

Abstract
Seasonal Influenza infections are associated with an estimated 250–500,000 deaths annually. Resistance to the antiviral M2 ion-channel inhibitors has largely invalidated their clinical utility. Resistance to neuraminidase inhibitors has also been observed in several influenza A strains. These data have prompted research on inhibitors that target the cap-snatching endonuclease activity of PA. Xofluza®, baloxavir marboxil, recently approved for clinical use, inhibits cap-snatching endonuclease. Resistance to Xofluza® has been reported in both in vitro systems and in the clinic. A X-ray crystallographic screening campaign of a fragment library targeting IAV endonuclease enzyme identified 5-chloro-3-hydroxypyridin-2(1H)-one as a bimetal chelating agent at the active site We have reported the structure-activity relationships for 3-hydroxypyridin-2(1H)-ones and 3-hydroxyquinolin-2(1H)-ones as endonuclease inhibitors. These studies identified two distinct binding modes associated with inhibition of this enzyme that are influenced by the presence of substituents at the 5- and 6-positions of 3-hydroxypyridin-2(1H)-ones We report herein on the structure-activity relationships associated with various para-substituted 5-phenyl derivatives of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-ones and the effect of using naphthyl, benzyl, and naphthylmethyl groups as alternatives to the p-fluorophenyl substituent on their activity as endonuclease inhibitors.
Keywords: Antiviral, Endonuclease, Influenza, Inhibitors, Pyridine-2-ones
Graphical Abstract

Influenza RNA-dependent RNA polymerase cap snatching endonuclease is an attractive target for anti-influenza drug development. Here we report the further development of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one, previously described, with various para-substituted 5- or 6-phenyl derivatives including naphthyl, benzyl, and naphthylmethyl.
Introduction
An estimated 250–500,000 deaths annually are associated with seasonal influenza viral infection.[1] While vaccines as a prophylactic have been available, because of the continual antigenic drift variation of influenza viruses, their effectiveness is highly dependent upon the correct prediction of the dominant infectious strain for any given year.[2] Moreover, it is not possible to generate prophylactic options against potential pandemic virus strains. There are three classes of FDA-approved clinical agents that have been developed for the treatment and prophylaxis of influenza infection. These consist of agents that targeted the matrix 2 (M2) ion-channel, neuraminidase (NA), and recently the cap-snatching endonuclease activity of the polymerase acidic protein (PA). Resistance to the M2 ion-channel inhibiting drugs, amantadine and rimantadine, has largely invalidated their clinical utility.[3,4] Resistance to NA inhibitors, including oseltamivir (Tamiflu®), has also been observed in several seasonal influenza A strains.[5,6] There has been intensive research on inhibitors of the cap-snatching endonuclease activity of PA. Structural studies on influenza A endonuclease and on the binding modes of various inhibitors of influenza virus PA endonuclease have been recently reported.[7–10] In addition, advances have been made in understanding the structure-activity relationships associated with metal-binding pharmacophores to influenza endonuclease.[11] Evidence has also been provided that suggest that resistance to experimental endonuclease inhibitors, such as L-742,001 can occur through specific mutations highlighting this potential and concern in the development of clinical endonuclease inhibitors.[12] Xofluza®, baloxavir marboxil, is a prodrug that has been recently approved to target the cap-snatching endonuclease. However, resistance to Xofluza® has been reported in both clinical and in vitro systems, but with significant decrease in viral fitness.[13,14]
Influenza A virus (IAV) has eight negative-stranded RNA genomic segments. The three largest genomic RNA segments encode the viral RNA-dependent RNA polymerase (RdRp) subunits, which consist of the polymerase acidic protein (PA) and the two polymerase basic proteins 1 (PB1) and 2 (PB2). The PA subunit has endonuclease activity, is involved in viral RNA (vRNA)/complementary RNA (cRNA) promoter binding, and interacts with the PB1 subunit.[15] PA has two domains, PAN and PAC. Crystal structures of PAC have been elucidated in complexes with N-terminal fragments of PB1.[16] The structure of PAN has been solved both unliganded and with various ligands in several crystal forms.[17–22]
Influenza RdRp is essential for the replication and transcription of the segmented viral RNA genes. Viral mRNA transcription involves a cap-snatching mechanism wherein the polymerase binds to the host cellular mRNA via the 5’-cap and cleaves the mRNA 12–13 nucleotides downstream. This cleaved host mRNA fragment, which contains the 5’ cap, then acts as a primer for viral mRNA synthesis.[23] Cap-snatching is a critical event in the life cycle of all members of the Orthomyxoviridae family of viruses, including influenza A, B, and C viruses.
As mammalian cells do not participate in an analogous activity, inhibitors of cap-snatching can be selective against multiple influenza types, subtypes and strains, including Tamiflu®-resistant IAV, as well as against IBV and subtypes resistant to M2 inhibitors, without interfering with function of the host cell (for example Xofluza).[24]
In addition to Xofluza and related compounds several different classes of influenza endonuclease inhibitors have been described. These include 2,4-dioxobutanoic acid derivatives,[19,20,25,26] 5-hydroxy-1,6-dihydropyrimidine-4-carboxylic acid derivatives,[20] flutimide and its derivatives,[27] 2-hydroxyphenyl amide derivatives,[28] salicylaldehyde thiosemicabazones,[29] various types of catechins,[30,31] pyromeconic acid and pyridinone deriviatives,[32] N-acylhydrazone derivatives,[33] 5-hydrox-4-pyridone-3-carboxy acid derivatives,[34] 4,5-dihydroxypyrimidine-6-carboxamide derivatives,[35] as well as tetramic acid derivatives.[36]
From an X-ray crystallographic screening campaign of a fragment library targeting the IAV endonuclease enzyme, we identified the 5-chloro-3-hydroxypyridin-2(1H)-one as a bimetal chelating ligand at the active site of the enzyme.[22] Using this information, we have reported the structure-activity relationships for two new series of compounds; 3-hydroxypyridin-2(1H)-ones and 3-hydroxyquinolin-2(1H)-ones, as endonuclease inhibitors.[37,38] We also examined the relative activity of various aza-analogs of 3-hydroxypyridin-2(1H)-one.[39]
Substituted 3-hydroxypyridin-2(1H)-ones can be effective inhibitors of IAV endonuclease (Figure 1). Studies from our laboratories have demonstrated that there are two distinct binding metal modes associated with inhibition of this enzyme that can be influenced by the presence of substituents on the phenyl groups at the 5- and 6-positions of 3-hydroxypyridin-2(1H)-ones. The data suggest the presence of a 5-tetrazoyl moiety at the para position of the 5-phenyl substituent of 2 is associated with enhanced activity relative to the 4-(p-cyanophenyl) derivative, 1. It is also more active than similar 5-phenyl-6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-ones wherein the 5-phenyl moiety is substituted at the meta-position with a cyano, 5-tetrazolyl, or carboxyl group, 3–5. A similar, but less notable effect was observed in examining the relative inhibitory activity of 5-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-ones with a 6-phenyl group where 7 is more active than 6 as an inhibitor.
Figure 1.
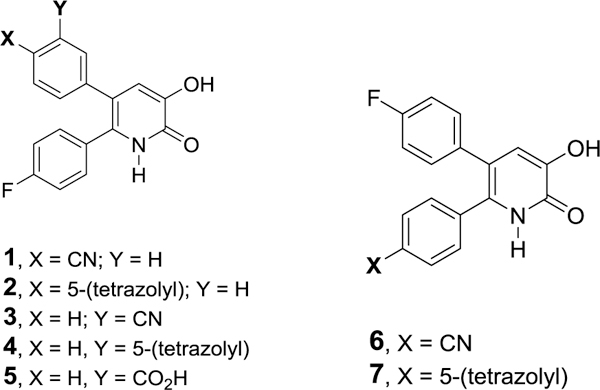
Previously reported 3-hydroxypyridin-2(1H)-ones as inhibitors of IAV endonuclease.
The present study examines the structure-activity relationships associated with various para-substituted 5-phenyl derivatives of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one. In addition, this investigation includes an assessment of the effect of using naphthyl, benzyl, and naphthylmethyl groups as alternatives to the p-fluorophenyl substituent at either the 5- or 6-position of these 3-hydroxypyridin-2(1H)-ones in both of these two sets of compounds on their activity as endonuclease inhibitors. The insertion of a methylene group between phenyl or naphthyl substituents at either the 5 or 6 positions of these 3-hydroxypyridin-2(1H)-ones significantly decreases rigidity, permitting greater molecular flexibility. Such modifications could permit enhanced inhibitory activity by permitting such analogs to adopt a greater binding affinity at the active site. The effect of these structural variations on inhibitory activity was evaluated.
Results and Discussion
The synthetic approach used in the preparation of 8 and 10, as well as the tetrazole derivative 9 was identical to that previously used for the preparation of 1 to 4 (Scheme 1).25 Commercially-available 2,3-dimethoxypyridine was treated with N-bromosuccinimide to provide 5-bromo-2,3-dimethoxypyridine. Suzuki-coupling using p-cyanophenylboronic acid provided 5-(p-cyanophenyl)-2,3-dimethoxypyridine, which was converted as previously described to 6-bromo-5-(p-cyanophenyl)-2,3-dimethoxypyridine, intermediate A. This intermediate was then used to prepare 8 and 10. Compound 8 was then converted to the tetrazole derivative 9 using sodium azide in DMF with a catalytic amount of acetic acid.[38] Compound 10 was treated under similar conditions. While the tetrazole derivative of 10 was formed under these conditions, it proved to be unexpectedly unstable. An alternative synthetic route that employed 4-(5-tetrazoyl)phenylboronic acid also gave the desired tetrazole derivative, but again the isolated product proved to be unstable.40
Scheme 1.

Synthesis of compounds 8–10: (a) NBS (1.1 mol equiv), AcCN, r.t. under Ar; (b) 4-cyanophenylboronic acid, Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (c) NBS (2.0 mol equiv), AcOH, 80 °C under Ar; (d) 1-naphthylboronic acid (for 8), 2-naphthylboronic acid (for 10), Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (e) BBr3 in CH2Cl2, 0 °C to r.t. under Ar; (f) NaN3 (4.0 mol equiv), cat. AcOH, DMF, 130 °C under Ar.
The synthesis of various para-substituted 5-phenyl derivatives structurally similar to 1 and 2 were synthesized as outlined in Scheme 2. The use intermediate B allowed for the preparation of additional analogs that would not survive the more harsh conditions required for efficient cleavage of the dimethoxy ethers intermediates that had been previously synthesized from intermediate A. This intermediate was prepared from 2-fluoro-3-hydroxypyridine by formation of its 6-iodo derivative and subsequent formation of the 3-((2-methoxyethoxy)methoxy) derivative using MEM chloride. The 2-fluoro substituent was selectively displaced with t-butoxide to form the t-butyl ether intermediate. Subsequent reaction of 2-(t-butoxy)-3-((2-methoxyethoxy)methoxy)-6-iodopyridine with 4-fluorophenyl-boronic acid provided the 6-(p-fluorophenyl)- 2-(t-butoxy)-3-((2-methoxyethoxy)methoxy)pyridine, which could be efficiently brominated using NBS to provide intermediate B. Using this intermediate together with the appropriate boronic acid or boronate under Suzuki coupling conditions provide the desired 5-phenyl derivatives of 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine. Treatment of these intermediates with trifluoracetic acid in dichloromethane at room temperature under argon provided the desired para-substituted 5-phenyl derivatives of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one, 11–15. Acetylation of the 5-(p-sulfonamidophenyl)-2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-pyridine intermediate with acetic anhydride in pyridine with subsequent removal of the removal of the protecting groups was used to prepare 16.
Scheme 2.

Synthesis of Compounds 11–16: (a) I2 (2.0 mol equiv), K2CO3 (2.0 mol equiv), water, r.t.; (b) NaH 60% dispersed in oil (2.0 mol equiv), THF, −15 °C followed by MEM-Cl (2.0 mol equiv), r.t. under Ar; (c) KOt-Bu (2.0 mol equiv), AcCN, r.t. under Ar; (d) 4-fluorophenylboronic acid, Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (e) NBS (2.0 mol equiv), AcCN, r.t. under Ar; (f) 4-carboxyphenylboronic acid (for 11), 4-(carboxymethyl)phenylboronic acid (for 12), 4-(1-methyl-tetrazol-5-yl)phenylboronic acid pinacol ester (for 13), 4-(2-methyl-tetrazol-5-yl)phenylboronic acid pinacol ester (for 14), 4-sulfonamidephenylboronic acid (for 15 and 16), Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (g) Ac2O, DMAP, pyridine, r.t., under Ar; (h) TFA: CH2Cl2 (1:2), r.t., under Ar.
The preparation of 17 was accomplished as outlined in Scheme 3. Using intermediate B together with 4-hydroxyphenylboronic acid under Suzuki coupling conditions, the 5-(p-hydroxyphenyl) intermediate was formed. Conversion of this phenol to its triflate followed by Sonogashira coupling with (trimethylsilyl)acetylene and removal of the TMS-protecting group provided the acetylene intermediate, which was converted to the triazole derivative. Removal of the t-butyl and MEM-protecting groups provided 17. The methods used for the preparation of 18–23 are outlined in Scheme 4. The preparation of the 5-(p-cyanophenyl)-6-arylakyl-3-hydroxypyridin-2(1H)-ones 18–20 was performed using a Negishi coupling reaction which employed intermediate A together with various organozinc intermediates. Excellent yields were obtained (80–87%) of the desired 5-(p-cyanophenyl)-6-arylakyl-2,3-dimethoxypyridines, which upon treatment with BBr3 in methylene chloride yielded 18-20. Treatment of 18–20 with sodium azide in DMF with a catalytic amount of acetic acid provided the tetrazolyl derivatives 20–23.
Scheme 3.

Synthesis of compound 17: (a) 4-hydroxyphenylboronic acid, Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (b) N-phenyl-bis(trifluoromethanesulfonamide) (1.2 mol equiv), Et3N (1.8 mol equiv.), CH2Cl2, 0 °C to r.t. under Ar; (c) (trimethylsilyl)acetylene, Pd(PPh3)2Cl2, CuI, Et3N, DMF, 100 °C under Ar; (d) K2CO3 (2.0 mol equiv.), MeOH, r.t. under Ar; (e) TMSN3 (4.0 mol equiv.), CuI (0.1 mol equiv), DMF:MeOH (9:1), 100 °C under Ar; (f) TFA: CH2Cl2 (1:2), r.t., under Ar.
Scheme 4.

Synthesis of compounds 18–23: (a) (4-fluorobenzyl)zinc(II) chloride (for 18 and 21), (1-naphthylmethyl)zinc(II) chloride (for 19 and 12), (2-naphthylmethyl)zinc(II) bromide (for 20 and 23), JohnPhos, Pd(OAc)2, K2CO3, dioxane under Ar; (b) BBr3 in CH2Cl2, 0 °C to r.t. under Ar; (c) NaN3 (4.0 mol equiv.), cat. AcOH, DMF, 130 °C under Ar.
Scheme 5 summarizes the method used for the preparation of 24. Treatment of commercially-available 5-bromo-3-hydroxypyridin-2(1H)-one with sodium hydride in DMF followed by MOM-chloride resulted in N,O-protected 5-bromo-3-(methoxy methoxy)-1-(methoxymethyl)pyridin-2(1H)-one intermediate as a major product. Suzuki coupling with 4-fluorophenylboronic acid followed by chlorination by Palau’s chlor gave intermediate 6-chloro-5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one. Suzuki coupling together with 4-carboxyphenylboronic acid followed by removal of protective groups provided compound 24.
Scheme 5.

Synthesis of compound 24: (a) NaH 60% dispersed in oil (5.0 mol equiv.), DMF, 0 °C followed by MOM-Cl (5.0 mol equiv.), r.t. under Ar; (b) 4-fluorophenylboronic acid, Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (c) Palau’s chlor, CH2Cl2 under Ar; (d) 4-carboxyphenylboronic acid, Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (e) 2N HCl/dioxane (1:1), 100 °C.
The method used for the preparation of 5-arylakyl-6-(p-cyanophenyl)-3-hydroxypyridin-2(1H)-ones, 25-27, and their tetrazole derivatives, 28–30 is outlined in Scheme 6. 2,3-Dimethoxypyridine was converted to its 5,6-dibromo derivative using a slight excess of N-bromosuccinimide in acetic acid in a modification of the previously reported procedure.[38]
Scheme 6.

Synthesis of compounds 25–30: Reagents and conditions: (a) NBS (2.2 mol equiv.), AcOH, 80 °C under Ar; (b) trimethylsilylmethylithium (1.1 mol equiv.), toluene, 0 °C followed by 4-fluoro-N-methoxy-N-methylbenzamide (2.0 mol equiv.) (for 25) (2.0 mol equiv.), N-mehoxy-N-methyl-1-naphthamide (2.0 mol equiv.) (for 26), N-methoxy-N-methyl-2-naphthamide (for 27), 0 °C under Ar; (c) NaBH4 (2.0 mol equiv.), THF under Ar; (d) Et3SiH (3.0 mol equiv), TFA (4.5 mol equiv.), 1,2-dichloroethane, 50 °C under Ar; (e) 4-cyanophenylboronic acid, Pd(PPh3)4, Na2CO3, dioxane/H2O (3:1), 100 °C under Ar; (f) BBr3 in CH2Cl2, 0 °C to r.t. under Ar; (g) NaN3 (4.0 mol equiv.), cat. AcOH, DMF under Ar.
Lithiation of this intermediate with trimethylsilylmethylithium and subsequent reaction with the appropriate Weinreb amide provided the aryloyl intermediates, which could be converted to 5-(arylmethyl)-6-bromo-2,3-dimethoxypyridine derivatives using a two-step reduction using NaBH4 to form the alcohol with subsequent treatment with triethylsilane and trifluoroacetic acid. The formation of 5-arylakyl-6-(p-cyanophenyl)-2,3-dimethoxypyr-idine derivatives was accomplished by Suzuki-coupling of these bromopyridines with p-cyanophenylboronic acid. The approach as outlined in Scheme 6 proved to be much more efficient than earlier attempts, which attempted to explore the utility of lithiating intermediate A, and then coupling this intermediate with either an arylaldehyde or a Weinreb amide derivative of p-fluorobenzamide and the requisite naphthylamides.
Formation of the desired 5-arylakyl-6-(p-cyanophenyl)-3-hydroxypyridin-2(1H)-ones 25–27 was accomplished by treatment of these 2,3-dimethoxypyridines intermediates with BBr3 in methylene chloride. The tetrazoyl derivatives 28-30 were prepared by treatment of 25-27 with sodium azide in DMF with a catalytic amount of acetic acid. The Weinreb amides that were employed in these syntheses were prepared by reacting N,O-dimethylhydroxylamine with the requisite acid chloride under conditions similar to those previously reported.41
The various aryl and arylalkyl substituted 3-hydroxypyridin(1H)-2-ones that were synthesized were evaluated for the ability to inhibit IAV endonuclease activity. The relative activity of these compounds was compared to that observed for several analogous compounds that had been previously synthesized and evaluated.[38] Their endonuclease inhibitory activities are provided in Table 1 and Table 2. The established influenza endonuclease inhibitor L-742,001 has an IC50 of 7 nM and ligand efficiency of 0.361 kcal/(mol·NHA) while the green tea catechin epigallocatechin gallate has an IC50 of 1 μM and a ligand efficiency of 0.249 kcal/(mol·NHA) in this assay.[36,31,]
Table 1.
Inhibition assay of IAV endonuclease by 6-aryl or 6-arylmethyl 5-(p-substituted phenyl)-3-hydroxypyridin-2(1H)-ones.
| Compd | 6-Aryl or 6-Arylmethyl |
 |
IC50 (nM) | LE kcal/(mol·NHA) |
|---|---|---|---|---|
| 1 |  |
C≡N | 136[a] | 0.409 |
| 2 |  |
 |
11[a] | 0.420 |
| 8 |  |
C≡N | 220 | 0.351 |
| 9 |  |
 |
18 | 0.354 |
| 10 |  |
C≡N | 544 | 0.330 |
| 11 |  |
CO2H | 11 | 0.455 |
| 12 |  |
COCH3 | 125 | 0.326 |
| 13 |  |
 |
298 | 0.331 |
| 14 |  |
 |
138 | 0.348 |
| 15 |  |
SO2NH2 | 347 | 0.354 |
| 16 |  |
SO2NHCOCH3 | 145 | 0.335 |
| 17 |  |
 |
25 | 0.401 |
| 18 |  |
C≡N | 507 | 0.360 |
| 19 |  |
C≡N | 114 | 0.353 |
| 20 |  |
C≡N | 660 | 0.314 |
| 21 |  |
 |
26 | 0.385 |
| 22 |  |
 |
15 | 0.358 |
| 23 |  |
 |
67 | 0.328 |
Data were previously reported.[38]
Table 2.
Inhibition assay of IAV endonuclease by 5-aryl or 5-arylmethyl 6-(p-substituted phenyl)-3-hydroxypyridin-2(1H)-ones.
| Compd | 5-Aryl or 5-Arylmethyl |
 |
IC50 (nM) | LE kcal/(mol·NHA) |
|---|---|---|---|---|
| 6 |  |
C≡N | 54[a] | 0.433 |
| 7 |  |
 |
23[a] | 0.403 |
| 24 |  |
COOH | 55 | 0.415 |
| 25 |  |
C≡N | 484 | 0.361 |
| 26 |  |
C≡N | 173 | 0.343 |
| 27 |  |
C≡N | 250 | 0.335 |
| 28 |  |
 |
54 | 0.369 |
| 29 |  |
 |
12 | 0.362 |
| 30 |  |
 |
81 | 0.324 |
Data were previously reported.[38]
Significant enhancement in inhibition was observed in earlier studies as one converted a p-cyanophenyl substituent to a p-(5-tetrazoyl)phenyl at the 5-position of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one, with 2 being a more potent inhibitor than 1. This trend proved consistent in when comparing the relative inhibitory activities of 6-(1-naphthyl) (8 and 9), the 6-(p-fluorobenzyl) (18 and 21), 6-[(1-naphthyl)methyl] (19 and 22) and 6-[(2-naphthyl)methyl] (20 and 23) derivatives.
The assessment of varied para-substituents on the phenyl substituted at the 5-position of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one provides insight into the structure-activity relationships at this site. Identical activity between 5-(p-(tetrazol-5-yl)phenyl, 2, and the 5-(p-carboxyphenyl) derivative, 11, of 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one indicates the importance of having an acidic functional group at this site. An X-ray crystal structure of 11 bound to the endonuclease (Figure 2) showed a similar binding mode (orientation of the pyridinone ring) of 11 compared to the previously published 2.[38] The 5-(p-carboxyphenyl) is situated in a similar position but makes additional interactions Arg124 through two bridging water molecules.
Figure 2.

X-ray crystal structure stereoview images of 11 and 29 with IAV endonuclease domain. Bound protein is coloured purple and apo residues orange. Chelation indicated by black dashes and hydrogen bonds as blue dashes. Compound omit maps are contoured at 3.0σ (blue). (A) Compound 11 (blue) bound at the IAV endonuclease active site. (B) Compound 29 (yellow) bound and 7 (green).
Replacement of the carboxylic acid of 11 with the methyl ester, 12, resulted in a dramatic loss in activity. Similarly, loss of the acidic proton on the tetrazole moiety by methylation at either the 1- or 2- position of 2 as in the case of 13 and 14, respectively, also resulted in a decrease in activity. Neither replacing the carboxylate with a sulfonamide, as in 15, or an acylsulfonamide, as in 16, resulted in analogs with similar potency as either 2 or 11. Only in the case of the 5-(4-triazolylphenyl) derivative, 17, was significant potency restored, relative to either 2 or 11.
For these 5-(p-cyanophenyl)- or 5-[p-(5-tetrazoyl)phenyl]-3-hydroxypyridin-2(1H)-ones, the presence of a (1-naphthyl)methyl substituent at the 6-position (19 and 22) was associated with greater potency than the similarly substituted p-fluorobenzyl derivatives 18 and 21 or the 2-naphthylmethyl derivative 10. Both the 6-(1-naphthyl)- and the 6-(2-naphthyl)-5-(p-cyanophenyl derivatives (19 and 20) were also much less active than their analogous tetrazole derivatives (22 and 23). These 6-(1-naphthyl)methyl derivatives, however, had similar activity to the p-fluorophenyl derivatives 1 and 2, as well as the naphthyl analogs, 8 and 9, taking into account the inherent relative impact of either a p-cyano or a p-5-tetrazoyl moiety.
The structure-activity relationships for aryl and arylalkyl groups at the 5-position of 3-hydroxypyridin-2(1H)-ones substituted with either 6-(p-cyanophenyl)- or 6-[p-(5-tetrazoyl)phenyl] were also evaluated. As previously observed for 6 and 7, the tetrazole derivatives were again more consistently active in this series than the cyano derivatives.[38] This can also be seen in a comparison of the relative inhibitory activities of the 6-(p-fluorobenzyl) (25 and 28), 6-[(1-naphthyl)methyl] (26 and 29) and 6-[(2-naphthyl)methyl] (27 and 30) derivatives. The synthesis and evaluation of 6-(4-carboxyphenyl)-5-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one 24 did indicate that this derivative, however, was less active as an inhibitor than previously observed for the structurally-related 6-(p-(5-tetrazoyl)phenyl) analog, 7.
There was another consistency noted in the structure-activity relationships observed for these 5-(p-cyanophenyl)- or 5-[p-(5-tetrazoyl)phenyl]-3-hydroxypyridin-2(1H)-ones and that observed for these 6-(p-cyanophenyl)- or 6-[p-(5-tetrazoyl)phenyl]-3-hydroxypyridin-2(1H)-ones. Within this series the presence of a (1-naphthyl)methyl substituent at the 5-position (26 and 29) was also associated with greater potency than the similarly substituted p-fluorobenzyl derivatives 25 and 28 or the 2-naphthylmethyl derivatives 27 and 30.
A X-ray crystal structure of 29 bound to the IAV endonuclease active site (Figure 2B) shows a similar binding mode as the previously published 7 for the tetrazole but there is significant rearrangement of the pocket to accommodate the (1-naphthyl)methyl substituent. The tetrazole placement, as seen previously with compounds 2 and 7, causes a flip in the pyrdinone ring to the less favorable orientation seen here.[38 ] Although the pyridinone ring in this orientation is less favorable for 2 and 7, in the case of either 29 or 22 it appears to be slightly more favorable perhaps due to changes in the water structure near the (1-naphthyl)methyl substituent that the pyridinone nitrogen forms hydrogen bonds to. The sidechain of Tyr24 rotates 90 degrees and has 1.5 Å backbone shift to stabilize an edge-to-face stacking interaction with the naphthyl. The 5-phenyl also has enhanced interaction with Ile38 due to decreased steric clash with the (1-naphthyl)methyl relative to a fluorophenyl.
Antiviral activity of the compounds was determined using a previously described PR8-mCherry assay. [42,43] Unfortunately, no antiviral activity except the 11 μM EC50 previously reported for compound 2 was detected using this cellular assay.[22]
Conclusions
Our studies on 4-hydroxypyridazin-3(2H)-ones, 5-hydroxypyrimidin-4(3H)-ones, and 3-hydroxyquinoline-2(1H)-ones failed to identify major advantages in potency as IAV endonuclease inhibitors over the phenyl substituted 3-hydroxypyridin-2(1H)-ones that had been studied earlier. The intent of the present study was to further examine aryl and arylalkyl substituted 3-hydroxypyridin-2(1H)-ones to see whether further improvement in potency with this series of compounds could be achieved. The increased hydrophobic nature of naphthyl analogs and the increased structural flexibility associated with methylene-linked analogs of both select phenyl and these naphthyl analogs was viewed as one possible approach wherein both an increase in activity as IAV endonuclease inhibitors and an improvement in cellular efficacy of these potential antiviral agents could be achieved. The compounds selected for synthesis, although synthetically more challenging, were among the more promising based upon our earlier structure-activity studies. While similar trends in terms of structure-activity could be observed, no pronounced improvement in potency was realized. More significantly, these select aryl and arylalkyl substituted 3-hydroxypyridin-2(1H)-ones failed to be efficacious in cellular antiviral assays.
Experimental Section
Chemistry: General Methods.
All reactions, unless otherwise stated, were done under nitrogen atmosphere. Reaction monitoring and follow-up were done using aluminum backed Silica G TLC plates with UV254 (Sorbent Technologies), visualizing with ultraviolet light. Flash column chromatography was done on a Combi Flash Rf Teledyne ISCO using hexane, ethyl acetate, dichloromethane, and methanol. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were done in CDCl3, Methanol-d4, and DMSO-d6 and recorded on a Bruker Avance III (400 MHz) Multinuclear NMR Spectrometer. Data is expressed in parts per million relative to the residual nondeuterated solvent signals, spin multiplicities are given as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), dt (doublet of triplets), q (quartet), m (multiplet), and bs (broad singlet), and coupling constants (J) are reported in Hertz. Melting points were determined using Mel-temp II apparatus and are uncorrected. Analytical HPLC was performed on a Shimadzu LC-20AT Prominence liquid chromatograph using a 150 × 4.6 mm Princeton SPHER-100 RP C18 1000A 5 u column using 0% water for 2 minutes and a 0–100% water/methanol gradient over a 5 minute period and 5 minutes at 100% methanol at a 2.0 ml/minute flow rate monitoring UV absorbance at 254 and 296 nm. Using this method of analysis, the purity of all compounds used in bioassays was determined to be ≥ 95%. HRMS experiments were conducted by Washington University Resource for Biomedical and Bioorganic Mass Spectrometry Department of Chemistry.
4-(5-Hydroxy-2-(naphthalen-1-yl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (8)
4-(5,6-Dimethoxy-2-(naphthalen-1-yl)pyridin-3-yl)benzonitrile (80 mg, 0.22 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (2.2 mL, 2.2 mmol) was added. It was then allowed to warm to room temperature and stirred for 24 hours. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10% MeOH/DCM to provide 4-(5-hydroxy-2-(naphthalen-1-yl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrileas a white solid (32 mg, 45%); m.p. 288–290 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.11 (bs, 1H), 9.47 (bs, 1H), 7.96 – 7.93 (m, 2H), 7.60 (d, J = 8Hz, 1H), 7.52 – 7.47 (m, 5H), 7.42 (d, J = 7 Hz, 1H), 7.13 (d, J = 8 Hz, 2H), 6.97 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 158.0, 146.9, 143.2, 132.9, 132.6, 131.7, 131.5, 131.2, 129.31, 129.25, 129.2, 128.3, 126.8, 126.1, 125.2, 124.8, 118.5, 117.5, 117.2, 108.8; HRMS (ESI) calculated for C22H15N2O2 (M+H)+339.1128, found 339.1136.
4-(5,6-Dimethoxy-2-(naphthalen-1-yl)pyridin-3-yl)benzonitrile
4-(2-Bromo-5,6-dimethoxypyridin-3-yl)benzonitrile (293 mg, 0.92 mmol), naphthalene-1-boronic acid (190 mg, 1.10 mmol), Pd(PPh3)4 (106 mg, 0.092 mmol) and Na2CO3 (292 mg, 2.75 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 18 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(5,6-dimethoxy-2-(naphthalen-1-yl)pyridin-3-yl)benzonitrile as a white solid (220 mg, 65%); m.p. 226–228 °C; 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J = 8 Hz, J = 1 Hz, 1H), 7.81 (d, J = 8 Hz, 2H), 7.48 (td, J = 7 Hz, J = 1 Hz, 1H), 7.42 –7.39 (m, 1H), 7.37 – 7.32 (m, 3H), 7.21 (s, 1H), 7.17 – 7.14 (m, 3H), 4.06 (s, 3H), 4.03 (s, 3H);13C NMR (100 MHz, CDCl3) δ 153.3, 144.6, 143.4, 136.9, 133.7, 132.9, 132.1, 131.8, 129.7, 129.2, 128.6, 128.4, 127.9, 126.1, 125.82, 125.77, 125.0, 119.1, 118.7, 110.3, 56.0, 54.2; HRMS (ESI) calculated for C24H19N2O2 (M+H)+ 367.1441, found 367.1450.
4-(2-Bromo-5,6-dimethoxypyridin-3-yl)benzonitrile
To a solution of 4-(5,6-dimethoxypyridin-3-yl)benzonitrile (603 mg, 2.51 mmol) in AcOH (20 mL) under nitrogen, NBS (893 mg, 5.02 mmol) was added. The reaction mixture was then stirred overnight at 80 °C. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NaHCO3 followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 20% EtOAc/Hexane. This afforded 4-(2-bromo-5,6-dimethoxypyridin-3-yl)benzonitrile as a white solid (588 mg, 73%); m.p. 151–153 °C; 1H NMR (400 MHz, CDCl3) δ 7.72 (dd, J = 9 Hz, 2H), 7.54 (d, J = 8 Hz, 2H), 6.96 (s, 1H), 4.06 (s, 3H), 3.88 (s, 3H);13C NMR (100 MHz, CDCl3) δ 153.4, 143.8, 143.7, 132.1, 130.4, 129.9, 125.9, 120.4, 118.6, 111.8, 56.3, 54.7.
4-(5,6-Dimethoxypyridin-3-yl)benzonitrile
5-Bromo-2,3-dimethoxypyridine (692 mg, 3.17mmol), 4-cyanophenyl boronic acid (699 mg, 4.76mmol), Pd(PPh3)4 (370 mg, 0.32 mmol) and Na2CO3 (1.01 g, 9.51mmol) were dissolved in a mixture of dioxane (60 mL) and water (20 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 18 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(5,6-dimethoxypyridin-3-yl)benzonitrile as a white solid (603 mg, 79%); m.p. 109–111 °C; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 2 Hz, 1H), 7.73 (d, J = 8 Hz, 2H), 7.64 (d, J = 9 Hz, 2H), 7.22 (d, J = 2 Hz, 1H), 4.07 (s, 3H), 3.96 (s, 3H);13C NMR (100 MHz, CDCl3) δ 154.8, 144.3, 142.6, 135.5, 132.7, 128.7, 127.3, 118.7, 115.8, 111.0, 55.8, 54.0.
5-Bromo-2,3-dimethoxypyridine
2,3-Dimethoxypyridine (2 g, 14.37 mmol) and NBS (3.84 g, 21.56 mmol) were dissolved in AcCN (100 mL). It was then stirred for 48 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with sat. NaHCO3 followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 50% DCM/Hexane. This afforded 5-bromo-2,3-dimethoxypyridine as colorless oil (1.36 g, 43%);1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 2 Hz, 1H), 7.13 (d, J = 2 Hz, 1H), 3.99 (s, 3H), 3.87 (s, 3H);13C NMR (100 MHz, CDCl3) δ 153.5, 144.6, 137.4, 120.2, 111.1, 56.0, 53.9.
4-(5-Hydroxy-2-(naphthalen-2-yl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (9)
4-(5,6-Dimethoxy-2-(naphthalen-2-yl)pyridin-3-yl)benzonitrile (100 mg, 0.27 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (2.7 mL, 2.7 mmol) was added. It was then allowed to warm to room temperature and stirred for 24 hours. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10 % MeOH/DCM to provide 4-(5-hydroxy-2-(naphthalen-2-yl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrileas a white solid (32 mg, 35%); m.p. 262–264; 1H NMR (400 MHz, DMSO-d6) δ 12.13 (bs, 1H), 9.49 (bs, 1H), 7.88 – 7.85 (m, 3H), 7.77 (d, J = 8 Hz, 1H), 7.64 – 7.62 (m, 2H), 7.55 – 7.54 (m, 2H), 7.25 (d, J = 8 Hz, 2H), 7.12 (d, J = 8 Hz, 1H), 6.92 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 158.2, 146.6, 143.5, 132.4, 132.0, 131.5, 131.4, 130.4, 129.5, 128.8, 128.7, 128.2, 127.5, 127.43, 127.37, 126.9, 126.5, 118.7, 117.9, 109.0; HRMS (ESI) calculated for C22H15N2O2 (M+H)+ 339.1128, found 339.1139.
4-(5,6-Dimethoxy-2-(naphthalen-2-yl)pyridin-3-yl)benzonitrile
4-(2-Bromo-5,6-dimethoxypyridin-3-yl)benzonitrile(300 mg, 0.97mmol), naphthalene-2-boronic acid (251 mg, 1.46 mmol), Pd(PPh3)4 (112 mg, 0.097 mmol) and Na2CO3 (310 mg, 2.92 mmol) were dissolved in a mixture of dioxane (30 mL) and water (10 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 18 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(5,6-dimethoxy-2-(naphthalen-2-yl)pyridin-3-yl)benzonitrile as a white solid (357 mg, 100%); m.p. 182–184 °C; 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 1 Hz, 1H), 7.82 – 7.80 (m, 1H), 7.73 – 7.70 (m, 2H), 7.55 (d, J = 8 Hz, 2H), 7.49 – 7.46 (m, 2H), 7.41 (dd, J = 9 Hz, J = 2 Hz, 1H), 7.35 (d, J = 8 Hz, 2H), 7.11 (s, 1H), 4.18 (s, 3H), 3.99 (s, 3H);13C NMR (100 MHz, CDCl3) δ 153.6, 145.2, 144.4, 143.3, 136.5, 133.1, 132.7, 132.2, 129.5, 128.3, 127.7, 127.6, 127.5, 127.4, 126.4, 126.2, 119.8, 118.8, 110.6, 56.1, 54.0; HRMS (ESI) calculated for C24H19N2O2 (M+H)+ 367.1441, found 367.1449.
5-(4-(1H-Tetrazol-5-yl)phenyl)-3-hydroxy-6-(naphthalen-1-yl)pyridin-2(1H)-one (10)
4-(5-Hydroxy-2-(naphthalen-1-yl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (60 mg, 0.18 mmol) and NaN3 (46 mg, 0.71 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was re- moved under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (37 mg, 55% yield); m.p. 223–225 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.07 (bs, 1H), 9.43 (bs, 1H), 7.95 – 7.92 (m, 2H), 7.65 (d, J = 8 Hz, 2H), 7.62 (d, J = 2Hz, 1H), 7.50 – 7.45 (m, 4H), 7.18 (d, J = 8 Hz, 2H), 7.01 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 158.0, 146.7, 141.2, 132.9, 132.2, 131.6, 129.4, 129.2, 129.1, 128.3, 126.8, 126.4, 126.1, 125.2, 124.9, 121.8, 118.1, 117.6; HRMS (ESI) calculated for C22H16N5O2 (M+H)+ 382.1299, found 382.1300.
4-(2-(4-Fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzoic acid (11)
To a solution of 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzoic acid (75 mg, 0.16 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 3 hours at room temperature. After removal of solvent, 4-(2-(4-fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzoic acid was crystallized as a white solid in EtOH:Et2O (1:2) (39 mg, 75%); dec. 301–303 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.82 (bs, 1H), 12.00 (bs, 1H), 9.37 (bs, 1H), 7.76 (d, J = 8 Hz, 2H), 7.22 – 7.18 (m, 2H), 7.15 – 7.10 (m, 4H), 6.85 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 167.0, 161.9 (JC,F= 245 Hz), 158.0, 146.4, 142.9, 133.0, 132.2 (JC,F= 9 Hz), 130.1 (JC,F= 3 Hz), 129.6, 129.1, 128.6, 118.1, 117.0, 115.0 (JC,F= 22 Hz);19F NMR (376 MHz, DMSO-d6) δ −113.0; HRMS (ESI) calculated for C18H13FNO4 (M+H)+ 326.0823, found 326.0812.
4-(6-(t-Butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzoic acid
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (200 mg, 0.47 mmol), 4-(carboxyphenyl) boronic acid (118 mg, 0.71 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and Na2CO3 (149 mg, 1.41 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 6 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzoic acid as a white solid (109 mg, 49%); m.p. 142–144 °C; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8 Hz, 2H), 7.44 (s, 1H), 7.31 – 7.29 (m, 4H), 6.93 (t, J = 9 Hz, 2H), 5.36 (s, 2H), 3.96 – 3.94 (m, 2H), 3.63 – 3.61 (m, 2H), 3.40 (s, 3H), 1.71 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 171.4, 162.2 (JC,F= 246 Hz), 154.1, 145.8, 145.5, 140.9, 135.7, 131.6 (JC,F= 8 Hz), 130.3, 129.9, 127.61, 127.57 127.4, 114.8 (JC,F= 21 Hz), 94.8, 80.8, 71.6, 68.0, 59.0, 28.9; 19F NMR (376 MHz, CDCl3) δ −115.1; HRMS (ESI) calculated for C26H29FNO6 (M+H)+ 470.1973, found 470.1974.
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine
To a solution of 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine (4 g, 11.45 mmol) in AcCN (100 mL) under nitrogen, NBS (4.08 g, 22.90 mmol) in AcCN (100mL) was added over 30 minutes. The reaction mixture was then stirred for 3 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with sat. NaHCO3followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 3-bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine as a white solid (3.92 g, 80%); m.p. 67–69 °C 1H NMR (400 MHz, CDCl3) δ 7.70 – 7.66 (m, 2H), 7.61 (s, 1H), 7.10 (t, J = 9Hz, 2H), 5.28 (s, 2H), 3.90 – 3.88 (m, 2H), 3.61 – 3.58 (m, 2H), 3.41 (s, 3H), 1.60 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.5 (JC,F= 245 Hz), 153.3, 145.8, 141.4, 135.5, 131.3 (JC,F= 8 Hz), 129.6, 114.6 (JC,F= 22 Hz), 108.4, 94.8, 81.0, 71.5, 68.2, 59.1, 28.7; 19F NMR (376 MHz, CDCl3)δ −113.8; HRMS (ESI) calculated for C19H24BrFNO4 (M+H)+ 428.0867, found 428.0867.
2-(t-Butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine
2-(t-Butoxy)-6-iodo-3-((2-methoxyethoxy)methoxy)pyridine (5.10 g, 13.38 mmol), 4-fluorophenyl boronic acid (2.81 g, 20.07 mmol), Pd(PPh3)4 (1.55 g, 1.34 mmol) and Na2CO3 (4.25 g, 40.14 mmol) were dissolved in a mixture of dioxane (45 mL) and water (15 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 18 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine as yellowish oil (4.00 g, 86%); 1H NMR (400 MHz, CDCl3) δ 7.92 – 7.89 (m, 2H), 7.38 (d, J = 8 Hz, 1H), 7.19 (d, J = 8 Hz, 1H), 7.11 (t, J = 9 Hz, 2H), 5.29 (s, 2H), 3.90–3.88 (m, 2H), 3.59 – 3.57 (m, 2H), 3.38 (s, 3H), 1.69 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.9 (JC,F= 246 Hz), 154.5, 146.3, 141.3, 135.3 (JC,F= 3 Hz), 127.9 (JC,F= 9 Hz), 125.5, 115.4 (JC,F = 22 Hz), 112.2, 94.7, 80.2, 71.6, 68.0, 59.0, 28.8; 19F NMR (376 MHz, CDCl3)δ −114.7; HRMS (ESI) calculated for C19H24FNO4Na (M+Na)+ 372.1582, found 372.1581.
2-(t-Butoxy)-6-iodo-3-((2-methoxyethoxy)methoxy)pyridine
To a solution of 2-fluoro-6-iodo-3-((2-methoxyethoxy)methoxy)pyridine (5.31 g, 16.23 mmol) in THF (160 mL) under nitrogen, potassium t-butoxide (3.64 g, 32.46 mmol) in DMF (10 mL) was added over 30 min. The reaction mixture was then stirred for 15 minutes at room temperature. After the reaction was complete, it was diluted with EtOAc and washed with water followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 2-(t-butoxy)-6-iodo-3-((2-methoxyethoxy)methoxy)pyridineas colorless oil (5.10 g, 82%);1H NMR (400 MHz, CDCl3) δ 7.13 (d, J = 8 Hz, 1H), 6.99 (d, J = 8 Hz, 1H), 5.20 (s, 2H), 3.84 – 3.81 (m, 2H), 3.54 – 3.52 (m, 2H), 3.36 (s, 3H), 1.58 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 154.1, 142.2, 126.74, 126.68, 101.6, 94.6, 81.5, 71.5, 68.0, 59.0, 28.3; HRMS (ESI) calculated for C13H21INO4 (M+H)+ 382.0510, found 382.0510.
2-Fluoro-6-iodo-3-((2-methoxyethoxy)methoxy)pyridine
To a solution of 2-fluoro-6-iodopyridin-3-ol (4.01 g, 16.78 mmol) in THF (170 mL) under nitrogen, NaH, 60% dispersion in mineral oil (1.34 g, 33.56 mmol) was added at −15 °C (ice/acetone). The reaction mixture was then stirred for 30 minutes at 0 °C. 2-Methoxyethoxymethyl chloride (3.83 mL, 33.56 mmol) was added to the mixture, and it was stirred for 3 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with water followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 2-fluoro-6-iodo-3-((2-methoxyethoxy)methoxy)pyridineas colorless oil (5.31 g, 97%);1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 8 Hz, 1H), 6.90 (dd, J = 10 Hz, J = 8 Hz, 1H), 4.90 (s, 2H), 3.46 – 3.44 (m, 2H), 3.15 – 3.13 (m 2H), 2.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.7 (JC,F= 244 Hz), 140.4 (JC,F= 24 Hz), 132.9 (JC,F= 5 Hz), 128.3 (JC,F= 4 Hz), 100.1 (JC,F= 12 Hz), 94.5, 71.4, 68.4, 59.0; 19F NMR (376 MHz, CDCl3)δ −80.5; HRMS (ESI) calculated for C9H12IFNO3 (M+H)+ 327.9840, found 327.9833.
2-Fluoro-6-iodopyridin-3-ol
To a solution of 2-fluoro-3-hydroxypyridine (2.5 g, 22.10 mmol) in water (125 mL), I2 (5.61g, 44.20 mmol) followed by K2CO3 (6.11 g, 44.20 mmol) were added. The reaction mixture was then stirred for 3 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with 2N HCl, 10% sodium thiosulfate followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 2-fluoro-6-iodopyridin-3-ol as a white solid (4.01 g, 76%); m.p. 117–119 °C; 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8 Hz, 1H), 7.01 (dd, J = 10 Hz, J = 8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 151.7 (JC,F= 239 Hz), 139.9 (JC,F= 27 Hz), 133.2 (JC,F= 5 Hz), 128.6 (JC,F= 5 Hz), 96.7 (JC,F= 12 Hz);19F NMR (376 MHz, CDCl3)δ −85.8; HRMS (ESI) calculated for C5H4FINO (M+H)+ 239.9316, found 239.9316.
Methyl 4-(2-(4-fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzoate (12)
To a solution of methyl 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzoate (75 mg, 0.16 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 3 hours at room temperature. After removal of solvent, methyl 4-(2-(4-fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzoate was crystallized as a white solid in EtOH:Et2O (1:2) (54 mg, 100%); m.p. 160–162 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.03 (bs, 1H), 9.42 (bs, 1H), 7.78 (d, J= 8 Hz, 2H), 7.22 – 7.18 (m, 2H), 7.16 (d, J = 8 Hz, 2H), 7.12 (t, J = 9 Hz, 2H), 6.86 (s, 1H), 3.82 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 165.9, 161.9 (JC,F= 245 Hz), 158.0, 146.4, 143.3, 133.1, 132.2 (JC,F= 8 Hz), 130.1, 129.8, 129.0, 127.5, 118.0, 116.9, 115.1 (JC,F= 21 Hz), 52.0;19F NMR (376 MHz, DMSO-d6) δ−112.9;HRMS (ESI) calculated for C19H15FNO4 (M+H)+ 340.0980, found 340.0965.
Methyl 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzoate
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (200mg, 0.47 mmol), 4-(methoxycarbonylphenyl) boronic acid (128 mg, 0.71 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and Na2CO3 (149 mg, 1.41 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 6 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded methyl 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzoate as colorless oil (116 mg, 51%); 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8 Hz, 2H), 7.42 (s, 1H), 7.29 – 7.24 (m, 4H), 6.91 (t, J = 9 Hz, 2H), 5.34 (s, 2H), 3.94 – 3.75 (m, 5H), 3.61 – 3.59 (m, 2H), 3.39 (s, 3H), 1.70 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 166.9, 162.2 (JC,F= 246 Hz), 154.0, 145.5, 144.8, 140.9, 137.3 (JC,F= 3 Hz), 131.6 (JC,F= 8 Hz), 129.7, 129.6, 128.5, 127.7, 127.5, 114.7 (JC,F= 21 Hz), 94.8, 80.7, 71.6, 68.0, 59.0, 52.1, 28.0; 19F NMR (376 MHz, CDCl3) δ −114.7; HRMS (ESI) calculated for C27H31FNO6 (M+H)+ 484.2130, found 484.2130.
6-(4-Fluorophenyl)-3-hydroxy-5-(4-(1-methyl-1H-tetrazol-5-yl)phenyl)pyridin-2(1H)-one (13)
To a solution of 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-(1-methyl-1H-tetrazol-5-yl)phenyl)pyridine (100 mg, 0.20 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 2 hours at room temperature. After removal of solvent, 6-(4-fluorophenyl)-3-hydroxy-5-(4-(1-methyl-1H-tetrazol-5-yl)phenyl)pyridin-2(1H)-one was crystallized as a white solid in Water:MeOH (2:1) (66 mg, 90%);m.p. 137–139 °C; 1H NMR (400 MHz, CDCl3+MeOD) δ 7.62 (d, J = 7 Hz, 2H), 7.26 (d, J = 8 Hz, 2H), 7.20 – 7.17 (m, 2H), 7.05 (s, 1H), 6.99 (t, J = 8 Hz, 2H), 4.16 (s, 3H); 13C NMR (100 MHz, CDCl3+MeOD) δ 162.9 (JC,F= 250 Hz), 158.6, 154.0, 145.9, 141.1, 132.9, 131.4 (JC,F= 8 Hz), 130.4, 129.6, 128.7, 122.2, 118.9, 118.8, 116.0 (JC,F= 22 Hz), 35.1; 19F NMR (376 MHz, CDCl3+MeOD) δ −111.0;HRMS (ESI) calculated for C19H15FN5O2 (M+H)+ 364.1204, found 364.1195.
2-(t-Butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-(1-methyl-1H-tetrazol-5-yl)phenyl)pyridine
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (197 mg, 0.46 mmol), 1-methyl-5-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl-1H-tetrazole (146 mg, 0.51 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and Na2CO3 (146 mg, 1.38 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 3 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-(1-methyl-1H-tetrazol-5-yl)phenyl)pyridine as a white solid (100 mg, 43%); m.p. 108–110 °C; 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8 Hz, 2H), 7.20 (d, J = 8 Hz, 2H), 7.14 – 7.11 (m, 2H), 7.10 (s, 1H), 6.75 (t, J = 9 Hz, 2H), 5.17 (s, 2H), 4.02 (s, 3H), 3.77 – 3.75 (m, 2H), 3.44 – 3.41 (m, 2H), 3.21 (s, 3H), 1.52 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.2 (JC,F= 246 Hz), 154.2, 154.1, 145.5, 143.4, 141.1, 135.7 (JC,F= 3 Hz), 131.6 (JC,F= 8 Hz), 130.6, 128.6, 127.4, 126.9, 122.0, 114.8 (JC,F= 21 Hz), 94.8, 80.9, 71.6, 68.0, 59.0, 35.1, 28.9;19F NMR (376 MHz, CDCl3)δ −114.5; HRMS (ESI) calculated for C27H31FN5O4 (M+H)+ 508.2355, found 508.2355.
6-(4-Fluorophenyl)-3-hydroxy-5-(4-(2-methyl-2H-tetrazol-5-yl)phenyl)pyridin-2(1H)-one (14)
To a solution of 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-(2-methyl-2H-tetrazol-5-yl)phenyl)pyridine (109 mg, 0.21 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 3 hours at room temperature. After removal of solvent, 6-(4-fluorophenyl)-3-hydroxy-5-(4-(2-methyl-2H-tetrazol-5-yl)phenyl)pyridin-2(1H)-one was crystallized as a white solid in Water:MeOH (2:1) (64 mg, 84%); m.p. 244–245 °C; 1H NMR (400 MHz, CDCl3+MeOD) δ 7.94 (d, J = 8 Hz, 2H), 7.17 – 7.13 (m, 4H), 7.04 (s, 1H), 6.94 (t, J = 9 Hz, 2H), 4.35 (s, 3H); 13C NMR (100 MHz, CDCl3+MeOD) δ 164.8, 162.8 (JC,F= 248 Hz), 158.6, 145.7, 139.8, 132.6, 131.4 (JC,F= 8 Hz), 130.1, 129.8, 126.8, 126.0, 119.6, 119.4, 115.8 (JC,F= 21 Hz), 39.4;19F NMR (376 MHz, CDCl3+MeOD) δ −111.4; HRMS (ESI) calculated for C19H15FN5O2 (M+H)+ 364.1204, found 364.1197.
2-(t-Butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-(2-methyl-2H-tetrazol-5-yl)phenyl)pyridine
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (193 mg, 0.45 mmol), 2-methyl-5-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl-2H-tetrazole (195 mg, 0.68 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and Na2CO3 (143 mg, 1.35 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 3 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-(2-methyl-2H-tetrazol-5-yl)phenyl)pyridine as a white solid (199 mg, 87%); m.p. 97–99 °C; 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8 Hz, 2H), 7.44 (s, 1H), 7.35 – 7.29 (m, 2H), 6.91 (t, J = 9 Hz, 2H), 5.35 (s, 2H), 4.41 (s, 3H), 3.95 – 3.93 (m, 2H), 3.62 – 3.59 (m, 2H), 3.39 (s, 3H), 1.70 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 165.0, 162.1 (JC,F= 246 Hz), 153.9, 145.4, 142.1, 140.9, 135.9 (JC,F= 3 Hz), 131.6 (JC,F= 8 Hz), 130.3, 127.82, 127.79, 126.8, 122.0, 114.7 (JC,F= 21 Hz), 94.8, 80.9, 71.6, 68.0, 59.0, 35.1, 28.9;19F NMR (376 MHz, CDCl3)δ −115.0; HRMS (ESI) calculated for C27H31FN5O4 (M+H)+ 508.2355, found 508.2355.
4-(2-(4-Fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzenesulfonamide (15)
To a solution of 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzenesulfonamide (100 mg, 0.20 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 3 hours at room temperature. After removal of solvent, 4-(2-(4-fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzenesulfonamide was crystallized as a white solid in Water:MeOH (2:1) (62 mg, 87%); m.p. 176–178 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.02 (bs, 1H), 9.42 (bs, 1H), 7.53 (d, J = 8 Hz, 2H), 7.21 – 7.17 (m, 2H), 7.14 (d, J = 8 Hz, 2H), 7.10 (t, J = 9 Hz, 2H), 6.82 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 161.9 (JC,F= 245 Hz), 157.9, 146.3, 142.3, 139.3, 133.1, 132.2 (JC,F= 8 Hz), 129.8, 126.2, 125.5, 118.0, 116.4, 115.1 (JC,F= 21 Hz);19F NMR (376 MHz, DMSO-d6) δ −112.8; HRMS (ESI) calculated for C17H14FN2O4S (M+H)+ 361.0653, found 361.0645.
4-(6-(t-Butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzenesulfonamide
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (200 mg, 0.47 mmol), 4-boronbenzene sulfonamide (143 mg, 0.71 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and Na2CO3 (149 mg, 1.41 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 3 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzenesulfonamide as colorless oil (225 mg, 95%); 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8 Hz, 2H), 7.37 (s, 1H), 7.29 (d, J = 9 Hz, 2H), 7.27 – 7.23 (m, 2H), 6.91 (t, J = 9 Hz, 2H), 5.22 (s, 2H), 3.92 – 3.90 (m, 2H), 3.59 – 3.57 (m, 2H), 3.35 (s, 3H), 1.68 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.2 (JC,F= 246 Hz), 154.1, 145.5, 144.8, 141.0, 140.3, 135.5 (JC,F= 4 Hz), 131.6 (JC,F= 8 Hz), 130.3, 127.4, 126.7, 126.5, 114.9 (JC,F= 21 Hz), 94.8, 81.0, 71.5, 68.0, 58.9, 28.8; 19F NMR (376 MHz, CDCl3)δ −114.3; HRMS (ESI) calculated for C25H30FN2O6S (M+H)+ 505.1803, found 505.1803.
N-((4-(2-(4-Fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)phenyl)sulfonyl)acetamide (16)
To a solution of N-((4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenyl)sulfonyl)acetamide (50 mg, 0.09 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 3 hours at room temperature. After removal of solvent, N-((4-(2-(4-fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)phenyl)sulfonyl)acetamidewas crystallized as a white solid in EtOH:Et2O (1:2) (33 mg, 92%); m.p. 291–293 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.04 (bs, 2H), 9.45 (bs, 1H), 7.73 (d, J = 8 Hz, 2H), 7.26 (d, J = 8 Hz, 2H), 7.22 – 7.19 (m, 2H), 7.12 (t, J = 8 Hz, 2H), 6.87 (s, 1H), 1.92 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.7, 162.0 (JC,F= 244 Hz), 158.0, 146.5, 143.7, 137.1, 133.4, 132.2 (JC,F= 8 Hz), 123.0, 129.6, 127.3, 117.9, 116.4, 115.1 (JC,F= 21 Hz), 23.2; 19F NMR (376 MHz, DMSO-d6) δ −112.7;HRMS (ESI) calculated for C19H16FN2O5S (M+H)+ 403.0758, found 403.0743.
N-((4-(6-(t-Butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenyl)sulfonyl)acetamide
To a solution of 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)benzenesulfonamide (150 mg, 0.30 mmol) in pyridine (1 mL) under nitrogen, acetic anhydride (0.1 mL, 1.06 mmol) followed by DMAP (11 mg, 0.09 mmol) were added. It was stirred for 18 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded N-((4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenyl)sulfonyl)acetamide as a white solid (116 mg, 71%); m.p. 162–164 °C; 1H NMR (400 MHz, CDCl3) δ 8.81 (br s, 1H), 7.95 (d, J = 8 Hz, 2H), 7.39 (s, 1H), 7.35 (d, J = 8 Hz, 2H), 7.27 – 7.24 (m, 2H), 6.92 (t, J = 9 Hz, 2H), 5.34 (s, 2H), 3.94 – 3.92 (m, 2H), 3.62 – 3.60 (m, 2H), 3.38 (s, 3H), 2.10 (s, 3H), 1.69 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 168.0, 162.3 (JC,F= 246 Hz), 154.3, 146.3, 145.6, 141.0, 136.6, 135.4 (JC,F= 4 Hz), 131.6 (JC,F= 8 Hz), 128.3, 127.3, 126.4, 122.3, 114.9 (JC,F= 22 Hz), 94.8, 81.0, 71.6, 68.0, 59.0, 28.8, 23.5; 19F NMR (376 MHz, CDCl3)δ −114.2; HRMS (ESI) calculated for C27H32FN2O7S (M+H)+ 547.1909, found 547.1909.
5-(4-(1H-1,2,3-Triazol-5-yl)phenyl)-6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one (17)
To a solution of 3-(4-(1H-1,2,3-triazol-5-yl)phenyl)-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (50 mg, 0.10 mmol) in anhydrous DCM (2 mL) under nitrogen, TFA (1 mL) was added. It was then stirred for 3 hours at room temperature. After removal of solvent, 5-(4-(1H-1,2,3-triazol-5-yl)phenyl)-6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one was crystallized as a white solid in EtOH:Et2O (1:2) (27 mg, 77%); dec.142–144 °C; 1H NMR (400 MHz, CDCl3+MeOD) δ 7.84 (s, 1H), 7.62 (d, J = 8 Hz, 2H), 7.17 – 7.14 (m, 2H), 7.08 (d, J = 8 Hz, 2H), 7.04 (s, 1H), 6.94 (t, J = 8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 161.8 (JC,F= 245 Hz), 158.0, 146.3, 138.0, 132.6, 132.2 (JC,F= 8 Hz), 132.0, 130.5, 130.0, 129.9, 125.4, 125.3, 118.3, 117.4, 115.0 (JC,F= 21 Hz); 19F NMR (376 MHz, DMSO-d6) δ −113.3; HRMS (ESI) calculated for C19H14FN4O2 (M+H)+ 349.1084, found 349.1095.
3-(4-(1H-1,2,3-Triazol-5-yl)phenyl)-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine
To a solution of 2-(t-butoxy)-5-(4-ethynylphenyl)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine (120 mg, 0.27 mmol) in a mixture of DMF (0.9 mL) and methanol (0.1 mL) under nitrogen, TMSN3 (0.15 mL, 1.08 mmol) followed by CuI (6 mg, 0.03 mmol) were added. The reaction mixture was then stirred overnight at 100 °C. After the reaction was completed, it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 3-(4-(1H-1,2,3-triazol-5-yl)phenyl)-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine as a white solid (39 mg, 30%); m.p. 154–156 °C;1H NMR (400 MHz, CDCl3) δ 12.34 (br s, 1H), 7.73 (s, 1H), 7.51 (d, J = 8 Hz, 2H), 7.14 – 7.11 (m, 2H), 7.07 (s, 1H), 7.02 (d, J = 8 Hz, 2H), 6.70 (t, J = 9 Hz, 2H), 5.16 (s, 2H), 3.75–3.73 (m, 2H), 3.43–3.40 (m, 2H), 3.19 (s, 3H), 1.49 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.1 (JC,F= 245 Hz), 162.7, 153.7, 146.8, 145.3, 140.9, 140.3, 136.1, 136.0, 131.6 (JC,F= 8 Hz), 130.2, 128.4, 128.0, 127.6, 126.0, 114.7 (JC,F= 22 Hz), 94.8, 80.6, 71.6, 68.0, 59.0, 28.9; 19F NMR (376 MHz, CDCl3) δ −115.1;HRMS (ESI) calculated for C27H30FN4O4 (M+H)+ 493.2245, found 493.2246.
2-(t-Butoxy)-5-(4-ethynylphenyl)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine
To a solution of 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-((trimethylsilyl)ethynyl)phenyl)pyridine (90 mg, 0.17 mmol) in methanol (5 mL) under nitrogen, K2CO3 (47 mg, 0.34 mmol) was added. The reaction mixture was then stirred for 5 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 2-(t-butoxy)-5-(4-ethynylphenyl)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)pyridine as yellow oil (76 mg, 100%);1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8 Hz, 2H), 7.29 (s, 1H), 7.21 – 7.18 (m, 2H), 7.04 (d, J = 8 Hz, 2H), 6.82 (t, J = 9 Hz, 2H), 5.23 (s, 2H), 3.84 – 3.82 (m, 2H), 3.51 – 3.49 (m, 2H), 3.29 (s, 3H), 3.02 (s, 1H), 1.59 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.1 (JC,F= 246 Hz), 153.8, 145.3, 140.9, 140.6, 135.9, 132.1, 131.6 (JC,F= 8 Hz), 129.7, 127.7, 120.5, 114.7 (JC,F= 21 Hz), 94.9, 83.5, 80.6, 77.6, 71.6, 68.0, 59.0, 28.9; 19F NMR (376 MHz, CDCl3) δ −115.0;HRMS (ESI) calculated for C27H29FNO4 (M+H)+ 450.2075, found 450.2075.
2-(t-Butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-((trimethylsilyl)ethynyl)phenyl)pyridine
4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenyl trifluoromethanesulfonate (223 mg, 0.39 mmol), (trimethylsilyl) acetylene (0.17 ml, 1.17 mmol), Pd(PPh3)2Cl2 (56 mg, 0.08 mmol), CuI (15 mg, 0.08 mmol) and Et3N (0.16 ml, 1.17mmol) were dissolved in DMF (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was stirred for 6 hours at 100 °C. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 10% EtOAc/Hexane. This afforded 2-(t-butoxy)-6-(4-fluorophenyl)-3-((2-methoxyethoxy)methoxy)-5-(4-((trimethylsilyl)ethynyl)phenyl)pyridine as colorless oil (84 mg, 41%);1H NMR (400 MHz, CDCl3) δ 7.385 (d, J = 8 Hz, 2H), 7.383 (s, 1H), 7.30 –7.27 (m, 2H), 7.11 (d, J = 8 Hz, 2H), 6.91 (t, J = 9 Hz, 2H), 5.33 (s, 2H), 3.94 – 3.92 (m, 2H), 3.61 – 3.59 (m, 2H), 3.39 (s, 3H), 1.69 (s, 9H), 0.28 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.2 (JC,F= 246 Hz), 153.9, 145.4, 140.9, 140.3, 136.0, 132.0, 131.6 (JC,F= 8 Hz), 129.6, 127.9, 127.8, 121.6, 114.7 (JC,F= 22 Hz), 105.0, 94.9, 80.7, 71.6, 59.0, 28.9, 0.0; 19F NMR (376 MHz, CDCl3) δ −115.0; HRMS (ESI) calculated for C30H37FNO4Si (M+H)+ 522.2470, found 522.2470.
4-(6-(t-Butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenyl trifluoromethanesulfonate
To solution of 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenol (100 mg, 0.23 mmol) in anhydrous DCM (10 mL) under nitrogen, N-phenyl-bis(trifluoromethanesulfonamide) (100 mg, 0.28 mmol) was added. It was cooled to 0 °C, and then Et3N (0.06 mL, 0.41 mmol) was added. The reaction mixture was allowed warm to room temperature, and it was stirred overnight. After the reaction was completed, it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenyl trifluoromethanesulfonate as colorless oil (121 mg, 92%);1H NMR (400 MHz, CDCl3) δ 7.40 (s, 1H), 7.28 – 7.19 (m, 6H), 6.92 (t, J = 9 Hz, 2H), 5.34 (s, 2H), 3.95 – 3.93 (m, 2H), 3.62 – 3.60 (m, 2H), 3.38 (s, 3H), 1.70 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 162.2 (JC,F= 246 Hz), 154.1, 148.4, 145.4, 141.0, 140.6, 135.6 (JC,F= 3 Hz), 131.6 (JC,F= 8 Hz), 131.5, 127.5, 126.5, 121.3, 114.8 (JC,F= 22 Hz), 94.8, 80.8, 71.6, 68.0, 59.0, 28.8; 19F NMR (376 MHz, CDCl3)δ −72.8, −114.6; HRMS (ESI) calculated for C26H28F4NO7S (M+H)+ 574.1518, found 574.1517.
4-(6-(t-Butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenol
3-Bromo-6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridine (200 mg, 0.47 mmol), 4-hydroxyphenylboronic acid (100 mg, 0.71 mmol), Pd(PPh3)4 (58 mg, 0.05 mmol) and Na2CO3 (150 mg, 1.42 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 8 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(6-(t-butoxy)-2-(4-fluorophenyl)-5-((2-methoxyethoxy)methoxy)pyridin-3-yl)phenol as a white solid (200 mg, 96%); m.p. 102–104 °C;1H NMR (400 MHz, MeOD) δ 7.38 (s, 1H), 7.33 – 7.29 (m, 2H), 6.96 (d, J = 9 Hz, 2H), 6.92 (t, J = 9 Hz, 2H), 6.71 (d, J = 9 Hz, 2H), 5.29 (s, 2H), 3.88 – 3.86 (m, 2H), 3.58 – 3.55 (m, 2H), 3.32 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, MeOD) δ 163.4 (JC,F= 244 Hz), 157.7, 154.5, 146.3, 142.4, 138.0, 132.8 (JC,F= 8 Hz), 132.4, 131.9, 130.5, 129.1, 116.3, 115.3 (JC,F= 21 Hz), 95.8, 81.4, 72.8, 69.1, 59.1, 29.3; 19F NMR (376 MHz, MeOD) δ −117.3;HRMS (ESI) calculated for C25H29FNO5 (M+H)+ 442.2024, found 442.2025.
4-(2-(4-Fluorobenzyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (18)
4-(2-(4-Fluorobenzyl)-5,6-dimethoxypyridin-3-yl)benzonitrile (146 mg, 0.42 mmol) was dissolved in anhydrous DCM (10 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (4.2 mL, 4.2 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 hours. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10% MeOH/DCM to provide 4-(2-(4-fluorobenzyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzonitrileas a white solid (63 mg, 53%); m.p. 191–193 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.95 (bs, 1H), 9.26 (bs, 1H), 7.83 (d, J = 8 Hz, 2H), 7.44 (d, J = 8 Hz, 2H), 7.06 (t, J = 9 Hz, 2H), 6.96 (dd, J = 8 Hz, J = 6 Hz, 2H), 6.70 (s, 1H), 3.79 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.8 (JC,F= 241 Hz), 158.4, 145.7, 143.2, 134.7, 132.7, 132.4, 130.0, 129.6 (JC,F= 8 Hz), 118.7, 117.7, 116.8, 115.1 (JC,F= 21 Hz), 109.7, 34.1; 19F NMR (376 MHz, DMSO-d6) δ −116.7; HRMS (ESI) calculated for C19H14FN2O2 (M+H)+ 321.1034, found 321.1019.
4-(2-(4-Fluorobenzyl)-5,6-dimethoxypyridin-3-yl)benzonitrile
4-(2-Bromo-5,6-dimethoxypyridin-3-yl)benzonitrile (300 mg, 0.94 mmol), 0.5 M in THF (4-fluorophenyl)zinc(II) chloride (4.7 mL, 2.35 mmol), Johnphos (56 mg, 0.19 mmol), Pd(OAc)2 (60 mg, 0.09 mmol) and K2CO3 (325 mg, 2.35 mmol)were dissolved in dioxane (20 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 8 hours. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(2-(4-fluorobenzyl)-5,6-dimethoxypyridin-3-yl)benzonitrile as a white solid (263 mg, 80%); m.p. 112–114 °C; 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 8 Hz, 2H), 7.34 (d, J = 8 Hz, 2H), 7.02 (dd, J = 9 Hz, J = 6 Hz, 2H), 6.89 (t, J = 9 Hz, 2H), 6.87 (s, 1H), 4.03 (s, 3H), 3.88 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.4 (JC,F= 243 Hz), 153.5, 144.7, 144.4, 142.3, 135.8 (JC,F= 3 Hz), 132.2, 130.2, 130.0 (JC,F= 8 Hz), 128.1, 119.1, 118.6, 114.9 (JC,F= 21 Hz), 111.3, 55.9, 53.9, 39.4; 19F NMR (376 MHz, CDCl3) δ −117.3; HRMS (ESI) calculated for C21H18FN2O2 (M+H)+ 349.1437, found 349.1347.
4-(5-Hydroxy-2-(naphthalen-1-ylmethyl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (19)
4-(5,6-Dimethoxy-2-(naphthalen-1-ylmethyl)pyridin-3-yl)benzonitrile (116 mg, 0.31 mmol) was dissolved in anhydrous DCM (10 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (3.05 mL, 3.05 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 hours. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10% MeOH/DCM to provide 4-(5-hydroxy-2-(naphthalen-1-ylmethyl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrileas a white solid (37 mg, 35%); m.p.133–135 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.89 (bs, 1H), 9.29 (bs, 1H), 7.94 – 7.89 (m, 2H), 7.80 (d, J = 8 Hz, 1H), 7.71 (d, J = 8 Hz, 2H), 7.55 – 7.49 (m, 2H), 7.44 (d,J = 8 Hz, 1H), 7.40 (d, J = 8 Hz, 2H), 6.98 (d, J = 7 Hz, 1H), 6.81 (s, 1H), 4.23 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ158.5, 145.9, 143.1, 135.0, 133.2, 132.3, 132.0, 131.0, 129.4, 128.5, 126.9, 126.2, 125.9, 125.6, 124.7, 123.2, 118.6, 117.6, 109.6, 32.5; HRMS (ESI) calculated for C23H16N2O2Na (M+Na)+ 375.1104, found 375.1107.
4-(5,6-Dimethoxy-2-(naphthalen-1-ylmethyl)pyridin-3-yl)benzonitrile
4-(2-Bromo-5,6-dimethoxypyridin-3-yl)benzonitrile (200 mg, 0.63 mmol), 0.25 M in THF (naphthalen-1-ylmethyl)zinc(II) chloride (6.27 mL, 1.57 mmol), Johnphos (37 mg, 0.13 mmol), Pd(OAc)2 (42 mg, 0.06 mmol) and K2CO3(217mg, 1.57 mmol) were dissolved in dioxane (20 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 2 hours. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(5,6-dimethoxy-2-(naphthalen-1-ylmethyl)pyridin-3-yl)benzonitrile as a white solid (202 mg, 85%); m.p. 173–175 °C; 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8 Hz, 1H), 7.84 (d, J = 8 Hz, 1H), 7.71 (d, J = 8 Hz, 1H), 7.65 (dd, J = 7 Hz, J = 2 Hz, 2H), 7.49 – 7.43 (m, 2H), 7.40 (dd, J = 7 Hz, J = 2 Hz, 2H), 7.32 (dd, J = 8 Hz, J = 7 Hz, 1H), 6.98 (dd, J = 7 Hz, J = 1Hz, 1H), 6.94 (s, 1H), 4.43 (s, 2H), 3.94 (s, 3H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.4, 144.8, 144.3, 142.3, 136.4, 133.8, 132.2, 132.0, 130.1, 128.6, 128.5, 126.8, 126.6, 125.7, 125.5, 125.3, 124.2, 119.1, 118.7, 111.2, 55.9, 53.9, 37.6; HRMS (ESI) calculated for C25H20N2O2Na (M+Na)+ 403.1417, found 403.1423.
4-(5-Hydroxy-2-(naphthalen-2-ylmethyl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (20)
4-(5,6-Dimethoxy-2-(naphthalen-2-ylmethyl)pyridin-3-yl)benzonitrile (139 mg, 0.37 mmol) was dissolved in anhydrous DCM (10 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (3.65 mL, 3.65 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 hours. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10% MeOH/DCM to provide 4-(5-hydroxy-2-(naphthalen-2-ylmethyl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrileas a white solid (44 mg, 34%); m.p. 167–169 °C;1H NMR (400 MHz, DMSO-d6) δ 12.00 (bs, 1H), 9.26 (bs, 1H), 7.86 – 7.78 (m, 5H), 7.48 – 7.44 (m, 4H), 7.41 (s, 1H), 7.14 (d, J = 8 Hz, 1H), 6.76 (s, 1H), 3.98 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 158.4, 145.8, 143.3, 136.3, 132.9, 132.6, 132.4, 131.6, 129.9, 128.0, 127.4, 126.5, 126.2, 125.9, 125.6, 118.7, 117.7, 117.1, 109.7, 35.1;HRMS (ESI) calculated for C23H16N2O2Na (M+Na)+ 375.1104, found 375.1106.
4-(5,6-Dimethoxy-2-(naphthalen-2-ylmethyl)pyridin-3-yl)benzonitrile
4-(2-Bromo-5,6-dimethoxypyridin-3-yl)benzonitrile (200 mg, 0.63 mmol), 0.5 M in THF (naphthalen-2-ylmethyl)zinc(II) bromide (3.14 mL, 1.57 mmol), Johnphos (37 mg, 0.13 mmol), Pd(OAc)2 (42 mg, 0.06 mmol) and K2CO3 (217mg, 1.57 mmol) were dissolved in dioxane (20 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 8 hours. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(5,6-dimethoxy-2-(naphthalen-2-ylmethyl)pyridin-3-yl)benzonitrile as a white solid (207 mg, 87%); m.p. 151–153 °C; 1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J = 7 Hz, J = 2 Hz, 1H), 7.75 – 7.68 (m, 4H), 7.48 – 7.44 (m, 3H), 7.39 (dd, J = 7 Hz, J = 2 Hz, 2H), 7.33 (dd, J = 8 Hz, J = 2 Hz, 1H), 6.93 (s, 1H), 4.12 (s, 2H), 4.09 (s, 3H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.5, 144.8, 144.5, 142.3, 138.0, 133.5, 132.1, 132.0, 130.3, 128.3, 127.8, 127.6, 127.5, 127.4, 126.8, 126.0, 125.4, 119.1, 118.7, 111.2, 55.9, 53.9, 40.5; HRMS (ESI) calculated for C25H21N2O2 (M+H)+ 381.1598, found 381.1596.
5-(4-(1H-Tetrazol-5-yl)phenyl)-6-(4-fluorobenzyl)-3-hydroxypyridin-2(1H)-one (21)
4-(2-(4-Fluorobenzyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (70 mg, 0.22 mmol) and NaN3 (57 mg, 0.88 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was re- moved under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (33 mg, 41%); dec. 154–156 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.91 (bs, 1H), 9.26 (bs, 1H), 8.03 (d, J = 8 Hz, 2H), 7.49 (d, J = 8 Hz, 2H), 7.07 (t, J = 9 Hz, 2H), 7.01 – 6.97 (m 2H), 6.74 (s, 1H), 3.83 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.8 (JC,F = 241 Hz), 158.4, 145.6, 141.1, 134.9, 132.4, 130.0, 129.6 (JC,F = 8 Hz), 127.1, 118.0, 117.3, 115.1 (JC,F = 21 Hz), 34.1;19F NMR (376 MHz, DMSO-d6) δ −116.7; HRMS (ESI) calculated for C19H15FN5O2 (M+H)+ 364.1204, found 364.1182.
5-(4-(1H-Tetrazol-5-yl)phenyl)-3-hydroxy-6-(naphthalen-1-ylmethyl)pyridin-2(1H)-one (22)
4-(5-Hydroxy-2-(naphthalen-1-ylmethyl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (25 mg, 0.07mmol) and NaN3 (18 mg, 0.28 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was re- moved under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (25 mg, 89%); dec. 156–158 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.87 (bs, 1H), 9.27 (bs, 1H), 7.96 – 7.90 (m, 4H), 7.82 (d, J = 8 Hz, 1H), 7.56 – 7.50 (m, 2H), 7.48 – 7.44 (m, 3H), 7.04 (d, J = 8 Hz, 1H), 6.87 (s, 1H), 4.29 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 158.5, 145.8, 141.0, 135.1, 133.2, 131.7, 131.0, 129.4, 128.5, 127.0, 126.9, 126.2, 125.9, 125.6, 124.8, 123.2, 118.0, 117.9, 32.6; HRMS (ESI) calculated for C23H18FN5O2 (M+H)+ 396.1455, found 396.1455.
5-(4-(1H-Tetrazol-5-yl)phenyl)-3-hydroxy-6-(naphthalen-2-ylmethyl)pyridin-2(1H)-one (23)
4-(5-Hydroxy-2-(naphthalen-2-ylmethyl)-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile (35 mg, 0.10 mmol) and NaN3 (26 mg, 0.40 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was re- moved under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (15 mg, 38%); dec. 64–66 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.97 (bs, 1H), 9.25 (bs, 1H), 8.01 (d, J = 8 Hz, 2H), 7.96 (s, 1H), 7.86 – 7.84 (m, 1H), 7.82 – 7.77 (m, 2H), 7.52 (d, J = 8 Hz, 2H), 7.47 – 7.45 (m, 2H), 7.18 – 7.16 (m, 1H), 6.73 (s, 1H), 4.02 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 158.4, 145.6, 141.2, 136.5, 132.9, 132.3, 131.6, 130.0, 127.9, 127.42, 127.40, 127.1, 126.5, 126.2, 125.9, 125.6, 118.1, 117.6, 35.1; HRMS (ESI) calculated for C23H18FN5O2 (M+H)+ 396.1455, found 396.1463.
4-(3-(4-Fluorophenyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-2-yl)benzoic acid (24)
4-(3-(4-Fluorophenyl)-5-(methoxymethoxy)-1-(methoxymethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzoic acid (62 mg, 0.15 mmol) was dissolved in a mixture of 2N HCl (5 mL) and dioxane (5 mL). It was refluxed for 6 hours, and it was cooled to room temperature. The white suspension was filtered, and it gave the pure product (49 mg, 100%); dec.324–325 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.01 (bs, 1H), 12.04 (bs, 1H), 9.45 (bs, 1H) 7.79 (d, J = 8 Hz, 2H), 7.35 (d, J = 8 Hz, 2H), 7.06 – 7.04 (m, 4H), 6.81 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 166.8, 161.0 (JC,F = 242 Hz), 157.9, 146.5, 138.2, 134.5 (JC,F = 3 Hz), 132.6, 131.4 (JC,F = 8 Hz), 130.1, 128.6, 118.5, 117.6, 115.1 (JC,F = 21 Hz); 19F NMR (376 MHz, DMSO-d6) δ −115.8;HRMS (ESI) calculated for C18H13FNO4 (M+H)+ 326.0823, found 326.0814.
4-(3-(4-Fluorophenyl)-5-(methoxymethoxy)-1-(methoxymethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzoic acid
6-Chloro-5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one (100 mg, 0.31 mmol), 4-carboxyphenyl boronic acid (76 mg, 0.46 mmol), Pd(PPh3)4 (36 mg, 0.03 mmol) and Na2CO3 (97mg, 0.92 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 3 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 100% EtOAc/Hexane. This afforded 5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one as yellowish oil (62 mg, 49%); m.p. 188–190 °C;1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8 Hz, 2H), 7.26 (d, J = 8 Hz, 2H), 7.03 (s, 1H), 6.88 – 6.84 (m, 2H), 6,75 (t, J = 9 Hz, 2H), 5.23 (s, 2H), 5.12 (s, 2H), 3.47 (s, 3H), 3.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.2, 161.6 (JC,F = 246 Hz), 158.8, 146.2, 138.2, 137.6, 134.0 (JC,F = 3 Hz), 131.3, 131.2, 130.6, 129.5, 121.9, 119.1, 115.1 (JC,F = 22 Hz), 95.1, 75.7, 57.3, 56.5; 19F NMR (376 MHz, CDCl3) δ −114.8; HRMS (ESI) calculated for C22H20FNO6Na (M+Na)+ 436.1167, found 436.1151.
6-Chloro-5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one
To a stirred solution of 5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one (212 mg, 0.72 mmol) in anhydrous DCM (10 mL) under nitrogen, Palau’s chlor (181 mg, 0.86 mmol) was added. The reaction mixture was stirred overnight. After the reaction was completed, it was filtered. The filtrate was purified by flash chromatography on silica gel eluting with 0 to 50% EtOAc/Hexane. This afforded 6-chloro-5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-oneas yellowish oil (100 mg, 42%);1H NMR (400 MHz, CDCl3) δ 7.34 – 7.30 (m, 2H), 7.10 (t, J = 9 Hz, 2H), 7.01 (s, 1H), 5.74 (s, 2H), 5.22 (s, 2H), 3.50 (s, 3H), 3.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.3 (JC,F =247 Hz), 158.8, 145.7, 133.1 (JC,F = 4 Hz), 131.2 (JC,F =8 Hz), 126.7, 121.7, 117.7, 115.5 (JC,F =21 Hz), 95.2, 76.6, 57.9, 56.6; 19F NMR (376 MHz, CDCl3) δ −113.6; HRMS (ESI) calculated for C15H16ClFNO4 (M+H)+ 328.0746, found 328.0739.
5-(4-Fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one
5-Bromo-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one (150mg, 0.54mmol), 4-fluorophenyl boronic acid (113 mg, 0.81 mmol), Pd(PPh3)4 (58 mg, 0.05mmol) and Na2CO3 (172 mg, 1.62 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 3 hours. After the reaction was completed, it was cooled to room temperature. It was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 100% EtOAc/Hexane. This afforded 5-(4-fluorophenyl)-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one as yellowish oil (116 mg, 73%); 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.38 (m, 2H), 7.28 – 7.26 (m, 2H), 7.10 (t, J= 9 Hz, 2H), 5.42 (s, 2H), 5.29 (s, 2H), 3.52 (s, 3H), 3.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 163.0 (JC,F = 245 Hz),157.9, 147.3, 132.9 (JC,F = 3 Hz), 127.7 (JC,F = 8 Hz), 125.4, 119.0, 118.7, 115.9 (JC,F = 22 Hz), 95.2, 78.6, 57.3, 56.5; 19F NMR (376 MHz, CDCl3) δ−115.0;HRMS (ESI) calculated for C15H17FNO4 (M+H)+ 294.1136, found 294.1136.
5-Bromo-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one
To a solution of 5-bromopyridin-2,3-diol (500 mg, 2.63 mmol) in DMF (25 mL) under nitrogen, NaH, 60% dispersion in mineral oil (1.05 g, 26.3 mmol) was added at 0 °C. The reaction mixture was then stirred for an hour at room temperature. Chloromethyl methyl ether (1.0 mL, 13.15 mmol) was added to the mixture, and it was stirred for 3 hours at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with water followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 100% EtOAc/Hexane. This afforded 5-bromo-3-(methoxymethoxy)-1-(methoxymethyl)pyridin-2(1H)-one as colorless oil (321 mg, 44%);1H NMR (400 MHz, CDCl3) δ 7.20 (d, J = 2Hz, 1H), 7.05 (d, J = 2Hz, 1H), 5.31 (s, 2H), 5.21 (s, 2H), 3.48 (s, 3H), 3.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.3, 147.6, 127.9, 121.8, 97.6, 95.3, 78.5, 57.4, 56.6.
4-(3-(4-Fluorobenzyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-2-yl)benzonitrile (25)
4-(3-(4-Fluorobenzyl)-5,6-dimethoxypyridin-2-yl)benzonitrile (145 mg, 0.42 mmol) was dissolve in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (4.2 mL, 4.2 mmol) was added. It was then allowed to warm to room temperature and stirred for overnight. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10 % MeOH/DCM to provide 4-(3-(4-fluorobenzyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-2-yl)benzonitrileas a white solid (70 mg, 53%);m.p. 225–227 °C; 1H NMR (400 MHz, MeOD) δ 7.81 (d, J = 8 Hz, 2H), 7.55 (d, J = 8 Hz, 2H), 7.02 (dd, J = 8 Hz, J = 6 Hz, 2H), 6.96 (t, J = 9 Hz, 2H), 6.75 (s, 1H), 3.66 (s, 2H); 13C NMR (100 MHz, MeOD) δ 162.9 (JC,F= 242 Hz), 159.9, 148.4, 139.8, 137.4, (JC,F= 3 Hz), 133.8, 133.5, 131.6, 131.1 (JC,F= 8 Hz), 120.9, 119.3, 119.1, 116.2 (JC,F= 21 Hz), 113.8, 36.2; 19F NMR (376 MHz, MeOD) δ −119.0; HRMS (ESI) calculated for C19H14FN2O2 (M+H)+ 321.1034, found 321.1046.
4-(3-(4-Fluorobenzyl)-5,6-dimethoxypyridin-2-yl)benzonitrile
2-Bromo-3-(4-fluorobenzyl)-5,6-dimethoxypyridine(190 mg, 0.58 mmol), 4-cyanophenylboronic acid (129 mg, 0.88 mmol), Pd(PPh3)4 (67 mg, 0.058 mmol) and Na2CO3 (185 mg, 1.75 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 18 hours. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(3-(4-fluorobenzyl)-5,6-dimethoxypyridin-2-yl)benzonitrile as a white solid (203 mg, 100%); m.p. 141–143 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 8 Hz, 2H), 7.61 (d, J = 8 Hz, 2H), 7.01 – 6.99 (m, 4H), 6.88 (s, 1H), 4.05 (s, 3H), 3.99 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 160.5 (JC,F= 243 Hz), 152.6, 144.4, 144.0, 143.5, 136.1 (JC,F= 4 Hz), 131.9, 129.8 (JC,F= 7 Hz), 136.4, 120.0, 118.9, 115.5 (JC,F= 22 Hz), 111.2, 55.9, 53.8, 39.1; 19F NMR (376 MHz, CDCl3) δ −116.5; HRMS (ESI) calculated for C21H18FN2O2 (M+H)+ 349.1347, found 349.1356.
2-Bromo-3-(4-fluorobenzyl)-5,6-dimethoxypyridine
(2-Bromo-5,6-dimethoxypyridin-3-yl)(4-fluorophenyl)-methanol (221 mg, 0.65 mmol) was dissolved in 1,2-dichloroethane (5 mL). The reaction mixture was treated with TFA (0.2 mL, 2.91 mmol) followed by triethylsilane (0.3 mL, 1.94 mmol). It was then heated up to 50 °C and stirred for 3 hours at the temperature. Then, it was cooled to room temperature and it was diluted with EtOAc. The organic layer was washed with sat. NaHCO3 followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 2-bromo-3-(4-fluorobenzyl)-5,6-dimethoxypyridine as a colorless oil (211 mg, 91%); 1H NMR (400 MHz, CDCl3) δ 7.13 (dd, J = 9 Hz, J = 5 Hz, 2H), 6.97 (t, J = 9 Hz, 2H), 6.77 (s, 1H), 3.99 (s, 3H), 3.96 (s, 2H), 3.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.6 (JC,F= 243 Hz), 152.3, 143.6, 134.8 (JC,F= 3 Hz), 130.1 (JC,F= 8 Hz), 129.2, 128.1, 120.8, 115.4 (JC,F= 21 Hz), 56.1, 54.4, 39.4; 19F NMR (376 MHz, CDCl3) δ −116.4; HRMS (ESI) calculated for C14H14BrFNO2 (M+H)+ 326.0186, found 326.0194.
(2-Bromo-5,6-dimethoxypyridin-3-yl)(4-fluorophenyl)methanol
(2-Bromo-5,6-dimethoxypyridin-3-yl)(4-fluorophenyl)methanone (378 mg, 1.11 mmol) was dissolved in THF (10 mL) and it was cooled to 0 °C. The reaction mixture was treated with NaBH4 (63 mg, 1.67 mmol), which was then allowed to warm to room temperature. The reaction mixture was stirred overnight at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with water followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded (2-bromo-5,6-dimethoxypyridin-3-yl)(4-fluorophenyl)-methanol as a colorless oil (269 mg, 71%); 1H NMR (400 MHz, CDCl3) δ 7.35 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.19 (s, 1H), 7.01 (t, J = 9 Hz, 2H), 6.04 (s, 1H), 3.99 (s, 3H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.3 (JC,F= 245 Hz), 153.0, 144.0, 137.8 (JC,F= 3 Hz), 131.9, 128.3 (JC,F= 9 Hz), 126.1, 118.1, 115.4 (JC,F= 21 Hz), 73.0, 56.1, 54.5; 19F NMR (376 MHz, CDCl3) δ −114.3; HRMS (ESI) calculated for C14H14BrFNO3 (M+H)+ 342.0136, found 342.0143.
(2-Bromo-5,6-dimethoxypyridin-3-yl)(4-fluorophenyl)methanone
2,3-Dibromo-5,6-dimethoxypyridine (546 mg, 1.84 mmol) was dissolved in toluene (5 mL) and it was cooled to −10 °C (acetone/ice). Then, it was treated with 1M in pentane trimethylsilylmethylithium (2.02 mL, 2.02 mmol). It was stirred for 30 minutes at the temperature. 4-Fluoro-N-methoxy-N-methylbenzamide (674 mg, 3.68 mmol) in toluene (5 mL) was slowly added to the reaction mixture. The reaction mixture was stirred an hour at −10 °C, which was stopped by adding 10 mL of water. The reaction mixture was diluted with EtOAc, which was washed with water followed by brine. The organic layer was concentrated under the reduced pressure. The resulting residue was purified by flash chromatography on silica gel eluting with 0 to 20% EtOAc/Hexane. This afforded(2-bromo-5,6-dimethoxypyridin-3-yl)(4-fluorophenyl)methanone as a white solid (379mg, 60%); m.p. 131–133 °C; 1H NMR (400 MHz, CDCl3) δ 7.86 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.16 (t, J = 9 Hz, 2H), 7.06 (s, 1H), 4.09 (s, 3H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.8, 166.2 (JC,F= 255 Hz), 154.6, 143.6, 132.8 (JC,F= 9 Hz), 129.4, 119.1, 116.0 (JC,F= 22 Hz), 56.3, 55.0; 19F NMR (376 MHz, CDCl3) δ −103.6; HRMS (ESI) calculated for C14H12BrFNO3 (M+H)+ 339.9979, found 339.9984.
4-(5-Hydroxy-3-(naphthalen-1-ylmethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzonitrile (26)
4-(5,6-Dimethoxy-3-(naphthalen-1-ylmethyl)pyridin-2-yl)benzonitrile (250 mg, 0.66 mmol) was dissolve in anhydrous DCM (10 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (6.57 mL, 6.57 mmol) was added. It was then allowed to warm to room temperature and stirred for overnight. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10 % MeOH/DCM to provide 4-(5-hydroxy-3-(naphthalen-1-ylmethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzonitrileas a white solid (92 mg, 40%);m.p. 289–291 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.91 (bs, 1H), 9.29 (bs, 1H), 7.93 – 7.89 (m, 3H), 7.91 (d, J = 8 Hz, 1H), 7.73 (d, J = 8 Hz, 1H), 7.64 (d, J = 7 Hz, 2H), 7.52 – 7.44 (m, 3H), 7.24 (d, J = 7 Hz, 1H), 6.49 (s, 1H), 4.07 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.7, 146.8, 138.4, 136.1, 133.4, 132.2, 131.3, 130.2, 128.5, 126.9, 126.3, 126.1, 125.7, 125.6, 123.4, 118.5, 118.0, 115.2, 111.1, 33.0; HRMS (ESI) calculated for C23H17N2O2 (M+H)+ 353.1285, found 353.1284.
4-(5,6-Dimethoxy-3-(naphthalen-1-ylmethyl)pyridin-2-yl)benzonitrile
2-Bromo-5,6-dimethoxy-3-(naphthalen-1-ylmethyl)pyridine (720 mg, 2.01 mmol), 4-cyanophenylboronic acid (443 mg, 3.01 mmol), Pd(PPh3)4 (232 mg, 0.20 mmol) and Na2CO3 (639 mg, 6.03 mmol) were dissolved in a mixture of dioxane (30 mL) and water (10 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for overnight. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 20% EtOAc/Hexane. This afforded 4-(5,6-dimethoxy-3-(naphthalen-1-ylmethyl)pyridin-2-yl)benzonitrile as a white solid (250 mg, 33%); m.p. 172–174 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8 Hz, 1H), 7.86 (d, J = 8 Hz, 1H), 7.80 (d, J = 8 Hz, 2H), 7.71 (d, J = 8 Hz, 2H), 7.53 – 7.46 (m, 2H), 7.42 (t, J = 8 Hz, 1H), 7.14 (s, 1H), 7.09 (d, J = 7 Hz, 1H), 4.43 (s, 2H), 3.92 (s, 3H), 3.68 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ152.1, 144.1, 143.6, 142.7, 136.5, 133.3, 312.0, 131.2, 129.6, 128.6, 126.9, 126.3, 126.2, 126.0, 125.8, 125.6, 123.3, 121.1, 118.7, 110.1, 55.6, 53.0, 34.5; HRMS (ESI) calculated for C25H21N2O2 (M+H)+ 381.1598, found 381.1597.
2-Bromo-5,6-dimethoxy-3-(naphthalen-1-ylmethyl)pyridine
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-1-yl)methanol (752 mg, 2.01 mmol) was dissolved in 1,2-dichloroethane (20 mL). The reaction mixture was treated with TFA (0.69 mL, 9.04 mmol) followed by triethylsilane (0.96 mL, 6.03 mmol). It was then heated up to 50 °C and stirred for 3 hours at the temperature. Then, it was cooled to room temperature and it was diluted with EtOAc. The organic layer was washed with sat. NaHCO3 followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 2-Bromo-5,6-dimethoxy-3-(naphthalen-1-ylmethyl)pyridine as a colorless oil (720 mg, 100%); 1H NMR (400 MHz, CDCl3) δ 7.93 – 7.87 (m, 2H), 7.79 (d, J = 8 Hz, 1H), 7.51 – 7.48 (m, 2H), 7.43 (t, J = 8 Hz, 1H), 7.22 (d, J = 7 Hz, 1H), 6.59 (s, 1H), 4.43 (s, 2H), 4.03 (s, 3H), 3.55 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.3, 143.6, 134.8, 134.0, 132.0, 128.9, 128.8, 128.1, 127.6, 127.0, 126.3, 125.8, 125.5, 123.9, 120.7, 55.9, 54.4, 37.6; HRMS (ESI) calculated for C18H17BrNO2 (M+H)+ 358.0437, found 358.0438.
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-1-yl)methanol
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-1-yl)methanone(927 mg, 2.49 mmol) was dissolved in THF (10 mL) and it was cooled to 0 °C. The reaction mixture was treated with NaBH4 (188 mg, 4.88 mmol), which was then allowed to warm to room temperature. The reaction mixture was stirred overnight at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with water followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded (2-bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-1-yl)methanol as a colorless oil (752 mg, 81%); 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 8 Hz, 1H), 7.96 (d, J = 8 Hz, 1H), 7.87 (d, J = 8 Hz, 1H), 7.60 (t, J = 7 Hz, 1H), 7.54 (t, J = 7 Hz, 1H), 7.46 (t, J = 8 Hz, 1H), 7.40 (s, 1H), 7.28 (d, J = 7 Hz, 1H), 6.52 (d, J = 6 Hz, 1H), 6.25 (d, J = 6 Hz, 1H), 3.89 (s, 3H), 3.74 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 152.4, 143.5, 138.5, 133.4, 132.6, 130.9, 128.6, 128.0, 126.2, 125.7, 125.6, 125.3, 124.3, 123.7, 119.9, 69.4, 55.8, 53.9; HRMS (ESI) calculated for C18H17BrNO3 (M+H)+ 374.0386, found 374.0388.
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-1-yl)methanone
2,3-Dibromo-5,6-dimethoxypyridine (546 mg, 1.84 mmol) was dissolved in toluene (5 mL) and it was cooled to −10 °C (acetone/ice). Then, it was treated with 1M in pentane trimethylsilylmethylithium (2.02 mL, 2.02 mmol). It was stirred for 30 minutes at the temperature. N-methoxy-N-methyl-1-naphthamide (792 mg, 3.68 mmol) in toluene (5 mL) was slowly added to the reaction mixture. The reaction mixture was stirred an hour at −10°C, which was stopped by adding 10 mL of water. The reaction mixture was diluted with EtOAc, which was washed with water followed by brine. The organic layer was concentrated under the reduced pressure. The resulting residue was purified by flash chromatography on silica gel eluting with 0 to 20% EtOAc/Hexane. It gave (2-bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-1-yl)methanone as a white solid (226 mg, 33%); m.p. 115–117 °C; 1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 8 Hz, 1H), 8.12 (d, J = 8 Hz, 1H), 8.00 (d, J = 8 Hz, 1H), 7.75 – 7.64 (m, 4H), 7.34 (s, 1H), 7.53 (t, J = 8 Hz, 1H), 4.17 (s, 3H), 3.95 (s, 3H);13C NMR (100 MHz, CDCl3) δ 196.0, 154.7, 143.5, 134.6, 134.0, 133.6, 131.1, 131.0, 128.5, 128.3, 126.7, 126.0, 125.8, 124.4, 124.2, 120.1, 56.3, 55.0; HRMS (ESI) calculated for C18H15BrNO3 (M+H)+ 372.0230, found 372.0233.
4-(5-Hydroxy-3-(naphthalen-2-ylmethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzonitrile (27).
4-(5,6-Dimethoxy-3-(naphthalen-2-ylmethyl)pyridin-2-yl)benzonitrile (133 mg, 0.35 mmol) was dissolve in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C and the 1M in DCM BBr3 (3.5 mL, 3.5mmol) was added. It was then allowed to warm to room temperature and stirred for overnight. Then, the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2N HCl followed by brine. The organic layer was dried over Na2SO4 followed by concentration under the vacuum. The residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane followed by 0 to 10 % MeOH/DCM to provide 4-(5-hydroxy-3-(naphthalen-2-ylmethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzonitrileas a white solid (89 mg, 73%);m.p.194–196 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.91 (d, J = 7 Hz, 2H), 7.86 – 7.80 (m, 3H), 7.61 (d, J = 7 Hz, 2H), 7.52 (s, 1H), 7.50 – 7.44 (m, 2H), 7.18 (d, J = 8 Hz, 1H), 6.64 (s, 1H), 3.77 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 157.6, 146.8, 138.5, 138.1, 133.1, 132.4, 132.2, 131.6, 130.5, 128.0, 127.41, 127.40, 127.03, 126.3, 126.1, 125.5, 118.5, 118.4, 115.6, 111.1, 35.6; HRMS (ESI) calculated for C23H17N2O2 (M+H)+ 353.1285, found 353.1295.
4-(5,6-Dimethoxy-3-(naphthalen-2-ylmethyl)pyridin-2-yl)benzonitrile
2-Bromo-5,6-dimethoxy-3-(naphthalen-2-ylmethyl)pyridine (137 mg, 0.38 mmol), 4-cyanophenylboronic acid (84 mg, 0.57 mmol), Pd(PPh3)4 (44 mg, 0.04 mmol) and Na2CO3 (121 mg, 1.15 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for overnight. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4Cl followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 4-(5,6-dimethoxy-3-(naphthalen-2-ylmethyl)pyridin-2-yl)benzonitrile as a white solid (133 mg, 92%); m.p. 61–63 °C; 1H NMR (400 MHz, CDCl3) δ 7.86 – 7.76 (m, 4H), 7.68 – 7.67 (m, 3H), 7.50 – 7.48 (m, 3H), 7.24 (dd, J = 8 Hz, J = 1 Hz, 1H), 6.98 (s, 1H), 4.20 (s, 2H), 4.11 (s, 3H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.6, 144.5, 144.1, 143.6, 138.1, 133.6, 132.9, 132.2, 131.9, 129.9, 128.5, 127.9, 127.7, 127.6, 127.0, 126.8, 126.3, 125.7, 120.3, 119.0, 111.1, 55.9, 53.8, 38.1; HRMS (ESI) calculated for C25H21BrN2O2 (M+H)+ 381.1598, found 381.1606.
2-Bromo-5,6-dimethoxy-3-(naphthalen-2-ylmethyl)pyridine
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-2-yl)methanol (153 mg, 0.41 mmol) was dissolved in 1,2-dichloroethane (5 mL). The reaction mixture was treated with TFA (0.20 mL, 1.23 mmol) followed by triethylsilane (0.14 mL, 1.84 mmol). It was then heated up to 50 °C and stirred for 3 hours at the temperature. Then, it was cooled to room temperature and it was diluted with EtOAc. The organic layer was washed with sat. NaHCO3 followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded 2-Bromo-5,6-dimethoxy-3-(naphthalen-2-ylmethyl)pyridine as a colorless oil (137 mg, 93%); 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.77 (m, 4H), 7.60 (s, 1H), 7.48 – 7.45 (m, 2H), 7.34 (dd, J = 8 Hz, J = 2 Hz, 1H), 4.18 (s, 2H), 4.03 (s, 3H), 3.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.3, 143.7, 136.7, 133.6, 132.3, 129.3, 128.34, 128.29, 127.7, 127.6, 127.2, 127.1, 126.2, 125.6, 121.0, 56.1, 54.4, 40.4; HRMS (ESI) calculated for C18H17BrNO2 (M+H)+ 358.0437, found 358.0347.
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-2-yl)methanol
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-2-yl)methanone(1.0 g, 2.69 mmol) was dissolved in THF (10 mL) and it was cooled to 0 °C. The reaction mixture was treated with NaBH4 (203 mg, 5.37 mmol), which was then allowed to warm to room temperature. The reaction mixture was stirred overnight at room temperature. After the reaction was completed, it was diluted with EtOAc and washed with water followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel eluting with 0 to 30% EtOAc/Hexane. This afforded (2-bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-2-yl)methanol as a colorless oil (869 mg, 86%); 1H NMR (400 MHz, CDCl3) δ 7.90 (s, 1H), 7.85 – 7.80 (m, 3H), 7.50 – 7.46 (m, 2H), 7.44 (d, J= 2 Hz, 1H), 7.24 (s, 1H), 6.27 (s, 1H), 4.02 (s, 3H), 3.80 (s, 3H), 2.57 (bs, 1H); 13C NMR (100 MHz, CDCl3) δ 153.0, 144,0, 139.4, 133.2, 132.9, 132.0, 128.4, 128.2, 127.7, 126.4, 126.3, 126.2, 125.2, 124.5, 118.5, 73.7, 56.1, 54.6; HRMS (ESI) calculated for C18H17BrNO3 (M+H)+ 374.0386, found 374.0389.
(2-Bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-2-yl)methanone
2,3-Dibromo-5,6-dimethoxypyridine (1.64g, 5.52 mmol) was dissolved in toluene (10 mL) and it was cooled to 0 °C. Then, it was treated with 1M in pentane trimethylsilylmethylithium (6.07 mL, 6.07 mmol). It was stirred for 30 minutes at the temperature. N-methoxy-N-methyl-2-naphthamide (2.38g, 11.04 mmol) in toluene (10 mL) was slowly added to the reaction mixture. The reaction mixture was stirred an hour at 0 °C, which was stopped by adding 10 mL of water. The reaction mixture was diluted with EtOAc, which was washed with water followed by brine. The organic layer was concentrated under the reduced pressure. The resulting residue was purified by flash chromatography on silica gel eluting with 0 to 20% EtOAc/Hexane. It gave (2-bromo-5,6-dimethoxypyridin-3-yl)(naphthalen-2-yl)methanone as a white solid (1.51 g, 73%); m.p. 152–154 °C; 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.90 (dd, J = 9 Hz, J = 2 Hz, 1H), 7.84 – 7.79 (m, 3H), 7.52 (t, J = 7 Hz, 1H), 7.45 (t, J = 8 Hz, 1H), 7.05 (s, 1H), 4.02 (s, 3H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 194.2, 154.5, 143.5, 135.9, 133.7, 132.9, 132.5, 129.9, 129.8, 129.0, 128.7, 127.9, 127.0, 124.9, 119.3, 56.3, 54.9; HRMS (ESI) calculated for C18H15BrNO3 (M+H)+ 372.0230, found 372.0239.
6-(4-(1H-Tetrazol-5-yl)phenyl)-5-(4-fluorobenzyl)-3-hydroxypyridin-2(1H)-one (28)
4-(3-(4-Fluorobenzyl)-5-hydroxy-6-oxo-1,6-dihydropyridin-2-yl)benzonitrile (140 mg, 0.44 mmol) and NaN3 (114 mg, 1.76 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was re- moved under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (73 mg, 46%); dec. 181–183 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.83 (bs, 1H), 9.23 (bs, 1H), 8.09 (d, J = 7 Hz, 2H), 7.60 (d, J = 7 Hz, 2H), 7.08 – 7.06 (m 4H), 6.60 (s, 1H), 3.64 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.7 (JC,F = 241 Hz), 157.7, 146.6, 136.7 (JC,F = 3 Hz), 136.4, 132.7, 130.5, 130.0 (JC,F = 8 Hz), 126.8, 124.2, 118.1, 115.1 (JC,F = 21 Hz), 34.6; 19F NMR (376 MHz, DMSO-d6) δ −117.1; HRMS (ESI) calculated for C19H15FN5O2 (M+H)+ 364.1204, found 364.1202.
6-(4-(1H-Tetrazol-5-yl)phenyl)-3-hydroxy-5-(naphthalen-1-ylmethyl)pyridin-2(1H)-one (29)
4-(5-Hydroxy-3-(naphthalen-1-ylmethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzonitrile (40 mg, 0.11 mmol) and NaN3 (30 mg, 0.46 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was removed under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (20 mg, 44%); dec. 179–181 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.93 (bs, 1H), 8.08 (d, J = 7 Hz, 2H), 7.92 (d, J = 8 Hz, 1H), 7.81 (d, J = 8 Hz, 1H), 7.69 (d, J = 7 Hz, 2H), 7.51 – 7.43 (m, 3H), 7.28 (d, J = 7 Hz, 1H), 6.50 (s, 1H), 4.11 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 158.3, 157.9, 157.8, 146.5, 136.5, 136.3, 133.4, 132.7, 131.3, 130.3, 128.5, 126.89, 126.85, 126.4, 126.1, 125.7, 125.6, 123.4, 118.0, 114.7, 33.1; HRMS (ESI) calculated for C23H18FN5O2 (M+H)+ 396.1455, found 396.1456.
6-(4-(1H-Tetrazol-5-yl)phenyl)-3-hydroxy-5-(naphthalen-2-ylmethyl)pyridin-2(1H)-one (30)
4-(5-Hydroxy-3-(naphthalen-2-ylmethyl)-6-oxo-1,6-dihydropyridin-2-yl)benzonitrile (50 mg, 0.14mmol) and NaN3 (37 mg, 0.57mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with catalytic amount of acetic acid. It was sealed and then it was heated at 130 °C for overnight. The reaction was cooled to room temperature and gave brownish suspension. After the completion of the reaction, the solvent was re- moved under vacuum. Addition of 2N HCl and stirring produced a solid, which was filtered to give the pure product (16 mg, 28%); dec. 197–199 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.85 (bs, 1H), 9.20 (bs, 1H), 8.09 (d, J = 8 Hz, 2H), 7.86 – 7.80 (m, 3H), 7.65 (d, J = 8 Hz, 2H), 7.56 (s, 1H), 7.47 – 7.45 (m, 2H), 7.21 (d, J = 9 Hz, 2H), 6.64 (s, 1H), 3.82 (bs, 1H); 13C NMR (100 MHz, DMSO-d6) δ 157.8, 146.6, 138.3, 136.5, 133.1, 131.6, 130.5, 128.0, 127.42, 127.39, 127.1, 126.8, 126.3, 126.1, 125.4, 124.2, 118.3, 35.7; HRMS (ESI) calculated for C23H18FN5O2 (M+H)+ 396.1455, found 396.1462.
Expression, Purification and Crystallization
Pandemic influenza A/California/04/09 H1N1 PA endonuclease motif (residues 1–204) was expressed and purified as described previously.[22] BL21 (RIL) cells (Stratagene) were grown to an OD600 of 0.8 and induced with 0.15 mM IPTG at 17 °C for 17 h. Cells were harvested by centrifugation and purified on Ni-NTA (Qiagen) according to manufacturer’s recommendations except the final elution was completed with 500 mM imidazole-containing buffer. The dual hexa-histidine (6xHis) tag was then removed by HRV14 3C protease cleavage. The endonuclease was further purified by size exclusion chromatography using HiLoad 26/60 Superdex 75 (GE Healthcare). The buffer used for size exclusion and the final buffer for storage of the purified protein was 100 mM NaCl and 20 mM Tris pH 8.0. The protein was concentrated to 5 mg/ml using an Ultrafree 10K (Millipore), aliquoted and stored at −80 °C.
Crystals were formed by mixing in a 1:1 ratio the purified endonuclease protein (5 mg/ml) with crystallization buffer containing 200 mM MES pH 6.7, 27% PEG8000, 200 mM ammonium sulfate, 1 mM manganese chloride, 10 mM magnesium acetate, 10 mM taurine, and 50 mM sodium fluoride. Crystals matured to full size in one to two weeks at 20 °C.
Compound Soaking, Data Collection and Processing
Crystal structures of compounds 11 and 29 were determined in complex with endonuclease as previously described.15 Compounds were soaked into previously formed crystals by stepwise gradient shifting the surrounding crystallization solution from starting well solution to 1 mM manganese sulfate, 200 mM HEPES pH 7.7, 25% (w/v) PEG 8000, 50 mM ammonium sulfate, 5 mM magnesium acetate, 80 mM L-arginine, 10% (v/v) DMSO containing 100 mM 11 or 29 and 10% (v/v) ethylene glycol. Crystals were then soaked with the ligand for 2 h at 20 °C before being flash-cooled and placed into liquid nitrogen for storage and transport. X-ray diffraction data collection was performed at the Cornell High Energy Synchrotron Source (CHESS) F1 beamline or the Advanced Photon Source (APS) 23-ID-D beamline. The diffraction data were indexed, processed, scaled, and merged using HKL2000.[44] The structure was solved and refined using the PHENIX software package.[45]
Endonuclease Assay
A high throughput endonuclease assay was performed as described previously.15 The endonuclease assay used a single-stranded DNA probe with the sequence (6-FAM)TGGCAATATCAGCTCCACA(MGBNFQ) (Applied Biosystems). Reaction buffer contained 50 mM Tris pH 7.5, 50 mM sodium chloride, 1 mM DTT, 5 mM magnesium chloride, 0.5 mM manganese sulfate, and 1 mM CHAPS. DNA probe (50 nM), endonuclease (25 nM), and compound were mixed on ice and then placed in a Varioskan Fluorometer (Thermo Scientific) preincubated at 37 ˚C to detect fluorescence with an excitation of 488 nm and emission of 518 nm. Fluorescence is measured at several time points (5, 60, and 90 minutes) and activity/inhibition is calculated based on the change in fluorescence over time using Prism Graphpad (6.0b) non-linear regression analysis. Ligand efficiency was calculated using the following equation: LE (in kcal/(mol·NHA)) = −RTln(IC50)/NHA.
Fluorescent-based antiviral assay
Canine Madin-Darby canine kidney (MDCK; ATCC CCL-34) cells were grown at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and 1% PSG (Penicillin, 100 units/ml; Streptomycin 100 μg/ml; Glutamine, 2 mM).[42,43] The replication-competent influenza A/Puerto Rico/08/1934 H1N1 expressing mCherry fluorescent protein (PR8-mCherry) used in this assay has been previously described.[42]
Antiviral-mediated inhibition of PR8-mCherry was evaluated as previously described.[42,43] Briefly, confluent monolayers of MDCK cells (96-well plate format; 5 × 104 cells/well; triplicates) were infected with 100 plaque-forming units (PFU). After 1 hour infection at room temperature, infectious media was supplemented with 25 μM or 1 μM of the indicated compounds and cell were incubated at 37°C. At 24–36 hours post-infection, cells were washed with PBS prior to mCherry quantification using a fluorescence plate reader (DTX-880, Becton Dickenson) as previously described.[42,43] Fluorescence values of PR8 mCherry-infected cells in the absence of drug were used to calculate 100% viral infection. Cells in the absence of viral infection were used to calculate the fluorescence background. Ribavirin, a compound with antiviral activity against influenza virus was used as internal control for all the assays. [42,43]
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from NIH R21 AI119321 (award to EA, EJL, and LMS). The Bruker Avance III 400 MHz NMR spectrometer used for this study was purchased with funds from NCRR Grant No. 1S10RR23698–1A1. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource with support from the NIH National Center for Research Resources Grant No. P41RR0954. We thank the laboratories of Ann Stock and Gaetano Montelione for access to equipment used in this study. X-ray data collection was conducted at the Cornell High Energy Synchrotron Source (CHESS) and the Advanced Photon Source (APS). CHESS is supported by the NSF & NIH/NIGMS via NSF award DMR-0225180, and the MacCHESS resource is supported by NIH/NCRR award RR-01646. The Advanced Photon Source GM/CA 23-ID beam line is funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References:
- [1].Dunning J, Baillie JK, Cao B, Hayden FG, International Severe Acute Respiratory and Emerging Infection Consortium (ISARIC), Lancet Infect Dis 2014, 14, 1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mills CE, Robins JM, Lipsitch M, Nature 2004, 432, 904–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hayden FG, Hay AJ, Curr. Top. Microbiol. Immunol. 1992, 176, 119–130. [DOI] [PubMed] [Google Scholar]
- [4].Bright RA, Medina M, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI, Lancet 2005, 366, 1175–1181. [DOI] [PubMed] [Google Scholar]
- [5].Moscona A, Engl N J. Med. 2005, 353, 2633–2636. [DOI] [PubMed] [Google Scholar]
- [6].Bloom JD, Gong LI, Baltimore D, Science 2010, 328, 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bauman JD, Harrison JJEK, Arnold E, IUCrJ 2016, 3, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, Hoshino T, Biochemistry 2016, 55, 2646–2660. [DOI] [PubMed] [Google Scholar]
- [9].Carcelli M, Rogolino D, Gatti A, De Luca L, Sechi M, Kumar G, White SW, Stevaert A, Naesens L, Sci Rep 2016, 6, 31500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beylkin D, Kumar G, Zhou W, Park J, Jeevan T, Lagisetti C, Harfoot R, Webby RJ, White SW, Webb TR, Sci Rep 2017, 7, 17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Credille CV, Dick BL, Morrison CN, Stokes RW, Adamek RN, Wu NC, Wilson IA, Cohen SM, J. Med. Chem. 2018, 61, 10206–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Song M-S, Kumar G, Shadrick WR, Zhou W, Jeevan T, Li Z, Slavish PJ, Fabrizio TP, Yoon S-W, Webb TR, et al. , Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 3669–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Omoto S, Speranzini V, Hashimoto T, Noshi T, Yamaguchi H, Kawai M, Kawaguchi K, Uehara T, Shishido T, Naito A, et al. , Sci Rep 2018, 8, 9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jones JC, Kumar G, Barman S, Najera I, White SW, Webby RJ, Govorkova EA, MBio 2018, 9, DOI 10.1128/mBio.00430-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Das K, Aramini JM, Ma L-C, Krug RM, Arnold E, Nat. Struct. Mol. Biol. 2010, 17, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, et al. , Nature 2008, 454, 1123–1126. [DOI] [PubMed] [Google Scholar]
- [17].Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, et al. , Nature 2009, 458, 909–913. [DOI] [PubMed] [Google Scholar]
- [18].Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RWH, Nature 2009, 458, 914–918. [DOI] [PubMed] [Google Scholar]
- [19].DuBois RM, Slavish PJ, Baughman BM, Yun M-K, Bao J, Webby RJ, Webb TR, White SW, PLoS Pathog. 2012, 8, e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kowalinski E, Zubieta C, Wolkerstorfer A, Szolar OHJ, Ruigrok RWH, Cusack S, PLoS Pathog. 2012, 8, e1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tefsen B, Lu G, Zhu Y, Haywood J, Zhao L, Deng T, Qi J, Gao GF, Virol J 2014, 88, 1935–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bauman JD, Patel D, Baker SF, Vijayan RSK, Xiang A, Parhi AK, Martínez-Sobrido L, LaVoie EJ, Das K, Arnold E, ACS Chem. Biol. 2013, 8, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Plotch SJ, Bouloy M, Ulmanen I, Krug RM, Cell 1981, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- [24].Takashita E, Morita H, Ogawa R, Nakamura K, Fujisaki S, Shirakura M, Kuwahara T, Kishida N, Watanabe S, Odagiri T, Front Microbiol 2018, 9, 3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu X, Seno K, Nishizawa Y, Hayashi F, Yamazaki A, Matsumoto H, Wakabayashi T, Usukura J, Exp. Eye Res. 1994, 59, 761–768. [DOI] [PubMed] [Google Scholar]
- [26].Hastings JC, Selnick H, Wolanski B, Tomassini JE, Antimicrob. Agents Chemother. 1996, 40, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tomassini JE, Davies ME, Hastings JC, Lingham R, Mojena M, Raghoobar SL, Singh SB, Tkacz JS, Goetz MA, Antimicrob. Agents Chemother. 1996, 40, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Carcelli M, Rogolino D, Bacchi A, Rispoli G, Fisicaro E, Compari C, Sechi M, Stevaert A, Naesens L, Mol. Pharm. 2014, 11, 304–316. [DOI] [PubMed] [Google Scholar]
- [29].Rogolino D, Bacchi A, De Luca L, Rispoli G, Sechi M, Stevaert A, Naesens L, Carcelli M, J. Biol. Inorg. Chem. 2015, 20, 1109–1121. [DOI] [PubMed] [Google Scholar]
- [30].Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, Neya S, Hoshino T, Bioorg. Med. Chem. 2015, 23, 5466–5475. [DOI] [PubMed] [Google Scholar]
- [31].Kuzuhara T, Iwai Y, Takahashi H, Hatakeyama D, Echigo N, PLoS Curr 2009, 1, RRN1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Credille CV, Chen Y, Cohen SM, J. Med. Chem. 2016, 59, 6444–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Carcelli M, Rogolino D, Gatti A, De Luca L, Sechi M, Kumar G, White SW, Stevaert A, Naesens L, Sci Rep 2016, 6, 31500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miyagawa M, Akiyama T, Mikamiyama-Iwata M, Hattori K, Kurihara N, Taoda Y, Takahashi-Kageyama C, Kurose N, Mikamiyama H, Suzuki N, et al. , Bioorg. Med. Chem. Lett. 2016, 26, 4739–4742. [DOI] [PubMed] [Google Scholar]
- [35].Beylkin D, Kumar G, Zhou W, Park J, Jeevan T, Lagisetti C, Harfoot R, Webby RJ, White SW, Webb TR, Sci Rep 2017, 7, 17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Parkes KEB, Ermert P, Fässler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K, J. Med. Chem. 2003, 46, 1153–1164. [DOI] [PubMed] [Google Scholar]
- [37].Sagong HY, Parhi A, Bauman JD, Patel D, Vijayan RSK, Das K, Arnold E, LaVoie EJ, ACS Med Chem Lett 2013, 4, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Parhi AK, Xiang A, Bauman JD, Patel D, Vijayan RSK, Das K, Arnold E, LaVoie EJ, Bioorg. Med. Chem. 2013, 21, 6435–6446. [DOI] [PubMed] [Google Scholar]
- [39].Sagong HY, Bauman JD, Patel D, Das K, Arnold E, LaVoie EJ, J. Med. Chem. 2014, 57, 8086–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Using 2-(t-Butyoxy-6-iodo-3-((2-methoxyethoxy)methoxy)pyridine, we prepared the 6-(2-naphthyl derivative by Suzuki coupling. This intermediated was converted with NBS in acetonitrile to its 5-bromo derivative, which was subsequently treated under Suzuki coupling conditions to provide the 5-(p-(5-tetrazoyl)phenyl) derivative. Removal of the t-butoxy and MEM protecting groups using TFA in dichloromethane, followed by 2N HCl in dioxane (1:1) provided the 6-(2-naphthyl)-5-(p-(5-tetrazoyl)phenyl)-3-hydroxypyridin-2(1H)-one. This compound prepared by this alternative route proved unstable and significantly decomposed in DMSO within a 1 hour time period as observed by LCMS.
- [41].Wendt MD, Rockway TW, Geyer A, McClellan W; Weitzberg M, Zhao X, Mantei R, Nienaber VL, Stewart K, Klinghofer V,;Giranda VL, J. Med. Chem. 2004, 47, 303–324. [DOI] [PubMed] [Google Scholar]
- [42].Nogales A, Baker SF, Martínez-Sobrido L, Virology 2015, 476, 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nogales A, Rodríguez-Sánchez I, Monte K, Lenschow DJ, Perez DR, Martínez-Sobrido L, Virus Res. 2016, 213, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Arnold E, Himmel DM, Internationale Union für Kristallographie, Eds., Crystallography of Biological Macromolecules, Wiley, Chichester, 2011. [Google Scholar]
- [45].Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. , Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.