

Abstract
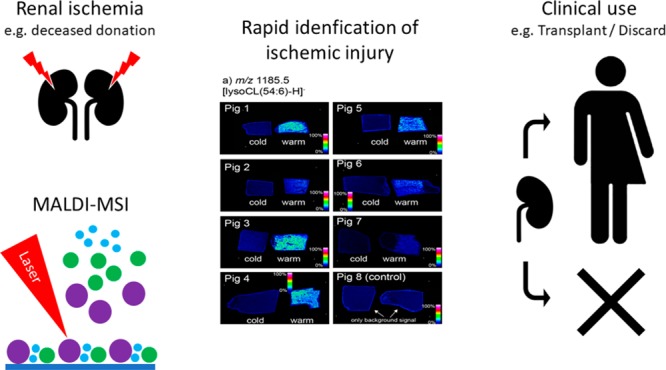
The increasing analytical speed of mass-spectrometry imaging (MSI) has led to growing interest in the medical field. Acute kidney injury is a severe disease with high morbidity and mortality. No reliable cut-offs are known to estimate the severity of acute kidney injury. Thus, there is a need for new tools to rapidly and accurately assess acute ischemia, which is of clinical importance in intensive care and in kidney transplantation. We investigated the value of MSI to assess acute ischemic kidney tissue in a porcine model. A perfusion model was developed where paired kidneys received warm (severe) or cold (minor) ischemia (n = 8 per group). First, ischemic tissue damage was systematically assessed by two blinded pathologists. Second, MALDI-MSI of kidney tissues was performed to study the spatial distributions and compositions of lipids in the tissues. Histopathological examination revealed no significant difference between kidneys, whereas MALDI-MSI was capable of a detailed discrimination of severe and mild ischemia by differential expression of characteristic lipid-degradation products throughout the tissue within 2 h. In particular, lysolipids, including lysocardiolipins, lysophosphatidylcholines, and lysophosphatidylinositol, were dramatically elevated after severe ischemia. This study demonstrates the significant potential of MSI to differentiate and identify molecular patterns of early ischemic injury in a clinically acceptable time frame. The observed changes highlight the underlying biochemical processes of acute ischemic kidney injury and provide a molecular classification tool that can be deployed in assessment of acute ischemic kidney injury.
Acute kidney injury (AKI) is associated with high morbidity and mortality and is most often caused by hypotension or hypoxia in cardiovascular or septic shock.1 AKI also occurs after kidney transplantation and its extent is associated with later graft function.2 After successful recovery of AKI there is an increasing incidence of chronic kidney disease (CKD), with increased risk for cardiovascular disease.3 At present, the pathophysiology of AKI is not fully understood, and good biomarkers to predict the severity of AKI are lacking.1
The aim of this study was to investigate the value of mass-spectrometry imaging (MSI) in the assessment of the extent of acute ischemic kidney damage. MSI is an innovative approach enabling the unlabeled molecular imaging of hundreds of molecules in a single experiment and can allow the correlation of localized molecular profiles to tissue state and morphology.4−7 MSI is now finding its way into clinical analyses because of its ability to reveal localized chemical profiles throughout tissue sections and discriminate between diseased and healthy tissue by virtue of these molecular patterns that are characteristic of cell and tissue types.4,8,9,7 Recent advances in instrumentation enable high-throughput tissue imaging and analysis times on a scale compatible with conventional histological analysis.10 This now opens up the possibility of using localized molecular information to aid in tissue diagnosis. Lipids present an exciting molecular class to target with MSI because of the relative ease of detection of many abundant lipid classes and their relatively simple and rapid sample preparation.11 In renal research, MSI has been applied to study localized sphingolipid changes in sphingomyelin-synthase-2-deficient mice and lipidomic changes after cisplatin chemotherapy. A recent study has also highlighted lipidomic changes associated with acute kidney injury.12−14
In this study, we confirm earlier findings that conventional histopathologic examination is not sufficient to accurately discriminate the extent of acute ischemic renal injury.15,16 In contrast, studying localized lipid profiles with matrix-assisted laser desorption/ionization–mass-spectrometry imaging (MALDI-MSI) provided accurate discrimination of cold and warm ischemia via observation of dramatically elevated levels of lysophospholipids in the case of severe ischemia, with a total analysis time of 1 h per tissue, including sample preparation. We propose this discriminative capacity opens up the door for molecular-guided quality assessment and can provide new insight into the pathophysiologic processes of ischemic kidney injury.
Experimental Section
Kidney Procurement and Ischemia Model
We used a paired porcine-kidney model to distinguish between ischemic injury in kidneys (see study design in Supporting Information Figure S1). We aimed to develop a system, that can be extrapolated to clinical transplantation, where in light of the increasing need of donor organs, kidneys of deceased donors with warm ischemia (i.e., in the donor) and cold ischemia (i.e., during transportation) are increasingly used. Kidneys were procured from a local slaughterhouse from eight pigs. Both kidneys of one pig were simultaneously procured 15 min after death by electroshock and exsanguination. Perirenal fat was removed and renal arteries were dissected, cannulated, and attached to an infusion bag. Kidneys were flushed 25 min after death with cold histidine–tryptophan–ketoglutarate preservation solution at 0–4 °C for 20 min. The start of perfusion was documented as the end of the warm-ischemia time (WIT) and start of cold-ischemia time (CIT). Simultaneously, topical cooling was performed using cold sodium chloride solution. Thereafter, both kidneys were stored in bags with sodium chloride solution and placed in a Styrofoam box with ice for transport to the laboratory (according to regular organ-transport protocols).
In our laboratory, kidneys were removed from cold storage, and after exactly 3 h of CIT, simultaneous hypothermic machine perfusion (HMP) was started using two Lifeport Kidney Transporters (model no. LKT-100-P; Organ Recovery Systems, Des Plaines, IL) and 0.5 L of Kidney Perfusion Solution 1 (KPS-1; Organ Recovery Systems, Des Plaines, IL) per kidney at a perfusion pressure of 40 mmHg, as is currently used in clinical-transplantation practice. Kidneys were hypothermically perfused for 1 h at 4 °C. Thereafter, they were flushed for 15 min to remove all intravascular content and Lifeport perfusion cassettes, and KPS-1 was discarded. Kidneys of seven of eight pairs were then randomly assigned to one of two type of perfusion: HMP for 4 h at 4 °C (cold or mild ischemia) or subnormothermic machine perfusion (SNMP) for 4 h at 28 °C (warm or severe ischemia). One pair was not perfused thereafter and directly biopsied (as described below) to serve as control tissue.
HMP was performed as described above but with the use of 1 L of KPS-1 with a mean temperature of 4 °C. For SNMP, a Lifeport Kidney Transporter was used. The reservoir, normally filled with ice water for HMP, was filled with water and continuously heated with a heat exchanger at a constant temperature of 35 °C. This led to a constant temperature of 28 °C for the KPS-1, in which the kidney was placed and perfused. Higher (normothermic) temperatures were not possible, as that would stop the perfusion machine from pumping. After 4 h of simultaneous machine perfusion (HMP or SNMP), wedge biopsies were taken. For the two kidneys that were not subject to the two groups, this was done directly after their first hour of machine perfusion (Supporting Information Figure S1). Two large (>1 cm2) wedges were cut in a manner such that both medullar and cortical renal tissue was included. One wedge biopsy was formalin fixed and paraffin embedded (FFPE) and stored for histopathologic examination. The other wedge biopsy was snap frozen using a 2-methylbutane solution and stored at −80 °C for MSI analysis (without the use of the OCT compound).
To test our presumption of different metabolic activity between groups and subsequent ischemic injury, machine perfusate was gathered during machine perfusion and tested for lactate and lactate dehydrogenase (LDH) as markers of metabolic activity and cellular injury. During the 4 h of HMP and SNMP, machine perfusate samples were taken at 10, 60, 120, 180, and 240 min. After withdrawal from the Lifeport sampling port, samples were centrifuged at 3000g for 3 min, aliquoted, and stored at −80 °C. After the samples were thawed, lactate was measured (mmol/L) using a blood-gas analyzer (GEM Premier 4000, Instrumentation Laboratory, Lexington, MA). Lactate dehydrogenase was measured (in U/L), using a Roche Cobas 8000 system (Roche Diagnostic International Ltd., Rotkreuz, Switzerland).
Histopathologic Scoring of Tissue Biopsies
FFPE tissues were sectioned at 4 μm, twice for every kidney. Subsequently they were stained with hematoxylin and eosin (H&E) or periodic acid–Schiff–diastase (PAS-D) according to standard protocols.17 Two nephropathologists (C.J.P.K. and L.M.H.), blinded for ischemia groups, performed histopathological examination of all biopsies using light microscopy. For each tissue section, 10 random fields of renal cortex containing at least 1 glomerulus were assessed at 200× magnification and scored (0–3 point scale: none, 0; mild, 0–30%; moderate, 30–60%; and severe, >60%) for five parameters associated with ischemic injury: tubular-cell necrosis, loss of brush border, edema, tubular casts, and blebs in tubular epithelial cells. A total injury score was calculated by adding the scores of all parameters.
Mass-Spectrometry Imaging (MSI)
Tissue Preparation
Wedge biopsies were collected from −80 °C storage, mounted on the specimen stage with H2O, sectioned at 12 μm at −21 °C using a Cryostat HM525 (Microm, Walldorf, Germany), thaw-mounted onto conductive indium–tin-oxide (ITO)-coated glass slides purchased from Delta Technologies (Loveland, CO, 4–8 Ω resistance), and dried under vacuum. On each slide, both kidneys of the same pig were mounted to ensure similar and comparable analysis during MSI. Norharmane matrix (Sigma-Aldrich, Zwijndrecht, The Netherlands) at 7 mg/mL in chloroform/methanol (2:1, v/v) was applied to sections using a SunCollect automatic pneumatic sprayer (Sunchrom GmbH, Friedrichsdorf, Germany). Fifteen layers were applied at the following flow rates: 10 μL/min for the first layer, 20 μL/min for the second layer, 30 μL/min for the third layer, and 40 μL/min for the fourth and subsequent layers.
Mass-Spectrometry Analysis
MALDI-MSI data were acquired on a Bruker rapifleX MALDI Tissuetyper (Bruker Daltonik GmbH, Bremen, Germany) in reflectron mode using a nominal acceleration potential of ±20 kV.10 Data were acquired in the m/z ranges of 400–1000 and 600–1600 for positive- and negative-ion mode, respectively. Mass calibration was performed before each analysis using red phosphorus clusters in positive- and negative-ion mode. Each tissue was imaged sequentially in positive- and negative-ion mode as recently described.18 Briefly, positive-ion data were first acquired using a 20 × 20 μm2 laser scan area and a 50 × 50 μm2 step size of the stage. After that, negative-ion data were acquired with identical parameters but with a 25 × 25 μm2 offset in stage position applied to produce an interlaced acquisition pattern. For each slide, the laser-focus position was optimized and 200 laser shots were acquired at each position. The total analysis time was approximately 1 h per tissue in both polarities, including sample preparation time. The identification of selected lipid species was based on both accurate-mass measurements and tandem-mass-spectrometry data using collision-induced dissociation acquired from adjacent tissues using an Orbitrap Elite mass spectrometer (Thermo Fisher, Bremen, Germany) coupled to a reduced-pressure MALDI-MSI source, as recently described.19 Data visualization and analysis was performed using FlexImaging 5.0 (Bruker Daltonik GmbH, Bremen, Germany), SCiLS lab 2016a (Bruker, Bremen, Germany), and Xcalibur 2.3 (Thermo Fisher, Bremen, Germany).
Histological Imaging of Tissue-Section Slides after MSI Analysis
After MSI, matrix was washed off using two 30 s submersions in 100% EtOH. Thereafter, H&E staining was performed using a rapid staining protocol with the use of a Leica linear stainer (Leica ST4020); the protocol included 10 s submersions in solutions in the following order: 2 × formaldehyde, 1 × H2O, 3 × hematoxylin, 1 × H2O, 1 × ammoniac water, 1 × tap water, 1 × eosin, 1 × tap water, 2 × 100% EtOH, and 2 × Xylene.
Statistical Analysis
Numeric variables are presented as means ± standard deviations (SDs) if approximately normally distributed and as medians with interquartile ranges (IQRs) otherwise. To measure the differences among perfusate concentrations at the measured time points (10, 60, 120, 180, and 240 min), we used the Wilcoxon signed-rank test. To test for differences in cold- and warm-ischemic injury by histological parameters, we used a Mann–Whitney-U test for all parameters scored, including the total score. All statistical analysis of MSI data was performed using SCiLS lab 2016a software. For ROC analysis, the mean spectrum from all 16 tissues was exported to mMass software.20 Peak picking was performed using a signal-to-noise threshold of 5 with no baseline corrections or smoothing applied. The generated peak list was then imported into SCiLS lab 2016a for all subsequent analysis using an interval width of ±0.25 Da. This was repeated for both positive- and negative-ion mode. ROC analysis was performed using the warm- and cold-ischemia tissues (seven tissues each per polarity) as two groups, and peaks were assigned as being discriminatory with AUCs >0.8 or <0.2.
Results and Discussion
Association of Ischemia Severity with Machine-Perfusate Markers and No Association with Histology
Ten minutes after the start of perfusion, lactate levels (mmol/L) in the perfusate were not different between HMP and SNMP kidneys (i.e., cold versus warm ischemia; median = 0.6 mmol/L (IQR, 0.3–0.7 mmol/L) vs 1.0 mmol/L (IQR, 0.4–1.0 mmol/L), respectively, P = 0.293). As sign of a higher metabolic activity, the flush-out of lactate was significantly higher in SNMP kidneys compared with in HMP kidneys over time (median lactate at 240 min HMP vs SNMP: 1.0 mmol/L (IQR, 0.9–1.1 mmol/L) vs 7.4 mmol/L (IQR, 5.2–9.0 mmol/L), P = 0.018). As sign of increased ischemic injury in SNMP kidneys, LDH concentrations (U/L) at baseline were not significantly different (median LDH at 10 min HMP vs SNMP: 5 U/L (IQR, 4–11 U/L) vs 11 U/L (IQR, 8–23 U/L), P = 0.236), but after 4 h of perfusion, they were (median LDH at 240 min HMP vs SNMP: 56 U/L (IQR, 41–62 U/L) vs 118 U/L (IQR, 61–130 U/L), P = 0.028). These results are also depicted in Supporting Information Figure S2.
A systematic histopathologic examination of the renal tissue sections by two blinded renal pathologists did not discriminate between the different types of ischemia. There were no significant differences found for scoring of tubular-cell necrosis, edema, tubular casts, or blebs in tubular epithelial cells, nor were differences found when combining these scores. One pathologist was able to find a significant difference between ischemic groups by scoring the loss of brush border (scores of HMP vs SNMP = 18 (IQR, 16–25) vs 25 (IQR, 23–29), P = 0.024). The results of this pathologist are depicted in Figure 1.
Figure 1.
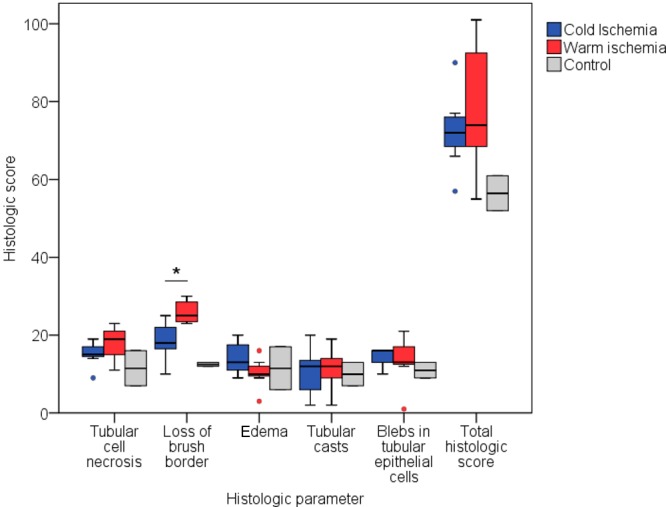
Comparison of histologic scores of cold and warm ischemia by a blinded pathologist. Five parameters were scored, and a total-histologic-score parameter was calculated by the sum of all five scored parameters. The Mann–Whitney-U test was applied to test for differences between groups. A P-value ≤0.05 was considered significant and annotated with an asterisk (*). *Median score for HMP vs SNMP: 18 (IQR, 16–25) vs 25 (IQR, 23–29), P = 0.024 for the loss of brush border.
We have tested the extent of ischemic kidney injury especially in a preclinical porcine model mimicking transplantation. At present, it is not possible to determine the exact viability of an ischemically damaged kidney and to estimate whether transplantation will lead to a successful outcome. There is no consensus on how to select kidneys suitable for transplantation, because many of the used selection criteria are only poor or moderate predictors of graft outcome.15,16,21−23 Normothermic machine perfusion, a promising organ-preservation strategy, is also used to assess kidneys before transplantation and is increasingly performed to assess AKI.24−26 In normothermic machine perfusion, extracorporeal membrane oxygenation is utilized to avoid warm ischemia. Preliminary results show that NMP may even recondition and repair ischemically damaged tissue.27 We used machine-perfusate markers to validate the presumed ischemic injury in our perfusion model. Lactate and LDH confirm ischemic injury and may therefore also have potential as viability markers. However, these two markers are often studied for their potential roles as viability markers and are indeed associated with ischemic injury and functional outcome, but they do not have predictive value to be used as clinical markers, as with most perfusate biomarkers thus far.28 At present, renal biopsies are increasingly used to assess the quality of the potential donor kidney, in which the extent of chronic histological damage is associated with long-term graft function.15−19 Assessment of acute ischemic damage in pretransplant procurement biopsies does not have clear additional value.16 This emphasizes the need for additional viability tests for acute kidney injury that can be used in transplantation. A possible test may be MSI.
Discrimination between Severe and Mild Ischemic Injury by MSI via Lipid-Degradation Products
In positive-ion-mode analysis of porcine tissues, phosphatidylcholines (PC) and sphingomyelins are mostly observed as mixtures of protonated, sodiated, and potassiated forms, a common observation in MSI. Negative-ion mode allows the detection of a wide variety of acidic lipids, including phosphatidylserines (PS), phosphatidylethanolamines (PE), phosphatidylglycerols (PG), phosphatidylinositols (PI), sulfatides (ST), and cardiolipins as predominantly deprotonated ([M–H]−) ions. The average positive- and negative-ion spectra acquired from the warm-ischemia, cold-ischemia, and control kidneys are provided as Supporting Information Figures S3 and S4. We performed a receiver–operator-characteristic (ROC) analysis to elucidate the discriminatory lipid MSI signals for ischemic injury in negative-ion mode between m/z 600 and 1600. In total, three monoisotopic m/z signals were found with area-under-the-curve (AUC) values greater than 0.8 (elevated in warm ischemia) which were attributed to the three monoisotopic lipids described below and listed in Table 1.
Table 1. Discriminatory m/z Values That Are Significantly Elevated (AUC > 0.8) in Ischemically Damaged Renal Tissuea.
| MALDI-ToF m/z | AUC | orbitrap m/z | lipid-sum composition | PPM m/z error |
|---|---|---|---|---|
| Negative-Ion Mode | ||||
| 619.3 | 0.85 | 619.28815 | [LPI(20:4)–H]− | –1.2 |
| 1185.8 | 0.89 | 1185.73349 | [MLCL(54:6)–H]− | –1.5 |
| 1207.8 | 0.85 | 1207.71537 | [MLCL(54:6)–2H+Na]− | –1.5 |
| Positive-Ion Mode | ||||
| 480.8 | 0.87 | 480.34436 | [LPC(O-16:1)+H]+ | –1.0 |
| 496.4 | 0.81 | 496.33935 | [LPC(16:0)+H]+ | –0.8 |
| 520.4 | 0.84 | 520.33924 | [LPC(18:2)+H]+ | –1.0 |
| 522.4 | 0.82 | 522.35495 | [LPC(18:1)+H]+ | –0.9 |
| 524.4 | 0.82 | 524.37059 | [LPC(18:0)+H]+ | –0.9 |
| 544.4 | 0.80 | 544.33805 | [LPC(18:1)+Na]+ | 1.3 |
| 546.4 | 0.82 | 546.35264 | [LPC(18:0)+Na]+ | –0.7 |
Only monoisotopic lipids are shown. Lipids were identified following ROC analysis of tissues exposed to cold and warm ischemia. No discriminative ions elevated in the cold-ischemic tissue were observed.
In negative-ion mode, an increase in ion signal between m/z 1180 and 1240 was observed for all tissues with induced warm-ischemic damage (Supporting Information Figure S5). In particular, ions observed at m/z 1185.8 and 1207.8 were elevated in the warm-ischemia tissue (Figure 2a). The discriminatory m/z 1185.8 ion was assigned by accurate mass to monolysocardiolipin [MLCL(54:6)–H]−, with the primary molecular species assigned to [MLCL(18:2_18:2_18:2)–H]− using tandem mass spectrometry. The m/z 1207.8 is assigned to the analogous sodium adduct [MLCL(18:2_18:2_18:2)–2H+Na]−. Interestingly, no corresponding decrease in the most abundant potential precursor to these MLCL species, namely, CL(72:8) as [CL(72:8)–H]− and [CL(72:8)–2H+Na]− at m/z 1447.9 and 1669.9, respectively, was observed, although we note that in the absence of internal standards, quantitative comparisons of ion abundance are difficult (Supporting Information Figure S6). Furthermore, in negative-ion mode, lysophosphatidylinositol (LPI) as [LPI(20:4)–H]− at m/z 619.3 was also elevated in tissues exposed to warm ischemia (Supporting Information Figure S7). No ions elevated in cold-ischemic or control tissues relative to in warm-ischemic tissues were observed. Ischemia-related elevation of lysophosphatidylcholine (LPC) lipids was also observed in positive-ion mode (Table 1 and Supporting Information Figure S8). The distribution of the most abundant LPC, which was observed at m/z 496.3 ([LPC(16:0)+H]+), throughout all 16 tissues is provided in Figure 2.
Figure 2.

MS images of discriminatory lipids: mass-spectrometry-imaging data acquired at 50 μm resolution revealing spatial distributions of the lysolipids (a) [MLCL(54:6)–H]− and (b) [LPC(16:0)+H]+ through all 16 tissues (7 warm ischemia, 7 cold ischemia, and 2 control). All images are normalized to the total ion count.
MSI results also suggested a spatial heterogeneity in the degradation of cardiolipins in ischemic tissue. To further investigate this we imaged a warm-ischemic tissue at 20 μm spatial resolution (Figure 3). The distribution of the [M–H]− ions of CL(72:8), MLCL(54:6), and the sulfatide ST(d42:1(2OH)) are shown in red, green, and blue, respectively. Coregistration of the post-MSI H&E-stained tissue (Figure 3b–d) revealed the sulfatide signal at m/z 906.5 to be localized in the area of the medullary rays (arrows in Figure 3a) and in the juxtaglomerular areas (asterisks in Figure 3e,f). Interestingly, the ischemia-related MLCL(54:6)-degradation product is not exclusively colocalized with its most probable precursor, CL(72:8), suggesting that the ischemia-related breakdown of mitochondrial-specific cardiolipin occurs to greater extent in distinct tubular areas of the renal cortex (Figure 3b–d).
Figure 3.
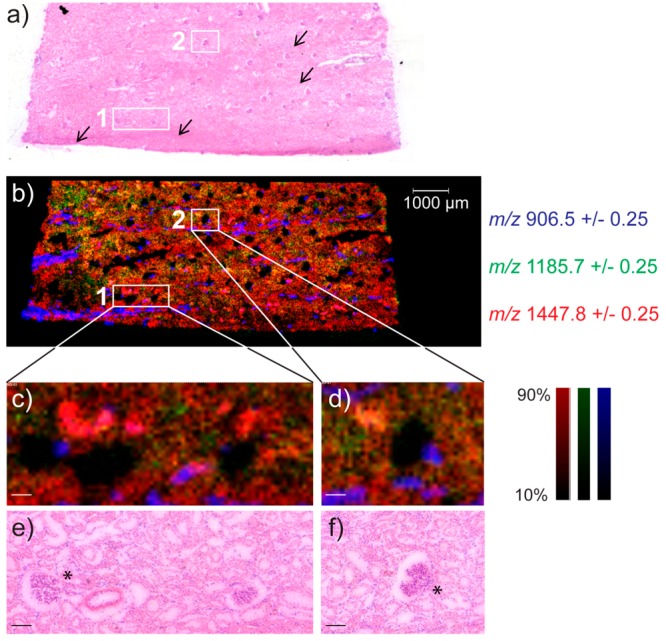
Lipid degradation and distribution in renal tissue after warm-ischemic injury. The post-MSI H&E-stained tissue (a,e,f) was used to generate the MSI data (b–d). (b) Mass-spectrometry-imaging data acquired at 20 μm resolution revealing spatial distributions of [ST(d42:1(2OH))–H]− (m/z 906.5, blue), [MLCL(54:6)–H]− (m/z 1185.7, green), and [CL(72:8)–H]− (m/z 1447.8, red). (c,d) Two regions of interest, marked as (c) 1 and (d) 2, enlarged to show the heterogeneous MLCL(54:6) distribution, relative to that of its main expected precursor, CL(72:8), in distinct tubular cortical areas. All images are normalized to the total ion count. Medullary rays are indicated with black arrows in (a). Asterisks (*) in (e) and (f) indicate juxtaglomerular areas.
Potential of MSI as a Viability Test and as a Way To Gain New Insights into the Pathophysiology of Ischemic Injury
In a porcine donor-kidney model, we show that it is feasible to rapidly visualize ischemic damage in renal tissue on the basis of local lipid profiles. We are able to visualize and identify alterations in the lipidome between kidneys with mild and severe ischemic injury using MSI. The advantage of such molecular analysis likely arises by detection of the rapid biochemical alterations occurring during acute tissue injury. This short time frame may not allow significant histological changes to be induced, leading to the poor accuracy of histological identification of acute ischemia (e.g., Figure 1). Using MALDI-MSI in this context has two distinct advantages. First, by rapid assessment of ischemically damaged kidneys using MSI, it may provide a practical tool to assist in viability assessment, and therefore, it holds the potential to improve donor-kidney selection. With the development of rapid MALDI-MSI instrumentation and automated matrix application, such as those employed in this study, a single tissue could be analyzed with 50 μm spatial resolution in 30 min in both positive- and negative-ion modes, which is within the critical time frame needed for graft-viability assessments in clinical transplantation, for example. The total analysis time, including matrix application, is approximately 1 h. Second, in this study, we have identified multiple discriminative molecules of ischemic injury not yet described in AKI. In particular, a suite of lysophospholipids have been identified as significantly elevated in severe ischemia compared with in mild ischemia. The source of these lysolipids is likely lipid degradation by increased activation of phospholipase A1 (PLA1) or A2 (PLA2) enzymes, with the latter in particular having been previously shown to exhibit elevated activation during ischemia.29−33 Although much more is known about the PLA2 family of enzymes, the differentiation of PLA1- and PLA2-driven hydrolysis is not generally possible using MSI or indeed conventional MS-based lipidomics.34 MSI typically lacks the isomeric specificity required to localize the sn positions of the acyl chains on both the precursor and the lyso-lipid products, which is essential for differentiating PLA1- and PLA2-based processes. Thus, our results likely reflect the cumulative effects of all PLA forms. The observation of degradation of both mitochondrial lipids (cardiolipin) and membrane lipids (PC, PI, and PE; also observed in the high-resolution Orbitrap analysis) may be reflective of different forms and activations of PLA1–PLA2 in renal ischemia that lead to the disruption of cell membranes.35−37 Therefore, MSI shows potential in identifying new molecules, which may play an important role in AKI assessment. Furthermore, it offers new insights in molecular pathways of ischemic injury and may even reveal novel targets for diagnostics and therapeutics that reduce ischemic injury.
Identification of Lipid-Degradation Products as Ischemic Markers by MSI
Using MSI, we were able to identify surrogate markers of ischemic processes such as MLCL, LPC, and LPI in kidney tissue with severe ischemia. Interference with cardiolipin activity has already shown to be beneficial in chronic renal damage in tubular and endothelial cells in the context of ischemia reperfusion as well as in the context of revascularization in renal-artery stenosis.38,38 Cardiolipins are located in the inner membranes of mitochondria and are needed for protection of mitochondrial cristae and ATP generation. Ischemia reperfusion-induced mitochondrial injury plays a pivotal role in organ viability in transplantation40 and is associated with kidney injury.41,42 Next to the kidney, cardiolipins are also involved in ischemia reperfusion injury in other organs, like the brain,32 heart,43 and liver.44 Moreover, the action of mitochondrial-specific PLA2 forms has been shown to result in further lipid mediators in ischemia that have further detrimental effects on tissue quality.45 We now are the first to rapidly measure and visualize elevated levels of MLCL in ischemic renal tissue in a clinically acceptable time frame, which may pave to the way to cardiolipin measurements in, for instance, assessment of graft viability.
Other important lysolipids that we identified in association with severe renal ischemia are LPCs and LPIs. Increased expression of LPCs has been described in other ischemic tissue, like liver tissue (including graft biopsies of DCD donors)46 and heart tissue.47 In renal ischemia, proximal tubules show increased expression of PI on their apical membranes.48,49 Elevated LPC levels were also found in renal ischemia.50,51 The function of LPCs is not fully understood, but in ischemic conditions they can, for example, increase tubular secretion of the pro-fibrotic factors PDGF-B and CTGF.52 LPCs are hydrolyzed from PCs following removal of fatty acyl chains by PLA1 or PLA2 from the sn-1 and sn-2 positions, respectively. PLA2 activity has been described as being altered in renal ischemia reperfusion injury, which can lead to differential cytotoxic or cytoprotective effects on renal tubular cells.30,35−37 Thus, we have visualized expression of compounds in ischemic processes, which are confirmed by the literature,38,39,48−51 in an early stage of loss of organ viability within a very short time frame in which morphological changes are difficult to observe. This holds the potential to aid in assessment of AKI, risk prediction for CKD, and ultimately the improvement of outcomes.
Limitations of the Study Model
Within the current porcine model, we compared two degrees of ischemic injury that were based on clinical transplantation: 4 h of cold perfusion (mild ischemia) versus 4 h of subnormothermic (warm) perfusion (severe ischemia). The difference in metabolic activity and cell injury is shown by our perfusate analyses. The clinical value of the current study needs further investigation in animal models of AKI in relation to outcome and in human kidney biopsies. Thus, future studies are needed to study alterations in human biopsies in recipients for whom the transplant outcome is known. Furthermore, in human biopsies, chronic histological damage in relation to inflammation and fibrosis may also be present (in contrast to the young pigs we studied), and thus lipid patterns may differ. Also, studies with MSI in relation to the pathophysiology of the AKI-to-CKD transition will increase our pathophysiological insights herein and provide novel diagnostic strategies to decrease kidney failure.
Conclusions
In conclusion, we have shown that MSI shows great potential as new tool to assess ischemic injury. In this study, we were able to discriminate moderately and severely ischemically injured kidneys for every pair analyzed. MSI has the capacity of identifying discriminatory molecular patterns of ischemic kidney injury in a clinically acceptable time frame. In addition to histopathological examination, it provides useful information on the biochemical changes during ischemia relative to tissue morphology. Furthermore, we have identified lipid molecules using MSI, which may potentially serve as markers for acute kidney injury and transplant-outcome predictions.
Acknowledgments
This work has been made possible with the financial support of the Dutch province of Limburg through the LINK program.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b05521.
Study time-line and setup; renal flush-out of lactate and LDH during machine perfusion; average positive-ion spectra from warm-ischemia, cold-ischemia, and control tissues; average negative-ion spectra from warm-ischemia, cold-ischemia, and control tissues; average negative-ion MALDI mass spectra in the m/z ranges 1180–1300, 1420–1540, and 610–630; and average positive-ion MALDI mass spectra in the m/z range 490–530 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Parikh C. R.; Puthumana J.; Shlipak M. G.; Koyner J. L.; Thiessen-Philbrook H.; McArthur E.; Kerr K.; Kavsak P.; Whitlock R. P.; Garg A. X.; Coca S. G. J. Am. Soc. Nephrol. 2017, 28, 3699–3707. 10.1681/ASN.2017010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kox J.; Moers C.; Monbaliu D.; Strelniece A.; Treckmann J.; Jochmans I.; Leuvenink H.; Van Heurn E.; Pirenne J.; Paul A.; Ploeg R. Transplantation 2018, 102, 1344–1350. 10.1097/TP.0000000000002188. [DOI] [PubMed] [Google Scholar]
- Basile D. P. Kidney Int. 2007, 72, 151–156. 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- Aichler M.; Walch A. Lab. Invest. 2015, 95, 422–431. 10.1038/labinvest.2014.156. [DOI] [PubMed] [Google Scholar]
- Lalowski M.; Magni F.; Mainini V.; Monogioudi E.; Gotsopoulos A.; Soliymani R.; Chinello C.; Baumann M. Nephrol., Dial., Transplant. 2013, 28, 1648–1656. 10.1093/ndt/gft008. [DOI] [PubMed] [Google Scholar]
- Chughtai K.; Heeren R. M. Chem. Rev. 2010, 110, 3237–3277. 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell L. A.; Heeren R. M. Mass Spectrom. Rev. 2007, 26, 606–643. 10.1002/mas.20124. [DOI] [PubMed] [Google Scholar]
- Schwamborn K.; Caprioli R. M. Nat. Rev. Cancer 2010, 10, 639–646. 10.1038/nrc2917. [DOI] [PubMed] [Google Scholar]
- Addie R. D.; Balluff B.; Bovee J. V.; Morreau H.; McDonnell L. A. Anal. Chem. 2015, 87, 6426–6433. 10.1021/acs.analchem.5b00416. [DOI] [PubMed] [Google Scholar]
- Ogrinc Potocnik N.; Porta T.; Becker M.; Heeren R. M.; Ellis S. R. Rapid Commun. Mass Spectrom. 2015, 29, 2195–2203. 10.1002/rcm.7379. [DOI] [PubMed] [Google Scholar]
- Zemski Berry K. A.; Hankin J. A.; Barkley R. M.; Spraggins J. M.; Caprioli R. M.; Murphy R. C. Chem. Rev. 2011, 111, 6491–6512. 10.1021/cr200280p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M.; Wakabayashi M.; Shimizu Y.; Yoshioka T.; Higashino K.; Numata Y.; Okuda T.; Zhao S.; Sakai S.; Igarashi Y.; Kuge Y. PLoS One 2016, 11, e0152191 10.1371/journal.pone.0152191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gordaliza E.; Esteban-Fernandez D.; Lazaro A.; Humanes B.; Aboulmagd S.; Tejedor A.; Linscheid M. W.; Gomez-Gomez M. M. Talanta 2017, 164, 16–26. 10.1016/j.talanta.2016.11.026. [DOI] [PubMed] [Google Scholar]
- Rao S.; Walters K. B.; Wilson L.; Chen B.; Bolisetty S.; Graves D.; Barnes S.; Agarwal A.; Kabarowski J. H. American journal of physiology. Renal physiology 2016, 310, F1136–1147. 10.1152/ajprenal.00100.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. J.; Wetmore J. B.; Crary G. S.; Kasiske B. L. Am. J. Transplant. 2015, 15, 1903. 10.1111/ajt.13213. [DOI] [PubMed] [Google Scholar]
- Hall I. E.; Reese P. P.; Weng F. L.; Schroppel B.; Doshi M. D.; Hasz R. D.; Reitsma W.; Goldstein M. J.; Hong K.; Parikh C. R. Clin. J. Am. Soc. Nephrol. 2014, 9, 573–582. 10.2215/CJN.08270813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijnen B. F.; Nelissen J.; van Essen H.; Fazzi G. E.; Cohen Tervaert J. W.; Peutz-Kootstra C. J.; Mullins J. J.; Schalkwijk C. G.; Janssen B. J.; Struijker-Boudier H. A. PLoS One 2013, 8, e57815 10.1371/journal.pone.0057815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S. R.; Cappell J.; Potocnik N. O.; Balluff B.; Hamaide J.; Van der Linden A.; Heeren R. M. Analyst 2016, 141, 3832–3841. 10.1039/C6AN00169F. [DOI] [PubMed] [Google Scholar]
- Belov M. E.; Ellis S. R.; Dilillo M.; Paine M. R. L.; Danielson W. F.; Anderson G. A.; de Graaf E. L.; Eijkel G. B.; Heeren R. M. A.; McDonnell L. A. Anal. Chem. 2017, 89, 7493–7501. 10.1021/acs.analchem.7b01168. [DOI] [PubMed] [Google Scholar]
- Strohalm M.; Hassman M.; Kosata B.; Kodicek M. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. 10.1002/rcm.3444. [DOI] [PubMed] [Google Scholar]
- Hoogland E. R.; de Vries E. E.; Christiaans M. H.; Winkens B.; Snoeijs M. G.; van Heurn L. W. Transplantation 2013, 95, 603–610. 10.1097/TP.0b013e31827908e6. [DOI] [PubMed] [Google Scholar]
- Doshi M. D.; Reese P. P.; Hall I. E.; Schroppel B.; Ficek J.; Formica R. N.; Weng F. L.; Hasz R. D.; Thiessen Philbrook H.; Parikh C. Transplantation 2017, 101, 1125. 10.1097/TP.0000000000001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh C. R.; Hall I. E.; Bhangoo R. S.; Ficek J.; Abt P. L.; Thiessen-Philbrook H.; Lin H.; Bimali M.; Murray P. T.; Rao V.; Schroppel B.; Doshi M. D.; Weng F. L.; Reese P. P. Am. J. Transplant. 2016, 16, 1526. 10.1111/ajt.13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochmans I.; Nicholson M. L.; Hosgood S. A. Curr. Opin. Organ Transplant. 2017, 22, 260–266. 10.1097/MOT.0000000000000405. [DOI] [PubMed] [Google Scholar]
- De Deken J.; Kocabayoglu P.; Moers C. Curr. Opin. Organ Transplant. 2016, 21, 294. 10.1097/MOT.0000000000000306. [DOI] [PubMed] [Google Scholar]
- Hosgood S. A.; Thompson E.; Moore T.; Wilson C. H.; Nicholson M. L. Br. J. Surg. 2018, 105, 388–394. 10.1002/bjs.10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRito J. R.; Hosgood S. A.; Tietjen G. T.; Nicholson M. L. Am. J. Transplant. 2018, 18, 2400–2408. 10.1111/ajt.14963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Smaalen T. C.; Hoogland E. R.; van Heurn L. W. Curr. Opin. Organ Transplant. 2013, 18, 168–173. 10.1097/MOT.0b013e32835e2a1b. [DOI] [PubMed] [Google Scholar]
- Liapis H.; Gaut J. P.; Klein C.; Bagnasco S.; Kraus E.; Farris A. B.; Honsova E.; Perkowska-Ptasinska A.; David D.; Goldberg J.; Smith M.; Mengel M.; Haas M.; Seshan S.; Pegas K. L.; Horwedel T.; Paliwa Y.; Gao X.; Landsittel D.; Randhawa P. Am. J. Transplant. 2017, 17, 140. 10.1111/ajt.13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre J. V. Kidney Int. 1993, 43, 1160–1178. 10.1038/ki.1993.163. [DOI] [PubMed] [Google Scholar]
- Adibhatla R. M.; Hatcher J. F.; Dempsey R. J. Antioxid. Redox Signaling 2003, 5, 647–654. 10.1089/152308603770310329. [DOI] [PubMed] [Google Scholar]
- Ji J.; Baart S.; Vikulina A. S.; Clark R. S.; Anthonymuthu T. S.; Tyurin V. A.; Du L.; St Croix C. M.; Tyurina Y. Y.; Lewis J.; Skoda E. M.; Kline A. E.; Kochanek P. M.; Wipf P.; Kagan V. E.; Bayir H. J. Cereb. Blood Flow Metab. 2015, 35, 319–328. 10.1038/jcbfm.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg J. M. Kidney Int. 1991, 39, 476–500. 10.1038/ki.1991.58. [DOI] [PubMed] [Google Scholar]
- Porta Siegel T.; Ekroos K.; Ellis S. R.. Angew. Chem., in press, 2019, 10.1002/ange.201812698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H.; Nemenoff R. A.; Gronich J. H.; Bonventre J. V. J. Clin. Invest. 1991, 87, 1810–1818. 10.1172/JCI115202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zager R. A.; Schimpf B. A.; Gmur D. J.; Burke T. J. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 8297–8301. 10.1073/pnas.90.17.8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V. D.; Cieslinski D. A.; Humes H. D. J. Clin. Invest. 1988, 82, 1098–1105. 10.1172/JCI113666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirin A.; Li Z.; Zhang X.; Krier J. D.; Woollard J. R.; Zhu X. Y.; Tang H.; Herrmann S. M.; Lerman A.; Textor S. C.; Lerman L. O. Hypertension 2012, 60, 1242–1249. 10.1161/HYPERTENSIONAHA.112.199919. [DOI] [PubMed] [Google Scholar]
- Liu S.; Soong Y.; Seshan S. V.; Szeto H. H. American journal of physiology. Renal physiology 2014, 306, F970–980. 10.1152/ajprenal.00697.2013. [DOI] [PubMed] [Google Scholar]
- Jassem W.; Heaton N. D. Kidney Int. 2004, 66, 514–517. 10.1111/j.1523-1755.2004.761_9.x. [DOI] [PubMed] [Google Scholar]
- Hall A. M.; Schuh C. D. Curr. Opin. Nephrol. Hypertens. 2016, 25, 355–362. 10.1097/MNH.0000000000000228. [DOI] [PubMed] [Google Scholar]
- Ralto K. M.; Parikh S. M. Semin. Nephrol. 2016, 36, 8–16. 10.1016/j.semnephrol.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran S.; Kurian G. A. Biomed. Pharmacother. 2017, 92, 7–16. 10.1016/j.biopha.2017.05.042. [DOI] [PubMed] [Google Scholar]
- Martens J. C.; Keilhoff G.; Halangk W.; Wartmann T.; Gardemann A.; Page I.; Schild L. Mol. Cell. Biochem. 2015, 400, 253–263. 10.1007/s11010-014-2282-1. [DOI] [PubMed] [Google Scholar]
- Tyurina Y. Y.; Poloyac S. M.; Tyurin V. A.; Kapralov A. A.; Jiang J.; Anthonymuthu T. S.; Kapralova V. I.; Vikulina A. S.; Jung M. Y.; Epperly M. W.; Mohammadyani D.; Klein-Seetharaman J.; Jackson T. C.; Kochanek P. M.; Pitt B. R.; Greenberger J. S.; Vladimirov Y. A.; Bayir H.; Kagan V. E. Nat. Chem. 2014, 6, 542–552. 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Casas-Ferreira A. M.; Ma Y.; Sen A.; Kim M.; Proitsi P.; Shkodra M.; Tena M.; Srinivasan P.; Heaton N.; Jassem W.; Legido-Quigley C. Sci. Rep. 2016, 5, 17737. 10.1038/srep17737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Bai N.; So J.; Laher I.; MacLeod K. M.; Rodrigues B. J. Mol. Cell. Cardiol. 2009, 47, 112–120. 10.1016/j.yjmcc.2009.03.026. [DOI] [PubMed] [Google Scholar]
- Molitoris B. A.; Falk S. A.; Dahl R. H. J. Clin. Invest. 1989, 84, 1334–1339. 10.1172/JCI114302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitoris B. A.; Meyer C.; Dahl R.; Geerdes A. American journal of physiology 1993, 264, F907–916. 10.1152/ajprenal.1993.264.5.F907. [DOI] [PubMed] [Google Scholar]
- Matthys E.; Patel Y.; Kreisberg J.; Stewart J. H.; Venkatachalam M. Kidney Int. 1984, 26, 153–161. 10.1038/ki.1984.149. [DOI] [PubMed] [Google Scholar]
- Cho K.; Min S. I.; Ahn S.; Min S. K.; Ahn C.; Yu K. S.; Jang I. J.; Cho J. Y.; Ha J. J. Proteome Res. 2017, 16, 2877–2886. 10.1021/acs.jproteome.7b00167. [DOI] [PubMed] [Google Scholar]
- Geng H.; Lan R.; Singha P. K.; Gilchrist A.; Weinreb P. H.; Violette S. M.; Weinberg J. M.; Saikumar P.; Venkatachalam M. A. Am. J. Pathol. 2012, 181, 1236–1249. 10.1016/j.ajpath.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.