

Abstract
Summary
Over the last two decades, we have observed an exponential increase in the number of generated array or sequencing-based transcriptomic profiles. Reverse engineering of biological networks from high-throughput gene expression profiles has been one of the grand challenges in systems biology. The Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe) represents one of the most effective and widely-used tools to address this challenge. However, existing ARACNe implementations do not efficiently process big input data with thousands of samples. Here we present an improved implementation of the algorithm, SJARACNe, to solve this big data problem, based on sophisticated software engineering. The new scalable SJARACNe package achieves a dramatic improvement in computational performance in both time and memory usage and implements new features while preserving the network inference accuracy of the original algorithm. Given that large-sampled transcriptomic data is increasingly available and ARACNe is extremely demanding for network reconstruction, the scalable SJARACNe will allow even researchers with modest computational resources to efficiently construct complex regulatory and signaling networks from thousands of gene expression profiles.
Availability and implementation
SJARACNe is implemented in C++ (computational core) and Python (pipelining scripting wrapper, ≥3.6.1). It is freely available at https://github.com/jyyulab/SJARACNe.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Due to the power of technologies in transcriptome profiling from microarray to next-generation sequencing, we have observed a tremendous increase in the number of gene expression profiles from normal to malignant samples over the past two decades. For example, The Cancer Genome Atlas project has profiled over 30 000 human adult cancer patients. The Gene Expression Omnibus accumulated array—or sequence-based transcriptomic profiles of over 2.5 million samples from bacteria to humans by July 2018. Reverse engineering of gene regulatory networks from transcriptomic profiles has been proven to be powerful in discovering hidden drivers and master regulators of disease phenotypes including cancer (Rodriguez-Barrueco et al., 2015), immunology (Du et al., 2018), drug resistance (Piovan et al., 2013) and drug response (Woo et al., 2015). Various computational algorithms have been developed to reconstruct gene regulatory networks from large-scaled gene expression data. Among these, Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe) (Margolin et al., 2006) represents one of the most efficient and widely-used network reconstruction methods based on mutual information (MI) that captures non-linear relationships between two variables. ARACNe-adaptive partitioning (AP) (Lachmann et al., 2016) improved MI estimation using an AP approach. However, neither ARACNe nor ARACNe-AP can handle big input data with thousands of samples. For example, the original ARACNe fails when the sample size is over 1500 and ARACNe-AP requires too much memory to be runnable on a standard computer. Here we present a scalable solution, SJARACNe, to address the big data problem by optimizing the depth of AP and redesigning the data structure. SJARACNe dramatically improves the computational performance, especially on the memory usage to allow even researchers with modest computational power to generate networks from thousands of samples. For example, SJARACNe can process data with 2000 samples on a laptop with only 8 GB RAM while ARACNe would fail and ARACNe-AP would require a supercomputer with at least 10 times more memory. We benchmarked the performance improvements of SJARACNe with datasets of various sizes compared to ARACNe and ARACNe-AP.
2 SJARACNe features and functions
2.1 Efficient data structures
Data structures alongside algorithms are critical to efficient computing. Here we replaced an inefficient and inextensible data structure used in ARACNe with a pointer-based and flexible data structure in SJARACNe, to enhance the computational power by reducing the access time and, therefore, the overall run time.
2.2 Optimization of the depth of adaptive partitioning
AP is an efficient solution for MI estimation. ARACNe forced a fixed convergence point in its AP implementation which limited its scalability; ARACNe-AP used a high threshold which results in high memory problem. SJARACNe solves both problems by utilizing a flexible convergence point.
2.3 New features and functions
SJARACNe provides enhanced annotations of network output including annotations of nodes, and extra statistics such as Spearman and Pearson correlation and regression coefficients. In addition, SJARACNe generates the network in various formats that can be taken by visualization tools.
3 Datasets
To benchmark the performance of SJARACNe in comparison with ARACNe and ARACNE-AP, we have chosen a large breast cancer dataset with 1981 samples (Curtis et al., 2012) and sampled the data into four datasets with different sample sizes: small (N = 100), medium (N = 500), large (N = 1000) and very large (N = 1981) while fixing the gene dimension (28 278 genes).
4 Results and discussion
We compared SJARACNe with ARACNe and ARACNe-AP on both runtime (Fig. 1 A) and memory usage (Fig. 1B) using four different datasets, from a small to very large number of samples with 100 bootstraps (the same seeds were used across all three methods). The performance results show that ARACNe-AP is the most memory consuming method among the three, while being close to SJARACNe in terms of run time. SJARACNe and ARACNe are in the same level in terms of memory consumption but SJARACNe is 2–2.5 times faster than its competitor as the number of samples increases. Further, ARACNe is unable to handle a dataset with a very large number of samples (it failed at N = 1981), while SJARACNe and ARACNe-AP successfully completed the job.
Fig. 1.
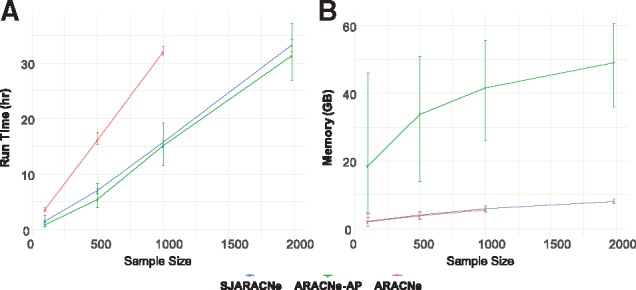
Performance comparison of SJARACNe (blue), ARACNe-AP (green) and ARACNe (red). (A) run time and (B) memory. No results of ARACNe in very large dataset (N = 1981) is due to its failure in handling big input data
We have performed the network similarity analysis on gene regulatory networks generated by the different methods for 10 680 transcripts representing 6 458 signaling factors in all four benchmark datasets with 100 bootstraps. Then we performed Fisher’s exact test to measure the significance of overlaps of targets for each isoform generated by the three algorithms. SJARACNe and ARACNe construct exactly the same networks with the same initial seeds. SJARACNe and ARACNe-AP produce highly similar networks: For each of the 10 680 signaling factor isoforms, the targets predicted by SJARACNe and ARACNe-AP overlap significantly (P < 10−9) (Supplementary Fig. S1).
In summary, SJARACNe addresses the pressing issue of reconstructing gene networks from big data and will have broad applications.
Supplementary Material
Acknowledgements
We would like to thank Stephen V. Rice, Manjunath Kustagi for code contributions, Jinghui Zhang and Michael Rusch for resource support and members in the Yu and Califano labs for testing and improving SJARACNe.
Conflict of Interest: Dr Califano is founder, equity holder, consultant and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc. The other authors declare no competing interests.
Funding
This work was supported by St. Jude Comprehensive Cancer Center Developmental Fund [to J.Y.); and by the National Institutes of Health [R35CA197745 to A.C. (Outstanding Investigator Award), U54CA209997 (Center for Cancer Systems Therapeutics), S10OD012351, S10OD021764).
References
- Curtis C., et al. (2012) The genomic and transcriptomic architecture of 2, 000 breast tumours reveals novel subgroups. Nature, 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., et al. (2018) Hippo/Mst signalling couples metabolic state and immune function of CD8alpha(+) dendritic cells. Nature, 558, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A., et al. (2016) ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics, 32, 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin A.A., et al. (2006) ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics, 7 (Suppl. 1), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovan E., et al. (2013) Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell, 24, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Barrueco R., et al. (2015) Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR-/HER2+ breast cancers. Genes Dev., 29, 1631–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J.H., et al. (2015) Elucidating compound mechanism of action by network perturbation analysis. Cell, 162, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.