies were implicated in the regulation of migration (Geisbrecht and Montell, 2004). This prompted us to test if B cell migration is regulated by mammalian BIRC3. Compared with ctrl cells, the migratory capacity of BIRC3 KO cells was severely impaired (Figure 2C). Strikingly, migration was also impaired in LU KD cells (Figure 2C). As LU KD cells still express BIRC3-SU (Figure S2B), this experiment showed that only BIRC3-LU, but not BIRC3-SU, is required for B cell migration. Taken together, our data indicate that BIRC3 protein has 3′UTR-independent functions, including the control of cell death (Figure 2B), but it also has 3′UTR-dependent functions, such as the regulation of B cell migration (Figure 2C).
BIRC3-LU regulates surface expression of CXCR4
We next asked how BIRC3-LU regulates B cell migration. The migration assay was performed in the presence of the cytokine CXCL12. CXCR4, the receptor for CXCL12, is expressed on normal and malignant B cells, and is known as an important survival factor in CLL as it allows CLL cells to migrate to protective bone marrow niches (Burger et al., 2005).
To elucidate how BIRC3-LU regulates CXCR4 expression, we investigated if BIRC3 regulates CXCR4 mRNA levels. Endogenous CXCR4 mRNA and protein levels were not affected in Raji cells lacking BIRC3 or lacking BIRC3-LU (Figures S2D-I). Instead, compared with ctrl B cells, we observed significantly lower surface expression of CXCR4 in B cells lacking BIRC3 or lacking BIRC3-LU (2D, 2E, S2J, S2K).
These results suggested that BIRC3-LU, but not BIRC3-SU, regulates surface CXCR4 expression. To corroborate this finding, we performed a rescue experiment. We used the BIRC3 KO3 cell line and re-expressed GFP-tagged BIRC3-SU, GFP-BIRC3-LU, or GFP alone. Despite similar protein expression of BIRC3-SU and BIRC3-LU, only the presence of GFP-BIRC3-LU was able to significantly increase surface expression of endogenous CXCR4 (Figures 2F, S2L).
Regulation of surface CXCR4 expression has been widely studied. Binding of the ligand CXCL12 to CXCR4 results in receptor internalization and partial re-expression at the plasma membrane to allow receptor re-sensitization (Grundler et al., 2009; Marchese, 2016). We performed a recycling assay to test if BIRC3-LU regulates CXCR4 receptor trafficking (Figure S2M). Absence of BIRC3-LU did not influence internalization of CXCR4, but it significantly reduced re-expression at the surface after ligand binding (Figure 2G). This suggests that BIRC3-LU regulates the recycling of CXCR4 back to the plasma membrane after receptor activation.
Despite similar CXCR4 mRNA levels, surface CXCR4 expression is increased in CLL cells compared to normal B cells
The relationship between BIRC3-LU and CXCR4 was also true in primary cancer cells derived from patients with CLL. Despite similar CXCR4 mRNA levels in normal and CLL B cells (Figure 2H), we observed increased surface CXCR4 expression in CLL (Figure 2I). The BIRC3 gene is located on chromosome 11q, a region that is heterozygously deleted in 15-20% of CLL patients (Rose-Zerilli et al., 2014). Therefore, currently, BIRC3 is regarded as a tumor-suppressor gene in CLL. In our patient cohort, overall BIRC3 mRNA expression was reduced in samples with 11q deletions, but they still had a relative increase in expression of the long BIRC3 3′UTR isoform (Figures S1E, S1F) (Lee et al., 2018). Importantly, even in CLL samples with 11q deletions, surface CXCR4 levels were upregulated and were expressed at comparable levels as in the remaining CLL samples (Figures S1G, S1H). This shows that in primary CLL cells, increased levels of BIRC3-LU correlate with upregulated surface CXCR4 expression. As BIRC3-LU is responsible for surface CXCR4 expression in malignant B cells (Fig. 2F), our results indicate that BIRC3-LU has a cancer-promoting role in CLL (Figure 2J).
Identification of 3′UTR-independent and 3′UTR-dependent interaction partners of BIRC3 protein
As the long BIRC3 3′UTR did not regulate BIRC3 protein abundance or localization, we hypothesized that the 3′UTR may facilitate formation of alternative protein complexes. Through participation in different protein complexes, BIRC3-SU and BIRC3-LU could accomplish different protein functions. To test this hypothesis, we determined the protein interaction partners of BIRC3 using quantitative mass spectrometry (MS; Figure 3A). We expressed GFP-BIRC3-SU and GFP-BIRC3-LU in cells grown in light and heavy SILAC media, respectively. This was followed by co-immunoprecipitation (co-IP) of GFP and analysis of the proteins bound to either GFP-BIRC3-SU or GFP-BIRC3-LU protein. We calculated the log2-based enrichment ratio of each protein interaction partner obtained from the heavy versus the light fraction and ranked them (Figure S3A, Table S2). We defined 3′UTR-independent interactors as proteins that bind equally well to BIRC3-SU and BIRC3-LU. They have LU/SU log2-enrichment ratios of approximately zero (N = 520, Figures 3B, grey, S3A). In contrast, interaction partners with higher LU/SU ratios are regarded as long 3′UTR-dependent or BIRC3-LU-enriched interactors (N = 394, Figures 3B, dark green, S3A).
Figure 3. Identification and validation of 3′UTR-independent and 3′UTR-dependent BIRC3 protein interactors.

(A) Experimental set-up: HEK293T cells are grown in heavy and light SILAC media, transfected with GFP-BIRC3-LU and GFP-BIRC3-SU, pooled, immunoprecipitated using GFP-trap, and co-IPed proteins are analyzed by MS.
(B) Interactors are classified as 3′UTR-independent (shared by BIRC3-SU and BIRC3-LU; log2 LU/SU ratio <0.585; grey) and long 3′UTR-dependent (BIRC3-LU-enriched; log2 LU/SU ratio >0.585). See also Table S2.
(C) Gene ontology enrichment score from interactors identified in (B). Red dotted line is cut-off for enrichment.
(D) Western blot validation of endogenous BIRC3 interactors. 3′UTR-independent (grey bar) and 3′UTR-dependent (green bar) BIRC3 protein interactors identified by GFP co-IP in HEK293T cells after transfection of constructs from Figure 1E. 1% of input was loaded.
BIRC3-LU-dependent interactors are involved in mitochondrial biology, chromatin regulation, and protein trafficking
Gene ontology analysis revealed that the 3′UTR-independent BIRC3 interactors were significantly enriched in factors that participate in ubiquitin-dependent regulation of NF-κB (Figures 3C, S3B). This is consistent with the known function of BIRC3 as E3 ubiquitin ligase involved in NF-κB pathway regulation (Beug et al., 2012). The BIRC3-LU-enriched interactors clustered into three major functional categories and localize to mitochondria, are chromatin regulators, or are involved in protein trafficking. These functions were largely not found among the 3′UTR-independent interactors (Figures 3C, S3B).
We then validated the candidates using GFP co-IP followed by western blot analysis. We were able to validate 8/10 candidates which indicates a high validation rate (80%). Among the validated BIRC3 interactors were two known interaction partners (DIABLO, XIAP) as well as six previously unknown interactors (Figures 3D, S3C). 5/6 of the previously unknown interactors were BIRC3-LU-enriched and included the chromatin regulator SMARCA4, the mitochondria-localized chaperone HSPA9 and the mitochondrial inner membrane translocase TIMM44. They also consisted of factors involved in protein trafficking, including the IQ motif containing GTPase activating protein 1 (IQGAP1) and the Ras GTPase RALA (Figure 3D). Taken together, the validated interactors of BIRC3 confirm the different functional gene classes obtained by gene ontology analysis.
We were surprised to find a large number of interactors. However, BIRC3 protein has six protein interaction domains (three BIR domains, a UBA, CARD, and RING domain (Figure S3D) (Beug et al., 2012). This may give BIRC3 ample opportunity to bind proteins in a 3′UTR-independent or 3′UTR-dependent manner.
Mechanism of 3′UTR-dependent interaction between BIRC3-LU, IQGAP1, and RALA
As our goal was to elucidate the molecular mechanism of CXCR4 trafficking regulation by BIRC3-LU, we focused on the interaction between known trafficking factors with BIRC3-LU. IQGAP1 was previously shown to regulate CXCR4 recycling in T cells. It is a large protein that provides a scaffold for several signaling pathways but also acts as cytoskeleton-binding protein with important roles in migration (Bamidele et al., 2015). The Ras GTPase RALA plays crucial roles in the regulation of actin dynamics, migration, and membrane protein trafficking (Neyraud et al., 2012).
Our MS validation results indicated that the long 3′UTR of BIRC3 is required for the protein-protein interaction between BIRC3 and IQGAP1 or RALA. There are two possible models for 3′UTR-dependent protein complex formation (Figure 4A). In model 1, BIRC3 acts as an RNA-binding protein that only binds to the long, but not the short BIRC3 3′UTR. 3′UTR-bound BIRC3 interacts in an RNA-dependent manner either directly with IQGAP1 and RALA or indirectly through other RNA-binding proteins.
Figure 4. The long BIRC3 3′UTR of is required for establishment but not maintenance of 3′UTR-dependent BIRC3 protein complexes.

(A) Models for 3′UTR-dependent protein complex formation. See text for details.
(B) Experimental set-up for RNA affinity pull-down to identify RNA-binding proteins that bind to the alternative 3′UTRs of BIRC3. Biotinylated uridines (red dots) are incorporated during in vitro transcription of the 3′UTRs. After incubation with Raji cell lysate, the RNAs together with bound RNA-binding proteins are pulled-down using streptavidin beads. Visualization of endogenously expressed RNA-binding proteins by western blot.
(C) RNA affinity pull-down described in (B). Western blot of endogenous BIRC3 and HuR after pull-down of the short BIRC3 3′UTR (SU), the long BIRC3 3′UTR (LU), the antisense (as) sequence of LU, and of beads only. 2% of input was loaded. The same number of RNA molecules was incubated with lysate. HuR serves as positive control (see Figure 5A).
(D) As in Figure 3D. GFP co-IP was performed in the presence or absence of RNase A.
To test if BIRC3 is an RNA-binding protein, we performed RNA affinity pull-down experiments. We in vitro transcribed and biotinylated the short (SU) and long (LU) BIRC3 3′UTRs and incubated them with cell lysate prepared from Raji cells that endogenously express BIRC3 protein. Using streptavidin beads, we pulled-down proteins bound to the 3′UTRs and found that BIRC3 protein is not an RNA-binding protein as it did not interact with the 3′UTRs (Figures 4B, 4C).
Model 1 predicts that the protein-protein interaction between BIRC3 and IQGAP1/RALA is RNA-dependent and would still occur when a BIRC3 construct lacking the coding region is being used for co-IP as the protein complex assembles on the long BIRC3 3′UTR. Whereas we disproved the predictions of model 1 (Figures 4D, S4A, S4B), all observations are consistent with model 2, where the long BIRC3 3′UTR recruits IQGAP1 and RALA to the site of BIRC3 protein translation (Figure 4A). We showed that only the long but not the short BIRC3 3′UTR is able to assemble the BIRC3/IQGAP1/RALA complex, indicating that the long 3′UTR is required for the establishment of the interaction (Figures S4C, S4D). However, the long 3′UTR was not required for maintaining the complex after its formation, as the addition of RNase A during co-IP did not disassemble previously formed complexes (Figure 4D). As only the long and not the short 3′UTR recruits the interactors, only the protein encoded from the long transcript can establish the 3′UTR-dependent complex.
Differential binding of RNA-binding proteins to the alternative 3′UTRs of BIRC3
According to our current model (Figure 4A, model 2), IQGAP1 and RALA are recruited by the long BIRC3 3′UTR. As the recruited proteins lack RNA-binding domains, we expect them to associate with the 3′UTR through RNA-binding proteins. We set out to identify the RNA-binding proteins that are responsible for the recruitment of IQGAP1 and RALA to BIRC3-LU. We used the in vitro transcribed biotinylated 3′UTRs from above (Figure 4B), followed by pull-down of the bound RNA-binding proteins and western blot analysis as read-out. To evaluate binding specificity, we used the antisense sequences of each 3′UTR.
The RNA-binding protein candidates for testing were obtained from the MS experiment (Figure S3A, Table S2). We had found a large number of RNA-binding proteins as potential interactors of BIRC3 in the MS experiment, but we were not able to validate them by western blot analysis as true BIRC3 protein interactors (Figure S5A). This suggests that RNA-binding proteins are not being transferred from the 3′UTR to the newly made protein, but that they are part of the mRNP complex during translation of BIRC3 protein. We chose RNA-binding proteins that were enriched in the BIRC3-LU sample (Figure S5B) and tested if they interacted better with the long than the short BIRC3 3′UTR, when equal numbers of RNA molecules were used for RNA affinity pull-down (Figures 5A, S5C). Out of 14 tested RNA-binding proteins, six were involved in splicing and mRNA processing. Five of them did not bind to the BIRC3 3′UTRs and one of them bound rather unspecifically (Figures 5A, see legend, S5C). From the remaining eight RNA-binding proteins, FUS did not bind, but seven of them bound better to the long than the short BIRC3 3′UTR (Figure 5A).
Figure 5. The RNA-binding proteins STAU1 and HuR recruit the 3′UTR-dependent protein interactors IQGAP1 and RALA.

(A) As in Figure 4C, but performed in HEK293T cells. LU1, fragment of the long BIRC3 3′UTR (see (G)). ARE is a repeated AU-rich element that serves as positive control for HuR. RNA-binding proteins involved in mRNA processing are SF3B1, KHSRP, NUDT21, hnRNPC, DDX17, and snRNP70 (shown in Figure S5C).
(B) As in (A), but indicated amounts of biotinylated RNAs were incubated with lysate.
(C) As in Figure 4D, but after stable expression of ctrl shRNAs or shRNAs against STAU1 (STAU-1, STAU-3).
(D) As in (C), but after stable expression of shRNAs against HuR (HuR-2, HuR-3).
(E) Model how different RNA-binding proteins recruit different 3′UTR-dependent interactors. IQGAP1 and RALA are recruited by STAU1 (S) and HuR (H) and not by IGF2BP1 (I). The RNA-binding proteins that recruit HSPA9, TIMM44, and SMARCA4 are unknown (indicated by ‘?’).
(F) In vivo reconstitution of IQGAP1/RALA recruitment to BIRC3 protein by STAU1 and HuR tethered to the short 3′UTR. See text and methods for details. GFP co-IP of IQGAP1 and RALA after transfection of GFP-BIRC3-SU (SU), GFP-BIRC3-LU (LU), or GFP-BIRC3-MS2-SU (MS2-SU) into HEK293T cells. MCP-mC-STAU1 and MCP-mC-HuR were cotransfected in the indicated lanes (see Figure S5H). They recruit endogenously expressed IQGAP1 and RALA. 1 % of input was loaded.
(G) AU-rich elements (AREs, red stars) located in the alternative BIRC3 3′UTRs. LU1 is a 647 nucleotide fragment derived from the long BIRC3 3′UTR that contains five AREs.
(H) The ratio of the RNA-binding proteins STAU1 and HuR bound to the alternative BIRC3 3′UTRs is important for the BIRC3-LU-specific recruitment of IQGAP1 and RALA. STAU1 binds to double-stranded RNA. The model is based on the data shown in (B).
As LU is approximately three-times longer than SU, LU is expected to contain three-times more biotinylated nucleotides than SU (Figure 4B). To control for the number of biotins in the RNAs used for RNA affinity pull-down, we repeated the experiment and added three- or six-fold more SU than LU to the cell lysates. Higher amounts of SU increased the binding of several RNA-binding proteins to SU. However, even after normalization to the number of biotinylated nucleotides, HuR, IGF2BP1, and hnRNPA1 still bound better to LU than to SU (Figures 5B, S5C).
Staufen and HuR are necessary for the recruitment of RALA and IQGAP1 to BIRC3-LU
In order to identify the RNA-binding proteins that are responsible for the recruitment of IQGAP1 and RALA, we used shRNAs to knock-down specific RNA-binding proteins. We chose the double-stranded RNA-binding protein Staufen homolog 1 (STAU1) as it is a known interactor of IQGAP1 (Villace et al., 2004). The interaction between STAU1 and IQGAP1 was confirmed by us using MS (Table S3). We stably knocked-down STAU1 (Figure S5D) and repeated the GFP co-IP after expression of GFP-BIRC3-SU or GFP-BIRC3-LU. This allowed us to examine if lack of STAU1 influences 3′UTR-dependent protein complex formation between BIRC3-LU and IQGAP1/RALA. KD of STAU1 did not change protein levels of IQGAP1, RALA or BIRC3 (Figure 5C, input panel). However, it abrogated the 3′UTR-mediated binding of IQGAP1 and RALA to BIRC3-LU, indicating that STAU1 is necessary for the recruitment of both BIRC3-LU-specific interactors.
We next tested if the RNA-binding protein HuR was necessary for IQGAP1/RALA recruitment, as HuR bound better to LU than to SU (Figure 5B). KD of HuR did not affect protein expression levels of STAU1, IQGAP1, RALA or BIRC3 (Figures 5D, input panel, S5E). However, it prevented the 3′UTR-mediated binding of IQGAP1 and RALA to BIRC3-LU (Figure 5D). In contrast, KD of IGF2BP1, another RNA-binding protein that bound better to LU than to SU did not affect the binding of several 3′UTR-dependent interactors (Figures S5F, S5G). Taken together, these experiments revealed that not all RNA-binding proteins that interact with the long 3′UTR are required for the recruitment of specific BIRC3 interaction partners. These data suggest that different RNA-binding proteins may recruit different groups of interactors. Whereas STAU1 and HuR recruit IQGAP1 and RALA, IGF2BP1 is likely to recruit other interactors of BIRC3-LU (Figure 5E).
We then asked if STAU1 and HuR are sufficient for the recruitment of IQGAP1 and RALA to BIRC3 protein. We used the MS2 system to tether STAU1 and HuR to BIRC3-SU (Figure 5F). MS2-binding sites (MS2) form RNA hairpins that specifically bind the MS2 coat protein (MCP) (Berkovits and Mayr, 2015). We added MS2 to GFP-BIRC3-SU to obtain GFP-BIRC3-MS2-SU.
We further fused either STAU1 or HuR to an mCherry (mC)-tagged MCP (MCP-mC-STAU1, MCP-mC-HuR). Co-expression of all three constructs tethers STAU1 and HuR to the short 3′UTR of BIRC3 mRNA. We performed GFP co-IP and observed 3′UTR-dependent recruitment of endogenous IQGAP1 and RALA to GFP-BIRC3-MS2-SU in the presence of STAU1 and HuR (Figures 5F, S5H).
These experiments demonstrated that in the context of a cell lysate the RNA-binding proteins STAU1 and HuR are necessary and sufficient for the 3′UTR-mediated binding of IQGAP1 and RALA to BIRC3. Our results further indicate that mere overexpression of STAU1 and HuR without their tethering to the 3′UTR is not sufficient for 3′UTR-dependent protein complex assembly as binding of IQGAP1/RALA was not observed in the SU sample or in the MCP sample (Figure 5F). This demonstrates that 3′UTR tethering of the RNA-binding proteins to the site of translation is required for 3′UTR-dependent protein complex formation.
The stoichiometry of 3′UTR-bound HuR and STAU1 is important for the recruitment of IQGAP1 and RALA
We then tried to identify the binding sites of STAU1 and HuR in the long BIRC3 3′UTR that are responsible for the recruitment of IQGAP1 and RALA. STAU1 does not bind to specific motifs, but is known to bind to double-stranded RNA (Sugimoto et al., 2015). In our RNA affinity pull-down assay STAU1 bound to both BIRC3 3′UTRs (Figure 5B), whereas HuR bound better to LU than to SU (Figures 5A, 5B, S5C). The sequence of LU that is not present in SU contains seven AREs (Figure 5G), which are known binding sites of HuR (Mayr, 2018; Ma and Mayr, 2018). As 5/7 LU-unique AREs clustered within a fragment of LU, called LU1, we tested if this fragment is able to recapitulate the HuR binding pattern observed with full-length LU. This experiment showed that despite the presence of five AREs, HuR did not bind to LU1 (Figures 5A, 5B). In contrast, the five AREs present in SU bind to HuR in a concentration-dependent manner (Figures 5A, 5B). These observations indicate that STAU1 and HuR bind to both LU and SU, especially if SU is present at high concentrations.
Importantly, we observed a quantitative difference in HuR binding: HuR binds better than STAU1 to the full-length long 3′UTR than to SU or LU1 alone, indicated by an increased ratio in the western blot signal for HuR compared with STAU1 in the LU lanes of Figures 5B and S5C. This result suggests that HuR binds to several sites located throughout the entire long 3′UTR (including the sequence of SU) to achieve proper HuR binding. Despite binding of both STAU1 and HuR to the short 3′UTR, our results from above showed that the short 3′UTR was unable to mediate recruitment of the trafficking factors IQGAP1 and RALA. This suggests that the ratio of bound HuR to STAU1 is important for the BIRC3-LU-specific protein trafficking function (Figure 5H).
Remarkably, our results indicate that in order to achieve a specific function of the long BIRC3 3′UTR, exclusive binding of RNA-binding proteins to LU is not required. Instead, through cooperative binding to sites located throughout the entire long 3′UTR a proper stoichiometry of the RNA-binding proteins HuR and STAU1 is achieved which is necessary for the recruitment of IQGAP1 and RALA.
BIRC3-LU recruits the protein complex consisting of IQGAP1 and RALA to CXCR4 receptor
As our goal was to investigate how BIRC3-LU regulates CXCR4 recycling, we examined if BIRC3 interacts with CXCR4 receptor. We transfected into HEK293T cells CXCR4 tagged with myc at the C-terminus (CXCR4-myc) and either GFP-BIRC3-SU, GFP-BIRC3-LU or GFP-BIRC3-NU (without 3′UTR, but with a polyadenylation signal) (Figure 6A). Co-IP of CXCR4-myc showed interaction between CXCR4 and BIRC3 protein translated from any of the three constructs, indicating that BIRC3 protein interacts with CXCR4 in a 3′UTR-independent manner (Figure 6B). However, importantly, the interaction of endogenously expressed IQGAP1 and RALA with CXCR4 required the presence of BIRC3-LU (Figures 6B, S6A). This suggested that the protein complex consisting of BIRC3-LU, IQGAP1 and RALA was recruited to CXCR4 upon BIRC3-LU binding (Figure 6C). As BIRC3-SU was not able to bind to IQGAP1 and RALA (Figures 3D, 4D, 5C-F), BIRC3-SU was incapable to deliver IQGAP1 and RALA to CXCR4, when it interacted with CXCR4 (Figures 6B, 6C).
Figure 6. IQGAP1 and RALA are recruited to CXCR4 in a BIRC3-LU-dependent manner and regulate surface CXCR4 expression and B cell migration.

(A) Constructs as in Figure 1E and GFP fusion with BIRC3 coding region, followed by no 3′UTR (NU; but the construct includes a polyadenylation signal).
(B) Myc co-IP of endogenous IQGAP1 and RALA after transfection of CXCR4-myc and the constructs from (A) into HEK293T cells. 4.5 % of input was loaded. Quantification in Figure S6A.
(C) Model of 3′UTR-independent interaction between BIRC3 and CXCR4, which can be accomplished by BIRC3-NU, BIRC3-SU or BIRC3-LU. The 3′UTR-dependent interaction happens between BIRC3-LU, IQGAP1, RALA, and CXCR4 and can only be accomplished by BIRC3-LU.
(D) FACS analysis of endogenous surface CXCR4 levels in Raji cells stably expressing a ctrl shRNA or shRNAs against IQGAP1 (IQ KD). LU KD is shown for comparison reasons. MFI ± SD from biological replicates. Mann Whitney test, ctrl KD vs each sample, **, p = 0.002. Representative example in Figure S6D.
(E) As in (D), but cells with stable expression of shRNAs against RALA (RALA KD). Mean MFI ± SD from biological replicates. Mann Whitney test, ctrl KD vs each sample, *, p = 0.037. Representative example in Figure S6E.
(F) B cell migration as in Figure 2C. Kruskal Wallis test, p = 0.009. Mann Whitney test, ctrl KD vs each sample, *, p = 0.05.
IQGAP1 and RALA are required for surface CXCR4 expression and B cell migration
So far, our data showed that the cytoskeleton-associated factors IQGAP1 and RALA only interact with CXCR4 in the presence of BIRC3-LU. We next tested if the two factors are necessary for surface expression of CXCR4. KD of IQGAP1 or RALA in Raji B cells significantly decreased surface expression of endogenous CXCR4 without affecting total CXCR4 expression and mimicked the effect observed in LU KD cells (Figures 6D, 6E, S6B-E), suggesting that IQGAP1 and RALA regulate CXCR4 trafficking. Moreover, both cytoskeleton-associated factors were necessary for B cell migration and again mimicked the effects of loss of BIRC3-LU protein (Figure 6F). Taken together, these results demonstrated that BIRC3-LU assembles a protein complex containing IQGAP1 and RALA and recruits these factors to CXCR4 to regulate surface levels of the receptor which is required for B cell migration towards the CXCL12 cytokine.
BIRC3-LU, IQGAP1, and RALA are required for trafficking of CD27 receptor
Lastly, we asked if the regulation of trafficking is specific to CXCR4 or if BIRC3-LU may regulate trafficking of additional surface receptors. We used FACS analysis to measure surface expression of various receptors important for B cell biology. We compared the surface expression of CD19, CD27, CD38, and CD47 between ctrl cells and LU KD cells. LU KD did not affect surface expression of CD19, CD38 or CD47 (Figures S7A-C), but it significantly reduced surface expression of CD27 (Figure S7D), a marker for memory B cells. A similar reduction in CD27 surface expression was observed in BIRC3 KO cells as well as after IQGAP1 KD or RALA KD (Figures S7E, 7F). In none of the conditions tested overall CD27 protein levels were affected (Figure S7G), suggesting that BIRC3-LU, through its interaction with IQGAP1 and RALA, regulates trafficking of at least two cell surface receptors that play important roles in B cell biology.
Gain of protein functions through 3′UTR-mediated protein complex formation has the potential to be widespread
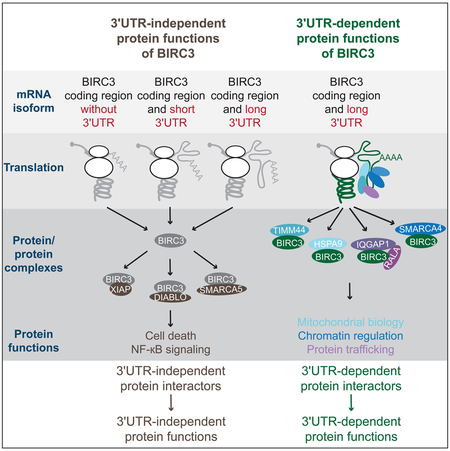
Figure 7A summarizes our findings on the 3′UTR-independent as well as 3′UTR-dependent functions of BIRC3. Through 3′UTR-independent interactors, BIRC3 protein regulates NF-κB signaling and cell death, whereas through 3′UTR-dependent interactors, BIRC3 regulates mitochondrial biology, chromatin, and protein trafficking. Whereas 3′UTR-independent functions can be achieved by BIRC3 protein that was synthesized from any construct that contains the BIRC3 coding region, 3′UTR-dependent functions can only be performed by BIRC3 protein that was translated from the BIRC3 mRNA with the long 3′UTR (Figure 7A).
Figure 7. 3′UTR-dependent protein interactors have the potential to be widespread.

(A) 3′UTR-independent BIRC3 functions are mediated by 3′UTR-independent protein interactors (brown), including XIAP, DIABLO, and SMARCA5. They can be accomplished when BIRC3 protein is expressed from transcripts containing no regulatory 3′UTR elements (NU), the short 3′UTR (SU) or the long 3′UTR (LU). 3′UTR-dependent BIRC3 roles include the regulation of mitochondrial biology (light blue), chromatin (dark blue), and protein trafficking (purple). These functions are mediated by BIRC3-LU and the 3′UTR-dependent protein interactors including HSPA9, TIMM44, SMARCA4, IQGAP1, and RALA.
(B) Change in mRNA pattern between CLL4 and NB. 3′UTR ratio change is shown on the y-axis and expression change is shown on the x-axis for genes with more than one 3′UTR. Each dot represents an mRNA. Color-coded upon significant difference between the two samples, calculated as in Figure S1A. Red/blue, increased/decreased expression of the short 3′UTR isoform in CLL; dark/light green, higher/lower expression of mRNA in CLL; black, significant change in mRNA levels and change in 3′UTR isoform ratio, and grey indicates no significant change.
We anticipate that BIRC3 is not the only protein that is able to gain additional functions through 3′UTR-dependent protein complex formation. When we compared 3′UTR isoform expression between normal and CLL B cells, we found that a number of mRNAs changed their mRNA abundance levels (Figure 7B, green) as expected. Other mRNAs changed their 3′UTR ratios (Figure 7B, red, blue). Strikingly, in 94% of the observed mRNA changes either 3′UTR ratios or mRNA levels were altered and in only 6% of changes both features were affected (Figure 7B, black). This means that the vast majority of alternative 3′UTR isoform changes between normal and CLL B cells do not alter the corresponding mRNA levels. Instead, it is likely that a sizable fraction of significant 3′UTR isoform changes are associated with changes in protein functions caused by 3′UTR-dependent protein complex formation.
Discussion
We chose to investigate the role of the long BIRC3 3′UTR isoform in CLL cells as it was consistently upregulated in malignant B cells compared with their normal B cell counterparts. This is an unusual feature, because CLL is a heterogenous disease and only very few markers are present in all CLL samples (Sutton and Rosenquist, 2015). Despite a significant BIRC3 3′UTR ratio change, the alternative 3′UTRs did not regulate BIRC3 protein localization or abundance. Instead, we show that BIRC3 protein can accomplish unique functions through 3′UTR-dependent protein complex formation. These functions can only be accomplished by the protein encoded from the mRNA with the long BIRC3 3′UTR. One of the newly discovered 3′UTR-dependent functions is the regulation of CXCR4-mediated B cell migration which is an important requirement for CLL cell survival in vivo (Burger et al., 2005), indicating that BIRC3-LU has a cancer-promoting role in CLL.
BIRC3-LU has cancer-promoting functions in CLL
Expression of CXCR4 on the surface of B cells mediates homing of cells to protective bone marrow niches, thus, enhancing CLL cell survival (Burger et al., 2005). The cancer-promoting function of BIRC3-LU is in contrast to BIRC3′s assumed role as a tumor-suppressor gene in CLL (Rossi et al., 2012). This view emerged because BIRC3 is part of a region located on chromosome 11q that is heterozygously deleted in 15-20% of CLL patients. In 83% of cases with 11q deletions, BIRC3 is deleted together with 40 other genes (Rose-Zerilli et al., 2014) which reduces overall BIRC3 mRNA levels by approximately 50%. Despite the reduction in overall BIRC3 mRNA levels, all our investigated CLL samples overexpressed the long BIRC3 3′UTR isoform. This correlated with increased expression of CXCR4 protein on the surface of CLL cells and with B cell migration. Together with the data obtained in a B cell line, this indicates that reduced BIRC3 levels are still sufficient for trafficking regulation of CXCR4 and suggests that for 3′UTR-dependent protein complexes protein abundance may not be the determining parameter.
3′UTR-dependent protein complex formation can change protein functions independently of protein localization
Many proteins form complexes to accomplish their cellular functions. So far, it has been largely thought that protein interactions are solely based on protein sequence and conformation as they can be accomplished using constructs that lack any 3′UTR regulatory sequences or can be recapitulated in vitro using purified proteins (Huttlin et al., 2017). We call these interactors and their mediated functions 3′UTR-independent. The formation of these complexes often increases with high abundance of the interactors. In contrast, for 3′UTR-dependent protein complex formation, the protein sequence is necessary but not sufficient as the establishment of the protein-protein interaction requires the presence of a specific 3′UTR (Berkovits and Mayr, 2015; Ma and Mayr, 2018).
Several scenarios can be envisioned how 3′UTRs assist in protein complex formation: They may provide proximity of the interactors or they may change the folding of the nascent peptide chain if the interactor binds in a co-translational manner (Shiber et al., 2018). It is also possible that the protein sequence of BIRC3-LU is post-translationally modified in a 3′UTR-dependent manner which could be the prerequisite for stable binding of some of the 3′UTR-recruited factors. Lastly, the 3′UTR may change the local environment at the site of translation by enabling translation within a specific RNA granule. This was recently shown to be the case for the interaction between CD47 and SET where instead of protein abundance driving the interaction, the local environment found at the site of translation seems to be the crucial determinant of the interaction (Ma and Mayr, 2018).
Interestingly, transfer of proteins from the 3′UTR to the nascent BIRC3 protein may also happen in RNA granules. This view is supported by our single molecule RNA-FISH experiments that showed that BIRC3 mRNA localizes to RNA foci in the cytoplasm. Furthermore, our data suggest that BIRC3 is translated in these foci as approximately 30% of MS-identified BIRC3 interactors are known components of RNA granules (Elvira et al., 2006). They include ribosomal proteins, translation factors, RNA-binding proteins, DEAD box helicases, and actin-related factors (Figure S3E, Table S2). However, it is currently unclear how these local RNA granules are formed, if their composition differs in a 3′UTR-dependent manner, and if the local RNA granules are required for 3′UTR-dependent protein complex formation.
Cooperative binding of RNA-binding proteins throughout the 3′UTR enables specific functions of the long 3′UTR
RNA-binding proteins mediate diverse functions of 3′UTRs (Mayr, 2017, 2018). A simple explanation how unique functions of long 3′UTRs can be accomplished is through RNA-binding proteins that only bind to the long, but not the short 3′UTR isoform. This was previously shown for the alternative 3′UTRs of CD47 (Berkovits and Mayr, 2015).
In the case of BIRC3, several RNA-binding proteins interacted with both the short and long 3′UTRs when the short 3′UTR was expressed at high levels. If the same RNA-binding proteins are able to bind both the short and the long 3′UTR, how can a specific function of the long 3′UTR be achieved? Our data indicate that what matters is the relative amount of HuR bound to the 3′UTR. Only when more HuR than STAU1 was bound, IQGAP1 was recruited by the 3′UTR. The higher HuR amount was achieved through cooperative binding of HuR to several binding sites distributed throughout the entire 3′UTR. This binding mode is especially powerful as a specific function of the long 3′UTR can be achieved, even when the short 3′UTR isoform is expressed at very high levels.
Regulation of protein functions by 3′UTR-mediated protein complex formation has the potential to be widespread
We provide here a strategy for the comprehensive identification of protein interactors using quantitative MS. We validated a number of 3′UTR-dependent BIRC3 interactors that are involved in diverse biological functions. Our data suggests that BIRC3-LU is part of many different stable substoichiometric complexes whose members interact with BIRC3-LU in a 3′UTR-dependent manner. These observations indicate that BIRC3-LU can exert a substantial number of different functions, depending on the nature of the interaction partner.
When comparing 3′UTR isoform ratios between normal and CLL cells, we detected a large number of genes whose mRNA transcripts with long 3′UTRs were upregulated in CLL. As the vast majority of alternative 3′UTR isoform changes did not alter their corresponding mRNA levels, it is likely that instead of protein abundance regulation many 3′UTR ratio changes will enable additional protein functions through 3′UTR-dependent protein complex formation. This suggests that BIRC3 is not the only protein that uses 3′UTR-dependent protein complex formation to gain additional protein functions.
Taken together, in addition to protein functions mediated by 3′UTR-independent interactors whose binding relies solely on features provided by the protein itself, our data reveal that a sizeable fraction of BIRC3 protein complexes require the presence of the long 3′UTR for their formation, but not for the continued maintenance of the complexes. This indicates that in addition to encoding the protein sequence, DNA contains information for protein functions that is transmitted through 3′UTR-dependent protein complex formation (Ma and Mayr, 2018). As 3′UTRs of mRNAs encoding membrane as well as non-membrane proteins are able to form 3′UTR-dependent protein complexes, it is likely that the protein recruitment function of 3′UTRs is widespread. Moreover, as 3′UTR length has expanded during the evolution of higher organisms, 3′UTRs may contribute to their increased biological complexity (Mayr, 2017). This may explain how a limited set of amino acid sequences can accomplish multi-functionality of proteins by using protein complex formation mediated by 3′UTRs.
STAR methods text
Samples for 3′-seq and APA analysis
The normal CD5+ B (NB) cell and the CLL B cell samples were previously published and the 3′-seq data can be accessed in the Gene Expression Omnibus database under the accession numbers GSE111310 and GSE111793 (Singh et al., 2018; Lee et al., 2018). 3/13 CLL samples had a 11q deletion as assessed by FISH. In one sample, the 11q deletion was subclonal, as only 25% of cells showed a heterozygous deletion of 11q. The data were processed as described previously and a significant difference in 3′UTR isoform ratio was determined using a generalized linear model with a false discovery rate (FDR)-adjusted P < 0.1. The ′short UTR index′ (SUI) of an mRNA is calculated as the ratio of reads that map to the proximal polyadenylation site within the 3′UTR divided by all reads that map to the 3′UTR of the mRNA (Lianoglou et al., 2013). A TPM difference (> log2 1.2) was used to identify differentially expressed mRNAs.
Cell lines
Raji cells are malignant B cells from lymphomas and were a gift from Dr. Hans-Guido Wendel (MSKCC). HEK293T cells (embryonic kidney) were purchased from ATCC. HeLa cells were provided by Dr. Jonathan Weissman (UCSF). Raji cells were cultured in RPMI with 20% FBS and 1% penicillin/streptomycin. HeLa and HEK293T cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin.
BIRC3 knock-out Raji cells
The pX458 vector, containing Cas9 from S. pyogenes with 2A-EGFP and a cloning site for guide RNAs was obtained from Addgene (# 48138) (Ran et al., 2013). The DNA oligonucleotides that served as guide RNAs to produce BIRC3 knock-out Raji cells are listed in Table S4 and were cloned into pX458 vector using the BbsI restriction site.
Raji cells were nucleofected using Nucleofactor V Kit with constructs containing guide RNAs targeting BIRC3. After 24 hours, GFP-positive cells were FACS-sorted into single wells and grown to obtain clones. Knock-out cells were screened using western blots and the positive as well as negative clones were validated using sequencing. The KO3 clone generates a truncated protein with 160 amino acids of BIRC3, followed by a frame-shift mutation that generates 6 additional amino acids followed by a stop codon from one allele and it generates 4 additional amino acids from the other allele (Figure S2A). The KO4 clone generates a truncated protein with 119 amino acids followed by a frame-shift mutation that generates 26 additional amino acids followed by a stop codon from one allele and it generates 8 additional amino acids from the other allele (Figure S2A).
Constructs
GFP-BIRC3-SU and GFP-BIRC3-LU were cloned into pcDNA3.1 using Gibson Assembly Cloning (New England Biolabs). The primers used for amplification of the pieces are listed in Table S4. The coding region of BIRC3 was amplified from human naïve B cell cDNA and the short or long 3′UTRs were amplified from genomic DNA of human peripheral blood mononuclear cells. GFP-BIRC3-NU does not contain any BIRC3 3′UTR, but contains the SV40 polyadenylation signal which is part of the pcDNA3.1 vector.
The GFP constructs that lack the BIRC3 coding region, but contain the 3′UTRs (ΔCDR-SU, ΔCDR-LU) were generated by replacing the eGFP-BIRC3 fragment in the GFP-BIRC3-SU and GFP-BIRC3-LU constructs by eGFP alone (obtained from pcDNA3.1-puro GFP construct (Ma and Mayr, 2018)).
Constructs for in vitro transcription.
All BIRC3 3′UTR fragments were cloned into pCR-Blunt II-TOPO using SacI and NotI. The antisense constructs were cloned by digesting the sense constructs with NsiI flanking both ends of the 3′UTR fragments. The LU1 fragment contains 647 nucleotides of the long BIRC3 3′UTR and is located between nucleotides 1138 and 1784 of the BIRC3 3′UTR. The primers used for PCR amplification are listed in Table S4. The sequence of the ARE was previously described (Ma and Mayr, 2018) and was cloned in the same way as the other 3′UTRs fragments. All the plasmids were linearized using NotI for sense in vitro transcription and HindIII for antisense transcription.
The CXCR4 coding region was cloned into pcDNA3.1/myc-His C using EcoRI and NotI sites. The MS2 sequence was described by us before and was inserted into GFP-BIRC3-SU after the stop codon using AgeI and HindIII sites to generate GFP-BIRC3-MS2-SU (Berkovits and Mayr, 2015). The MCP-mCherry (mC) and MCP-mC-HuR constructs were described previously and MCP-mC-STAU1 was generated by replacing HuR using BsrGI and XbaI sites (Berkovits and Mayr, 2015).
For shRNA knock-down experiments pSUPERretropuro (pSUPER) or pSUPER containing eGFP (pSUPER-GFP) was used as described previously (Berkovits and Mayr, 2015). The DNA oligonucleotides used as shRNA precursors are listed in Table S4 and were cloned into pSUPER-GFP or pSUPER. Retroviral particles were generated as described previously (Mayr and Bartel, 2009). For knock-down experiments of IGF2BP1 shRNA constructs were purchased from Sigma (MISSION® shRNA TRCN0000218799, TRCN0000230114). As control an shRNA against luciferase (MISSION® shRNA SHC007) was used.
To generate stable LU KD Raji cell lines, Raji cells were transduced with retrovirus, the culture plates were coated overnight with 5 μg/ml fibronectin. Raji cells were spin-infected containing 8 μg/ml polybrene at 992 g at 30°C for 45 min. The inf ection procedure was repeated after 48 hours. After cultivating for another two days, the infected cells were selected using 2 μg/ml puromycin. For knockdown of IQGAP1, RALA, STAU1 and HuR, stable cell lines were generated using nucleofection of shRNA constructs using Nucleofactor V Kit (Lonza, Program M-13). After 48 hours, 80% of cells were preserved for FACS analysis and the rest was selected using 2 μg/ml puromycin.
RNA-FISH
Custom Stellaris EGFP FISH probes were described previously (Berkovits and Mayr, 2015). HeLa cells were plated on 4-well Millicell EZ silde and transfected with GFP fusion constructs. 14 hours after transfection, cells were washed with PBS, fixed with 3.7% formaldehyde for 10 min at room temperature and washed twice for 5 min with PBS. PBS was discarded and 1 ml 70% ethanol was added. The slide was kept at 4°C fo r 8 hours. The 70% ethanol was aspirated, 1 ml wash buffer was added (2× SSC, 10% formamide in RNase-free water) and incubated at room temperature for 5 min. Hybridization mix was prepared by mixing 10% dextran sulfate, 10% formamide, 2 × SSC, 2 mM ribonucleoside vanadyl complex (NEB), 0.02% BSA, 200 μg/ml yeast tRNA, 200 μg/ml single stranded DNA and FISH probe (1:200). To each well 200 μl hybridization mix was added and hybridized at 37°C overnight. Slides were washed twice for 30 min each with pre-warmed wash buffer (1 ml, 37°C) in the dark, followed by one quick wash with PBST, and then mounted with mounting solution. Images were captured using confocal ZEISS LSM 880.
Confocal microscopy
Confocal imaging was performed using a Leica TCS SP8 microscope. HeLa cells were plated on 3.5 cm glass bottom dishes (Cellvis, D35-20-1-N) and allowed to grow overnight. On the next day, the HeLa cells were transfected with GFP-BIRC3-SU or GFP-BIRC3-LU plasmid using Lipofectamine 2000 and were imaged after 24 hours at the Sloan Kettering Institute Molecular Cytology Core Facility.
Northern blot
Northern blots were performed as previously described (Lianoglou et al., 2013). The template used for generating the BIRC3 probe was PCR amplified from cDNA of human peripheral blood naïve B cells. The primers are listed in Table S4. The levels of the short of long 3′UTR isoforms were quantified using the MultiGauge program (Fuji).
QRT-PCR
Total RNA was isolated by Tri reagent solution (Invitrogen #AM9738) and digested with DNase I (Invitrogen #AM1906). RNA was reverse transcribed using qScript cDNA SuperMix (Quanta Biosciences #101414-106) and quantitative PCR was performed using FastStart universal SYBR green master mix (Roche) on a 7900HT Fast Real-Time PCR System (Applied Biosystems). The qRT-PCR was performed in triplicates from three independent cDNA preparations.
Cell viability assay
Ctrl KD, LU KD and BIRC3 KO Raji cells were seeded into 96-well plates and treated with 4 μg/ml Fludarabine. On day 2, 10% (v/v) of Resazurin (R&D Systems, AR002) was added to each well and the plates were incubated for 4 hours at 37°C. The fluorescence in the wells was measured using a microplate reader (SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA) with excitation and emission of 560 nm and 590 nm, respectively. Results were normalized to wells containing media without cells. Each experiment was repeated at least three times with triplicates.
Migration assay
Raji cells were counted and 100 μl containing 8 × 105 cells were added to each transwell (Costar, diameter 6.5 mm, pore size 5 μm). The lower chambers were supplemented with conditioned media containing 50 ng/ml CXCL12 (Life technologies). After 3.5 hours of incubation at 37°C and 5% CO2, the migrated cells were collected from the lower chambers and counted manually using Trypan blue. Each biological replicate was performed in triplicates.
FACS
For surface expression, cells were washed with ice-cold PBS once, incubated with appropriate fluorochrome-conjugated antibodies for 30 min at 4°C and washed twice with ice-cold PBS containing 0.5% FCS. The following antibodies were used: anti-CD184-APC (CXCR4, 560936, BD Biosciences), anti-CD27-PE (555441, BD Biosciences), anti-CD19-APC (555415, BD Biosciences), anti-CD38-APC (555462, BD Biosciences), anti- CD47-APC (561261, BD Biosciences).
To measure total CXCR4 or CD27 protein level by FACS, Raji cells were fixed with 4% formaldehyde at room temperature for 15 min. After two washes with excess PBS, fixed cells were resuspended with ice-cold PBS and permeablized with 90% methanol for 10 min on ice. Cells were then washed with cold PBS twice and resuspended with the incubation buffer (PBS + 0.5% BSA). Cells were aliquoted and incubated with anti-CD184-APC or anti-CD27-PE for 20 min at 4°C. After two washes with the incubation buffer, cells were analyzed using a BD FACS Calibur (BD Biosciences) and data were analyzed using the FlowJo software.
Recycling assay
Raji cells were incubated with 50 ng/ml CXCL12 for 30 min at 37°C and 5% CO2. After washing with PBS once, 10% of cells were kept on ice for FACS analysis and the rest of the cells was resuspended in conditioned culture media and incubated at 37°C and 5% CO2 for an additional 3 hours. All cells were collected and surface CXCR4 expression was analyzed by FACS.
Co-immunoprecipitation
GFP-BIRC3-SU and GFP-BIRC3-LU were transfected into HEK293T cells using calcium phosphate. After 24 hours, transfected cells were collected and lysed on ice for 30 min with GFP-trap_A RIPA buffer (Chromotek) containing freshly added proteinase inhibitor cocktail (Thermo Scientific) and phosphatase inhibitors (Sigma). After centrifugation at 21,130 g for 10 min, GFP-trap_A immunoprecipitation was performed following the manufacturer′s instructions. GFP-trap_A beads were added, incubated with cell lysate for 2 hours at 4°C on a rotator, and washed three times with ice-cold GFP-trap dilution buffer. GFP-trap beads were mixed with 2x Laemmli sample buffer and boiled at 95°C for 5 min. The immunoprecipitates were run on 4-12% Bis-Tris NuPAGE gels using MES running buffer, followed by western blot analysis of the endogenously expressed candidate interactors.
GFP co-IP in the presence of RNase A was performed as described above. RNase A (R4642, Sigma) was added to dilution buffer (1:900) and mixed with cell lysate (final concentration is 1: 1500). The mixture was incubated at room temperature for 15 mins, then GFP-trap slurry was added and incubated for another 2.5 hours at 4°C.
MS2 tethering assay. The BIRC3-GFP co-IP was performed as described above, but different constructs were used: Instead of GFP-BIRC3-LU, GFP-BIRC3-MS2-SU (containing 24 MS2 binding sites (MS2) after the stop codon) was used. HEK293T cells were transfected with GFP-BIRC3-MS2-SU, MCP-mC-STAU1 and MCP-mC-HuR. This results in tethering of STAU1 and HuR to the short BIRC3 3′UTR (Figure 5F, right panel). Tethered STAU1 and HuR recruit endogenously expressed IQGAP1 and RALA to the site of BIRC3 translation, resulting in 3′UTR-dependent protein complex formation between BIRC3, IQGAP1 and RALA, shown by GFP co-IP in Figure 5F, bottom panel. Two control experiments were performed: GFP-BIRC3-MS2-SU was co-transfected with MCP-mC, followed by GFP co-IP (Figure 5F, left panel). And GFP-BIRC3-SU was co-transfected with MCP-mC-STAU1 and MCP-mC-HuR, followed by GFP- co-IP. In both control experiments IQGAP1 and RALA were unable to interact with BIRC3 protein (Figure 5F, bottom panel). This shows that overexpression of the RNA-binding proteins without tethering to the 3′UTR is not sufficient for 3′UTR-dependent protein complex assembly.
CXCR4-myc co-IP was performed as above with slight modifications. Cells were lysed on ice for 1 hour with CHAPS buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X, 1% Na-deoxycholate, 1 mM EDTA, 0.5% CHAPS) with freshly added proteinase inhibitor cocktail and phosphatase inhibitors. After centrifugation of the lysates at 21,130 g for 10 min at 4°C, the supernatant was collected and pre-cleared using 20 μl Protein A/G PLUS-Agarose beads (sc-2003, SCBT) for 1.5 hours at 4°C with rotation. The pre-cleared lysates were incubated with 1 μg of anti-MYC antibody (Sigma, M4439) or IgG (IgG, sc-2025, SCBT) for 2 hours at 4°C with rotation, then 20 μl of agarose beads were added and the reaction was rotated for an additional 45 min at 4°C. After washing the beads three times with wash buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, freshly added proteinase inhibitor cocktail), beads were mixed with 2x Laemmli sample buffer (Sigma), followed by western blot analysis.
Quantitative mass spectrometry
GFP-trap co-IP was performed with slight modifications. HEK293T cells were cultivated in DMEM medium supplemented with 10% FBS and 1% penicillin and containing either “light” (L-Arginine-HCL) or “heavy” (L- Arginine-HCL (13C6, 99%; 15N4, 99%; Cambridge Isotope Laboratories, CNLM-539-H-0.05) stable isotope labeled amino acids. Cells were cultivated for at least six passages before the incorporation efficiency was verified by mass spectrometry analysis. The ′light′ HEK293T cells were transfected with GFP-BIRC3-SU and the ′heavy′ cells were transfected with GFP-BIRC3-LU using calcium phosphate. After 24 hours, transfected cells were collected and lysed on ice for 30 min and lysates from the light and heavy samples were pooled, followed by GFP-trap co-IP. The beads were mixed with 2x Laemmli sample buffer followed by SDS-gel electrophoresis in MES running buffer using 4-12% Bis-Tris NuPAGE gels. The protein gels were stained with SimplyBlue (Life technologies) following manufacturer′s instructions and submitted to the MSKCC Proteomics Core facility for SILAC mass spectrometry analysis.
Mass spectrometry was performed as was described previously (Shevchenko et al., 2006). Gel slices were then washed with 1:1 (Acetonitrile: 100 mM ammonium bicarbonate) for 30 min, dehydrated with 100% acetonitrile for 10 min, excess acetonitrile was removed and slices were dried in speed-vac for 10 min without heat. Gel slices were reduced with 5 mM DTT for 30 min at 56°C in a thermomixer (Eppendorf) and chilled down to room temperature, and alkylated with 11 mM IAA for 30 min in the dark. Gel slices were washed with 100 mM ammonium bicarbonate and 100% acetonitrile for 10 min each. Excess acetonitrile was removed and dried in speed-vac for 10 min without heat and gel slices were rehydrated in a solution of 25 ng/μl trypsin in 50 mM ammonium bicarbonate on ice for 30 min. Digestions were performed overnight at 37°C in a thermomixer. Digested peptides were collected and further extracted from gel slices in extraction buffer (1:2 (v/v) 5% formic acid/acetonitrile) at high speed shaking in a thermomixer. Supernatant from both extractions was combined and dried in a vacuum centrifuge. Peptides were desalted with C18 resin-packed stage-tips, lyophilized and stored at −80°C until further use.
LC-MS/MS analysis:
Desalted peptides were dissolved in 3% acetonitrile/0.1% formic acid and were injected onto a C18 capillary column on a nano ACQUITY UPLC system (Water) which was coupled to the Q Exactive plus mass spectrometer (Thermo Scientific). Peptides were eluted with a non-linear 200 min gradient of 2-35% buffer B (0.1% (v/v) formic acid, 100% acetonitrile) at a flow rate of 300 nl/min. After each gradient, the column was washed with 90% buffer B for 5 min and re-equilibrated with 98% buffer A (0.1% formic acid, 100% HPLC-grade water). MS data were acquired with an automatic switch between a full scan and 10 data-dependent MS/MS scans (TopN method). Target value for the full scan MS spectra was 3 × 106 ions in the 380-1800 m/z range with a maximum injection time of 30 ms and resolution of 70,000 at 200 m/z with data collected in profile mode. Precursors were selected using a 1.5 m/z isolation width. Precursors were fragmented by higher-energy C-trap dissociation (HCD) with a normalized collision energy of 27 eV. MS/MS scans were acquired at a resolution of 17,500 at 200 m/z with an ion target value of 5 × 104, maximum injection time of 60 ms, dynamic exclusion for 15 s and data collected in centroid mode.
To generate protein IDs and SILAC ratios all .raw data were analyzed by using MaxQuant (Max Planck Institute of Biochemistry; version 1.5.1.0) at default settings with first search tolerance and main search tolerance of 20 ppm and 6 ppm, respectively. Data were further analyzed by Scaffold 4 version 4.5. Only proteins with a least two different peptides were considered for analysis and the log2 ratio of the heavy versus the light fraction (LU/SU) was calculated. This showed equal abundance of BIRC3 in the heavy versus the light fraction as the log2 enrichment ratio was 0.033 (which corresponds to a ratio of 1.02). A cut-off of log2 0.585 (1.5-fold) was used to identify the BIRC3-LU-enriched protein interaction partners. The 3′UTR-independent interactors had a log2 LU/SU ratio <∣0.585∣.
Gene ontology analysis
Gene ontology analysis was performed using DAVID 6.8 (Huang da et al., 2009). The enrichment scores and corresponding p values of the functional categories that were exclusively enriched in either the 3′UTR-independent or 3′UTR-dependent (BIRC3-LU-enriched) interactors are reported.
Western blot analysis
Cells were lysed on ice for 30 min with RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 1% Na-deoxycholate, 1 mM EDTA, 0.05% SDS). Lysates were cleared using centrifugation at maximum speed, mixed with 2x Laemmli sample buffer (Sigma), boiled at 95 °C for 5 min, and chilled on ice for 1 min before loading on gels. For CXCR4 analyses, cells were lysed on ice for 1 hour with CHAPS buffer containing proteinase inhibitor cocktail and phosphatase inhibitors. Lysates were cleared using centrifugation at max speed, mixed with 2x Laemmli sample buffer (Sigma) and incubated at 37°C for 1 hour. Lysates were run using 4-12% Bis-Tris NuPAGE gels (Life technologies) with MES running buffer (Natural Diagnostics or Life technologies). The separated proteins were transferred to nitrocellulose membranes (1620252, Bio-Rad), blocked with Odyssey Blocking Buffer (Li-Cor, 927-40000) for 1 hour at room temperature, followed by incubation with primary antibodies at 4°C overnight. After two washes using PBS and 0.1% Tween 20 (PBST), the blots were incubated with IRDye-conjugated secondary antibodies for 50 min at room temperature. After one wash with PBST and two washes with PBS, proteins were detected with Odyssey CLx imaging system (Li-Cor).
The following primary antibodies were used: anti-ACTIN (A4700, Sigma, A2066, Sigma), anti-GFP (ab13970, Abcam), anti-DIABLO (2954, Cell Signaling), anti-XIAP (2042, Cell Signaling), anti-SMARCA4 (BRG1, 3508, Cell Signaling), anti-SMARCA5 (SNF2H, 38410, Cell Signaling), anti-HSPA9 (SAB4100033, Sigma), anti-TIMM44 (ab194829, Abcam), anti-IQGAP1 (SAB4200079, Sigma), anti-RALA (ab126627, Abcam), anti-BAZ1B (sc-514287, SCBT), anti-ATP5B (ab14730, Abcam), anti-MYC (M4439, Sigma), anti-BIRC3 (3130P, Cell Signaling), anti-mCherry (ab125096, Abcam), anti-CXCR4 (4G10, sc-53534, SCBT), anti-ELAVL1 (HuR, 07-1735, Millipore), anti-STAU1 (14225-1-AP, Proteintech), anti-IGF2BP1 (ARP40658_P050, Aviva Systems Biology), anti-hnRNPA1 (sc-374526, SCBT), anti-hnRNPQ (R5653, Sigma), anti-CTR9 (SAB1100738, Sigma), anti-YBX1 (A303-231A-M, Bethyl Laboratories), anti-SF3B1 (D221-3, Medical & Biological Laboratories Co., LTD), anti-NUDT21 (sc-81109, SCBT), anti-FUS (SAB4200478, Sigma), anti-KHSRP (SAB4200566, Sigma), anti-hnRNPC (sc-32308, SCBT), anti-DDX17 (ab24601, Abcam), and anti-snRNP70 (sc-9571, SCBT).
The secondary antibodies used included anti-mouse IRDye 700 (610-730-002, Rockland Immunochemicals), anti-rabbit IRDye 680 (926-68073, Li-Cor Biosciences), anti-rabbit IRDye 800 (926-32213, Li-Cor Biosciences), and anti-mouse IRDye 800 (926-32212, Li-Cor Biosciences).
RNA affinity pull-down assay
This assay was performed as before with modifications as outlined below (Panda et al., 2016). After linearization of the plasmids the DNA templates were purified by Phenol:Chloroform extraction, followed by ethanol precipitation containing sodium acetate. In vitro transcription was performed according to the manufacturer′s instructions (MEGAscript T7, Invitrogen). In the mixture, 0.5 μg of linearized DNA template was used and Biotin-UTP (AM8450, Invitrogen) was added to biotin-label the RNAs. The ratio of the ribonucleotide solutions was ATP:CTP:GTP:UTP:Biotin-UTP =1: 0.9: 0.9: 0.945: 0.055). After labeling, the RNAs were recovered using Phenol:Chloroform extraction and isopropanol precipitation. The RNAs were stored at −80°C until further use.
The amount of RNAs to incubate with cell lysate was determined by first measuring the RNA concentrations using Nanodrop, followed by agarose gel analysis to confirm the concentrations. The same molar amount of RNA molecules were used for RNA affinity pull-down reactions, if not stated otherwise.
Biotinylated RNAs were thawed on ice and boiled at 95°C for 2 minutes, slowly cooled down to room temperature and then were kept on ice. HEK293T cells or Raji cells were lysed in lysis buffer (20 mM Tris-HCL pH 7.5, 250 mM NaCl, 50 mM KCl, 2.5 mM MgCl2, 0.25% NP-40, 0.5% Triton X-100, 1 mM EDTA pH 8 with 1x protease inhibitor). One 10 cm2 dish of cells (about 30 million of Raji cells) was used for one pull-down reaction. Each dish was washed with cold PBS twice and lysed with 500 μl lysis buffer on ice for 20 minutes. Cell lysate was collected after spinning at 21,000 × g for 10 minutes at 4°C. Pull-down mixture was prepared as follows: 450 μl of cell lysate (keep 30 μl as input for western blot), 1 μg of LU RNA (same molar amounts of the other 3′UTR RNAs), and 0.2 U/ml of RNaseOUT. The reactions were incubated at 4°C for 1.5 hours with rotation. Meanwhile, Dynabeads® MyOne™ Streptavidin C1 (65002, Invitrogen) were washed with wash buffer (10 mM Tris-HCL pH 7.5, 250 mM NaCl, 0.5% Triton X-100, 1 mM EDTA pH 8 with 1x protease inhibitor). After three washes, 50 μl of beads were added to each pull-down reaction and the mixtures were incubated for another 40 minutes. The pull-down beads were washed 5 times with wash buffer and resuspended with 2x sample buffer (Sigma). All samples were boiled at 95°C for 2 min, incubate d on ice for 1 min, and half of the sample was loaded onto NuPage gel for western blot analysis.
Statistical tests
To test for differences in cell viability a t-test for independent samples was used (equal variance was not assumed). For pairwise comparisons a Mann Whitney test was performed. If more than two samples were tested, first a Kruskal Wallis test was applied.
Supplementary Material
Table S1. Genes with significant 3′UTR isoform change between normal and CLL B cells, Related to Figures 7B and S1A.
Table S2. BIRC3 protein interactors determined by SILAC-based MS, Related to Figure 3B and S3A.
Table S3. STAU1 protein interactors determined by SILAC-based MS, Related to Figure 5C.
Table S4. Oligonucleotide sequences, Related to STAR methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ACTIN | Sigma-Aldrich | Cat# A4700, RRID:AB_476730 |
| ACTIN | Sigma-Aldrich | Cat# A2066, RRID:AB_476693 |
| GFP | Abcam | Cat# ab13970, RRID:AB_300798 |
| IQGAP1 | Sigma-Aldrich | Cat# SAB4200079, RRID:AB_10604111 |
| RALA | Abcam | Cat# ab126627, RRID:AB_11127656 |
| DIABLO | Cell Signaling | Cat# 2954, RRID:AB_2131196 |
| XIAP | Cell Signaling | Cat# 2042, RRID:AB_2214870 |
| SMARCA4 | Cell Signaling | Cat# 3508, RRID:AB_2193944 |
| SMARCA5 | Cell Signaling | Cat# 38410 |
| HSPA9 | Sigma-Aldrich | Cat# SAB4100033, RRID:AB_10743101 |
| TIMM44 | Abcam | Cat# ab194829 |
| BAZ1B | Santa Cruz Biotechnology | Cat# sc-514287 |
| ATP5B | Abcam | Cat# ab14730, RRID:AB_301438 |
| VDAC1 | Abcam | Cat# ab15895, RRID:AB_2214787 |
| MYC | Sigma-Aldrich | Cat# M4439, RRID:AB_439694 |
| BIRC3 (c-IAP2) | Cell Signaling | Cat# 3130P, RRID:AB_10693298 |
| mCherry | Abcam | Cat# ab125096, RRID:AB_11133266 |
| CXCR4 | Santa Cruz Biotechnology | Cat# sc-53534, RRID:AB_782002 |
| ELAVL1 (HuR) | Millipore | Cat# 07-1735, RRID:AB_1977173 |
| STAU1 | Proteintech | Cat# 14225-1-AP, RRID:AB_2302744 |
| hnRNPA1 | Santa Cruz Biotechnology | Cat# sc-374526, RRID:AB_10991524 |
| hnRNPQ (SYNCRIP) | Sigma-Aldrich | Cat# R5653, RRID:AB_261964 |
| CTR9 | Sigma-Aldrich | Cat# SAB1100738, RRID:AB_10013607 |
| SF3B1 | Gift from Omar Abdel-Wahab (MSKCC) | N/A |
| NUDT21 | Santa Cruz Biotechnology | Cat# sc-81109, RRID:AB_2153989 |
| FUS | Sigma-Aldrich | Cat# SAB4200478, RRID:AB_2737446 |
| KHSRP | Sigma-Aldrich | Cat# SAB4200566, RRID:AB_2737444 |
| YBX1 | Bethyl Laboratories | Cat# A303-231A-M, RRID:AB_2781069 |
| IGF2BP1 | Aviva Systems Biology | Cat# ARP40658_P050, RRID:AB_2122664 |
| DDX17 | Abcam | Cat# ab24601, RRID:AB_731871 |
| snRNP70 | Santa Cruz Biotechnology | Cat# sc-9571, RRID:AB_2193707 |
| hnRNPC | Santa Cruz Biotechnology | Cat# sc-32308, RRID:AB_627731 |
| IgG | Santa Cruz Biotechnology | Cat# sc-2025, RRID:AB_737182 |
| CD27 | BD Biosciences | Cat# 555441, RRID:AB_395834 |
| CD47 | BD Biosciences | Cat# 561261, RRID:AB_10611734 |
| CD38 | BD Biosciences | Cat# 555462, RRID:AB_398599 |
| CD19 | BD Biosciences | Cat# 555415, RRID:AB_398597 |
| CD184 (CXCR4) | BD Biosciences | Cat# 560936, RRID:AB_10563070 |
| anti-mouse IRDye 700 | Rockland Immunochemicals | Cat# 610-730-002, RRID:AB_1660934 |
| anti-rabbit IRDye 680 | Li-Cor Biosciences | Cat# 926-68073, RRID:AB_10954442 |
| anti-rabbit IRDye 800 | Li-Cor Biosciences | Cat# 926-32213, RRID:AB_621848 |
| anti-mouse IRDye 800 | Li-Cor Biosciences | Cat# 926-32212, RRID:AB_621847 |
| Bacterial and Virus Strains | ||
| E.coli DH5 alpha | Lab strain | N/A |
| Biological Samples | ||
| Human CLL cells | Lee, Singh et al., 2018 | N/A |
| Human normal CD5+B cells | Lee, Singh et al., 2018 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tri reagent solution | Invitrogen | Cat# AM9738 |
| Fibronectin | Invitrogen | Cat# 33016-015 |
| Polybrene (hexadimethrine bromide) | Sigma-Aldrich | Cat# H9268 |
| 16% Paraformaldehde, aqueous solution | Fisher scientific | Cat# 50-980-487 |
| Formamide | Sigma-Aldrich | Cat# F7503 |
| Ribonucleoside vanadyl complex | New England Biolabs | Cat# S1402 |
| Dextran sulfate sodium salt | Spectrum Chemical | Cat# DE131 |
| Fludarabine phosphate | Sigma-Aldrich | Cat# F9813 |
| Resazurin | R&D Systems | Cat# AR002 |
| CXCL12 | Life technologies | Cat# 10118-HNAE-25 |
| Trypan blue | Sigma-Aldrich | Cat# 93595 |
| CHAPS hydrate | Sigma-Aldrich | Cat# C3023 |
| Na-deoxycholate | Fisher scientific | Cat# BP349-100 |
| Triton X-100 | Fisher scientific | Cat# BP151-100 |
| Nonidet P-40 | Sigma-Aldrich | Cat# 74385 |
| Sodium chloride | Fisher scientific | Cat# S271-3 |
| Bovine serum albumin (BSA) | Fisher scientific | Cat# BP1605100 |
| Tris base | Fisher scientific | Cat# BP152-1 |
| Q5 High-fidelity DNA polymerase | New England Biolabs | Cat# M0491L |
| T4 DNA ligase | New England Biolabs | Cat# M0202L |
| Chloroform | Fisher scientific | Cat# C607-4 |
| Isopropanol | Fisher scientific | Cat# BP26184 |
| Ethanol | Fisher scientific | Cat# BP28184 |
| Methanol | Fisher scientific | Cat# A412-4 |
| SeeBlue® Plus2 pre-stained standard | Invitrogen | Cat# LC5925 |
| Biotin-UTP | Invitrogen | Cat# AM8450 |
| Phenol | Sigma-Aldrich | Cat# P4682 |
| Critical Commercial Assays | ||
| FastStart universal SYBR green master mix | Roche | Cat# 04913850001 |
| Experimental Models: Cell Lines | ||
| Raji | Gift from Hans-Guido Wendel (MSKCC) | |
| HEK293T | ATCC | ATCC Cat# CRL-3216, RRID:CVCL_0063 |
| HeLa | Gift from Jonathan Weissman (UCSF) | |
| Oligonucleotides | ||
| Oligonucleotides for PCR | This paper | Table S4 |
| Oligonucleotides for shRNA cloning | This paper | Table S4 |
| Oligonucleotides for construct cloning | This paper | Table S4 |
| Oligonucleotides for CRISPR guide cloning | This paper | Table S4 |
| Oligonucleotides for Northern blot probe | This paper | Table S4 |
| Recombinant DNA | ||
| pcDNA3.1-GFP-BIRC3-NU | This paper | N/A |
| pcDNA3.1-GFP-BIRC3-SU | This paper | N/A |
| pcDNA3.1-GFP-BIRC3-LU | This paper | N/A |
| pcDNA3.1-GFP | This paper | N/A |
| pcDNA3.1-GFP-ΔCDR-SU | This paper | N/A |
| pcDNA3.1-GFP-ΔCDR-LU | This paper | N/A |
| pcDNA3.1-CXCR4-myc | This paper | N/A |
| pcDNA3.1-GFP-BIRC3-MS2-SU | This paper | N/A |
| pcDNA3.1-MCP-mCherry-STAU1 | This paper | N/A |
| pcDNA3.1-MCP-mCherry-HuR | This paper | N/A |
| pcDNA3.1-MCP-mCherry | This paper | N/A |
| pSUPERretropuro-shRNA Control | Berkovits and Mayr, 2015 | N/A |
| pSUPERretropuro-shRNA HuR-2 | Berkovits and Mayr, 2015 | N/A |
| pSUPERretropuro-shRNA HuR-3 | Berkovits and Mayr, 2015 | N/A |
| pSUPERretropuro-shRNA STAU1-1 | This paper | N/A |
| pSUPERretropuro-shRNA STAU1-3 | This paper | N/A |
| pSUPERretropuro-shRNA BIRC3-1 | This paper | N/A |
| pSUPERretropuro-shRNA BIRC3-3 | This paper | N/A |
| pSUPERretropuro-shRNA RalA-2 | This paper | N/A |
| pSUPERretropuro-shRNA RalA-3 | This paper | N/A |
| pSUPERretropuro-shRNA IQGAP1-1 | This paper | N/A |
| pSUPERretropuro-shRNA IQGAP1-2 | This paper | N/A |
| pCR-Blunt-ARE | This paper | N/A |
| pCR-Blunt-SU | This paper | N/A |
| pCR-Blunt-LU | This paper | N/A |
| pCR-Blunt-LU1 | This paper | N/A |
| pCR-Blunt-antisense SU | This paper | N/A |
| pCR-Blunt-antisense LU | This paper | N/A |
| pCR-Blunt-antisense LU1 | This paper | N/A |
| pLKO.1-puro-shRNA IGF2BP1 | Sigma-Aldrich | MISSION® shRNA TRCN0000218799, TRCN0000230114 |
| pLKO.1-puro-shRNA Luciferase (control shRNA) | Sigma-Aldrich | MISSION® shRNA SHC007 |
| pX458-BIRC3 guide 3 | This paper | N/A |
| pX458-BIRC3 guide 4 | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | https://www.flowjo.com |
| Odyssey | LI-COR Biosciences | https://www.licor.com/bio/products/imaging_systems/odyssey/ |
| MaxQuant | Max Planck Institute of Biochemistry | version 1.5.1.0 |
| Scaffold 4 | Proteome Software | version 4.5, http://www.proteomesoftware.com/products/scaffold/ |
| MultiGauge | Fuji | |
| ZEN | ZEISS | https://www.zeiss.com/microscopy/int/downloads/zen.html |
| Other | ||
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Trypsin-EDTA (0.05%) | Fisher scientific | Cat# 25300062 |
| Opti-MEM I reduced-serum medium | Invitrogen | Cat# 31985-070 |
| Penicillin-streptomycin solution, 100X | Fisher scientific | Cat# 15070-063 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Protein A/G PLUS-agarose beads | Santa Cruz Biotechnology | Cat# sc-2003 |
| qScript cDNA SuperMix | Quanta Biosciences | Cat# 101414-106 |
| Nucleofactor V kit | Lonza Bioscience | Cat# VCA-1003 |
| Gibson assembly master mix | New England Biolabs | Cat# E2611L |
| GFP-trap_A beads | Chromotek | Cat# gta-100 |
| 2x Laemmli sample buffer | Sigma-Aldrich | Cat# S3401 |
| MES running buffer | Natural Diagnostics | Cat# NP0002 |
| RNase A | Sigma-Aldrich | Cat# R4642 |
| MES running buffer (20X) | Life technologies | Cat# NP0002 |
| MES running buffer (20X) | National Diagnostics | Cat# EC-868 |
| SILAC culture media | Cambridge Isotope Laboratories | Cat# CNLM-539-H-0.05 |
| SimplyBlue | Invitrogen | Cat# LC6065 |
| Odyssey blocking buffer | Li-Cor Biosciences | Cat# 927-40000 |
| MEGAscript T7 | Life technologies | Cat# AM1333 |
| RNaseOUT | Life technologies | Cat# 10777-019 |
| Dynabeads® MyOne™ Streptavidin C1 | Life technologies | Cat# 65002 |
| RNase A | Sigma-Aldrich | Cat# R4642 |
| Halt protease inhibitor cocktail | Thermo Fisher Scientific | Cat# 78439 |
| Phosphatase inhibitors 2 | Sigma-Aldrich | Cat# P5726 |
| Phosphatase inhibitors 3 | Sigma-Aldrich | Cat# P0044 |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and Virus Strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children's Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; and Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
Highlights:
The long BIRC3 3′UTR isoform is upregulated in leukemia
Quantitative mass spectrometry identifies 3′UTR-dependent BIRC3 protein interactors
The long 3′UTR is required for the formation of several BIRC3 protein complexes
BIRC3 is cancer-promoting through control of CXCR4 trafficking and B cell migration
Acknowledgements
We thank the MSKCC Proteomics Facility for performing the mass spectrometry experiments and the Molecular Cytology Facility for help with the imaging. We thank all members of the Mayr lab for helpful discussions and Dirk Remus and Sibylle Mitschka for critical reading of the manuscript. This work was funded by the NIH Director′s Pioneer Award (DP1-GM123454), the Damon Runyon Innovator Award, the Pershing Square Sohn Cancer Research Alliance, and the NCI Cancer Center Support Grant (P30 CA008748).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bamidele AO, Kremer KN, Hirsova P, Clift IC, Gores GJ, Billadeau DD, and Hedin KE (2015). IQGAP1 promotes CXCR4 chemokine receptor function and trafficking via EEA-1+ endosomes. The Journal of cell biology 210, 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovits BD, and Mayr C (2015). Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 522, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beug ST, Cheung HH, LaCasse EC, and Korneluk RG (2012). Modulation of immune signalling by inhibitors of apoptosis. Trends in immunology 33, 535–545. [DOI] [PubMed] [Google Scholar]
- Brumbaugh J, Di Stefano B, Wang X, Borkent M, Forouzmand E, Clowers KJ, Ji F, Schwarz BA, Kalocsay M, Elledge SJ, et al. (2018). Nudt21 Controls Cell Fate by Connecting Alternative Polyadenylation to Chromatin Signaling. Cell 172, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger M, Hartmann T, Krome M, Rawluk J, Tamamura H, Fujii N, Kipps TJ, and Burger JA (2005). Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood 106, 1824–1830. [DOI] [PubMed] [Google Scholar]
- Elvira G, Wasiak S, Blandford V, Tong XK, Serrano A, Fan X, del Rayo Sanchez-Carbente M, Servant F, Bell AW, Boismenu D, et al. (2006). Characterization of an RNA granule from developing brain. Molecular & cellular proteomics : MCP 5, 635–651. [DOI] [PubMed] [Google Scholar]
- Geisbrecht ER, and Montell DJ (2004). A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell 118, 111–125. [DOI] [PubMed] [Google Scholar]
- Gruber AR, Martin G, Muller P, Schmidt A, Gruber AJ, Gumienny R, Mittal N, Jayachandran R, Pieters J, Keller W, et al. (2014). Global 3' UTR shortening has a limited effect on protein abundance in proliferating T cells. Nature communications 5, 5465. [DOI] [PubMed] [Google Scholar]
- Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, Pogacic V, Villa A, Ehret S, Berridge G, et al. (2009). Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. The Journal of experimental medicine 206, 1957–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Yuan S, Wang Y, Fu Y, Ge Y, Ge Y, Lan X, Feng Y, Qiu F, Li P, et al. (2017). The role of alternative polyadenylation in the antiviral innate immune response. Nature communications 8, 14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Singh I, Tisdale S, Abdel-Wahab O, Leslie CS, and Mayr C (2018). Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 561, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lianoglou S, Garg V, Yang JL, Leslie CS, and Mayr C (2013). Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev 27, 2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, and Mayr C (2018). A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3'UTR-Mediated Protein-Protein Interactions. Cell 175, 1492–1506 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A (2016). Monitoring Chemokine Receptor Trafficking by Confocal Immunofluorescence Microscopy. Methods in enzymology 570, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C (2017). Regulation by 3'-Untranslated Regions. Annual review of genetics 51, 171–194. [DOI] [PubMed] [Google Scholar]
- Mayr C (2018). What Are 3' UTRs Doing? Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, and Bartel DP (2009). Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyraud V, Aushev VN, Hatzoglou A, Meunier B, Cascone I, and Camonis J (2012). RalA and RalB proteins are ubiquitinated GTPases, and ubiquitinated RalA increases lipid raft exposure at the plasma membrane. J Biol Chem 287, 29397–29405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Martindale JL, and Gorospe M (2016). Affinity Pulldown of Biotinylated RNA for Detection of Protein-RNA Complexes. Bio-protocol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-Zerilli MJ, Forster J, Parker H, Parker A, Rodriguez AE, Chaplin T, Gardiner A, Steele AJ, Collins A, Young BD, et al. (2014). ATM mutation rather than BIRC3 deletion and/or mutation predicts reduced survival in 11q-deleted chronic lymphocytic leukemia: data from the UK LRF CLL4 trial. Haematologica 99, 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, Chiaretti S, Del Giudice I, Fabbri G, Bruscaggin A, et al. (2012). Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood 119, 2854–2862. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, and Burge CB (2008). Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science 320, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature protocols 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Shiber A, Doring K, Friedrich U, Klann K, Merker D, Zedan M, Tippmann F, Kramer G, and Bukau B (2018). Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Lee SH, Sperling AS, Samur MK, Tai YT, Fulciniti M, Munshi NC, Mayr C, and Leslie CS (2018). Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nature communications 9, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Burge CB, and Bartel DP (2013). 3' UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res 23, 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D'Ambrogio A, Luscombe NM, and Ule J (2015). hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton LA, and Rosenquist R (2015). The complex interplay between cell-intrinsic and cell-extrinsic factors driving the evolution of chronic lymphocytic leukemia. Seminars in cancer biology 34, 22–35. [DOI] [PubMed] [Google Scholar]
- Villace P, Marion RM, and Ortin J (2004). The composition of Staufen-containing RNA granules from human cells indicates their role in the regulated transport and translation of messenger RNAs. Nucleic Acids Res 32, 2411–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KX, Tan L, Pellegrini M, Zipursky SL, and McEwen JM (2016). Rapid Changes in the Translatome during the Conversion of Growth Cones to Synaptic Terminals. Cell reports 14, 1258–1271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes with significant 3′UTR isoform change between normal and CLL B cells, Related to Figures 7B and S1A.
Table S2. BIRC3 protein interactors determined by SILAC-based MS, Related to Figure 3B and S3A.
Table S3. STAU1 protein interactors determined by SILAC-based MS, Related to Figure 5C.
Table S4. Oligonucleotide sequences, Related to STAR methods.