

Abstract
Mitochondrial Zn2+ accumulation, particularly in CA1 neurons, occurs after ischemia and likely contributes to mitochondrial dysfunction and subsequent neurodegeneration. However, the relationship between mitochondrial Zn2+ accumulation and their disruption has not been examined at the ultrastructural level in vivo. We employed a cardiac arrest model of transient global ischemia (TGI), combined with Timm’s sulfide silver labeling, which inserts electron dense metallic silver granules at sites of labile Zn2+ accumulation, and used transmission electron microscopy (TEM) to examine subcellular loci of the Zn2+ accumulation. In line with prior studies, TGI-induced damage to CA1 was far greater than to CA3 pyramidal neurons, and was substantially progressive in the hours after reperfusion (being significantly greater after 4- than 1-hour recovery). Intriguingly, TEM examination of Timm’s-stained sections revealed substantial Zn2+ accumulation in many postischemic CA1 mitochondria, which was strongly correlated with their swelling and disruption. Furthermore, paralleling the evolution of neuronal injury, both the number of mitochondria containing Zn2+ and the degree of their disruption were far greater at 4- than 1-hour recovery. These data provide the first direct characterization of Zn2+ accumulation in CA1 mitochondria after in vivo TGI, and support the idea that targeting these events could yield therapeutic benefits.
Keywords: Excitotoxicity, Hippocampus, Ischemia, Mitochondria, Rat, Stroke, Zinc
INTRODUCTION
Brain ischemia is a leading cause of death and disability worldwide, but there are as yet no effective neuroprotective therapies. Many studies implicate “excitotoxicity” caused by excessive glutamate release as an important contributor and have largely focused on consequences of rapid Ca2+ entry through NMDA receptors. However, NMDA targeted therapies have shown limited clinical efficacy (1, 2). Further studies have implicated contributions of another divalent cation, Zn2+ (3–7). Despite high total levels of brain Zn2+ (100–200 µM), almost all of it is bound or sequestered, and free cytosolic levels are generally subnanomolar. However, a small portion (∼10%) of brain Zn2+ is loosely bound (termed labile Zn2+); this pool largely comprises Zn2+ present in certain presynaptic vesicles (most conspicuously in the mossy fiber pathway) (8), from which it can be coreleased with glutamate upon synaptic activation (9–11), and can be visualized using a histochemical technique (Timm’s sulfide silver labeling), or with membrane permeant fluorescent indicators (8, 12, 13). Indeed, indicating its specificity, Timm’s labeling is completely absent in brains of knockout mice lacking the vesicular Zn2+ transporter, ZnT3 (14). In pathological conditions of ischemia or prolonged seizures associated with strong synaptic activation, there is release of this presynaptic Zn2+, along with appearance of new labile Zn2+ in somata of some hippocampal pyramidal neurons (and other forebrain neurons) (3–5, 11). This reflects a combination of synaptically released Zn2+ that can enter postsynaptic neurons through various routes (“Zn2+ translocation”), and Zn2+ mobilization from buffers (particularly metallothionein-III [MT-III]) already present in the postsynaptic neurons in response to oxidative stress and acidosis (15, 16).
However, the redistribution of labile Zn2+ has never been examined at the ultrastructural level. Multiple studies have indicated that Zn2+ can induce potent effects on mitochondria in vitro and after transient ischemia in vivo (6, 16–19). In addition, we have carried out studies using hippocampal slices subjected to prolonged oxygen glucose deprivation (OGD) to model acute ischemia, and found that early cytosolic Zn2+ rises and mitochondrial Zn2+ entry appears to contribute to the acute hippocampal pyramidal neuron injury (20–22). Furthermore, after shorter (sublethal) episodes of OGD there was a gradual recovery of the early cytosolic Zn2+ elevations accompanied by progressive and persistent Zn2+ accumulation in mitochondria of CA1 (but not CA3) neurons that appeared to contribute to delayed mitochondrial swelling (22), possibly consistent with the selective delayed degeneration of these neurons after transient ischemia (23–25). However, whereas these in vitro observations support the possibility that mitochondria are critical loci of early Zn2+ effects in vivo, this question has not as yet been directly examined in animals.
In the present study, we employ a rat asphyxial cardiac arrest (CA) model of transient global ischemia (TGI), which provides the advantage over focal ischemia that the entire brain is subjected to the same duration of ischemia, such that regional differences in outcome largely reflect differences in susceptibility. Using this model system, we aimed, for the first time, to examine the redistribution of Zn2+ into mitochondria and its potential contribution to mitochondrial disruption and neuronal injury. Such a study requires an ultrastructural assessment to correlate Zn2+ accumulation with morphology of individual mitochondria, and the presently employed technique of Timm’s labeling (which inserts electron dense silver deposits in situ at subcellular sites of labile Zn2+ accumulation) combined with transmission electron microscopy (TEM) would seem ideally suited for such an investigation. The present findings, which show for the first time progressive Zn2+ accumulation in CA1 mitochondria after in vivo TGI to be strongly correlated with their physical disruption, provide new support for a direct contributory role of the mitochondrial Zn2+ in the delayed and progressive mitochondrial damage and cell death of these neurons.
MATERIALS AND METHODS
Ethics Statement
This study was carried out in accordance with the recommendations from the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Animals
Experiments were performed using male Wistar rats (Charles River Laboratories, Wilmington, MA) weighing 300–350 g (∼8–12 weeks). After applying the inclusion criterion that chest compressions lasted shorter than 1 minute (as previously described) (26), the final number of rats included for data analysis was 25. Upon arrival, rats were maintained in a 12-hour light/12-hour dark (6:00 am/6:00 pm) cycle and fed standard rat chow. They are handled for at least 5 days for acclimation to the researchers and their new environment.
CA and Resuscitation
This study utilizes an asphyxial CA model largely as previously described (26, 27). Rats were calorically restricted to 25% of normal food intake 14 hours prior to CA experiment. Rats were intubated using a 14-gauge endotracheal tube (B. Braun Melsungen AG, Melsungen, Germany), connected to a TOPO mechanical ventilator (Kent Scientific, Torrington, CT) and isoflurane vaporizer for delivery of 2% isoflurane and 50% O2 and 50% N2 gas during surgical preparation for CA. Femoral artery and vein cannula allowed for monitoring of blood pressure and heart rate and administration of intravenous medication. Invasive arterial blood pressure was measured continuously using a transducer (CWE, Inc., Ardmore, PA).
CA experiments began at “minute 0,” when the isoflurane level was reduced to 1%–1.5% to prepare for anesthesia wash out and inhaled gas was switched to 100% O2. After 2 minutes, isoflurane was stopped to wash out anesthesia, neuromuscular blockade was initiated with 1 mL of intravenous Vecuronium (2 mg/kg) injected with 1 mL of heparinized saline, and the inlet was disconnected from oxygen to allow room air to be mechanically delivered to the rat. At minute 5, asphyxial CA was induced by turning the ventilator off and clamping the ventilator tubing. CA time was defined as systolic blood pressure <30 and pulse pressure of 10 or less. Baseline arterial blood gas measurements (Abaxis, Union City, CA) were obtained within 30 minutes prior to initiation of asphyxia. Durations of asphyxia were all between 8 and 9 minutes. In the last minute of asphyxia, as the ventilator was being reconnected and turned on, 0.4 mL epinephrine (0.01 mg/kg) and 0.5 mL bicarbonate (1.0 mmol/kg) were given to stimulate the sympathetic nervous system and manage acidosis, respectively. Of 17 rats subjected to asphyxial CA used in the data analysis, the mean duration of asphyxia was 8.5 ± 0.1 minutes, yielding CA durations of 6.8 ± 0.2 minutes (mean ± SEM). Cardiopulmonary resuscitation ([CPR], manual sternal compressions at 180–240 per minute) was performed and continued until return of spontaneous circulation (ROSC). Arterial blood gases were obtained 10 minutes postROSC to assess ventilation and modify ventilator settings as necessary. Over the next 1-hour postROSC, vessels were decannulated and rats were extubated.
Vanadium Acid Fuchsin/Toluidine Blue Staining and Confocal Imaging of Mitochondrial Morphology
One or 4 hours after CPR, animals were anesthetized with isoflurane and perfused transcardially with phosphate buffered saline (PBS) for 2 minutes, followed by 4% paraformaldehyde (PFA) in PBS for 10 minutes. The brains were postfixed in 4% PFA/1% glutaraldehyde for 72 hours, then PFA was exchanged for 30% sucrose (in PBS) for another 2 days and 30-μm brain sections containing hippocampus were cut using a cryostat (ThermoFisher Scientific, Waltham, MA).
To assess neuronal injury, sections were stained with vanadium acid fuchsin (VAF)/toluidine blue (TB) largely as described (28). In brief, slices were stained with VAF for 1–2 minutes, washed with PBS, incubated in 0.01% borax solution for 20–30 seconds, and rinsed in distilled water. Finally, brain slices were cleared by acetate buffer (PH 3.3) for 30 seconds and counterstained with 0.025% TB for 20–30 seconds. Stained slices were assessed using light microscopy.
To assess mitochondrial morphology, sections were labeled with primary antibodies against the mitochondrial outer membrane protein, translocase outer membrane-20 ([TOM20]; 1:200; Santa Cruz Biotechnology, Santa Cruz, CA) and secondary antirabbit fluorescent antibodies (1:200, DyLight 488; Jackson ImmunoResearch, West Grove, PA). The sections were imaged using an inverted stage Nikon Eclipse Ti chassis microscope with a Yokogawa CSUX spinning disk head and a 100× (1.49 numerical aperture) objective and images acquired using a Hamamatsu electromultiplying CCD camera. Excitation (488 nm) was via a Coherent sapphire laser source synchronized with the camera, emission was monitored with a 525 (50) nm filter, and images were acquired using MIcroManager ImageAcquisition software (version 1.4.16).
Cardiac Perfusion and Tissue Sections Preparation for Timm’s Labeling
In initial studies, brain sections were prepared in 2 ways (as outlined in Results). Our first attempts sought to maximize rapid Zn2+ fixation in situ; animals were perfused with 0.2% Na2S in Millonig’s buffer for 5 minutes to precipitate Zn2+, followed by 4% PFA/1% glutaraldehyde solution in PBS for the next 12 minutes. After perfusion the brains were removed and postfixed using 2% PFA/2% glutaraldehyde/0.2% Na2S, 4°C for 72 hours, then placed into 30% sucrose solution in PBS for 48–72 hours, followed by OCT embedding and cryostat sectioning (to 50 or 80 μm thickness). Subsequent studies sought to optimize tissue preservation and to that aim, rats were perfused transcardially with 4% PFA/1% glutaraldehyde solution in PBS for 5 minutes, followed by 0.4% Na2S/Millonig’s buffer (0.12 M; 0.002% CaCl2, 1.6% NaH2PO4, and 0.4% NaOH; PH 7.3) for the next 7 minutes to precipitate Zn2+ (29). Brains were removed and postfixed using 2% PFA/2% glutaraldehyde/0.2% Na2S, 4°C overnight, prior to vibratome sectioning (again to 50 or 80 μm thickness).
For Timm’s staining, slices were incubated in the dark in a solution containing 1 part solution A (1 M AgNO3), 20 parts solution B (2% hydroquinone and 5% citric acid in water), and 100 parts of solution C (30% gum arabic in water). Development was performed in the dark, was monitored by periodic evaluations under low light, and was terminated by washing in water. Eighty-micrometer-thick slices were placed into PBS and processed for electron microscopy; 50-μm-thick slices were analyzed under light microscopy.
Electron Microscopy
For TEM analysis, we utilized ultrathin sections (∼1 µm thickness) from vibratome- or cryostat-cut fixed brain slices, prepared largely as previously described (30), with small modifications. The sections were rinsed in PBS and postfixed with 1% osmium tetroxide in PBS for 1 hour, then dehydrated in increasing serial dilutions of ethanol (70%, 85%, 95%, 100%), put into intermediate solvent propylene oxide (2 times for 10 minutes), and incubated in 1:1 mixture of propylene oxide/Spurr's resin for 1 hour. Finally, slices were embedded in Spurr’s resin overnight. Ultrathin sections (∼70-nm thickness) were cut using a Leica Ultracut UCT ultramicrotome (Leica, Vienna, Austria) mounted on 150 mesh copper grids, stained with lead citrate, and viewed using a JEOL 1400 electron microscope (JEOL, Tokyo, Japan). Images were captured using a Gatan digital camera (Gatan, Pleasanton, CA).
Statistics and Data Analysis
All counts/ratings were carried out entirely by raters blinded to the experimental conditions (TEM, TOM-20), or verified via extensive sampling by blinded raters (VAF/TB). For assessment of neuronal injury (VAF/TB), photomicrographs (40× magnification) were obtained from each section. The rater examined the micrographs and judged each identifiable neuron in the section as healthy or injured based upon criteria as described. For mitochondrial ratings of TEM images of sections from CA1, all evident mitochondria in images from CA1 pyramidal neurons were rated as to the presence of Timm’s stain deposits (representing Zn2+) and the integrity of the mitochondria according to criteria as described. For measurement of TOM-20-labeled mitochondria, large neurons in the pyramidal cell layer were imaged using confocal microscopy. To control for differing behavior of mitochondria between cellular compartments, we focused our studies on mitochondria in the perinuclear region in the plane of sharp focus. Images were imported into ImageJ software and were adjusted to provide optimal discrimination of the apparent mitochondrial edges from background. Length and width measurements were obtained on all clearly demarcated mitochondria adjacent to and surrounding the nuclear circumference.
To assess significance, either two-tailed t-test or one-way ANOVA with Tukey posthoc analysis (indicated in each figure legend) was used, depending on the number of groups of comparison. All values are displayed as mean ± SEM). All experiments were repeated at least 3 times.
RESULTS
Hippocampal Pyramidal Neuron Injury After TGI
Rats were subjected to asphyxial CA as described (see Materials and Methods). After resuscitation followed by 1 hour or 4 hours of recovery, rats were perfused and their hippocampi processed for histological analysis.
To assess overall injury to CA1 and CA3 pyramidal neurons, sections were stained with a modified acid fuchsin labeling procedure to identify acidophilic neurons (termed VAF labeling) (28), and counterstained with TB to assess morphology. Sections were examined and each identifiable neuron rated as healthy or injured. Healthy neurons showed distinct round nuclei surrounded by TB-stained cytoplasm. Injured neurons were of 2 types: One subset that showed clear atrophy along with VAF (acidophilic) labeling in the cytoplasm indicative of early injury, whereas others showed severe swelling with loss of clear distinction between nucleus and surrounding cytoplasm, vacuolar changes, and absence of clear VAF labeling (Fig. 1A, left). We noted that in comparison to control, episodes of global ischemia caused distinct and substantial injury to CA1 pyramidal neurons, with relatively little injury in CA3. In addition, the CA1 injury was greater after 4 hours than after 1-hour recovery, indicating apparent progression of the injury in the hours after the ischemic episode (Fig. 1A, right).
FIGURE 1.

Global ischemia induces progressive neuronal injury and Zn2+ accumulation in CA1 pyramidal neurons. Rats were subjected to TGI followed by resuscitation as described. After 1- or 4-hour recovery, rats were killed, perfused, and brain tissue removed for histological examination as described. Injury was assessed on hippocampal slices subjected to a modified acid fuchsin labeling procedure (VAF; detailed in Materials and Methods) and counterstained with toluidine blue; Zn2+ accumulation was assessed via Timm’s labeling (see Materials and Methods). (A) Global ischemia induced injury to hippocampal CA1 pyramidal neurons. Left: Representative images. Hippocampal slices were photographed at low magnification (Bar = 500 µm), and CA1 and CA3 regions (indicated by blue and red rectangles, respectively) examined at higher magnification (Bar = 50 µm). While most neurons appear intact in control slices, note the increased numbers of VAF-stained neurons as well as of severely swollen neurons (with vacuolar changes and loss of distinct nuclear outlines) after ischemia. Further note that these changes are more prevalent in CA1 than in CA3 and appear to increase from 1 to 4 hours after ischemia (particularly in CA1). Examples of intact (arrow) and damaged (VAF+, triangle; swelling and vacuolar changes, circled) neurons are marked. Right: Quantitative assessment. All discernible neurons were rated as intact or injured, and the percentages of CA1 and CA3 neurons determined to be injured within each hippocampal section were calculated. Note that the extent of neuronal injury was far greater in CA1 that in CA3, and that in CA1 it was significantly greater after 4-hour than 1-hour recovery. Bars represent mean ± SEM from 4 to 7 independent animals (comprising ≥1000 cells from CA1; ≥500 from CA3; with ≥3 sections counted for each animal; ** indicates p < 0.01 by one-way ANOVA with Tukey post hoc). (B) Transient loss of mossy fiber Zn2+ and progressive Zn2+ accumulation in CA1 pyramidal neurons after transient global ischemia. Photomicrographs show low magnification images of Timm’s-labeled hippocampal sections (Bar = 500 µm), and greater magnification images of the CA1 regions from the same sections (indicated by rectangles; Bar = 50 µm). Note the robust stain in the mossy fibers (indicative of presynaptic vesicular Zn2+) in control, and the substantial loss of this Zn2+ at 1-hour recovery. Further note the appearance of some Zn2+ in the somata of CA1 pyramidal neurons that progresses from 1 hour to 4 hours after ischemia (a small amount of accumulation was also noted in some CA3 pyramidal neurons, not shown; Images are representative of ≥3 repetitions each condition).
To begin to address our questions concerning possible Zn2+ contributions to the injury, some sections were stained with a modified Timm’s procedure (see Materials and Methods) to assess the presence and localization of labile Zn2+, and examined under conventional brightfield microscopy (Fig. 1B). As discussed, the Timm’s technique detects labile or loosely bound Zn2+ in tissues by using sulfide ion to precipitate the Zn2+ and “developing” the label (much as with photographic prints) via reduction and deposition of metallic silver at loci of the ZnS precipitates, and in brain appears to be entirely selective for Zn2+ (8, 14). Note that in control slices, distinct Timm’s labeling, indicative of labile Zn2+, is most prominent in the mossy fiber boutons in the dentate gyrus, the hilus, and extending along the CA3 pyramidal cell layer, but that labeling is largely absent in neuronal somata. Further note the substantial loss of Timm’s label from the mossy fibers 1 hour after ischemia, with partial recovery of labeling after 4 hours. Finally, note the appearance of weak Timm’s label in somata of CA1 neurons after ischemia that is greater with 4- than with 1-hour recovery, indicative of some relocalization of labile Zn2+, paralleling the observed neuronal injury.
TGI Induces Mitochondrial Swelling
Previous studies have suggested that mitochondria are likely to be important targets of Zn2+-induced neuronal injury (19). In light of our recent observations of protracted Zn2+ accumulation in CA1 mitochondria after OGD in hippocampal slice (22), we next sought to examine mitochondrial morphology in CA1 neurons after TGI. Sections were immunostained for the mitochondrial outer membrane marker, TOM-20, examined under confocal microscopy (1000×), and images obtained in the CA1 and CA3 pyramidal layers, largely as previously described (22). For quantitative assessment, images were adjusted (using Image J software) to optimally discriminate mitochondrial borders from background, and perinuclear regions cropped from images and coded for blinded measurement of mitochondrial lengths and widths (see Materials and Methods). We found that OGD caused a marked “rounding-up” of the mitochondria with substantial decreases in their mean lengths, increases in their widths, and decreased length/width (L/W) ratios. Notably, whereas direct visual examination of the microscope fields yielded an impression that mitochondrial swelling and disruption were worse in CA1 that in CA3, and were greater after 4- than 1-hour recovery, the quantification procedure did not confirm these differences (showing substantially decreased L/W ratios in all ischemic conditions; Fig. 2). This was felt to reflect a limitation of the approach—as only mitochondria with distinct visualization of edges were amenable to measurement—and extreme swelling and consequent blurring of the borders of the most effected mitochondria precluded accurate assessment of levels of swelling towards the extreme end of the scale.
FIGURE 2.
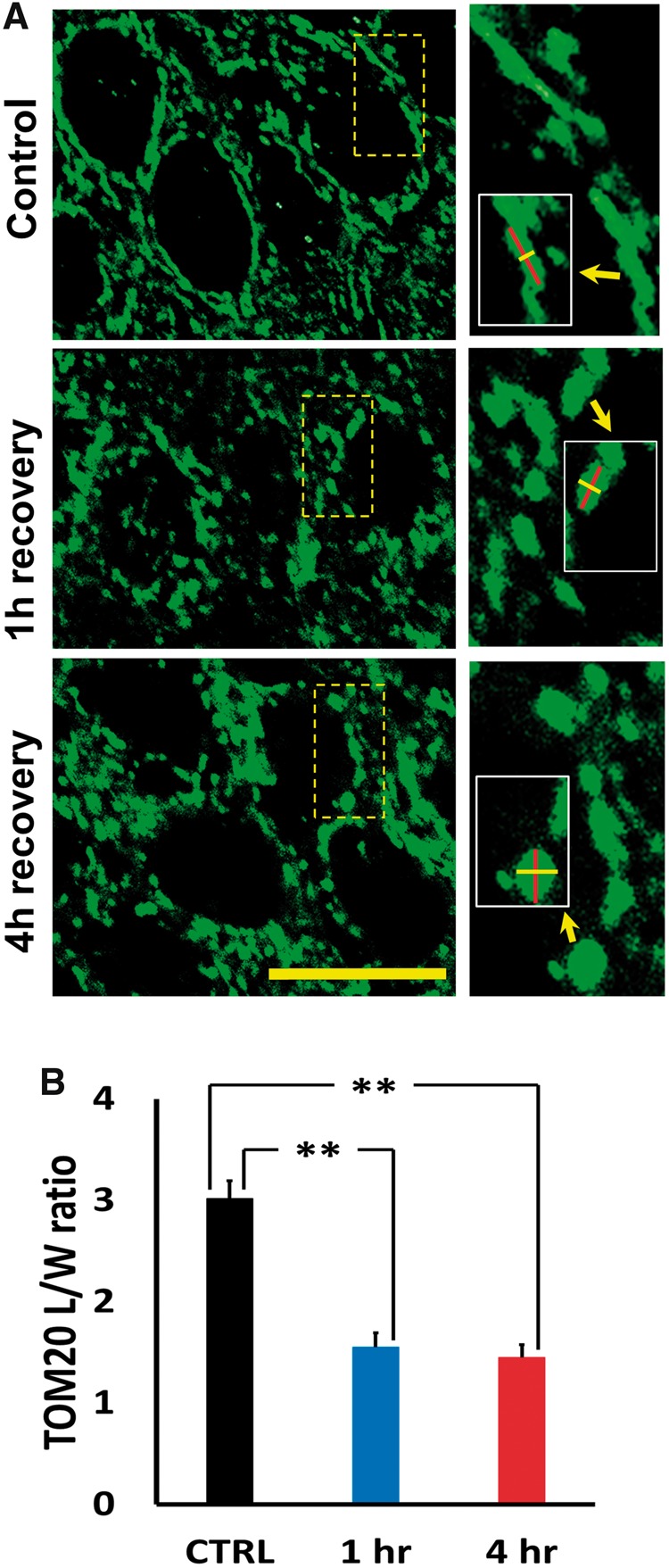
Ischemia disrupts mitochondrial morphology in CA1 pyramidal neurons. Rats were subjected to TGI followed by 1- or 4-hour recovery. Hippocampal slices were immunostained using antibody against the mitochondrial outer membrane marker, TOM20, and examined under confocal microscopy (1000×). (A) Representative images of TOM20-labeled CA1 mitochondria. Note the generally elongated mitochondria seen in perinuclear regions of control, in contrast to the abundant fragmented and swollen mitochondria 1 or 4 hours after ischemia. Small images of regions indicated show high magnifications of representative perinuclear mitochondria; inserts in small images indicate markings for length and width measurements of representative mitochondria (Bar = 10 µm). (B) Ischemia impacts mitochondrial morphology. Lengths and widths of individual mitochondria were measured blindly (using ImageJ software) and length/width (L/W) ratios calculated to quantify morphological change (lower L/W ratios indicates rounding of mitochondria). The L/W ratio for all mitochondria measured in neurons from a single animal was averaged to produce a single, mean L/W ratio for that animal. Note the significant decrease in L/W ratio after ischemia. Bars represent mean L/W ratio ± SEM from 4 to 6 independent animals each condition (comprising 125 mitochondria from 38 cells, control; ≥250 mitochondria, from ≥60 cells per group after TGI; ** indicates p < 0.01 by one-way ANOVA with Tukey post hoc).
Ischemia-Induced Mitochondrial Zn2+ Accumulation Correlates With Ultrastructural Disruption
We next set out to use the Timm’s sulfide silver labeling technique to localize sites of neuronal Zn2+ accumulation at an ultrastructural level. As indicated above, this technique inserts metallic silver deposits at sites of labile or loosely bound Zn2+ accumulation. Staining in neurons is considered to be quite specific for Zn2+, and was found to be entirely absent in mice lacking the vesicular Zn2+ transporter (ZnT3 knockout mice) (14). To further validate the Zn2+ specificity of the stain, we made use of the well characterized hippocampal slice OGD model of brain ischemia (20–22). In control slices, Timm’s staining was present in the mossy fiber pathway and was completely absent in the pyramidal cell layer and stratum radiatum, with distinct labeling appearing in these regions after a 10-minute episode of OGD (much as in Fig. 1B). However, this label was almost completely eliminated when the slices were perfused with the Zn2+-preferring chelator N, N, N′, N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) during OGD (data not shown), further supporting the contention that the Timm’s stain largely reflects accumulation of labile Zn2+ in postsynaptic neurons.
To investigate the redistribution of labile Zn2+ in the somata of CA1 pyramidal neurons after in vivo TGI, sections from postCA rats were Timm’s-labeled and subjected to TEM examination as described (see Materials and Methods). This approach is particularly well suited for the ultrastructural examination of Zn2+ localization because the silver deposit is electron-dense and thus easily visualized. Timm’s labeling and TEM examination are strongly dependent upon animal perfusion and tissue processing techniques. Prior studies have employed a number of variations to try to optimize both the visualization of the Zn2+ and the preservation of the tissue ultrastructure for TEM examination. A key component of Timm’s labeling is the early use of Na2S in the perfusion procedure to precipitate the labile Zn2+ in situ. In early trials we carried out 2 distinct variations (see Materials and Methods for full details) as follows: (i) initial perfusion with Na2S (0.2% in buffer, 5 minutes) prior to perfusion with PFA/glutaraldehyde in order to maximize Zn2+ detection by fixing it in situ at the earliest possible time point; or (ii) perfusion with PFA/glutaraldehyde (5 minutes) prior to Na2S (0.4%, 7 minutes) in order to optimize tissue preservation. In both cases perfusion was followed by brain removal and postfixation incorporating PFA, glutaraldehyde, and Na2S. In control slices subjected to these 2 perfusion protocols we noted no qualitative difference in the appearance or localization of the Timm’s label, and opted for the second approach (fixation first), which is the more conservative in terms of Zn2+ detection, in subsequent experiments.
In our ultrastructural studies, hippocampal CA1 sections from 10 animals (i.e. 3 controls, 4 ischemia followed by 1-hour recovery; and 3 ischemia followed by 4-hour recovery) were subjected to blinded quantitative examination. In agreement with prior studies (8, 14), in control rats we found electron-dense Zn2+ foci to be most evident in presynaptic mossy fiber boutons, with little evidence of Zn2+ at other loci or within neuronal somata. As somata of CA1 neurons were identified by their characteristic nuclei, high-magnification TEM images for mitochondrial analysis were obtained adjacent to nuclei and comprised mitochondria in perinuclear and proximal dendritic regions. In the controls, a substantial majority of mitochondria appeared healthy and intact; these generally were elongated and had distinct cristae structure visible along with an intact double outer membrane (OM; Figs. 3A and 4A). However, a minority (∼20%–25%) showed evidence of mild injury; these were generally rounder than elongated, suggestive of early swelling, and often showed some disruption or blurring of their cristae. Interestingly, close to 50% of these mildly damaged mitochondria contained apparent Zn2+ deposits, as indicated by the presence of electron-dense silver granules similar to those seen in presynaptic boutons. In contrast, Zn2+ deposits were only seen in a very small fraction (<1%) of intact appearing mitochondria in all conditions.
FIGURE 3.
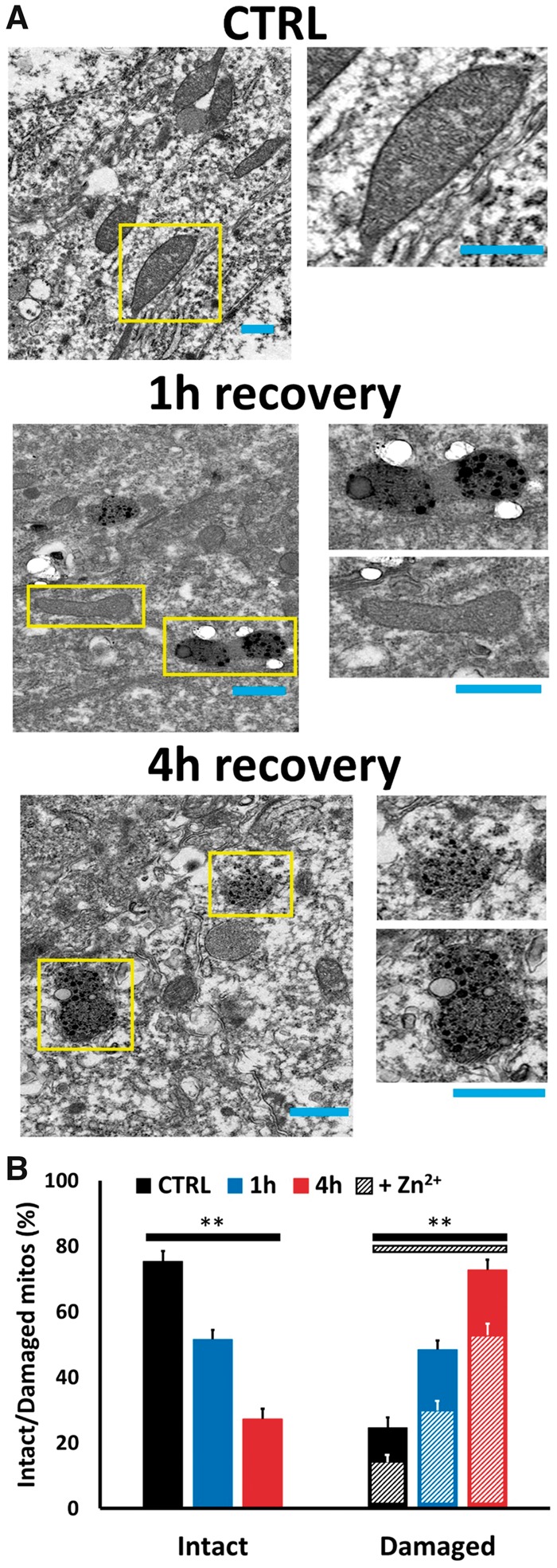
Global ischemia induces progressive Zn2+ accumulation and injury in CA1 pyramidal neuronal mitochondria. Rats were subjected to TGI followed by 1- or 4-hour recovery. To assess mitochondrial damage and Zn2+ accumulation in CA1 pyramidal neurons, Timm’s-labeled hippocampal slices were examined under TEM. (A) Representative electron micrographs. Representative TEM images (4000×) of Timm’s-labeled slices from a control rat (top), or rats subjected to ischemia followed by 1 hour (middle), or 4 hours (bottom) recovery (Bar = 0.5 µm; rectangles show regions displayed at greater magnification, right; Bar = 0.5 µm). Note the intact structure and absence of Timm’s precipitate in most mitochondria in control, the significant numbers of mitochondria showing early damage (with rounding) and distinct presence of Timm’s precipitate with 1-hour recovery, with greater numbers of mitochondria displaying Zn2+ accumulation and extensive injury after 4-hour recovery. (B) Quantitative evaluation. To assess mitochondrial Zn2+ accumulation and damage, all evident mitochondria in images from CA1 pyramidal neurons were rated by an observer blinded to the experimental condition. Bars display percentage of mitochondria appearing intact or showing evidence of damage (assessed as described), and hatchmarks indicate the presence of evident Zn2+ deposits in the damaged mitochondria (Zn2+ deposits were only present in <1% of intact appearing mitochondria in all conditions). Note the marked increase in numbers of damaged appearing mitochondria after ischemia, the marked increase in their numbers with increased recovery duration (from 1 to 4 hours), and the parallel increase in numbers containing distinct Zn2+ deposits, present in the majority of damaged mitochondria in each condition. Values represent mean ± SEM from 3 to 4 independent animals each condition (for each animal, ≥70 mitochondria from ≥10 sections were rated; ** indicates p < 0.01 by one-way ANOVA with Tukey post hoc).
In comparison to the controls, the numbers of damaged mitochondria were substantially increased after global ischemia. With 1-hour recovery, ∼50% of mitochondria were damaged. Interestingly, the damage appeared to be strongly progressive, with >75% of mitochondria showing damage after 4-hour recovery. Furthermore, at each of these recovery time points, the majority of damaged mitochondria had clear Zn2+ deposits (Fig. 3B) supporting a Zn2+ contribution to the mitochondrial damage. Yet, in each condition, a minority of damaged mitochondria lacked clear Zn2+ deposits. The reasons for this are not completely clear but may well reflect a number of factors, including: (i) threshold level of Zn2+ accumulation may be needed for detection by the Timm’s labeling procedure; (ii) the use of our conservative perfusion paradigm, which maximizes structural protection/integrity at the possible expense of some loss of Zn2+ from loci of intracellular accumulation; and (iii) contributions of Zn2+ independent mitochondrial swelling mechanisms.
We further sought to discriminate degrees of mitochondrial damage related to mitochondrial Zn2+ accumulation (Fig. 4). As noted above, all the damaged mitochondria showed evidence of mild swelling and early disruption of cristae structure. However, some showed more extreme disruption or even complete absence of evident cristae. In some there was apparent vacuole formation within mitochondria, whereas others showed varying degrees of OM rupture, progressing from a small area of rupture to complete loss of evident OM. In some mitochondria with apparent severe OM rupture there was apparent “leakage” of Zn2+ containing material into the surrounding cytoplasm (Fig. 4A). These characteristics are suggestive of progressive stages of mitochondrial damage.
FIGURE 4.

Discrimination of degrees of mitochondrial damage: Progression of mitochondrial disruption with time of recovery. (A) Graded degrees of mitochondrial structural disruption: Discriminating criteria and representative images. The table illustrates the spectrum of mitochondrial morphologies as described. To be rated as intact, mitochondria were generally elongated and had intact outer membrane and clear abundant cristae. Mitochondria were rated as showing mild damage if they showed evidence of early swelling and some loss of cristae but with an intact outer membrane (blue arrows highlight sparse cristae), and were considered to be severely damaged if there was apparent disruption of the outer membrane (red arrows); virtually all mitochondria with outer membrane rupture showed no discernible cristae. Such severely damaged mitochondria also displayed a spectrum of damage, ranging from early membrane rupture (left) to substantial rupture with loss of mitochondria contents (middle) or complete absence of membrane (right). (B) Mitochondrial damage progresses with increasing recovery time. To assess possible progression of mitochondrial disruption over time after ischemia, we re-examined the same postischemic images (as in Fig. 3), only evaluating the mitochondria that both appeared damaged and contained Zn2+ precipitates. Each mitochondrion was rated as mildly damaged if it was judged to have an intact outer membrane (OM; blue) or severely damaged if its OM was disrupted (red). Note the substantial progression of mitochondrial disruption over time, with a far greater percentage of mitochondria showing OM disruption after 4-hour than after 1-hour recovery. Bars show percentage of injured mitochondria in each category (intact or disrupted OM), and represent mean ± SEM from 3 to 4 independent animals (≥55 mitochondria counted from ≥10 sections, each animal; ** Indicates p < 0.01 by 2-tailed Student t-test).
To further address the possible progression of the mitochondrial damage and disruption, we carried out a second blinded count and rating of mitochondria from the same set of postischemic images, but only evaluating the mitochondria that both appeared damaged and contained Zn2+ precipitates. In this assessment, we separated the mitochondria on the basis of the integrity of their OMs, considering that a ruptured OM reflected relatively severe and likely irrecoverable damage. Interestingly, we found the distribution of the mitochondrial damage to differ substantially between 1- and 4-hour recovery, with the proportion of mitochondria with ruptured OMs being far greater with 4-hour recovery (Fig. 4B).
Thus, present observations reveal that both the presence of Zn2+ accumulation within mitochondria and the extent of damage of the Zn2+ containing mitochondria are progressive in the hours after an episode of TGI. Whereas this does not prove that the Zn2+ causes the mitochondrial damage, the data are certainly consistent with a model we have proposed based upon recent hippocampal slice studies (19, 22) wherein progressive Zn2+ accumulates in CA1 mitochondria both during and for a considerable period of time after the ischemic episode, contributing to their progressive damage and disruption.
DISCUSSION
Summary of Findings
Despite strong evidence for Zn2+ contributions to neuronal injury in ischemia and after prolonged seizures, with accumulation of labile Zn2+ in many degenerating neurons and neuroprotective effects of Zn2+ chelation as well as numerous clues that mitochondria are likely to be important sites of injurious Zn2+ effects, the relationship between mitochondrial Zn2+ accumulation, mitochondrial disruption and neuronal injury in vivo has not been previously examined. The present study is the first to use the Timm’s labeling approach—which yields electron-dense silver deposits at sites of labile Zn2+ accumulation—in order to examine the redistribution of Zn2+ in CA1 pyramidal neurons after in vivo TGI at the ultrastructural level. Notably, in these postischemic neurons, we found a strong relationship between the appearance of Timm’s deposit within mitochondria with their swelling and structural disruption. This observation provides new support to the idea that the Zn2+ contributes to the mitochondrial damage/dysfunction seen in these neurons. Furthermore, examination at 2 time points after TGI revealed a progression in both numbers of damaged and Zn2+ containing mitochondria as well as in the degree of their disruption. This delayed temporal evolution correlates with both the delayed Zn2+ accumulation we observed in CA1 but not in CA3 mitochondria after sublethal OGD in the hippocampal slice model of ischemia (22), and with the delayed degeneration associated with mitochondrial disruption that is characteristically seen in CA1 neurons after ischemia (23–25), and suggests the possibility that the mitochondrial Zn2+ accumulation and its consequences may be progressive events during the hours postischemia in CA1 neurons that are amenable to delayed therapeutic interventions.
Zn2+ in Ischemic Hippocampal Injury
As noted above, a small portion of brain Zn2+ is loosely bound and can be detected histochemically using Timm’s labeling or visualized with membrane permeable fluorescent markers; under normal conditions, this pool largely comprises Zn2+ present in presynaptic vesicles of some excitatory pathways (most prominently in large mossy fiber boutons) (8, 12). Thus, observations that labile Zn2+ accumulates in injured and degenerating neurons after prolonged seizures or ischemia and that these effects were attenuated by an extracellular Zn2+ chelator led to the presumption that they resulted from presynaptic Zn2+ release and its “translocation” into the postsynaptic neurons (3–5). This idea was tested via use of mice lacking the ZnT3 vesicular Zn2+ transporter, which are entirely lacking in presynaptic vesicular Zn2+ (14). Surprisingly, seizure-induced Zn2+ accumulation and injury to CA1 pyramidal neurons was actually increased in ZnT3 knockout mice, indicating a distinct nonsynaptic source of toxic Zn2+ accumulation in these neurons (31).
A likely candidate source was Zn2+ that is bound to Zn2+-buffering proteins like MT-III (the primary metallothionein isoform in neurons) (32). Indeed, studies in neuronal culture revealed that strong Zn2+ mobilization from these proteins could trigger Zn2+-dependent neuronal injury in the absence of extracellular Zn2+ entry (33, 34). The generation of MT-III knockouts provided a model to directly examine the contributions of Zn2+ release from this protein (35). In vivo seizure studies, as well as hippocampal slice OGD studies, using both ZnT3 and MT-III KO’s highlighted distinct contributions of these pools of Zn2+ to the Zn2+ accumulation and injury that occurred, with mobilization from MT-III predominating in CA1. Presynaptic release and translocation into post-synaptic neurons appears to predominate in CA3 (15, 20, 22), which receives strong synaptic input from the Zn2+-rich mossy fiber terminals, and (in contrast to CA1) is preferentially injured after prolonged limbic seizures. Interestingly, although CA3 neurons also contain considerable MT-III and like CA1 showed strong cytosolic Zn2+ loading during acute slice OGD, they appear to be better able to recover from the Zn2+ loads and did not show the progressive and persistent mitochondrial Zn2+ accumulation we observed in CA1 after sublethal episodes of OGD (22).
Mitochondria Are Likely to Be Important Targets of Zn2+ Effects
Despite the considerable evidence that “excitotoxic” mechanisms are important contributors to neurodegeneration occurring after acute brain insults including ischemia, trauma, and prolonged seizures, many downstream mechanisms have been implicated and there are as yet no neuroprotective treatments that have shown clear benefit in humans. As discussed, early studies focused on the key role of Ca2+ entry through highly Ca2+ permeable NMDA channels. Studies of relevant downstream mechanisms highlighted mitochondria as a likely important site of Ca2+ effects, with Ca2+ entering mitochondria through the mitochondrial Ca2+ uniporter (MCU), and with sufficiently large mitochondrial loads, triggering deleterious effects including reactive oxygen species generation and opening of the mitochondrial permeability transition pore (mPTP), leading to release of apoptotic mediators including cytochrome C (36). However, it has also become apparent that Zn2+ has very potent effects on mitochondria. This slow recognition of likely Zn2+ contributions reflects in part the relatively recent availability of Zn2+ selective indicators (37). Indeed, virtually all the available “Ca2+ indicators” respond to Zn2+ with greater potency, a factor that likely led to the mistaken attribution of some Zn2+ effects to Ca2+ (38, 39).
In many studies on isolated mitochondria and cultured neurons, application of Zn2+ appeared to disrupt mitochondrial function with a high degree of potency (40–44). These Zn2+ effects on mitochondria appeared to be dependent upon mitochondrial Zn2+ entry through the MCU, and comprise effects that had been previously attributed to Ca2+, including induction of prolonged reactive oxygen species generation, and swelling and release of cytochrome C resulting from opening of the mPTP (18, 42–46). However, some studies have found Zn2+ to induce effects on mitochondria with more modest potency, and clues have emerged to factors underlying these divergent results (for review see [19]). One key variable concerns the levels of free Zn2+ achieved in intact neurons upon exogenous exposure. This depends both upon the route of Zn2+ entry (44) but also critically upon the integrity of Zn2+ buffering by MTs and related peptides (18, 43, 45). Another likely variable concerns Zn2+ interactions with Ca2+, with synergism between effects of these ions on mitochondria (18, 42, 46), and possible dependence upon Ca2+ for Zn2+ to enter mitochondria through the MCU (47–50). Thus, under pathophysiologically relevant conditions of ischemia, in which Zn2+ buffering by MT’s is impaired by acidosis and oxidative stress, and Ca2+ is present, in vitro studies would predict Zn2+ to impact mitochondria with a very high degree of potency.
Whereas above studies of Zn2+ effects on mitochondria applied Zn2+ to isolated mitochondria or cultured neurons, there is also evidence that endogenous Zn2+ accumulates in mitochondria, contributing to their dysfunction after in vivo ischemia (6, 17). In addition, studies in our acute hippocampal slice OGD ischemia model provide evidence that early Zn2+ uptake into mitochondria via the MCU occurs upstream from and contributes to the occurrence of a sharp Ca2+ rise (termed “Ca2+ deregulation”) that is a terminal event (20, 21). More recently, we have subjected slices to sublethal OGD, terminating the exposure shortly after the initial Zn2+ rise but before the terminal Ca2+ deregulation, in order to model events that may occur after transient in vivo ischemia. Interestingly, consistent with present observations after in vivo TGI, we found evidence for delayed and progressive Zn2+ uptake into mitochondria of CA1 pyramidal neurons after OGD termination which appeared to contribute to delayed mitochondrial swelling. Furthermore, delayed administration of the MCU blocker, ruthenium red prevented this late Zn2+ accumulation and diminished the subsequent mitochondrial swelling. In contrast, in CA3 neurons mitochondrial Zn2+ recovered rapidly after OGD termination (22).
Conclusions and Future Directions
The development of neuroprotective interventions for stroke has presented an extremely difficult challenge, for both logistical reasons and incomplete understanding of critical events in the injury cascade. Studies over the past 2 decades have made it progressively clear that Zn2+ is an important ionic mediator of the excitotoxic injury cascade in CA1 as well as in many forebrain neurons in which it accumulates. Paralleling the evolving evidence for Zn2+ contributions in ischemia, there has been a rapid increase in understanding of ways in which Zn2+ injures neurons, and, in particular, emerging clues that mitochondria might be important targets of its early effects, with compelling clues from both in vivo and slice models highlighting large and long lasting effects after transient ischemia in CA1 (19).
Yet, until now, no study has specifically examined the relationship between mitochondrial Zn2+ accumulation, mitochondrial disruption and neuronal injury after in vivo ischemia. Such a study requires an ultrastructural approach to correlate Zn2+ accumulation with organelle morphology at the individual mitochondrion level, and the presently employed Timm’s labeling (to fix Zn2+ in situ) combined with TEM would seem ideally suited for such an investigation. The present findings in a rat TGI model demonstrate, for the first time, progressive Zn2+ accumulation in CA1 mitochondria to be strongly correlated with their physical disruption. While this does not in itself indicate causation, it provide new support for a possible direct contributory role of the mitochondrial Zn2+ in the delayed mitochondria damage and cell death that are characteristic of CA1 (23–25).
Future aims will seek to assess potential therapeutic utility of targeting these early events. Relevant questions include the nature of interventions that may provide benefit, the temporal window of opportunity to intervene, and the definition of contributions of Zn2+/mitochondrial interactions in neuronal populations other than CA1 in which early Zn2+ accumulation occurs. Based upon our studies in the hippocampal slice OGD model of ischemia, we feel that the delayed administration of MCU blockers or Zn2+ chelators has potential to diminish the progressive Zn2+ uptake into mitochondria, thereby diminishing mitochondrial dysfunction and yielding neuroprotective benefit. Other potential interventions could target downstream consequences of the mitochondrial Zn2+ uptake, including reactive oxygen species generation and mPTP opening. We hope these insights will aid the development of therapeutic interventions that improve outcomes when delivered after ischemia.
ACKNOWLEDGMENTS
We wish to thank Oswald Steward and Ilse Sears-Kraxberger for excellent support and help with the electron microscopy.
Supported by NIH grants NS096987 (JHW), NS100494 (JHW), R21EB024793 (YA), a pilot grant to Y.A. via UL1 TR001414, the American Heart Association grants 17GRNT33410181 (JHW) and 16PRE29560003 (SGJ), and the Roneet Carmell Memorial Endowment Fund (YA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding sources.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Hoyte L, Barber PA, Buchan AM, et al. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med 2004;4:131–6 [DOI] [PubMed] [Google Scholar]
- 2. Ikonomidou C, Turski L.. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 2002;1:383–6 [DOI] [PubMed] [Google Scholar]
- 3. Frederickson CJ, Hernandez MD, McGinty JF.. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res 1989;480:317–21 [DOI] [PubMed] [Google Scholar]
- 4. Koh JY, Suh SW, Gwag BJ, et al. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996;272:1013–6 [DOI] [PubMed] [Google Scholar]
- 5. Tonder N, Johansen FF, Frederickson CJ, et al. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci Lett 1990;109:247–52 [DOI] [PubMed] [Google Scholar]
- 6. Calderone A, Jover T, Mashiko T, et al. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci 2004;24:9903–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin HZ, Sensi SL, Ogoshi F, et al. Blockade of Ca2+-permeable AMPA/kainate channels decreases oxygen-glucose deprivation-induced Zn2+ accumulation and neuronal loss in hippocampal pyramidal neurons. J Neurosci 2002;22:1273–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perez-Clausell J, Danscher G.. Intravesicular localization of zinc in rat telencephalic boutons. A histochemical study. Brain Res 1985;337:91–8 [DOI] [PubMed] [Google Scholar]
- 9. Assaf SY, Chung SH.. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984;308:734–6 [DOI] [PubMed] [Google Scholar]
- 10. Howell GA, Welch MG, Frederickson CJ.. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature 1984;308:736–8 [DOI] [PubMed] [Google Scholar]
- 11. Sloviter RS. A selective loss of hippocampal mossy fiber Timm stain accompanies granule cell seizure activity induced by perforant path stimulation. Brain Res 1985;330:150–3 [DOI] [PubMed] [Google Scholar]
- 12. Frederickson CJ, Rampy BA, Reamy-Rampy S, et al. Distribution of histochemically reactive zinc in the forebrain of the rat. J Chem Neuroanat 1992;5:521–30 [DOI] [PubMed] [Google Scholar]
- 13. Haug FM. Electron microscopical localization of the zinc in hippocampal mossy fibre synapses by a modified sulfide silver procedure. Histochemie 1967;8:355–68 [DOI] [PubMed] [Google Scholar]
- 14. Cole TB, Wenzel HJ, Kafer KE, et al. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci USA 1999;96:1716–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee JY, Kim JH, Palmiter RD, et al. Zinc released from metallothionein-III may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol 2003;184:337–47 [DOI] [PubMed] [Google Scholar]
- 16. Shuttleworth CW, Weiss JH.. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol Sci 2011;32:480–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonanni L, Chachar M, Jover-Mengual T, et al. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J Neurosci 2006;26:6851–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ji SG, Weiss JH.. Zn(2+)-induced disruption of neuronal mitochondrial function: Synergism with Ca(2+), critical dependence upon cytosolic Zn(2+) buffering, and contributions to neuronal injury. Exp Neurol 2018;302:181–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji SG, Medvedeva YV, Wang HL, et al. Mitochondrial Zn(2+) accumulation: A potential trigger of hippocampal ischemic injury. Neuroscientist 2019;25:126–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Medvedeva YV, Lin B, Shuttleworth CW, et al. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci 2009;29:1105–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Medvedeva YV, Weiss JH.. Intramitochondrial Zn(2+) accumulation via the Ca(2+) uniporter contributes to acute ischemic neurodegeneration. Neurobiol Dis 2014;68:137–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Medvedeva YV, Ji SG, Yin HZ, et al. Differential vulnerability of CA1 versus CA3 pyramidal neurons after ischemia: Possible relationship to sources of Zn2+ accumulation and its entry into and prolonged effects on mitochondria. J Neurosci 2017;37:726–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colbourne F, Sutherland GR, Auer RN.. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci 1999;19:4200–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 1982;239:57–69 [DOI] [PubMed] [Google Scholar]
- 25. Sugawara T, Fujimura M, Morita-Fujimura Y, et al. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 1999;19:RC39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang YJ, Tian G, Bazrafkan A, et al. Recovery from coma post-cardiac arrest is dependent on the orexin pathway. J Neurotrauma 2017;34:2823–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crouzet C, Wilson RH, Bazrafkan A, et al. Cerebral blood flow is decoupled from blood pressure and linked to EEG bursting after resuscitation from cardiac arrest. Biomed Opt Express 2016;7:4660–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Victorov IV, Prass K, Dirnagl U.. Improved selective, simple, and contrast staining of acidophilic neurons with vanadium acid fuchsin. Brain Res Brain Res Protoc 2000;5:135–9 [DOI] [PubMed] [Google Scholar]
- 29. Danscher G, Zimmer J.. An improved Timm sulphide silver method for light and electron microscopic localization of heavy metals in biological tissues. Histochemistry 1978;55:27–40 [DOI] [PubMed] [Google Scholar]
- 30. Park J, Trinh VN, Sears-Kraxberger I, et al. Synaptic ultrastructure changes in trigeminocervical complex posttrigeminal nerve injury. J Comp Neurol 2016;524:309–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee JY, Cole TB, Palmiter RD, et al. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: Evidence against synaptic vesicle origin. J Neurosci 2000;20:RC79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maret W. Metallothionein/disulfide interactions, oxidative stress, and the mobilization of cellular zinc. Neurochem Int 1995;27:111–7 [DOI] [PubMed] [Google Scholar]
- 33. Aizenman E, Stout AK, Hartnett KA, et al. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J Neurochem 2000;75:1878–88 [DOI] [PubMed] [Google Scholar]
- 34. Bossy-Wetzel E, Talantova MV, Lee WD, et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K(+) channels. Neuron 2004;41:351–65 [DOI] [PubMed] [Google Scholar]
- 35. Erickson JC, Hollopeter G, Thomas SA, et al. Disruption of the metallothionein-III gene in mice: Analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J Neurosci 1997;17:1271–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nicholls DG, Budd SL.. Mitochondria and neuronal survival. Physiol Rev 2000;80:315–60 [DOI] [PubMed] [Google Scholar]
- 37. Gee KR, Zhou ZL, Ton-That D, et al. Measuring zinc in living cells. A new generation of sensitive and selective fluorescent probes. Cell Calcium 2002;31:245–51 [DOI] [PubMed] [Google Scholar]
- 38. Cheng C, Reynolds IJ.. Calcium-sensitive fluorescent dyes can report increases in intracellular free zinc concentration in cultured forebrain neurons. J Neurochem 1998;71:2401–10 [DOI] [PubMed] [Google Scholar]
- 39. Stork CJ, Li YV.. Intracellular zinc elevation measured with a “calcium-specific” indicator during ischemia and reperfusion in rat hippocampus: A question on calcium overload. J Neurosci 2006;26:10430–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dineley KE, Richards LL, Votyakova TV, et al. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion 2005;5:55–65 [DOI] [PubMed] [Google Scholar]
- 41. Gazaryan IG, Krasinskaya IP, Kristal BS, et al. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J Biol Chem 2007;282:24373–80 [DOI] [PubMed] [Google Scholar]
- 42. Jiang D, Sullivan PG, Sensi SL, et al. Zn(2+) induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem 2001;276:47524–9 [DOI] [PubMed] [Google Scholar]
- 43. Sensi SL, Ton-That D, Sullivan PG, et al. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci USA 2003;100:6157–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sensi SL, Yin HZ, Carriedo SG, et al. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA 1999;96:2414–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clausen A, McClanahan T, Ji SG, et al. Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons. PLoS ONE 2013;8:e83347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sensi SL, Yin HZ, Weiss JH.. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur J Neurosci 2000;12:3813–8 [DOI] [PubMed] [Google Scholar]
- 47. Saris NE, Niva K.. Is Zn2+ transported by the mitochondrial calcium uniporter?. FEBS Lett 1994;356:195–8 [DOI] [PubMed] [Google Scholar]
- 48. De Stefani D, Patron M, Rizzuto R.. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta 2015;1853:2006–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamer KJ, Mootha VK.. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 2015;16:545–53 [DOI] [PubMed] [Google Scholar]
- 50. Marchi S, Pinton P.. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J Physiol 2014;592:829–39 [DOI] [PMC free article] [PubMed] [Google Scholar]