

Abstract
Motivation
Metagenomics is a powerful tool for assaying the DNA from every genome present in an environment. Recent advances in bioinformatics have enabled the rapid assembly of near-complete metagenome-assembled genomes (MAGs), and there is a need for reproducible pipelines that can annotate and characterize thousands of genomes simultaneously, to enable identification and functional characterization.
Results
Here we present MAGpy, a scalable and reproducible pipeline that takes multiple genome assemblies as FASTA and compares them to several public databases, checks quality, suggests a taxonomy and draws a phylogenetic tree.
Availability and implementation
MAGpy is available on github: https://github.com/WatsonLab/MAGpy.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Discovering and studying microbes in the environment have been a goal of genomic technologies for many years (Brodie et al., 2006; Watson et al., 2007), but advances in DNA sequencing (Goodwin et al., 2016; Loman and Watson, 2015; Watson, 2014) have enabled a revolution in metagenomics that has accelerated this area of research. Metagenomics refers to the whole-genome investigation of every organism within a particular environment, and is often used in microbiome studies to investigate changes in the taxonomic and functional profile of samples of interest. This method of simultaneously quantifying taxonomic and functional structure has been used in studies of age and geography in the human gut (Yatsunenko et al., 2012), release of carbon due to permafrost thawing (Mackelprang et al., 2011), the environmental impact and feed efficiency of animal agriculture (Roehe et al., 2016; Wallace et al., 2015), environmental characterization of Earth’s oceans (Venter et al., 2004) and the extraction of industrially and commercially relevant enzymes from environmental samples (Roumpeka et al., 2017).
Metagenomics also offers the ability to assemble near-complete and draft microbial genomes without the need for culture. Such ‘metagenomic binning’ approaches involve the assembly of metagenomic sequence reads into contigs followed by clustering, or binning, of contigs into putative genomes, called metagenome-assembled genomes (MAGs) (Bowers et al., 2017). Recently, we have used this technique to assemble complete and draft genomes from the cattle rumen (Stewart et al., 2018), and in this manuscript we present MAGpy, the pipeline we used to characterize the 913 genomes presented in that paper.
A major challenge in the analysis of MAGs is that researchers are often presented with hundreds or thousands of putative genomes, which need to be annotated, characterized, placed within a phylogenetic tree and assigned a putative function or role. This is further complicated by the fact that many of the putative genomes do not have close relatives with good quality reference genomes, making comparative genomics almost impossible.
Here we present MAGpy (pronounced ‘magpie’), a reproducible pipeline for the characterisation of MAGs using open source and freely available bioinformatics software. MAGpy is implemented as a Snakemake (Koster and Rahmann, 2012) pipeline, enabling reproducible analyses, extensibility, integration with high-performance-compute clusters and restart capabilities. MAGpy annotates the genomes, predicts putative protein sequences, compares the MAGs to multiple genomic, proteomic and protein family databases, produces several reports and draws a taxonomic tree. We demonstrate the utility of MAGpy on a subset of 800 bacterial and archaeal MAGs recently published by Parks et al. (Parks et al., 2017).
2 Comparison to other tools
The aim of MAGpy is to assist researchers in characterizing hundreds or thousands of MAGs, specifically to help researchers identify the likely taxonomy of each MAG, and it is the pipeline we use for characterization of rumen microbes assembled from metagenomes (Stewart et al., 2018). Few other tools have similar aims or scope. CheckM predicts the taxonomic lineage of each MAG as an initial step in testing MAG quality, and this evidence is incorporated into MAGpy. PhyloPhlAn enables researchers to place any genome(s) into the tree of life, which can assist in identification. Again, PhyloPhlAn is run as part of the MAGpy pipeline.
Generic genome and metagenome annotation tools exist: Prokka (Seemann, 2014) is a genome annotation tool that can be installed locally and which can annotate microbial genomes and prepare them for submission to GenBank; whereas PATRIC (Wattam et al., 2014), RAST (Aziz et al., 2008), MG-RAST (Keegan et al., 2016), Microscope (Vallenet et al., 2009) and IMG/M (Chen et al., 2017) are online tools that provide services such as genome and metagenome annotation. The focus of these tools is on annotation—i.e. identifying the location and likely function of genes and proteins. Whilst this information can be used to identify the likely taxonomy of a newly assembled genome or MAG, it is not their primary focus. The focus of MAGpy is not (meta)genome annotation per se; rather we wish to leverage genome sequence data and predicted protein sequences to help identify the closest sequenced relative to each MAG; we want to enable this as a local analysis and we want to do this at scale.
Proteins are more conserved and can provide matches to more distant relatives. MAGpy uses Prodigal to predict proteins, a similar approach to Prokka. Mash (Ondov et al., 2016) and Sourmash (Brown and Irber, 2016) are relatively new tools that use MinHash distances to compare massive sequence datasets rapidly. Both enable novel genomes to be compared to tens of thousands of existing genomes in the public databases. We integrate Sourmash into MAGpy to enable comparison of MAGs to over 100 000 public genomes in GenBank.
3 Materials and methods
MAGpy makes use of Snakemake to define an analysis workflow for MAGs based on open source and freely available bioinformatics software. First, CheckM (Parks et al., 2015) is run, which uses a set of pre-computed core genes to assess the completeness and contamination of MAGs. CheckM also attempts to assign a taxonomic level to the MAGs, though in our experience this is often a conservative estimate. In tandem, MAGpy predicts the protein coding sequences of MAGs using Prodigal (Hyatt et al., 2010). DIAMOND (Buchfink et al., 2015) BLASTP is used to compare the proteins to UniProt (UniProt Consortium, 2018). This has multiple purposes—the hits from UniProt provide a form of annotation of the putative proteins and may predict function; many MAGs may show little similarity to published genomes at the DNA level, but as proteins are more conserved, protein hits can help define the closest sequenced genome; and the length of the predicted protein and that protein’s hits can be used to detect truncated genes and proteins in the MAG annotation. Reports of the DIAMOND results at the level of the MAG and for each contig within each MAG are produced. The proteins are also compared to protein families in Pfam (Finn et al., 2014) using PfamScan; and to create a tree using PhyloPhlAn (Segata et al., 2013), which is subsequently visualised using GraPhlAn (Asnicar et al., 2015). The MAG genome sequences are also compared to over 100 000 public genomes using MinHash signatures as implemented in Sourmash (Brown and Irber, 2016).
4 Results and discussion
We applied MAGpy to 800 Bacterial and Archaeal MAGs from Parks et al. (Parks et al., 2017). The CheckM report [which uses Ete3 (Huerta-Cepas et al., 2016) to expand the taxonomic prediction] can be seen in Supplementary Table S1, the Sourmash report in Supplementary Table S2 and the Uniprot report in Supplementary Table S3. The PhyloPhlAn tree can be seen in Figure 1 . Specific examples reveal the strengths of each approach. Whilst CheckM predicts a lineage of s__algicola for UBA6511, the UniProt results show 3403 (86%) of that genome’s 3945 predicted proteins have a top hit to Maribacter dokdonensis DSW-8 with an average similarity of 95.78%. The Sourmash results (Supplementary Table S2) also predict a strong hit to Maribacter. On many occasisons, CheckM is only able to predict a lineage of k__Bacteria, as in the case of UBA3429. However, both the UniProt and Sourmash results show a strong similarity of this genome to Thermus thermophiles HB8, providing strain-level resolution where CheckM fails.
Fig. 1.
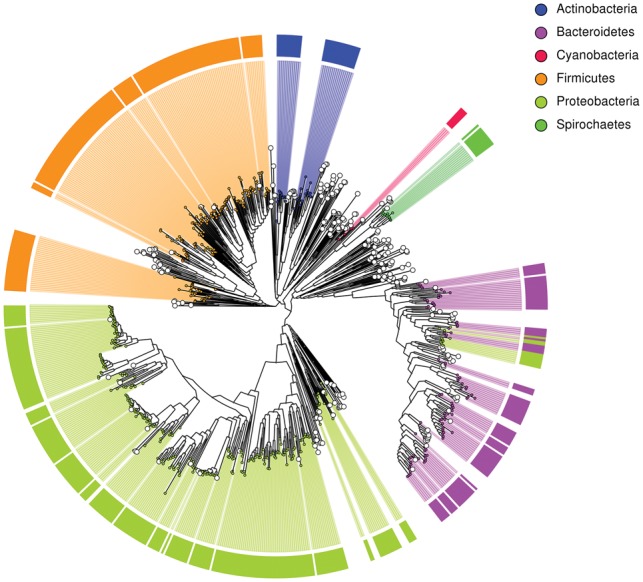
Phylogenetic tree of 800 MAGs created using PhyloPhlAn and produced by MAGpy
The outputs of MAGpy can also be used to identify potential chimeric MAGs. As well as producing a MAG-level report for the UniProt comparisons, a contig-level report is also produced for each MAG. This report includes the number of proteins predicted for each contig, and the most popular genus and species for those proteins from the diamond search. Supplementary Table S4 shows a contig-level report for UBA7370, a high quality MAG. Most contigs show very high protein-level similarity to the same genus (‘Synechococcus’) and species (‘Synechococcus sp. KORDI-49’). There are only two exceptions, with one contig showing similarity to ‘Synechococcus sp. (strain WH8103)’ and another hitting the genus ‘uncultured’. Deeper examination shows these to come from hits to ‘uncultured marine type-A Synechococcus’.
In comparison, Supplementary Table S5 shows a contig-level report from UBA6779. In this MAG, many of the contigs show high protein-level similarity to genus ‘Zunongwangia’ and species ‘Zunongwangia profunda (strain DSM 18752/CCTCC AB 206139/SM-A87)’; however, there are also contigs with high-similarity to Salegentibacter and Leeuwenhoekiella, and, towards the bottom of the table, multiple contigs with low protein-level similarity to Gramella, Mesonia, Legionella and various others. Whilst many of these are from the same family (Flavobacteriaceae), there are certainly signs this is a chimeric MAG. Researchers would be advised to remove these contigs from the MAG and re-analyze.
We conclude that MAGpy represents a novel, useful and reproducible workflow that enables researchers to predict the closest relative to newly sequenced and assembled MAGs. MAGpy carries out extensive comparative genomics at the DNA and protein-level, attempts to place MAGs within a phylogenetic tree, produces detailed reports and allows for the identification of potential chimeric MAGs.
Funding
This project was supported by the Biotechnology and Biological Sciences Research Council (BBSRC; BB/N016742/1, BB/N01720X/1), including institute strategic programme and national capability awards to The Roslin Institute (BBSRC: BB/P013759/1, BB/P013732/1, BB/J004235/1, BB/J004243/1); and by the Scottish Government (RESAS) as part of the 2016-2021 commission.
Conflict of Interest: none declared.
Supplementary Material
References
- Asnicar F., et al. (2015) Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ, 3, e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz R.K., et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers R.M., et al. (2017) Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol., 35, 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie E.L., et al. (2006) Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl. Environ. Microbiol., 72, 6288–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C.T., Irber L. (2016) sourmash: a library for MinHash sketching of DNA. J. Open Source Softw., 1, 27. [Google Scholar]
- Buchfink B., et al. (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Chen I.-M.A., et al. (2017) IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res., 45, D507–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., et al. (2014) Pfam: the protein families database. Nucleic Acids Res., 42, D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S., et al. (2016) Coming of age: ten years of next- generation sequencing technologies. Nat. Publ. Gr., 17, 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J., et al. (2016) ETE 3: reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol., 33, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D., et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan K.P., et al. (2016) MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol. Biol., 207–233. [DOI] [PubMed] [Google Scholar]
- Koster J., Rahmann S. (2012) Snakemake–a scalable bioinformatics workflow engine. Bioinformatics, 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Loman N.J., Watson M. (2015) Successful test launch for nanopore sequencing. Nat. Methods, 12, 303–304. [DOI] [PubMed] [Google Scholar]
- Mackelprang R., et al. (2011) Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature, 480, 368–371. [DOI] [PubMed] [Google Scholar]
- Ondov B.D., et al. (2016) Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol., 17, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H., et al. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res., 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H., et al. (2017) Recovery of nearly 8, 000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol., 2, 1533–1542. [DOI] [PubMed] [Google Scholar]
- Roehe R., et al. (2016) Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PLoS Genet., 12, e1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumpeka D.D., et al. (2017) A review of bioinformatics tools for bio-prospecting from metagenomic sequence data. Front. Genet., 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30, 2068–2069.24642063 [Google Scholar]
- Segata N., et al. (2013) PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun, 4, 2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R.D., et al. (2018) Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun., 9, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium,T. (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res., 46, 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet D., et al. (2009) MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford), 2009, bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C., et al. (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science, 304, 66–74. [DOI] [PubMed] [Google Scholar]
- Wallace R.J., et al. (2015) The rumen microbial metagenome associated with high methane production in cattle. BMC Genomics, 16, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M., et al. (2007) DetectiV: visualization, normalization and significance testing for pathogen-detection microarray data. Genome Biol., 8, R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M. (2014) Illuminating the future of DNA sequencing. Genome Biol., 15, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattam A.R., et al. (2014) PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res., 42, D581–D591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T., et al. (2012) Human gut microbiome viewed across age and geography. Nature, 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.