

Abstract
It has been asserted that chronic traumatic encephalopathy (CTE) pathology is only present in former athletes and others who have been exposed to repetitive concussions, subconcussive blows, or both. We hypothesized that CTE pathology would be present in men who had no known history of repetitive neurotrauma. Comprehensive medical record reviews and health surveys completed by a family member were available for the 8 men in this case series, none of whom had known exposure to repetitive neurotrauma but 2 of whom had a history of traumatic brain injury (TBI). Postmortem tissue was immunostained for hyperphosphorylated tau (p-tau) to assess for CTE pathology, Braak stage, and aging-related p-tau. The neuropathologist was blind to age, personal history, and clinical history. Six of the 8 cases (75%) showed p-tau in neurons, astrocytes, and cell processes around small blood vessels in an irregular pattern at the depths of the cortical sulci. The changes were focal and limited in terms of overall extent, and some of the cases had a clearer pattern of pathology and some could be considered equivocal. Two of the 8 cases had a history of TBI and one of them showed CTE pathology. Five of the 6 cases with no known history of neurotrauma appeared to meet consensus criteria for CTE. This study adds to the emerging literature indicating that CTE pathology is present in people not known to have experienced multiple concussions or subconcussive blows to the head.
Keywords: Brain injury, Chronic traumatic encephalopathy, Concussion, Sports
INTRODUCTION
Over the past decade, chronic traumatic encephalopathy (CTE) has been described as a delayed-onset and progressive neurodegenerative disease, with symptoms appearing in midlife (1, 2) or decades after exposure to concussions or other forms of repetitive neurotrauma (1–9). The theory is that CTE is a distinct (1, 3, 9–11) and unique (10) neurodegenerative disease (9–13). Prior to 2016, there were no agreed upon neuropathological criteria for CTE, and the criteria published by the 2 leading research groups in the United States differed substantially (7, 14). A consensus panel of 7 neuropathologists, convened by the National Institutes of Health (NIH), was tasked with developing preliminary pathological criteria for CTE. To do so, they were asked to blindly examine 10 cases of advanced CTE pathology (2 with Stage III and 8 with Stage IV pathology [14]) and 15 cases with primary tau-related neurodegenerative diseases (15). The virtual slides for these cases were selected and prepared by researchers from Boston University. The panel was provided an a priori presumptive definition and criteria for the neuropathology of CTE (found in Supplementary Data Material S1 of the original article [15]). The panel did not ultimately accept all of the a priori criteria for CTE; they did not include pathology known to be associated with aging (i.e. primary age-related tauopathy [PART] [16] and aging-related tau astrogliopathy [ARTAG] [17]). PART is characterized by hyperphosphorylated tau (p-tau) in neurons, with neurofibrillary degeneration (p-tau) in the brainstem, deep gray matter, basal forebrain, medial temporal lobe, and olfactory areas (bulb and cortex). PART is associated with sparse or absent Aβ, which distinguishes it from Alzheimer disease (AD) (16). ARTAG is characterized by subpial, subependymal, and perivascular accumulation of p-tau in astrocytes in older adults who may or may not have cognitive impairment (17). Prior to the consensus criteria definition published in 2016 (15), ARTAG pathology was described as characteristic of CTE (2–4, 6, 7, 11, 14, 18–21). More recently, researchers have begun to separate CTE pathology from ARTAG pathology (22).
It has been asserted repeatedly that CTE pathology is only found in people who have been exposed to repetitive neurotrauma, such as boxers, contact and collision sport athletes, and military veterans (2, 4, 9–11, 13). However, in recent years, CTE pathology has been discovered in women with no known exposure to multiple concussions or contact sports (23). In addition, it has been found in a small number of people with (1) substance abuse (24), (2) temporal lobe epilepsy (25), (3) amyotrophic lateral sclerosis (ALS) (26), (4) multiple system atrophy (22), and (5) other neurodegenerative diseases (23) who have no known participation in collision or contact sports and no known exposure to repetitive neurotrauma. When CTE pathology is found in studies that do not involve former collision or contact sport athletes, it is usually, but not always, reported that the subjects had a history of at least 1 brain injury (23), or the authors speculate that the subjects could have experienced injuries to their brains that were not documented, such as in falls (22).
The purpose of this study is to apply the consensus criteria for the neuropathology of CTE to an autopsy case series of men with no known history of participation in contact or collision sports. In addition, we carefully examined this case series for pathology associated with aging (PART and ARTAG) and AD. We hypothesized that CTE pathology would be present in some men who had no known history of repetitive neurotrauma.
MATERIALS AND METHODS
Case Ascertainment
This study was embedded in a larger ongoing research program entitled the Tampere Sudden Death Study (TSDS). We sought to identify a case series of middle-aged and older adult men from the general population for postmortem neuropathological analysis. For the present study, a consecutive sample of 12 men, between the ages of 49 and 82, who died out of the hospital, were collected to study (during 2014–2015). Of those 12 cases, family health surveys were returned from 8 (between the ages of 56 and 82). Therefore, those 8 cases were included in this study. This small sample was obtained as part of the ongoing TSDS series research program which included men and women who underwent medical-legal autopsy at the Department of Forensic Medicine at the University of Tampere between 2010 and 2015 (n = 700). The primary aim of the larger study was to obtain tissue samples to study the pathology as well as genetic and acquired risk factors of cardiovascular diseases. From every case, a comprehensive set of neuropathological samples was also taken to study the association between vascular dementia and AD. Indications for an autopsy were out-of-hospital death of a previously healthy person, accidental death, suspected intoxication, or suicide. In Finland, a medical-legal autopsy is performed in 16.5% of all deaths. The study was approved by the ethics committee of the Tampere University hospital (R09097) and by the Board of Medicolegal Affairs of Finland (Dnro 564/05.01.0 0.06/2010). Departing from the normal TSDS study protocol, the included consecutive male cases underwent more detailed data collection.
The following data were collected: (i) findings from the autopsy (including routine histopathology), (ii) large brain tissue samples, (iii) a centralized national electronic medical record review, (iv) a mail survey completed by a relative/family member, and (v) blood samples. According to the family surveys, none of the 8 men had a history of participation in contact or collision sports. Two of the 8 cases had a history of traumatic brain injury (TBI) documented in their medical records. The specific questions relating to sports and head injury history were as follows: (i) Did he ever fall or in some other way hit his head so that he was unconscious, amnestic, or confused? (ii) Has he ever been treated by a doctor or admitted to a hospital because of the aforementioned injury or some other head injury? (iii) How many head injuries did he sustain during his life? (iv) At what age did these head injuries occur? (v) Did he suffer from persistent symptoms or problems related to these head injuries? (vi) Did he ever take part in head injury-associated sports? The individuals answering the survey were spouses or first-degree relatives (e.g. father, mother, or children).
Autopsy Study Procedures
The cause of death was determined in a routine manner by the forensic pathologist (P.J.K.) who performed the autopsy using the same general rules for choosing the underlying cause of death that were used in autopsies not belonging to the study series. The medical-legal autopsy protocol included an external and internal examination. In addition to collecting tissue samples for the study purpose, the following organs were routinely assessed: Skin, eyes, brain (including arteries of the circle of Willis), lungs, heart, stomach, liver, gallbladder (including bile duct), pancreas, spleen, aorta, urinary tracts (including kidneys, bladder and prostate), and genitals. Histological (brain, lungs, heart, liver, pancreas, kidneys, prostate) and biochemical (blood, urine, vitreum) samples were taken in order to determine the cause of death. The full autopsy reports were reviewed, and all abnormal findings were recorded. The autopsy findings are presented in Table 1.
TABLE 1.
Review of Medical Records and Autopsy Findings
| Case 1 | Age: 56; Medical Records: Chronic alcohol abuse; Arthrosis; Neuroimaging: CT (age = 54): General brain atrophy, no posttraumatic, nor ischemic lesions; Medication: No regular medication |
| Autopsy Findings: Height/Weight: 171 cm, 60 kg; Brain: 1205 g, no signs of hemorrhage or trauma, cerebellar atrophy, slight signs of atherosclerosis in the main arteries (circle of Willis); Heart: Hypertrophy, cardiomyopathy, coronary heart disease; Liver: 2254 g, cirrhosis; Prostate: prostatitis, prostate stones; Aorta: Atherosclerosis; Pancreas: pancreatitis, necrosis | |
| Case 2 | Age: 82; Medical Records: Hypercholesterolemia; Reflux esophagitis; Diaphragmatic hernia; Prostate hyperplasia; Neurosensory hearing impairment; Neuroimaging: Not done. Medication: Lansoprazole, simvastatin |
| Autopsy Findings: Height/Weight: 165 cm, 67 kg; Brain: 1371 g, no signs of hemorrhage or trauma, slight signs of atherosclerosis in the main arteries (circle of Willis); Heart: Aortic valve stenosis, coronary heart disease; Aorta: Moderate atherosclerosis | |
| Case 3 | Age: 80; Medical Records: Bronchiectasis; Prostate hyperplasia; Acute biliary pancreatitis (age = 77); Obesity; Neuroimaging: Not done; Medication: 5-alfa reductase |
| Autopsy Findings: Height/Weight: 172 cm, 112 kg; Brain: 1541 g, no signs of hemorrhage or trauma, slight signs of atherosclerosis in the main arteries (circle of Willis); Heart: Hypertrophy, enlargement, coronary heart disease; Liver: 1077 g, steatohepatosis; Prostate: Prostate hyperplasia; Aorta: Atherosclerosis | |
| Case 4 | Age: 68; Medical Records: Heart failure with pleural effusion; Seropositive rheumatoid arthritis; Neuroimaging: Not done; Medication: Prednisolone, methotrexate, folic acid |
| Autopsy Findings: Height/Weight: 182 cm, 75 kg; Brain: 1273 g, no signs of hemorrhage or trauma; Heart: Hypertrophy, enlargement, coronary heart disease, amyloidosis, cardiac failure; Lungs: Pneumonitis, vasculitis; Liver: 1973 g, normal; Prostate: Prostate hyperplasia; Aorta: Atherosclerosis | |
| Case 5 | Age: 73; Medical Records: Neurosensory hearing impairment; Cataract; Neuroimaging: Not done; Medication: No regular medication |
| Autopsy Findings: Height/Weight: 170 cm, 90.5 kg; Brain: 1480 g, no signs of hemorrhage or trauma, slight signs of atherosclerosis in the main arteries (circle of Willis), 2 small meningiomas; Heart: Hypertrophy, enlargement, coronary heart disease, amyloidosis, cardiac failure; Lungs: Emphysema; Liver: 1636 g, normal; Prostate: Prostate hyperplasia; Aorta: Atherosclerosis | |
| Case 6 | Age: 78; Medical Records: Hypertension; Asthma; Prostate hyperplasia; Unknown dementia (MMSE 15/30, age = 78); Pulmonary fibrosis; Cardiac failure; Tricuspid valve regurgitation; Neuroimaging: Not done; Medication: Warfarin, furosemide, bisoprolol, salbutamol, ciclesonide |
| Autopsy Findings: Height/Weight: 160 cm, 67 kg; Brain: 1582 g, no signs of hemorrhage or trauma, slight signs of atherosclerosis in the main arteries (circle of Willis); Heart: Hypertrophy, coronary heart disease; Lungs: Pulmonary fibrosis; Liver: 1104 g, congestive hepatopathy; Prostate: Prostate hyperplasia; Aorta: Atherosclerosis | |
| Case 7 | Age: 70; Medical Records: Hypertension; Asthma; COPD; Diabetes, type II; Hypercholesterolemia; Chronic alcohol abuse; Resuscitation, ventricular tachycardia (age = 69); Prolonged QT-time; Coronary heart disease; Trifascicular block; Sick sinus syndrome; ICD pacemaker (age = 70); Dementia (MMSE 26/30, age = 69); Neuroimaging: CT (Mar 2014): Old cerebral infarction (caudate nucleus, corona radiata); Medication: Salbutamol, simvastatin, ASA, metoprolol, enalapril, magnesium hydroxide |
| Autopsy Findings: Height/Weight: 169 cm, 82 kg; Brain: 1482 g, no signs of hemorrhage or trauma, clear signs of atherosclerosis in the main arteries (circle of Willis); Heart: Hypertrophy, severe coronary heart disease; Lungs: Emphysema; Liver: 1254 g, necrosis, septicemia; Aorta: Atherosclerosis | |
| Case 8 | Age: 59; Medical Records: Chronic alcohol abuse; Alcohol abstinence convulsions; Delirium episodes; Traumatic brain contusion and ICH (age = 54); Neuroimaging: MRI (age = 54): SDH, contusions, temporal; Medication: Unknown |
| Autopsy Findings: Height/Weight: 170 cm, 70 kg; Brain: 1324 g, old traumatic contusion in the right frontal and temporal lobe, cerebellar atrophy; Heart: Hypertrophy, signs of earlier pericarditis; Lungs: Edema; Liver: 2546 g, steatohepatosis; Aorta: Normal |
For the present study, 2 ∼2-cm-thick coronal plane brain slices were collected for neuropathological examination. The first slice was taken anterior and the second slice posterior from the mammillary bodies. One axial slice containing the locus coeruleus was taken from the pons. The large coronal brain slices were anatomically mapped and cut into smaller subsamples. The subsamples were placed in Super Mega-Cassettes (Sakura Finetek USA, Torrance, CA) and fixed in phosphate-buffered 4% formaldehyde solution for at least 2 weeks. After fixation, these tissue blocks were embedded in paraffin and shipped to the senior author (R.J.C.). These paraffin mega-blocks were later melted and embedded in smaller blocks prior to conducting a neuropathological analysis. The neuropathologist did not know the personal or medical history of any of the cases, including the age of the decedent.
Medical Record Reviews and Family Surveys
The national centralized electronic medical records of all the patients were reviewed by one author (T.M.L.; see Table 1). Information on prior diagnosed diseases and medications was collected. A special interest and priority was given to psychiatric, neurological, and neurosurgical problems, and all neuroimaging (if performed) findings were also recorded. A questionnaire was sent to the relatives by mail. The questionnaire included questions on the following topics: Occupation, marital status, family background, diseases, head injury history, medication, certain psychiatric symptoms prior to death, dietary habits, smoking, alcohol consumption, drug/substance use, exercise habits, and sports history (especially contact sports). No information about the demographics or clinical features of the cases was provided to the neuropathologist prior to the macroscopic and microscopic analyses.
Genotyping
The salt precipitation method was used on frozen blood samples for DNA isolation. APOE genotyping was performed as described elsewhere (27).
Neuropathology
Macroscopic Examination
Two coronal slices of bilateral cerebral hemispheres and 1 section of pons were examined in each case. The coronal slices of cerebral hemisphere extended from frontal premotor cortex to parietal cortex, and included temporal and insular cortices, cingulate gyrus, basal ganglia, thalamus, amygdala, hippocampal formation, entorhinal cortex, midbrain (coronal plane), centrum semiovale, corona radiata, internal capsule, external capsule, claustrum, extreme capsule, and corpus callosum, among other structures. Additionally, all macroscopic findings (e.g. hemorrhage, contusions, ischemic lesions, anatomic variations) noted at autopsy were collected from the written routine autopsy reports.
Tissue Processing and Immunohistochemistry
In the lab of the senior author (R.J.C.), all tissue was fixed in buffered formalin, sectioned in the coronal plane, processed through graded ethanol and xylene solutions, and embedded in paraffin. Five-micrometer-thick paraffin sections were prepared from all blocks and stained with hematoxylin and eosin. All immunohistochemical stains were performed using an automated immunostainer and antigen retrieval, along with positive and negative (omission of primary antibody) controls. Antibodies included p-tau (AT8), amyloid-β, TAR-DNA binding protein 43 (TDP-43), a-synuclein, neurofilament protein, and amyloid-β protein precursor (Thermo Fisher, Waltham, MA). Antibody information, dilution, and antigen retrieval protocols are provided in Table 2. Bielschowsky silver impregnation was also performed on frontal cortex and hippocampus in each case.
TABLE 2.
Primary Antibodies, Pretreatment, and Dilution
| Antibody | Clone | Vendor | Pretreatment | Dilution |
|---|---|---|---|---|
| PHF-tau | AT8 | ThermoFisher | Heat-induced, citrate, 20 min | 1:250 |
| Amyloid-β | BAM01 | ThermoFisher | Formic acid, 30 min | 1:50 |
| TDP-43 | Polyclonal | ThermoFisher | Formic acid, 30 min | 1:50 |
| Neurofilament | 2F11 | ThermoFisher | Heat-induced, citrate, 20 min | 1:2000 |
| APP | Polyclonal | ThermoFisher | Heat-induced, citrate, 20 min | 1:200 |
| Alpha-synuclein | Polyclonal | ThermoFisher | None | 1:800 |
Paraffin sections from all blocks were immunostained for p-tau to assess for CTE pathology (15), Braak stage (28), and aging-related p-tau (16, 17). Slides from selected blocks were immunostained for amyloid-β and stained with Bielschowsky silver impregnation in order to approximate Thal amyloid phase (29), CERAD plaque score (30), and to apply the National Institute on Aging-Alzheimer's Association (NIA-AA) 2012 approach for documenting AD neuropathologic changes (31), recognizing that cerebellum, midbrain, medulla, and occipital cortex were not available for study. TDP-43 immunohistochemistry was performed on medial temporal lobe including hippocampus in all cases. Amyloid-β precursor protein (APP) and neurofilament protein immunostains were performed to evaluate for possible axonal trauma. α-Synuclein immunostains were performed on selected blocks to rule out synucleinopathy. In addition to the above studies, all cases were assessed for pathological processes in general, including any possible infectious, immune-mediated, metabolic, neoplastic, traumatic, malformative, neurodegenerative, or ischemic processes.
Preliminary Neuropathological Criteria for CTE
The consensus group defined the single pathognomonic criterion for CTE as “an accumulation of abnormal p-tau in neurons, astrocytes, and cell processes around small vessels in an irregular pattern at the depths of the cortical sulci” (15). They defined supportive criteria as follows: “(1) abnormal p-tau-immunoreactive pretangles and NFTs preferentially affecting superficial layers (layers II–III), in contrast to layers III and V as in AD; (2) in the hippocampus, pretangles, NFTs or extracellular tangles preferentially affecting CA2 and pretangles and prominent proximal dendritic swellings in CA4. These regional p-tau pathologies differ from the preferential involvement of CA1 and subiculum found in AD; (3) abnormal p-tau-immunoreactive neuronal and astrocytic aggregates in subcortical nuclei, including the mammillary bodies and other hypothalamic nuclei, amygdala, nucleus accumbens, thalamus, midbrain tegmentum, and isodendritic core (nucleus basalis of Meynert, raphe nuclei, substantia nigra and locus coeruleus); (4) p-tau-immunoreactive thorny astrocytes at the glial limitans most commonly found in the subpial and periventricular regions; and (5) p-tau-immunoreactive large grain-like and dot-like structures (in addition to some threadlike neurites)” (15).
RESULTS
General Macroscopic Assessment
General macroscopic assessment revealed no specific gross pathological processes in 7 of the 8 cases. Case 8 had a 4 cm remote contusion in the left temporal lobe. The posterior corpus callosum appeared marginally thin in cases 1, 3, and 7, possibly with some mild ventricular dilatation.
General Microscopic Assessment
The remote, gross left temporal lobe contusion in case 8 was confirmed microscopically. Routine hematoxylin and eosin stained sections showed some AD pathology as well as focal hippocampal sclerosis (circumscribed focus of pyramidal neuron loss involving a portion of CA-1) in case 2. No microhemorrhages were noted in the case series, although incidental, scant perivascular macrophages with hemosiderin were present in all cases. No infectious, immune-mediated, toxic/metabolic, neoplastic, or malformative processes were present. No recent or remote structural brain injury attributable to neurotrauma was noted in the tissue examined in the case series, with the exception of case 8.
Representative immunohistochemical stains for neurofilament protein highlighted some variability in axon diameter, which was interpreted as a variation of normal in all cases. There were no APP-positive axonal varicosities. Changes suggestive of traumatic axonal injury were generally absent, aside from encephalomalacia from the remote contusion in case 8.
Assessment for CTE Pathology per 2016 Consensus Recommendations
Assessment for required and supportive features of CTE pathology was undertaken (Table 3). With respect to CTE pathology, the presence or absence of the required criterion was assessed by histopathological examination and analysis of p-tau immunostains. The distinction between neuronal and astrocytic p-tau was based on the morphological appearance of the immunoreactivity per consensus recommendations. Six of the 8 cases (75%) showed p-tau in neurons, astrocytes, and cell processes around small blood vessels in an irregular pattern at the depths of the cortical sulci. The changes were focal and limited in terms of overall extent. Examples of this pathognomonic lesion are presented in Figure 1. Examples of supportive feature lesions are presented in Figures 2–4.
TABLE 3.
CTE Pathology in Case Series According to Consensus Recommendations
| Case | Required Criterion | Superficial Laminae NFT | Prominent CA-2 p-Tau | Subcortical p-Tau | Glia Limitans p-Tau | p-Tau Grains | TDP-43 |
|---|---|---|---|---|---|---|---|
| 1 | + | + | – | + | + | + | Minimal |
| 2 | + | +++ | – | + | – | + | ++ |
| 3 | + | +++ | – | ++ | + | + | ++ |
| 4 | – | ++ | – | + | – | – | Minimal |
| 5 | + | +++ | – | ++ | + | + | ++ |
| 6 | + | +++ | – | + | + | + | + |
| 7 | + | +++ | + | + | + | + | + |
| 8 | – | – | – | + | – | – | + |
Note: – = none, + = sparse, ++ = moderate, and +++ = abundant. NFT, neurofibrillary tangle.
FIGURE 1.

Examples of expression of p-tau in depths of sulci. Note: Case #1: Subpial astrocytic p-tau, scattered NFTs, and p-tau in scattered cell processes in an irregular distribution (500 µm and 300 µm). Case #2: Pathognomonic lesion, sulcal depth (200 µm). Case #3: Sulcal depth p-tau in cell processes and perikarya, blood vessel mid left (200 µm). Case #5: Subpial predominately astrocytic p-tau found at the glia limitans in the sulcal depths (400 µm and 500 µm) and a small amount of discontinuous p-tau in superficial laminae (bottom right corner) (500 µm); another area of predominately neuronal p-tau with NFT in sulcal depth around a small blood vessel (200 µm). Case #6: P-tau around a small blood vessel in sulcal depth (200 µm). Case #7: Irregular p-tau in sulcal depth around a small vessel (200 µm). Cases #4 and #8 did not have CTE pathology in depths of sulci.
FIGURE 2.

Examples of expression of p-tau in superficial laminae. Upper images: Case #5, Superficial lamina p-tau with NFT (300 µm and 800 µm). Lower images: Note: Case #3 Superficial lamina NFTs (200 µm) and deep lamina in same area lacking NFTs (200 µm).
FIGURE 3.

Examples of expression of p-tau in amygdala. Upper images: Case #3; Irregular patch of p-tau in amygdala (4 mm), mostly astrocytic but with some neuronal p-tau (400 µm). Lower images: Case #7: Low magnification amygdala with irregular p-tau (3 mm), and high magnification irregular neuronal and astrocytic p-tau in amygdala (300 µm).
FIGURE 4.
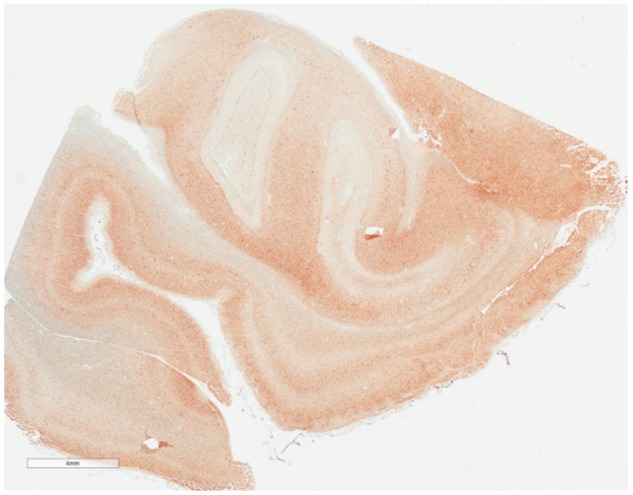
Abundant p-tau in medial temporal lobe (anterior hippocampus and part of amygdala) at low magnification (4 mm). Case #5. This case is Braak NFT stage IV, Aβ Plaque Score (Thal Phase) 1, with sparse CERAD Neuritic Plaques, and NIA-AA Designation = A1B2C1.
Among the supportive features, p-tau NFTs with a relative tendency to involve superficial laminae were present (+) to a varying extent in all cases, and at least focally abundant (+++) in the temporal lobe in cases 2, 3, 5, 6, and 7. p-Tau with relatively dense involvement of the CA-2 region of Ammon’s horn was seen in case 7, but it was not seen in the other cases. All cases showed p-tau in subcortical structures, varying from sparse (+) to moderate (++). Five of the 8 cases showed focal involvement (+) of the glia limitans, in subependymal and subpial areas. Grain and dot-like structures by p-tau immunohistochemistry were noted (+) in 6 of the 8 cases. TDP-43 immunostains showed grains, threads, and cytoplasmic inclusions including NFT in the medial temporal lobe in all cases, varying from minimal to sparse (+) to, moderate (++).
Assessment of AD Neuropathologic Change per NIA-AA 2012 Guidelines
Approximate Thal phase, Braak Stage, and CERAD Plaque Scores were obtained in each case per NIA-AA 2012 consensus guidelines (Table 4). Only 1 case showed changes that would be “sufficient explanation for dementia” (case 2) according to the guidelines. Variable tau burden was present, ranging from Braak stage I to Braak stage V. The 1 case with Braak stage V pathology also had CERAD moderate plaque pathology and Thal phase 3 (approximate, based Aβ involving striatum) amyloid deposits. In combination, this represents an intermediate degree of AD pathology that is a sufficient explanation for dementia according to 2012 NIA-AA guidelines (31). Four cases showed Braak III to IV neurofibrillary change with either no or at most sparse amyloid-β limited to neocortex, consistent with PART. Each of these 4 cases also showed variable astrocytic tau disposed in thorny astrocytes in subpial, subependymal, and/or perivascular areas, and occasional astrocytic plaques, consistent with ARTAG. An additional case showed astrocytic p-tau, with Braak stage II neurofibrillary change.
TABLE 4.
Assessment of Alzheimer Disease Neuropathologic Change per NIA-AA 2012 Consensus Recommendation, and Presence/Absence of PART, ARTAG, and CTE
| Case | Age | APOE | Aβ Plaque Score (Thal Phase) | NFT Stage (Braak; p-tau) | CERAD Neuritic Plaques | NIA-AA Designation | Level of AD Change | Sufficient for Dementia | PART | ARTAG | CTE |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 56 | e4–3 | 1 | II | Sparse | A1B1C1 | Low | No | – | + | + |
| 2 | 82 | e4–3 | 3–4 | V | Moderate | A3B3C2 | Intermediate | Yes | – | – | + |
| 3 | 80 | e3–3 | 0 | IV | None | A0B2C0 | Not | No | + | + | + |
| 4 | 68 | e3–3 | 0 | II | None | A0B1C0 | Not | No | – | – | – |
| 5 | 73 | e3–3 | 1 | IV | Sparse | A1B2C1 | Low | No | + | + | + |
| 6 | 78 | e3–3 | 2 | IV | Sparse | A1B2C1 | Low | No | + | + | + |
| 7 | 70 | e4–3 | 0 | III | None | A0B2C0 | Not | No | + | + | + |
| 8 | 59 | e3–3 | 0 | I | None | A0B1C0 | Not | No | – | – | – |
Aβ, Amyloid Beta (Aβ plaque score); Braak Stage: Neurofibrillary tangle stage (p-tau); CERAD Neuritic Plaque Score; NIA-AA: National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer disease; Levels of Alzheimer disease neuropathologic change = not, low, intermediate, or high. PART, Primary age-related tauopathy; ARTAG, Aging-related tau astrogliopathy; CTE, chronic traumatic encephalopathy neuropathologic changes.
Summary of Neuropathological Assessment
Two cases showed objective structural pathology. Case 8 showed a left temporal lobe remote contusion. Case 2 showed hippocampal sclerosis, in addition to amyloid deposits that exceeded the other cases. Findings in the remaining cases consisted of proteinopathy only. As might be expected, p-tau pathology was variable, both between cases and within individual cases. Required and supportive features for CTE pathology were identified in 6 of the 8 cases (75%). There was no obvious gradient to the required and supportive CTE changes, with the possible exception of marked subcortical p-tau pathology in cases 3 and 5, and superficial laminae p-tau that appeared pronounced in cases with PART pathology and the 1 case with intermediate AD pathology. The 1 case with intermediate AD pathology and the 4 cases with PART had the highest overall p-tau burden. TDP-43 burden tended to follow the overall p-tau burden, although lesser in extent.
DISCUSSION
The results of this study are striking in that 75% of our small case series met neuropathological criteria for CTE, but none of the men had a known history of participation in contact sports, collision sports, or multiple concussions (Table 5). Two of the 8 men had a history of a single TBI, and one of those men had CTE pathology. He also had PART and ARTAG pathology. Of the 6 men with no known history of neurotrauma, 5 met consensus criteria for having CTE (83%). The amount of CTE pathology in terms of the required criterion in these cases was limited, likely mostly stage I and stage II, although the staging is not entirely clear. In 2 of our cases, we noted abundant neurofibrillary degeneration in the medial temporal lobe and subcortical structures including diencephalon and brainstem. The images depicted for cases 1, 2, and 5 appear somewhat more compelling than the images for 3, 6, and 7. When only small amounts of pathology are present, there can be differing opinions on the extent to which it is truly patchy in a perivascular distribution at the depths of sulci. The results of this study should be considered preliminary. It will be necessary, moving forward, to carefully study and define the lower bounds of CTE pathology, and to examine the rates of agreement of neuropathologists for identifying small amounts of pathology.
TABLE 5.
Summary of the Clinical Features of the Case Series
| Case | Age | Cause of Death | TBI | Contact Sport History | Mental Health Problem | Alcohol Abuse | Cognitive Impairment | CTE Pathology |
|---|---|---|---|---|---|---|---|---|
| 1 | 56 | Hemorrhagic pancreatitis | Unknown | Unknown | Unknown | Yes | Unknown | Yes |
| 2 | 82 | Aortic valve stenosis | No | No | Possibly | No | No | Yes |
| 3 | 80 | Cardiomyopathy | No | No | No | No | No | Yes |
| 4 | 68 | Cardiac amyloidosis | No | No | No | No | No | No |
| 5 | 73 | Cardiomyopathy | No | No | Possibly | No | No | Yes |
| 6 | 78 | Pulmonary fibrosis | No | No | No | Unknown | Yes | Yes |
| 7 | 70 | Pancreatitis, alcohol poisoning | Yes | No | Yes | Yes | Yes | Yes |
| 8 | 59 | Alcoholism | Yes | No | Yes | Yes | Yes | No |
Some researchers have asserted that CTE pathology is only found in people who have been exposed to repetitive neurotrauma, such as contact and collision sport athletes and military veterans (2, 4, 9–11, 13), and it has not been found in control subjects or people with other health conditions. The present study, however, adds to a steadily growing number of studies showing that CTE pathology is not unique to contact sport athletes or other people exposed to repetitive neurotrauma. CTE pathology has been identified in a small number of people with a variety of neurological and psychiatric disorders, who have no known participation in collision or contact sports and no known exposure to repetitive neurotrauma, such as people with temporal lobe epilepsy (25), schizophrenia who have undergone leucotomy (32), ALS (26), multiple system atrophy (22), and other neurodegenerative diseases (23). CTE pathology has been identified in women (23) and men (23, 24) who do not have neurological disorders, and in people with substance abuse histories (24).
Noy et al examined 111 brains for CTE pathology, from adults between the ages of 18 and 60, in a routine neuropathology service in Canada (24). They attempted to use the staging system of McKee et al (14), and they identified 4.5% of their cases as having CTE (3 cases of Stage I and 2 cases of Stage II). However, they noted that there is no lower bound for classifying Stage I CTE pathology, so if one includes sparse pathology characteristic of Stage I, an additional 34 cases were identified (30.6% of their sample). Only 1 subject in their sample had a known history of contact or collision sports participation. Some of their cases had no known history of even a single injury to the brain. Similar to the Noy study, we found small amounts of CTE pathology in our cases. The sample studied by Noy et al was considerably younger than our case series. It is reasonable to assume that CTE-like pathology might be more common in older adult control subjects, such as our sample, than middle-aged and younger control subjects. There is a pressing need for more research on control subjects to better understand how common CTE-like pathology is in the general population, across the lifespan.
Leading CTE researchers have clearly indicated that a single concussion does not lead to CTE (33). Whether a single moderate or severe TBI can cause CTE pathology is not well understood. In a large-scale neuropathology study, a subgroup of 33 patients was identified as having at least 1 TBI from motor vehicle accidents, assaults, domestic violence, or falls, and none of these individuals had the neuropathology characteristic of CTE (34). In a more recent study, 2 out of 12 people with a history of severe TBI had neuropathology consistent with CTE (35). There are other studies, however, indicating that different types of neuropathology, such as neuroinflammation (36), β-amyloid deposition (37), and accumulation of TDP-43 (38) and p-tau (37, 39, 40) can and do arise following a single moderate-severe TBI (41–43), and there is emerging evidence that multiple types of neuropathology can be present following repetitive mild TBI (43, 44). The authors of the aforementioned studies have not, however, asserted that mild TBIs cause a unique and inexorably progressive neurodegenerative disease.
Our study has several important limitations. First, the most obvious limitation is that it is possible, in fact likely, that some of the men in our case series experienced one or more concussions during the course of their lives. Concussions are very common in men in the general population, and many people, especially in the past, do not seek medical attention following such an injury (45). Second, it is also possible that some of them played contact sports, at least briefly, during their lives and that their family members did not know about this. These limitations are inherent in virtually all neuropathology studies of this type. Third, there is an art and science to neuropathology, and there can be disagreements as to what constitutes a specific pattern of immunostaining, how big a “patch” of p-tau needs to be, and what demarcates the depth of a sulcus. The consensus group did not define a lower bound of pathology for classifying a person as having CTE, and the Canadian study by Noy et al found that small amounts of pathology were very common in their community sample (24). We too found that our subjects had small amounts of CTE pathology that we presumed to be clinically inconsequential. Finally, we did not perform sledge microtome, free-floating immunohistochemical preparations for this study. A sledge microtome has a moveable long blade capable of slicing large hemispheric tissue slabs, as opposed to a conventional microtome that has a moveable paraffin block sliced by a fixed blade. Sledge microtomes are not used in mainstream diagnostic neuropathology, although they do allow for large hemispheric slabs of brain to be sliced into 50-μm-thick sections, floated into reagent, and hand-immunostained. In doing so, the researcher immunostains a brain slice that is 10 times the thickness of standard immunohistochemistry, and many times the surface area. Such preparations are often depicted macroscopically, for illustrative purposes. It has been reported that this technique may detect CTE pathology in ∼20% of cases that is otherwise not detectable by routine histopathological and immunohistochemical methods for dementia diagnosis (15). Therefore, we cannot exclude the possibility that our negative cases would have been positive using this technique.
Quantitative analyses of pathology have recently been used in CTE studies (46–48). For example, Hsu et al (48) used immunohistochemistry for glial fibrillary acidic protein as a marker for astrogliosis and examined, using automated quantitative analysis, the characteristics and extent of gliosis in postmortem tissue from people with stage III and IV CTE (n = 14), AD (n = 3), frontotemporal dementia ([FTD], n = 3), and controls with no neurodegenerative diseases (n = 6). The authors reported that astrogliosis in the CTE tissue samples was more diffuse compared with that of AD and FTD patients, whereas the astrogliosis in AD and FTD tissue samples was more concentrated in the sulcal depths. Small amounts of the degeneration were also found in their healthy control cases leading the authors to surmise that there might be multiple pathways that result in the degeneration of astrocytes. The samples of tissue with CTE pathology showed evidence of diffuse degenerating astrocyte pathology, characterized by beaded GFAP-immunoreactive astrocytic processes. This degenerating astrocyte pathology was not related to the subjects’ age or stage of CTE pathology, and it was also present in the tissue from people with AD and FTD. Interestingly, there was no correlation between levels of p-tau in the sulcal depths and the extent of astrogliosis or astrocytic degeneration in the white matter adjacent to the sulcal depths, leading the authors to conclude that the astrogliosis might be a distinct process.
Another new method for quantifying CTE pathology involves using ultra high-resolution neuroimaging of postmortem tissue in comparison to histological evidence of cellular pathology. In 1 study (47), 10 ex vivo tissue samples of superior frontal cortex (Brodmann area 8/9) from neuropathologically confirmed cases of stage III and stage IV CTE were evaluated for radiological-pathological correlations. Diffusion MRI data were obtained using an 11.74 T MRI scanner, and white matter underlying sulci with high levels of tau pathology was imaged. They discovered that reduced axon integrity (as inferred from measures of fractional anisotropy) was related to the degree of p-tau pathology in directly adjacent gray matter, and that fractional anisotropy metrics were modestly correlated with histological evidence of axon disruption. The authors emphasized the importance of examining tissue with less severe p-tau pathology, such as stage I and II tissue to determine if the findings would remain consistent, and they also encouraged future studies with tissue from people with nonCTE tauopathies and other control cases to determine whether the relationship between axonal injury and tau pathology is unique to CTE. Applying some of these new quantitative methods might advance our understanding of CTE pathology and its association with age-related pathology.
There is an urgent need for more pathology studies involving control subjects from the general population, particularly those who have clinical conditions that resemble those of former athletes and military veterans whose brains are being donated for research—such as depression, substance abuse, sleep apnea, obesity, and cardiovascular disease. It will also be important for the research community to define the lower bound for identifying CTE pathology. There currently is no lower bound, which is problematic because it is thus possible to diagnose someone has having CTE when they have only a tiny amount of signature pathology—even late in life. The consensus panel was given 10 cases carefully selected to depict extensive pathology. Future researchers should use a similar approach but with cases depicting very small amounts of pathology.
In conclusion, the prevailing theory of CTE, often expressed in articles as fact, is that it is a distinct (1, 3, 9–11) and unique (10) neurodegenerative disease (9–13). It should be noted, however, that there are no longitudinal or epidemiological studies that support these assertions, and a number of reviews of the literature have questioned many of the basic tenets of the prevailing CTE theory (49–58). The present study adds to a steadily emerging literature indicating that CTE pathology (or more aptly CTE-like pathology) is present in people who have no known exposure to multiple concussions or subconcussive blows to the head. Given the current state of the science, it is not known whether, or the extent to which, the emergence, course, or severity of clinical symptoms experienced by former athletes are caused by, or are even modestly correlated with, CTE pathology.
Dr Grant Iverson has been reimbursed by the government, professional scientific bodies, and commercial organizations for discussing or presenting research relating to MTBI and sport-related concussion at meetings, scientific conferences, and symposiums. He has a clinical practice in forensic neuropsychology involving individuals who have sustained mild TBIs (including athletes). He has received honorariums for serving on research panels that provide scientific peer review of programs. He is a co-investigator, collaborator, or consultant on grants relating to mild TBI funded by the federal government and other organizations. He has received research support from test publishing companies in the past, including ImPACT Applications Systems, Psychological Assessment Resources, and CNS Vital Signs. He has received support from the Harvard Integrated Program to Protect and Improve the Health of NFLPA Members. He serves as a scientific advisor for BioDirection, Inc. He acknowledges unrestricted philanthropic support from the Mooney-Reed Charitable Foundation, Heniz Family Foundation, and ImPACT Applications, Inc. Dr. Castellani has been reimbursed for travel for speaking at educational programs and has received fees for medicolegal consulting. He has received intramural research funding at Western Michigan University School of Medicine, unrelated to this study. He is subcontracted to the NIH Neurobiobank at the University of Maryland and the Lieber Institute for Brain Development. The neuropathological examinations for this study were funded by Western Michigan University Homer Stryker MD School of Medicine.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Gavett BE, Stern RA, McKee AC.. Chronic traumatic encephalopathy: A potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 2011;30:179–88, xi [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stern RA, Riley DO, Daneshvar DH, et al. Long-term consequences of repetitive brain trauma: Chronic traumatic encephalopathy. PM R 2011;3:S460–7 [DOI] [PubMed] [Google Scholar]
- 3. McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009;68:709–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013;81:1122–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Montenigro PH, Baugh CM, Daneshvar DH, et al. Clinical subtypes of chronic traumatic encephalopathy: Literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther 2014;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Omalu BC. Chronic traumatic encephalopathy. Prog Neurol Surg 2014;28:38–49 [DOI] [PubMed] [Google Scholar]
- 7. Omalu B, Bailes J, Hamilton RL, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery 2011;69:173–83; discussion 183 [DOI] [PubMed] [Google Scholar]
- 8. Omalu BI, Bailes J, Hammers JL, et al. Chronic traumatic encephalopathy, suicides and parasuicides in professional American athletes. The role of the forensic pathologist . Am J Forensic Med Pathol 2010;31:130–2 [DOI] [PubMed] [Google Scholar]
- 9. Baugh CM, Stamm JM, Riley DO, et al. Chronic traumatic encephalopathy: Neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav 2012;6:244–54 [DOI] [PubMed] [Google Scholar]
- 10. Baugh CM, Robbins CA, Stern RA, et al. Current understanding of chronic traumatic encephalopathy. Curr Treat Opt Neurol 2014;16:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mez J, Stern RA, McKee AC.. Chronic traumatic encephalopathy: Where are we and where are we going? Curr Neurol Neurosci Rep 2013;13:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gavett BE, Cantu RC, Shenton M, et al. Clinical appraisal of chronic traumatic encephalopathy. Current perspectives and future directions. Curr Opin Neurol 2011;24:525–31 [DOI] [PubMed] [Google Scholar]
- 13. McKee AC, Daneshvar DH, Alvarez VE, et al. The neuropathology of sport. Acta Neuropathol 2014;127:29–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McKee AC, Stein TD, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKee AC, Cairns NJ, Dickson D, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016;131:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol 2014;128:755–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol 2016;131:87–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Montenigro PH, Corp DT, Stein TD, et al. Chronic traumatic encephalopathy: Historical origins and current perspective. Annu Rev Clin Psychol 2015;11:309–30 [DOI] [PubMed] [Google Scholar]
- 19. McKee AC, Robinson ME.. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement 2014;10:S242–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2010;69:918–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riley DO, Robbins CA, Cantu RC, et al. Chronic traumatic encephalopathy: Contributions from the Boston University Center for the Study of Traumatic Encephalopathy. Brain Inj 2015;29:154–63 [DOI] [PubMed] [Google Scholar]
- 22. Koga S, Dickson DW, Bieniek KF.. Chronic traumatic encephalopathy pathology in multiple system atrophy. J Neuropathol Exp Neurol 2016;75:963–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ling H, Holton JL, Shaw K, et al. Histological evidence of chronic traumatic encephalopathy in a large series of neurodegenerative diseases. Acta Neuropathol 2015;130:891–3 [DOI] [PubMed] [Google Scholar]
- 24. Noy S, Krawitz S, Del Bigio MR.. Chronic traumatic encephalopathy-like abnormalities in a routine neuropathology service. J Neuropathol Exp Neurol 2016;75:1145–54 [DOI] [PubMed] [Google Scholar]
- 25. Puvenna V, Engeler M, Banjara M, et al. Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res 2016;1630:225–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fournier CN, Gearing M, Upadhyayula SR, et al. Head injury does not alter disease progression or neuropathologic outcomes in ALS. Neurology 2015;84:1788–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hixson JE, Vernier DT.. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545–8 [PubMed] [Google Scholar]
- 28. Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59 [DOI] [PubMed] [Google Scholar]
- 29. Thal DR, Rub U, Orantes M, et al. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–800 [DOI] [PubMed] [Google Scholar]
- 30. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–86 [DOI] [PubMed] [Google Scholar]
- 31. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: A practical approach. Acta Neuropathol 2012;123:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shively SB, Edgerton SL, Iacono D, et al. Localized cortical chronic traumatic encephalopathy pathology after single, severe axonal injury in human brain. Acta Neuropathol 2017;133:353–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stein TD, Alvarez VE, McKee AC.. Concussion in chronic traumatic encephalopathy. Curr Pain Headache Rep 2015;19:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 2015;130:877–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zanier ER, Bertani I, Sammali E, et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain 2018;141:2685–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frugier T, Morganti-Kossmann MC, O'Reilly D, et al. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma 2010;27:497–507 [DOI] [PubMed] [Google Scholar]
- 37. Johnson VE, Stewart W, Smith DH.. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012;22:142–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson VE, Stewart W, Trojanowski JQ, et al. Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol 2011;122:715–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kondo A, Shahpasand K, Mannix R, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015;523:431–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shultz SR, Wright DK, Zheng P, et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 2015;138:1297–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blennow K, Hardy J, Zetterberg H.. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012;76:886–99 [DOI] [PubMed] [Google Scholar]
- 42. Smith DH, Uryu K, Saatman KE, et al. Protein accumulation in traumatic brain injury. Neuromol Med 2003;4:59–72 [DOI] [PubMed] [Google Scholar]
- 43. Smith DH, Johnson VE, Stewart W.. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat Rev Neurol 2013;9:211–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hay J, Johnson VE, Smith DH, et al. Chronic traumatic encephalopathy: The neuropathological legacy of traumatic brain injury. Annu Rev Pathol Mech Dis 2016;11:21–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sosin DM, Sniezek JE, Thurman DJ.. Incidence of mild and moderate brain injury in the United States, 1991. Brain Inj 1996;10:47–54 [DOI] [PubMed] [Google Scholar]
- 46. Armstrong RA, McKee AC, Stein TD, et al. A quantitative study of tau pathology in 11 cases of chronic traumatic encephalopathy. Neuropathol Appl Neurobiol 2017;43:154–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holleran L, Kim JH, Gangolli M, et al. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol 2017;133:367–80 [DOI] [PubMed] [Google Scholar]
- 48. Hsu ET, Gangolli M, Su S, et al. Astrocytic degeneration in chronic traumatic encephalopathy. Acta Neuropathol 2018;136:955–72 [DOI] [PubMed] [Google Scholar]
- 49. Solomon G. Chronic traumatic encephalopathy in sports: A historical and narrative review. Dev Neuropsychol 2018;43:279–311 [DOI] [PubMed] [Google Scholar]
- 50. Asken BM, Sullan MJ, DeKosky ST, et al. Research gaps and controversies in chronic traumatic encephalopathy: A review. JAMA Neurol 2017;74:1255–62 [DOI] [PubMed] [Google Scholar]
- 51. Ling H, Neal JW, Revesz T.. Evolving concepts of chronic traumatic encephalopathy as a neuropathological entity. Neuropathol Appl Neurobiol 2017;43:467–76 [DOI] [PubMed] [Google Scholar]
- 52. Iverson GL, Gardner AJ, McCrory P, et al. A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev 2015;56:276–93 [DOI] [PubMed] [Google Scholar]
- 53. Gardner A, Iverson GL, McCrory P.. Chronic traumatic encephalopathy in sport: A systematic review. Br J Sports Med 2014;48:84–90 [DOI] [PubMed] [Google Scholar]
- 54. Iverson GL, Keene CD, Perry G, et al. The need to separate chronic traumatic encephalopathy neuropathology from clinical features. J Alzheimers Dis 2018;61:17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schwab N, Hazrati LN.. Assessing the limitations and biases in the current understanding of chronic traumatic encephalopathy. JAD 2018;64:1067–76 [DOI] [PubMed] [Google Scholar]
- 56. Zuckerman SL, Brett BL, Jeckell A, et al. Chronic traumatic encephalopathy and neurodegeneration in contact sports and American football. J Alzheimers Dis 2018;66:37–55 [DOI] [PubMed] [Google Scholar]
- 57. Asken BM, Bauer RM.. Chronic traumatic encephalopathy: The horse is still chasing the cart. J Orthop Sports Phys Ther 2018;48:672–5 [DOI] [PubMed] [Google Scholar]
- 58. Randolph C. Chronic traumatic encephalopathy is not a real disease. Arch Clin Neuropsychol 2018;33:644–8 [DOI] [PubMed] [Google Scholar]