

Abstract
The prenyl fragment is the quintessential constituent of terpenoid natural products, a diverse family which contains numerous members with diverse biological properties. In contrast, fluorinated and multifluorinated arenes make up an important class of anthropogenic molecules which are highly relevant to material, agricultural, and pharmaceutical industries. While allylation chemistry is well developed, effective prenylation strategies hav e been less forthcoming. Herein, we describe the photocatalytic defluoroprenylation, a powerful method that provides access to “hybrid molecules” that possess both the functionality of a prenyl group and fluorinated arenes. This approach involves direct prenyl group transfer under very mild conditions, displays excellent functional group tolerance, and relatively short reaction times (<4 h), which is the fastest photocatalytic C–F functionalization developed to date. Additionally, the strategy can be extended to include allyl and geranyl (10 carbon fragment) transfers. Another prominent finding is a reagent dependent switch in regioselectivity of the major product from para to ortho C–F functionalization.
Graphical Abstract
The prenyl fragment is ubiquitous in a multitude of natural products, and is the fundamental building block of the biodiverse and efficacious terpenoids which present impressive and diverse biological activities.1 Consequently, the development of prenylation strategies within the context of natural products2 has been widely studied.2–3 Many prenylation strategies involve multistep procedures, and one-step prenylation has only been solved for a narrow subset of substrates.4 Even recently, Porco employed indirect allylation followed by cross-metathesis to affect prenylation to access a class of polyprenylated acylphloroglucinols.5
Meanwhile, fluorine substituents have been shown to impart a number of positive attributes, such as resistance to metabolic degradation, improved binding, enhanced lipophilicity, and passive diffusion of compounds across membranes.6 Not surprisingly, multifluorinated arenes make up an important class of molecules in pharmaceutical,7 material,8 and agricultural chemistry,9 but despite their importance, methods to build these molecules largely depends on just a handful of rather harsh reactions, such as the Balz-Schiemann decomposition of an aryl diazonium tetrafluoroborate salt,10 or the high temperature (ca 230 °C) halex process.11 Arguably, this limitation has led to an overreliance on commercially available pre-fluorinated building blocks which can simply be incorporated into other molecules. Almost certainly, this leads to incomplete structure activity relationships studies.
Building on the strategy promoted by Braun12, Richmond,13 Uneyama,14 and many others,15 we seek to address this limitation by developing reactions that start with inexpensive highly fluorinated arenes, and sculpt far more complex fluorinated arenes via C–F functionalization than have historically been synthetically accessible.16 We were particularly drawn to the prenyl motif due to its ubiquity within nature as well as its synthetic versatility. Accomplishing this goal would allow us to wed the natural prenyl group and the unnatural organofluorines to give hybrid molecules.
While allylation chemistry is relatively well developed, there are a number of serious challenges that arise as a consequence of the two additional methyl groups found within a prenyl group. As carbenium ions, they are prone to elimination,4a, 4c and as anions regioselectivity issues arise (Scheme 1A).17 The use of a prenyl radical might lead to selectivity in the addition, but addition to an arene is expected to be a highly endergonic step.
Scheme 1.
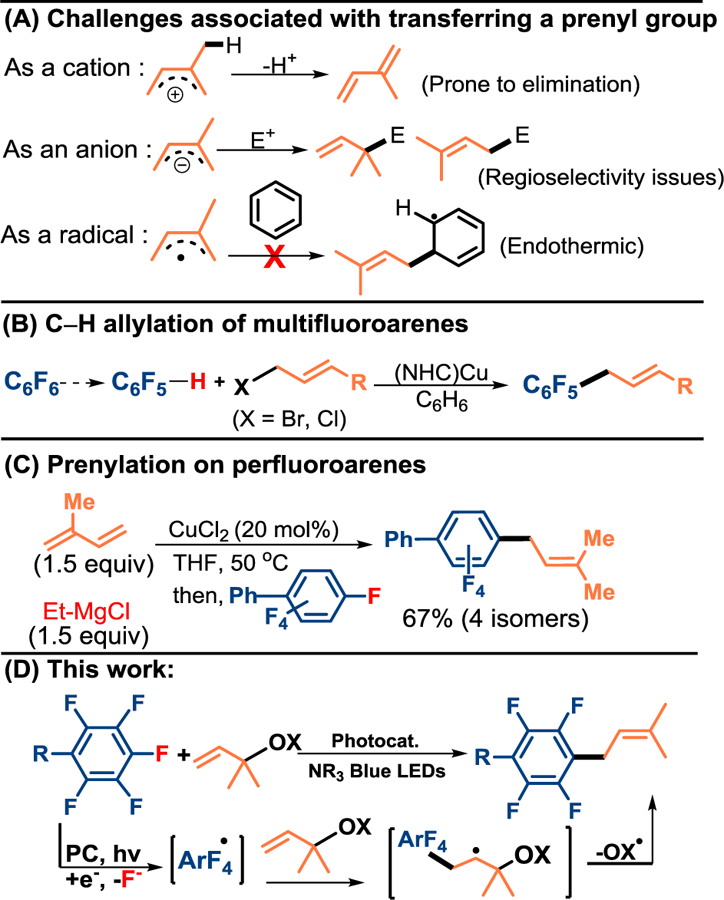
Allylation and Prenylation of electron deficient perfluoroarenes
Several strategies have been explored to achieve related allylation and isoprenylation on perfluoroarenes18 via C–F functionalization both directly and indirectly (Scheme 1B–C). Sukbok18c has shown that a Cu(NHC) catalyst is capable of C–H functionalization of highly fluorinated arenes (Scheme 1B), which has become increasingly relevant with recent improvements to hydrodefluorination technologies,15d, 19 but at the very least requires two steps. In an alternative approach, Kambe17 showed (Scheme 1C) that copper (II) chloride could facilitate formation of an prenyl magnesiate which would then undergo uncatalyzed addition to the perfluoroarene. The major product arises from linear addition of the most stable prenyl metal species, but gives rise to a total of 4 isomers in substantial quantities.
Inspired by the work of Zard who has popularized a number of allyl radicophiles,20 we envisioned that such a reagent could be designed to facilitate C–F prenylation. Previously, we have shown that upon photocatalytic electron transfer to a perfluoroarene, an unstable radical anion results, which undergoes mesolytic cleavage to generate a fluoride and a perfluoroaryl radical.19e, 21 Our hope was that the radicophile would intercept the perfluoroaryl radical to regioselectively generate the key C–C bond. Addition would result in an alkyl radical intermediate that would undergo homolytic fragmentation of the β-leaving group, unmasking the prenyl group. We anticipated that the nature of the leaving group would be key to preventing simple hydrogen atom transfer (HAT) to the radical.21e Vital to the rate of the β-fragmentation is the bond strength, and in this regard Zard has provided insight, demonstrating that the C–O bond of allyl alcohol (80.1 kcal/mol BDE)22 can be weakened by converting the hydroxy to an aryloxy group; rendering it susceptible to homolytic fragmentation.20, 23 This stabilization is mirrored by the phenoxy radical (PhO–H BDE = 87.3 kcal/mol) which is more stable than hydroxyl radical (HO–H BDE = 119.3 kcal/mol).24
Thus, we began our investigation with the hope of finding a group capable of activating the C–O bond towards homolytic fragmentation (Table 1). As expected, reaction with isoprenyl alcohol (2a) itself simply results in hydroarylation of the alkene,21b highlighting the need for an activating group that can increase the rate of fragmentation. Next, we assessed the 6-halopyridine motif (2b, 2c) previously explored by Zard23 in lauroyl peroxide mediated transfer of xanthates to olefins. The pyridyl group is conveniently introduced via SNAr addition of isoprenyl alcohol. We were delighted to see the desired prenylated product (3a) in 54% and 43% respectively. The mass balance was primarily comprised of hydrodefluorination (HDF) as well as an oxidized version of the desired product. However, reagents 2b and 2c both underwent competitive [3,3]-sigmatropic rearrangement, which further complicated the situation and necessitate higher reagent loading to compensate for this background reaction. By blocking the ortho position with an ester group and increasing the steric demand (2d), we hoped to curtail both the rearrangement and the oxidation. Indeed, we halted the rearrangement, but unfortunately this reagent did not deliver desired prenylated product, and instead yielded only hydrodefluorination (HDF) product. This behavior is perhaps due to competitive and unproductive electron transfer to 2d rather than the perfluoroarene.
Table 1.
Screening of Prenyl sources
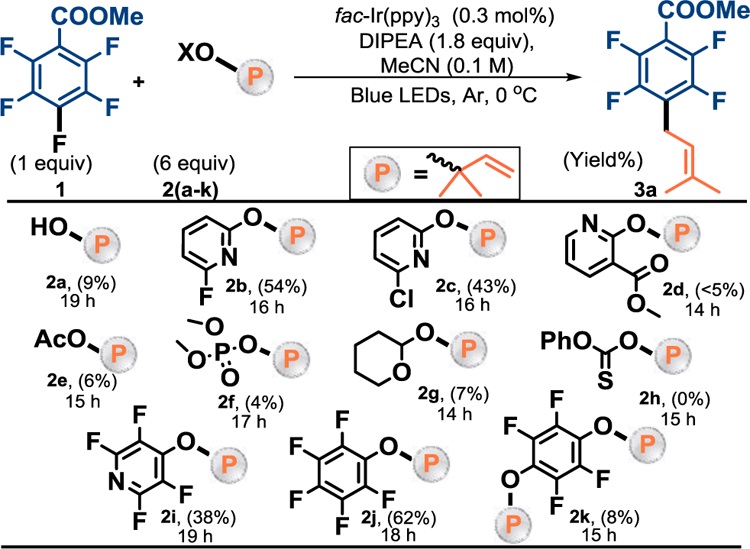 |
Observed: 2a - HDF, amine addition, C-O coupling, 2b and 2c - HDF, oxidized prenylation, [3,3]-rearrangement, 2d - >90% HDF, 2e - HDF, C-O coupling, multiple side products, 2f - mostly HDF, multiple side products, 2g - HDF, incomplete conversion, 2h - HDF, multiple side products, 2i - HDF, 2j - HDF, 2k - HDF. No further conversion to product over extended time.
The importance of a weak C–O bond can be observed in low yields of 2e and 2f, which would be expected to work better if the C–O bond were to break heterolytically. However, substrates 2g and 2h were expected to have a weaker C–O bond and still failed to give improved yields. We next evaluated aryl ethers formed from the perfluoroarene (2i and 2j). We were pleased to see that these prenyl transfer reagents afforded the desired product, with HDF as the only by-product.
It is possible that the fluorines on the reagent serve to prevent rearrangement and sterically reduce the activity of the resulting aryloxy radical, potentially involved in the formation of the previously observed oxidized product. However, 2k displayed significantly decreased activity even though it contained two prenyl groups. Given the positive results and the simple nature of 2j we opted to use it for further reaction development.
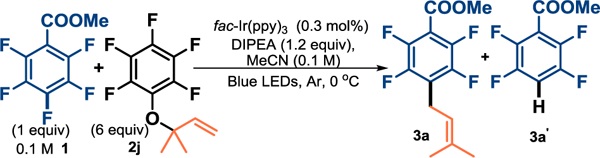
Using conditions that had facilitated C–F reductive alkylation,21b using Blue LEDs we irradiated methyl pentafluorobenzoate and diisopropylethyl amine (DIPEA) as the electron source, along with 6 equiv of 2j at 0 °C. Previously, we observed that lowering the temperature could reduce the amount of competitive HDF in the related C–F arylation reaction.21c We were pleased to see that the desired C−C-coupled product was formed as the major product in decent yield, albeit along with a significant amount of HDF product (Table 2, entry 1).25
Table 2.
Optimization of reactions
 | ||||
|---|---|---|---|---|
| Entry | Modifications | Yield of 3a(%)a | 3a:3a’d | Time (h) |
| 1 | none | 31/62 | 1.6:1 | 5/17 |
| 2 | no cataylst or in dark | 0 | na | 17b |
| 3 | without amine | 0 | na | 17b |
| 4 | −10 °C instead of 0°C | 29/64 | 1.7:1 | 5/25 |
| 5 | DIPEA (1.0 equiv) | 25/57 | 2:1 | 5/23b |
| 6 | DIPEA (1.8 equiv) | 36/64 | 1.9:1 | 5/18 |
| 7 | DIPEA (2.5 equiv) | 38/57 | 1.3:1 | 5/15 |
| 8 | 0.25 mol% catalyst, DIPEA (1.8 equiv) | 31/58 | 1.8:1 | 5/24 |
| 9 | 0.025 mol% catalyst, DIPEA (1.8 equiv) | 6/28 | 1.6:1 | 5/24b |
| 10 | H2O (10 equiv), DIPEA (1.8 equiv) | 57/65 | 1.9:1 | 3/4 |
| 11 | H2O (15 equiv), DIPEA (1.8 equiv) | 59/65 | 1.9:1 | 3/4 |
| 12 | Entry 10 with 0.4 equiv TEMPO | 39/68 | 2.1:1 | 4/10 |
a determined by 19F NMR analysis. Reaction complete unless otherwise noted.
bReaction did not go to completion over extended time, observed 0%, 0%, 83% and 44% conversion of 1 in entries 2, 3, 5 and 9 respectively.
dReported for the final time point.
Control studies demonstrated the necessity of light, catalyst, and amine (entry 2-3). Carrying out the reaction at lower temperature increased the reaction time with no significant improvement in 3a:3a’ ratio (entry 4). While decreasing the DIPEA loading slowed the reaction, increasing amine resulted in more HDF (entry 5–7); ultimately, 1.8 equivalents of DIPEA produced the optimal yield. There appears to be a rate dependency on catalyst concentration (entries 8 and 9). Next, we investigated the effect of water on the reaction. Unexpectedly, the presence of water significantly accelerated the reaction from 18 h to 4 h and improved product: HDF ratio (entry 10-11). One explanation could be the increase in exothermicity of the reaction due to hydration of fluoride.26 An alternative explanation is that water acidifies the pentafluorophenol generated in situ.27 By doping the reaction mixture with pentafluorophenol we observed its inhibitory effect on the reaction progress. This effect was diminished upon the addition of water.27 Recently, Wu showed that the competitive reduction pathway could be minimized by the addition of TEMPO,28 we found it initially helped but ultimately slowed the reaction.29
Ultimately, using 0.1 M substrate, 1.8 equiv DIPEA, 0.3 mol% fac-Ir(ppy)3 and 10 equiv H2O, we began evaluating substrate scope. Under these conditions a number of perfluoroarenes smoothly underwent C–F defluoroprenylation in good to modest yields. The reaction tolerates a number of functional groups including esters (Table 3, 3a-3c), nitriles (3d), CF3 (3e), and perfluoroheterocyclic arenes (3g). Hexafluorobenzene, devoid of any additional electron-withdrawing functional group also proceeded to form 3f, though it required the use of 2b as the prenyl source. 3h-l are noteworthy as they demonstrate the preference for chlorine fragmentation over that of fluorine despite the position on the ring. Given that many chlorofluoro-starting materials are commercially available, it provides a convenient strategy for accessing complementary regioisomers. When a chlorine was placed at the site of preferential fluorine fragmentation (4 position for pyridine), it displayed moderately shorter reaction time compared to pentafluoropyridine (i.e. 3h compared to 3g).
Table 3.
Scope of prenylation
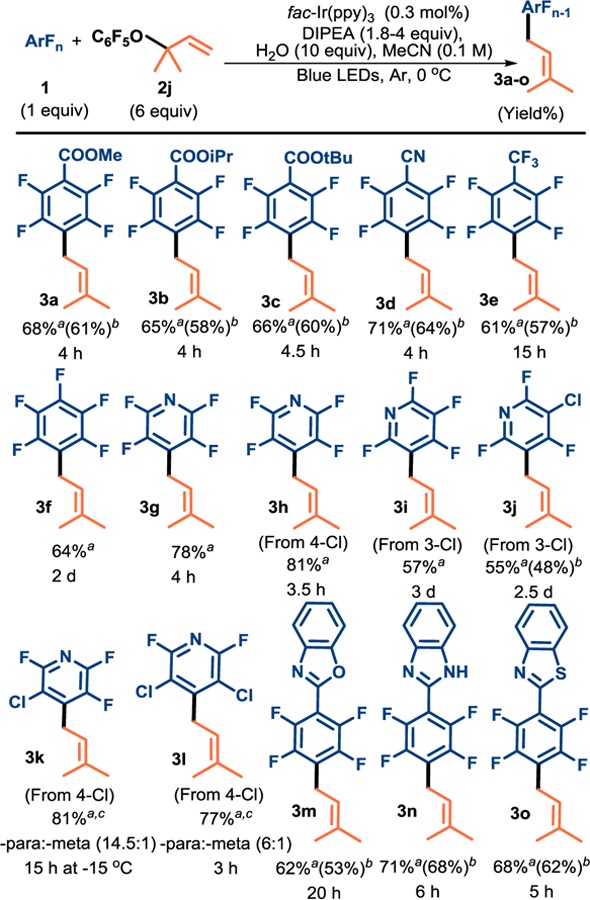 |
a19F NMR yield determined using monofluorobenzene as internal standard. bisolated yield. cComprises of both products 3f required 2b as the prenylating source.
This could be because fragmentation of chloride ion is more exothermic than fluoride, resulting in a faster fragmentation event. Furthermore, the presence of chlorine substituents create greater steric demand than fluorine substituents, and could result in a relative increase the ground state energy.30 Likewise, steric repulsion between the substituents in the radical anion may also accelerate its breakdown, leading to faster fragmentation. Interestingly, prenylation at the meta position was also possible but required longer reaction time (3i and 3j vs 3g). Whereas the mesolytic fragmentation of fluoride is highly regioselective, chloride is generally less regioselective and is temperature dependent (3g vs 3k and 3l), but still results in useful selectivities. This method can be used to access heterocyclic substituted perfluoroarenes like oxazoles (3m), benzimidazoles (3n) and benzothiazoles (3o). In all these reactions, HDF made up the mass balance.
Looking to expand the utility of the method, we applied this strategy towards allyl transfer to perfluoroarenes. As expected, allylation with transposition of the double bond proceeded smoothly to yield 5a-5f (Table 4) in good yield. Substitution at both the β-position (5c and 5d), and the γ-position were well tolerated (5e and 5f). While the yield was acceptable, in the case of unsymmetric 5f, the E/Z-selectivity was very modest (1.3:1) and did not display significant temperature dependence (See SI for details), which may limit its use in cases where the olefin geometry is essential.
Table 4.
Scope of allylation
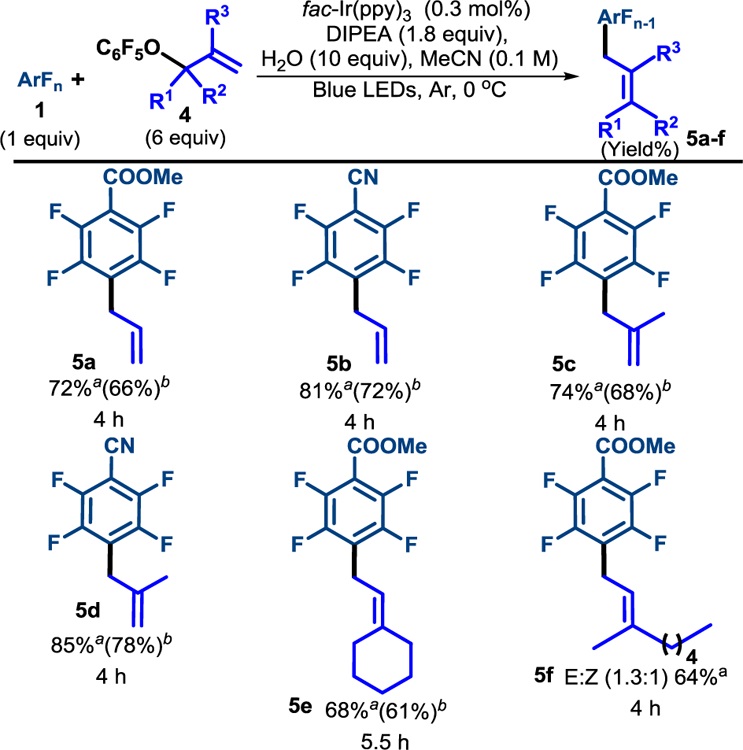 |
a19F NMR yield determined using monofluorobenzene internal standard. bisolated yield.
Encouraged by our success with prenyl transfer, we hoped to extend our strategy to geranyl transfer, adding a 10 carbon unit. Using the same optimized conditions, we reacted perfluoroarenes with pentafluoroisogeranyl ether (6, Table 5). We were concerned that the alkyl radical formed upon addition to the alkene would undergo intramolecular addition to the additional olefin (i.e. 5-exo-trig cyclization) faster than fragmentation of the pentafluorophenoxy radical.
Table 5.
Scope of geranylation
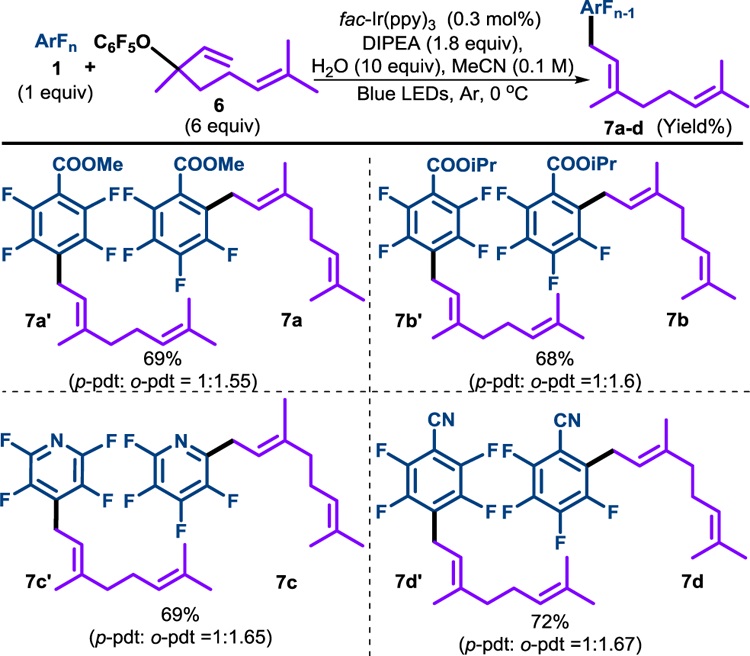 |
Determined by 19F NMR. Reactions completed in 6–8 h.
Thus, we were pleased to obtain geranylated product in similar yields to the prenylation reaction. A striking difference, however, was that the reaction took place preferentially at the position ortho to the substituent on the perfluoroaryl ring (c.a. ortho: para 1.6:1). This is particularly noteworthy as it is the first time that we have observed an external reagent capable of influencing the C–F regioselectivity in a photocatalytic C–F functionalization.31
Furthermore, while, 7a-7d were produced as ortho-para isomers, it demonstrated perfect diastereoselectivity, giving only the E-alkene.32 This selectivity is in stark contrast to the unsymmetric allyl derivative, 5f, which was produced as a mixture.
In conclusion, we have developed a strategy and reagents that enable photocatalytic defluoro-allylation, -prenylation, and -geranylation of perfluoroarenes. Further, as we moved from prenylation to geranylation, we observed a change in the regioselectivity of the major product from para-C–F functionalization to ortho-C–F functionalization. Our approach allows direct allyl, prenyl and geranyl substitution of C–F bonds using very mild conditions and short reaction times. This strategy should facilitate investigations involving synthesis of hybrid fluorinated analogs of natural products. Additionally, this reaction presents a number of interesting mechanistic facets which are currently being studied, and the findings will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We thank Jon Day for help in editing this manuscript and art-work.
Funding Sources
We gratefully acknowledge NIH NIGMS (5R01GM115697) for financial support of this work. Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Includes procedures, additional experiments, compound characterization, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1. (a).Randall SK; Marshall MS; Crowell DN, Protein isoprenylation in suspension-cultured tobacco cells. Plant Cell 1993, 5, 433; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zou Y; Zhan Z; Li D; Tang M; Cacho RA; Watanabe K; Tang Y, Tandem Prenyltransferases Catalyze Isoprenoid Elongation and Complexity Generation in Biosynthesis of Quinolone Alkaloids. J. Am. Chem. Soc 2015, 137, 4980; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lozinski O; Bennetau-Pelissero C; Shinkaruk S, The Synthetic and Biological Aspects of Prenylation as the Versatile Tool for Estrogenic Activity Modulation. Chemistry Select 2017, 2, 6577; [Google Scholar]; (d) Viliam B; Karel S; Jan H; Veronika T, Anti-inflammatory Natural Prenylated Phenolic Compounds - Potential Lead Substances. Curr. Med. Chem 2018, 25, 1094. [DOI] [PubMed] [Google Scholar]
- 2.Li D-F; Liu K; Jiang Y-X; Gu Y; Zhang J-R; Zhao L-M, Access to 3-Prenylated Oxindoles by α-Regioselective Prenylation: Application to the Synthesis of (±)-Debromoflustramine E. Org. Lett 2018, 20, 1122. [DOI] [PubMed] [Google Scholar]
- 3. (a).Leung JC; Geary LM; Chen T-Y; Zbieg JR; Krische MJ, Direct, Redox-Neutral Prenylation and Geranylation of Secondary Carbinol C–H Bonds: C4-Regioselectivity in Ruthenium-Catalyzed C–C Couplings of Dienes to α-Hydroxy Esters. J. Am. Chem. Soc 2012, 134, 15700; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang YJ; Skucas E; Krische MJ, Direct Prenylation of Aromatic and α,β-Unsaturated Carboxamides via Iridium-Catalyzed C−H Oxidative Addition−Allene Insertion. Org. Lett 2009, 11, 4248; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Luzung M; Lewis C; Baran P, Direct, Chemoselective N‐tert‐ Prenylation of Indoles by C-H Functionalization. Angew. Chem. Int. Ed 2009, 48, 7025; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lipshutz BH; Ellsworth EL; Dimock SH; Smith RAJ, New methodology for conjugate additions of allylic ligands to α,β-unsaturated ketones: synthetic and spectroscopic studies. J. Am. Chem. Soc 1990, 112, 4404; [Google Scholar]; (e) Hosomi A; Saito M; Sakurai H, 2-trimethylsilylmethyl-1,3-butadiene as a novel reagent for isoprenylation. New access to ipsenol and ipsdienol, pheromones of Ips paraconfusus. Tetrahedron Lett 1979, 20, 429; [Google Scholar]; (f) Jefford CW; Sledeski AW; Boukouvalas J, A direct synthesis of (±)-eldanolide via the highly regioselective prenylation of 2-trimethylsiloxyfuran. Tetrahedron Lett 1987, 28, 949; [Google Scholar]; (g) Zhao L-M; Zhang S-Q; Dou F; Sun R, Zinc-Mediated Highly α-Regioselective 1,4-Addition of Chalcones with Prenyl Bromide in THF. Org. Lett 2013, 15, 5154. [DOI] [PubMed] [Google Scholar]
- 4. (a).Marsden SP; Depew KM; Danishefsky SJ, Stereoselective Total Syntheses of Amauromine and 5-N-Acetylardeemin. A Concise Route to the Family of “Reverse-Prenylated” Hexahydropyrroloindole Alkaloids. J. Am. Chem. Soc 1994, 116, 11143; [Google Scholar]; (b) Schmitt M; Grenning AJ; Tunge JA, Intercepted decarboxylative allylations of nitroalkanoates. Tetrahedron Lett 2012, 53, 4494; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM; Malhotra S; Chan WH, Exercising Regiocontrol in Palladium-Catalyzed Asymmetric Prenylations and Geranylation: Unifying Strategy toward Flustramines A and B. J. Am. Chem. Soc 2011, 133, 7328; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Weaver JD; Recio A; Grenning AJ; Tunge JA, Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem. Rev 2011, 111, 1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grenning AJ; Boyce JH; Porco JA, Rapid Synthesis of Polyprenylated Acylphloroglucinol Analogs via Dearomative Conjunctive Allylic Annulation. J. Am. Chem. Soc 2014, 136, 11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. (a).Matsumori N; Okazaki H; Nomura K; Murata M, Fluorinated cholesterol retains domain-forming activity in sphingomyelin bilayers. Chem. Phys. Lipids 2011, 164, 401; [DOI] [PubMed] [Google Scholar]; (b) Maienfisch P; Hall RG, The Importance of Fluorine in the Life Science Industry. CHIMIA 2004, 58, 93; [Google Scholar]; (c) Jarman M, Perflouroarenes as novel and selective protecting reagents; applications in anticancer drug development. J. Fluor. Chem 1989, 42, 3; [Google Scholar]; (d) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA, Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315; [DOI] [PubMed] [Google Scholar]; (e) Hagmann WK, The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359. [DOI] [PubMed] [Google Scholar]
- 7. (a).Müller K; Faeh C; Diederich F, Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881; [DOI] [PubMed] [Google Scholar]; (b) Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114, 2432. [DOI] [PubMed] [Google Scholar]
- 8. (a).Hird M, Fluorinated liquid crystals - properties and applications. Chem. Soc. Rev 2007, 36, 2070; [DOI] [PubMed] [Google Scholar]; (b) Sakamoto Y; Suzuki T; Miura A; Fujikawa H; Tokito S; Taga Y, Synthesis, Characterization, and Electron-Transport Property of Perfluorinated Phenylene Dendrimers. J. Am. Chem. Soc 2000, 122, 1832. [Google Scholar]
- 9. (a).Theodoridis G, Chapter 4 Fluorine-Containing Agrochemicals: An Overview of Recent Developments. In Advances in Fluorine Science, Tressaud A, Ed. Elsevier: 2006; Vol. 2, pp121; [Google Scholar]; (b) Fujiwara T; O’Hagan D, Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem 2014, 167, 16. [Google Scholar]
- 10.Günther B; Günther S, Über aromatische Fluorverbindungen I: Ein neues Verfahren zu ihrer Darstellung. Chem. Ber 1927, 60, 1186. [Google Scholar]
- 11.Finger GC; Kruse CW, Aromatic Fluorine Compounds. VII. Replacement of Aromatic -Cl and -NO2 Groups by -F1,2. J. Am. Chem. Soc 1956, 78, 6034. [Google Scholar]
- 12.Ahrens T; Kohlmann J; Ahrens M; Braun T, Functionalization of Fluorinated Molecules by Transition-Metal-Mediated C–F Bond Activation To Access Fluorinated Building Blocks. Chem. Rev 2015, 115, 931. [DOI] [PubMed] [Google Scholar]
- 13.Kiplinger JL; Richmond TG; Osterberg CE, Activation of Carbon-Fluorine Bonds by Metal Complexes. Chem. Rev 1994, 94, 373. [Google Scholar]
- 14.Amii H; Uneyama K, C−F Bond Activation in Organic Synthesis. Chem. Rev 2009, 109, 2119. [DOI] [PubMed] [Google Scholar]
- 15. (a).Eisenstein O; Milani J; Perutz RN, Selectivity of C–H Activation and Competition between C–H and C–F Bond Activation at Fluorocarbons. Chem. Rev 2017, 117, 8710; [DOI] [PubMed] [Google Scholar]; (b) Whittlesey MK; Peris E, Catalytic Hydrodefluorination with Late Transition Metal Complexes. ACS Catal 2014, 4, 3152; [Google Scholar]; (c) Clot E; Eisenstein O; Jasim N; Macgregor SA; McGrady JE; Perutz RN, C−F and C−H Bond Activation of Fluorobenzenes and Fluoropyridines at Transition Metal Centers: How Fluorine Tips the Scales. Acc. Chem. Res 2011, 44, 333; [DOI] [PubMed] [Google Scholar]; (d) Sabater S; Mata JA; Peris E, Hydrodefluorination of carbon–fluorine bonds by the synergistic action of a ruthenium–palladium catalyst. Nat. Commun 2013, 4, 2553; [DOI] [PubMed] [Google Scholar]; (e) Chen K; Berg N; Gschwind R; König B, Selective Single C(sp3)–F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation. J. Am. Chem. Soc 2017, 139, 18444; [DOI] [PubMed] [Google Scholar]; (f) Wang H; Jui NT, Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc 2018, 140, 163. [DOI] [PubMed] [Google Scholar]
- 16. (a).Xie J; Rudolph M; Rominger F; Hashmi ASK, Photoredox-Controlled Mono- and Di-Multifluoroarylation of C(sp3)−H Bonds with Aryl Fluorides. Angew. Chem. Int. Ed 2017, 56, 7266; [DOI] [PubMed] [Google Scholar]; (b) Nicholls TP; Robertson JC; Gardiner MG; Bissember AC, Identifying the potential of pulsed LED irradiation in synthesis: copper-photocatalysed C–F functionalisation. Chem. Commun 2018, 54, 4589. [DOI] [PubMed] [Google Scholar]
- 17.Iwasaki T; Okamoto K; Kuniyasu H; Kambe N, Cucatalyzed Reductive Coupling of Perfluoroarenes with 1,3-Dienes. Chem. Lett 2017, 46, 1504. [Google Scholar]
- 18. (a).Vinogradov AS; Krasnov VI; Platonov VE, Organozinc reagents from polyfluoroarenes: Preparation and reactions with allyl halides. Synthesis of allylpolyfluoroarenes. Russ. J. Org. Chem 2008, 44, 95; [Google Scholar]; (b) Yu YB; Fan S; Zhang X, Copper‐ and Phosphine‐Ligand‐Free Palladium‐Catalyzed Direct Allylation of Electron‐Deficient Polyfluoroarenes with Allylic Chlorides. Chem. Eur. J 2012, 18, 14643; [DOI] [PubMed] [Google Scholar]; (c) Weilong X; Sukbok C, [Cu(NHC)]‐Catalyzed C−H Allylation and Alkenylation of both Electron‐Deficient and Electron‐Rich (Hetero)arenes with Allyl Halides. Angew. Chem. Int. Ed 2016, 55, 1876; [DOI] [PubMed] [Google Scholar]; (d) Yao T; Hirano K; Satoh T; Miura M, Stereospecific Copper‐Catalyzed C-H Allylation of Electron‐Deficient Arenes with Allyl Phosphates. Angew. Chem. Int. Ed 2011, 50, 2990; [DOI] [PubMed] [Google Scholar]; (e) Li Z; Zhang Y; Liu Z-Q, Pd-Catalyzed Olefination of Perfluoroarenes with Allyl Esters. Org. Lett 2012, 14, 74; [DOI] [PubMed] [Google Scholar]; (f) Fan S; Chen F; Zhang X, Direct Palladium‐Catalyzed Intermolecular Allylation of Highly Electron‐Deficient Polyfluoroarenes. Angew. Chem. Int. Ed 2011, 50, 5918. [DOI] [PubMed] [Google Scholar]
- 19. (a).Vela J; Smith JM; Yu Y; Ketterer NA; Flaschenriem CJ; Lachicotte RJ; Holland PL, Synthesis and Reactivity of Low-Coordinate Iron(II) Fluoride Complexes and Their Use in the Catalytic Hydrodefluorination of Fluorocarbons. J. Am. Chem. Soc 2005, 127, 7857; [DOI] [PubMed] [Google Scholar]; (b) Lv H; Zhan J-H; Cai Y-B; Yu Y; Wang B; Zhang J-L, π–π Interaction Assisted Hydrodefluorination of Perfluoroarenes by Gold Hydride: A Case of Synergistic Effect on C–F Bond Activation. J. Am. Chem. Soc 2012, 134, 16216; [DOI] [PubMed] [Google Scholar]; (c) Hongbin L; Yuan-Bo C; Jun-Long Z, Copper-Catalyzed Hydrodefluorination of Fluoroarenes by Copper Hydride Intermediates. Angew. Chem 2013, 125, 3285; [DOI] [PubMed] [Google Scholar]; (d) Senaweera SM; Singh A; Weaver JD, Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics. J. Am. Chem. Soc 2014, 136, 3002; [DOI] [PubMed] [Google Scholar]; (e) Khaled MB; El Mokadem RK; Weaver JD, Hydrogen Bond Directed Photocatalytic Hydrodefluorination: Overcoming Electronic Control. J. Am. Chem. Soc 2017, 139, 13092. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. (a).Quiclet-Sire B; Zard SZ, Fun with radicals. Some new perspectives for organic synthesis. Pure Appl. Chem 2011, 83, 519; [Google Scholar]; (b) Debien L; Quiclet-Sire B; Zard SZ, Allylic Alcohols: Ideal Radical Allylating Agents? Acc. Chem. Res 2015, 48, 1237. [DOI] [PubMed] [Google Scholar]
- 21. (a).Weaver J; Senaweera S, C–F activation and functionalization of perfluoro- and polyfluoroarenes. Tetrahedron 2014, 70, 7413; [Google Scholar]; (b) Singh A; Teegardin K; Kelly M; Prasad KS; Krishnan S; Weaver JD, Facile synthesis and complete characterization of homoleptic and heteroleptic cyclometalated Iridium(III) complexes for photocatalysis. J. Organomet. Chem 2015, 776, 51; [Google Scholar]; (c) Senaweera S; Weaver JD, Dual C–F, C–H Functionalization via Photocatalysis: Access to Multifluorinated Biaryls. J. Am. Chem. Soc 2016, 138, 2520; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Singh A; Fennell CJ; Weaver JD, Photocatalyst size controls electron and energy transfer: selectable E/Z isomer synthesis via C–F alkenylation. Chem. Sci 2016, 7, 6796; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Singh A; Kubik JJ; Weaver JD, Photocatalytic C–F alkylation; facile access to multifluorinated arenes. Chem. Sci 2015, 6, 7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanksby SJ; Ellison GB, Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255. [DOI] [PubMed] [Google Scholar]
- 23.Charrier N; Quiclet-Sire B; Zard SZ, Allylic Alcohols as Radical Allylating Agents. An Overall Olefination of Aldehydes and Ketones. J. Am. Chem. Soc 2008, 130, 8898. [DOI] [PubMed] [Google Scholar]
- 24.Wright JS; Ingold KU, Understanding Trends in C-H, N-H, and O-H Bond Dissociation Enthalpies. J. Chem. Educ 2000, 77, 1062. [Google Scholar]
- 25. No prenylation or HDF of reagent 2j was observed.
- 26.Zhan C-G; Dixon DA, Hydration of the Fluoride Anion: Structures and Absolute Hydration Free Energy from First-Principles Electronic Structure Calculations. J. Phys. Chem. A 2004, 108, 2020. [Google Scholar]
- 27. See SI for a working mechanism.
- 28.Xue F; Deng H; Xue C; Mohamed DKB; Tang KY; Wu J, Reaction discovery using acetylene gas as the chemical feedstock accelerated by the “stop-flow” micro-tubing reactor system. Chem. Sci 2017, 8, 3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. See SI for more details.
- 30. (a).Brown GM; Strydom OAW, Hexachlorobenzene, C6Cl6: the crystal and molecular structure from least-squares refinement with new X-ray data. Acta Crystallogr. A 1974, 30, 801; [Google Scholar]; (b) Intermolecular potential function model for crystalline hexachlorobenzene. J. Chem. Phys 1974, 60, 2414; [Google Scholar]; (c) Hexachlorobenzene is less aromatic than hexafluorobenzene due to non-planarity caused by the chloride substituents. [Google Scholar]
- 31. While we are uncertain about the cause of this change in regioselectivity, our working hypothesis is that there is an attractive interaction between 6 and the radical anion of 1. This results in an intermolecular complex which sterically perturbs the ortho C–F bond, presumably causing deviation from planarity, and resulting in a faster fragmentation. See SI for more discussion.
- 32. This selectivity did not display a significant temperature dependence (See SI for details).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.