

Abstract
The non-contractile cytoskeleton in cardiomyocytes is comprised of cytoplasmic actin, microtubules, and intermediate filaments. In addition to providing mechanical support to these cells, these structures are important effectors of tension-sensing and signal transduction and also provide networks for the transport of proteins and organelles. The majority of our knowledge on the function and structure of these cytoskeletal networks comes from research on proliferative cell types. However, in recent years, researchers have begun to show that there are important cardiomyocyte-specific functions of the cytoskeleton. Here we will discuss the current state of cytoskeletal biology in cardiomyocytes, as well as research from other cell types, that together suggest there is a wealth of knowledge on cardiac health and disease waiting to be uncovered through exploration of the complex signaling networks of cardiomyocyte non-sarcomeric cytoskeletal proteins.
1. Introduction
The vast majority of studies on the mammalian cytoskeleton are conducted on proliferative cell types due to the key roles of the cytoskeleton in cell division and migration. As such, studies on non-contractile cytoskeletal proteins such as microtubules, intermediate filaments, and cytoplasmic actin comprise a relatively small subset of cardiovascular research. Cardiomyocytes stand apart from other cells in that they experience constant cyclical contractile stress, despite having incredibly low replicative capacity. Furthermore, cardiomyocytes are densely packed with sarcomeres, mitochondria, nuclei, T-tubules, and the sarcoplasmic reticulum, leaving only a small portion of the cell’s volume for cytoskeletal proteins [1]. Such tight packing of organelles and myofibrils would then ostensibly require exquisite architectural and structural organization. It thus stands to reason that the cardiomyocyte cytoskeleton may function or is regulated differently to the cytoskeletons of the neuronal and proliferative cell types from which we have gleaned most of our knowledge of cytoskeletal proteins. From the current body of literature, it is evident that the non-sarcomeric cardiomyocyte cytoskeleton is critical for contractile function, tension sensing, and signal transduction. Although sarcomeric actin functions with myosin to generate muscle contractile force, neither myofibrillar protein extends all the way to the intercalated discs (ICDs) and costameres at the ends of the cardiomyocytes. Instead cytoplasmic actin, microtubules, and intermediate filaments act as bridges between the costameres and ICDs at membranes and the sarcomeres (Fig. 1). Cytoskeletal networks thus take on crucial roles in electrical and mechanical coupling in cardiomyocytes, facilitating inter- and intracellular signal transduction. With costameres and ICDs acting as hubs for all three of the cytoskeletal networks, it is easy to envision copious crosstalk between these proteins in cardiomyocytes. Indeed, the communication between actin and microtubules has been known for nearly 50 years, since researchers found in 1970 that the development of actinbased protrusions in fibroblasts depend on an intact microtubule network [2]. Throughout this review we will highlight research that suggests the cytoskeletal networks within cardiomyocytes are no less interdependent. Due to the intricacies of these networks, the dysregulation and dysfunction of cytoskeletal proteins can have dire consequences for heart function and culminate in various cardiomyopathies. Altogether, this review will discuss the current knowledge of how cytoplasmic actin, microtubules, and intermediate filaments affect healthy and diseased cardiac function (Table 1). Where there is a lack of cardiac-specific research on these proteins, we will discuss findings from skeletal muscle- and non-muscle-based studies.
Fig. 1.
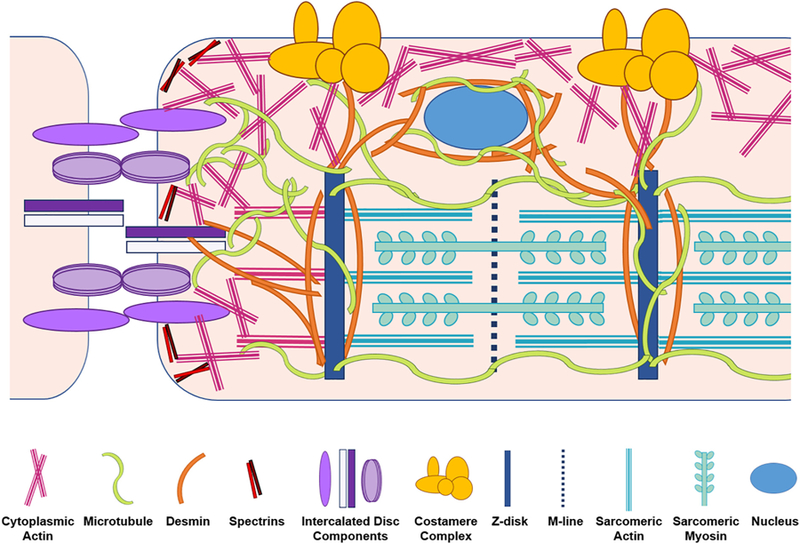
Interactions and localizations of the major cytoskeletal proteins with other structures in the cardiomyocytes. Non-sarcomeric actin, microtubules, and desmin intermediate filaments all serve as linkages running from sarcomeric structures to intercalated discs and costameres. Microtubules and desmin also link nuclei to the sarcomere.
Table 1.
A summary of cytoskeletal proteins discussed in this review along with their cardiomyocyte-specific functions, genetic manipulations, and roles in heart disease. Several of the proteins lack full characterization of cardiomyocyte-specific functions.
| Protein | Gene | Cardiomyocyte-specific Functions | Genetic Manipulations | Roles in Cardiovascular Disease |
|---|---|---|---|---|
| β-actin | ACTB | Poorly characterized. | Overexpression causes formation of cardiomyocyte lamellipodia. Knockout is embryonically lethal. | Upregulated in response to right ventricular pressure overload. |
| γ-actin | ACTG1 | Poorly characterized. | Overexpression causes formation of cardiomyocyte lamellipodia. Knockout mice are viable. | |
| Mena | ENAH | Maintains integrity of ICDs. | Overexpression or deletion in mice causes cardiomyopathy. | |
| Cofilin-2 | CFL2 | Regulates F-actin depolymerization. | Inactive form found in protein aggregates from human dilated cardiomyopathy samples. | |
| αII-spectrin | SPTAN1 | Organizes/stabilizes connections between sarcomeres and membrane. | Knockout causes cardiac defects and embryonic lethality. | |
| βII-spectrin | SPTBM2 | Interacts with ankyrin-B to localize key ion transporters. | Knockout is embryonically lethal and deficiency causes cardiac dysfunction. | Causes congenital ateriovenous malformations in Beckwith-Wiedmann syndrome. |
| βIV-spectrin | SPTBN4 | Crosslinks actin and interacts with ankyrin-G to facilitate CamKII localization to ICDs. | Targets STAT3 to ICD during pressure overload and is reduced during human heart failure. | |
| Ankyrin-B | ANK2 | Localizes various ion channels to regulate excitation-contraction coupling. | Heterozygous null mice have calcium handling defects and arrhythmias. | Human mutations cause “ankyrin-B syndrome” disorders. Downregulated after myocardial infarction and in atrial fibrillation. |
| Ankyrin-G | ANK3 | Localizes Nav1.5 to ICDs. | Loss of its interaction with Nav1.5 associated with Brugada syndrome. | |
| 4.1 proteins | EPB41 | Links spectrin-actin complexes. 4.1R isoform specifically colocalizes with Na+/K +−ATPase and NCX1. | 4.1R knockout reduces cardiac NCX1 current density and results in bradycardia. | |
| Non-muscle myosin II | MYH10 | Aids in tension-sensing processes. | NMII-B knockout is embryonically lethal and cardiac-specific deletion causes hypertrophy. | NMII deposition increases at costameres in diseased hearts. |
| Myosin VI | MYO6 | Poorly characterized. | Mutations linked with familial hypertrophic cardiomyopathy. | |
| Myosin XVIIIb | MYO18B | Poorly characterized. | Knockout is embryonically lethal due to significant cardiac defects. | |
| Obscurin | OBSCN | Regulates cardiomyocyte adhesion and cell size. | Mutations associated with dilated and hypertrophic cardiomyopathies. | |
| α/β-tubulin | TUBA/TUBB | Regulates cardiomyocyte response to shear stress. Provides track for delivery of cargo to intracellular compartments. | Heavily detyrosinated in failing human hearts | |
| Microtubule associated protein 4 | MAP4 | Regulates the density of the microtubule network and regulates speed of cargoes moving along microtubules. | Overexpression increases microtubule network density and reduces transport of mRNA along microtubules. | Phosphorylation changes and enhanced expression occur during cardiac hypertrophy. |
| Centromere-binding protein F | CENPF | Contributes to proper ICD structure and microtubule architecture. | Cardiomyocyte-specific deletion causes progressive dilated cardiomyopathy and reduced cardiomyocyte coupling. | Single nucleotide polymorphism associated with systolic heart failure. |
2. Cytoplasmic actin
There are several isoforms of actin that reside in cardiomyocytes. Within the sarcomere, thin filaments are primarily comprised of α-actin, and actively contribute to contraction via their interaction with myosin-based thick filaments. Three isoforms of α-actin exist: cardiac, skeletal, and smooth muscle. Outside of the sarcomere, actin microfilaments are made up of the cytoplasmic β and γ isoforms that provide a subcortical support network. A smooth muscle isoform of γ-actin also exists, but is only found in enteric smooth muscle. Knockout mice of any one actin isoform exhibit compensatory upregulation of other actin isoforms. Yet because the functional roles of each actin are different, these knockout mice still display phenotypes, most of which include cardiac dysfunction [3]. Between all mammalian actin isoforms there is at least 93% sequence identity. Given the sequence similarities and compensation, in addition to the fact that the vast majority of actin within a cardiomyocyte is cardiac α-actin, it has been technically challenging to observe and study cytoplasmic actin in these cells. As such, the most common reagent used to observe actin is fluorescently labeled phalloidin, a toxin that binds to all filamentous (F) actin, sarcomeric and cytoplasmic, making it difficult to delineate changes between the two within cardiomyocytes. It has also been challenging to define the differences in function between cytoplasmic β and γ isoforms themselves due to the fact that they differ by only 4 amino acids in their N-termini. Despite this, recent evidence suggests that there are differences in polymerization rates and stability between these two isoforms [4].
Actg1 (encoding γ-actin) knockout mice are viable and can survive into adulthood, but large numbers die as newborns due to developmental delays. Actg1−/−EIIa-cre conditional knockouts do survive beyond development, but then develop centronuclear myopathy [5,6]. Intriguingly, γ-actin can incorporate into sarcomeric thin filaments of skeletal muscle without significant contractile detriment in a γ-actin overexpression mouse model. However, the overexpression of γ-actin in skeletal α-actin null mice was not able to rescue their lethality [7]. Actb−/− mice display embryonic lethality [8,9], and this is attributed to the fact that Actb−/− mouse embryonic fibroblasts (MEFs) have severely hampered cellular growth and migration, which are both critical for development. This study also revealed that the loss of β-actin, unlike the loss of γ-actin, significantly decreases the ratio of globular (G) actin to F actin in MEFs, such that less actin is free to be polymerized into new filaments [10]. To-date, there has been no exploration of the functional or organizational effects when these proteins are genetically manipulated specifically in the heart in vivo.
2.1. Function and localization of cytoplasmic actin in cardiomyocytes
Studies on the localization and function of the two cytoplasmic actin isoforms in striated muscle are scarce in general. In skeletal muscle, a portion of the γ-actin population tethers at costameres, colocalizing perfectly with dystrophin [11]. In both cardiac and skeletal muscle cells, costameres are essentially specialized versions of the focal adhesion structures that non-contractile cells use as nodal points to transmit and receive tension-based signals. The protein networks of these structures span the sarcolemmal membrane to the Z-disks that provide structural integrity to the sarcomere [12]. γ-actin also co-localizes with mitochondria in diaphragm muscle [13]. Using an adenoviral construct to overexpress GFP-tagged β-actin in adult cardiomyocytes, it was found that this isoform colocalizes with α-actinin at the Z-disks and vinculin at costameres and ICDs [14]. Unlike costameres, ICDs are only present in cardiomyocytes, not skeletal myocytes, and serve as cell-to-cell contacts for tension-, calcium-, and growth-based signaling between cardiomyocytes [15].
Another group showed that transfection of neonatal rat ventricular myocytes (NRVMs) with fluorescently-tagged β-actin caused in-corporation of this isoform into sarcomeres like α-actin [16]. Given that the overexpression of γ-actin caused its aberrant sarcomeric incorporation [7], adenoviral overexpression may also cause mislocalization of β-actin. β-actin is thought to be the dominant isoform in the family that controls cell protrusions and migration [10]. Nonetheless, one study found that overexpression of either β- or γ-actin causes “extreme induction of filopodia” in adult rat cardiomyocytes [17]. Such leading edge dynamics are not well studied in the heart; however, in a mouse model of enhanced YAP activity that exhibits increased cardiac proliferation, cardiomyocytes also display cellular protrusions further demonstrating that this classic function of the non-sarcomeric actin cytoskeleton does exist in cardiomyocytes. ChIP-seq analysis of these YAP hearts revealed an upregulation of genes that control actin cytoskeletal dynamics and actin polymerization, including those encoding mammalian enabled (Mena), δ sarcoglycan—a key costameric protein, and the actin polymerization factor formin 2 [18].
The formation of actin microfilaments that make up the cytoskeleton and, within myocytes, the thin filaments of the sarcomere, occurs through polymerization of G actin into F actin. ATP-bound G actin monomers are added to the “barbed” or plus ends of filaments at a rate much faster than that which they are added to the “pointed” or minus ends [19]. The ratio of G to F actin in a cell, and even the micro-environments at the plus and minus ends of a filament, is critical to the rate of polymerization. A higher concentration of G actin at one end of the filament will facilitate polymerization at that location. An actin filament is said to be in a “treadmilling” state when the concentration of monomers is lower at the minus end, causing disassembly, and higher at the plus end, leading to polymerization [20]. Profilin is essential in essential in the polymerization process. It binds G actin and aids in the conversion of ADP to ATP and dissociates from actin upon ATP hy-drolysis that occurs as actin monomers are added to filaments [19].
Nucleation and elongation promoting factors (NEPFs) bind profilin-G-actin complexes to promote polymerization. These include Ena/VASP proteins (of which Mena is a member), Wickett-Aldrich syndrome proteins, and formins. Mena was originally found to localize with actin stress fibers in fibroblasts and act as a ligand for profilin [21]. Either overexpression or deletion of Mena in mice leads to cardiomyopathy [22,23]. In cardiomyocytes, Mena associates with αII-spectrin, another actin binding protein, and localizes to Z-discs and ICDs. Moreover, when Mena is genetically deleted along with VASP (vasodilator-stimulated phosphoprotein), the cardiac β-actin network becomes disrupted [24]. Formins are another family of NEPFs consisting of 15 mammalian proteins that bind the profilin-G-actin complex and accelerate ATP hydrolysis using the free energy to recruit and add more profilin-G-actin complexes to the filament [25]. As of late, there has been a wealth of studies on the roles of formins and profilin in sarcomeric thin filament maturation. Despite this, only one study has suggested a possible role for formins in regulating the cardiomyocyte cytoplasmic actin cytoskeleton, pointing to the localization of the formin FHOD1 at costameres and ICDs [26]. Interestingly, several in vitro studies have shown both formins and profilin can bind and affect the function of microtubules. Formins can stabilize microtubules and direct their orientation in migratory cells [27]. In the case of mDia1, this formin can actually cause bundling of actin and alignment of micro-tubules with those actin bundles [28]. Profilin can also interact with microtubules, such that when actin polymerization is blocked by cytochalasin D treatment, thus increasing the pool of free profilin, these proteins shift their localization to microtubules [29]. On the other side of actin polymerization are the actin depolymerizing factor (ADF)/cofilin group of proteins which associate with ADP-bound F-actin and sever the filaments. The resulting actin fragments can either be depolymerized or nucleated with the help of ADF/cofilins. ADF has slightly lower actin nucleating abilities compared to cofilin-2 (found in myo-cytes) and cofilin-1 (in all other cell types) [30]. Altogether, however, there is an unmistakable lack of data on how cytoplasmic actin microfilaments are constructed and depolymerized in cardiomyocytes and how these processes may contribute to overall heart function.
2.2. Functions of cytoplasmic actin-associated proteins
We know much more about the cardiac-specific functions of various cytoplasmic actin accessory proteins and non-muscle myosins than the cytoplasmic actin filaments themselves. Linking of the cytoplasmic actin filaments to the sarcolemma and membrane proteins, which likely facilitates localization and retention of key signaling platforms, pre-dominantly occurs through families of adaptor cytoskeletal regulators such as spectrins, ankyrins, and 4.1 proteins. Spectrins are constituents of the cytoskeleton that play a key role in providing structural support for the sarcolemma and contribute to the localization and anchoring of key signaling complexes in the cytoplasmic actin cytoskeleton. As long as ago as 1983, spectrins were found to associate with γ-actin at myo-cyte costameres [31]. However, only in recent years have researchers begun to truly understand their function in the heart. Members of this family, of which there are two α-spectrin and five β-spectrin genes, associate with actin via recruitment of accessory capping proteins such as adducin and tropomodulin. β-spectrins can also directly interact with the sarcolemmal membrane via their C-terminal pleckstrin homology domains. In erythrocytes, spectrins are known to associate with desmin intermediate filaments [32], but this function has not yet been reported in myocytes.
At least three splice variants of αII-spectrin have been described in the heart [33,34], including a cardiomyocyte-specific variant (αII-cardi +) that appears to be developmentally regulated and localizes to the Z-disks [35]. αII-spectrin also localizes to T-tubules, sarcoplasmic reticulum, and ICDs [34,36,37]. It may additionally play a role in coordinating the organization of proteins in the sarcolemmal membrane such that they are better positioned for signaling with the sarcomere [38]. Loss of αII-spectrin has been shown to cause cardiac defects and results in embryonic lethality [39], revealing an essential develop-mental role in the heart. Comparatively, we have a better understanding of the role of β-spectrins in the heart. In adult cardiomyocytes, βII-spectrin interacts with ankyrin-B to localize and position key ion transporters such as the sodium/potassium ATPase (Na+/K+-ATPase) and sodium/calcium exchanger (NCX1) [40]. Accumulating evidence indicates an important role for βIV-spectrin in cardiomyocytes as well; Hund and colleagues demonstrated that βIV-spectrin interacts with ankyrin-G at the ICD and that this interaction facilitates the targeting of calcium/calmodulin-dependent kinase II (CamKII) to the ICD. This, in turn, was demonstrated to regulate Nav1.5-mediated inward Na+ currents [37].
Closely associated functionally with spectrins are the ankyrins, Ank1 (encoding ankyrin-R) [41], Ank2 (encoding ankyrin-B) [42], and Ank3 (encoding ankyrin-G) [43], all of which have been identified in cardi-omyocytes [44–46]. Although these three proteins share a high level of sequence similarity, studies from knockout mouse models have revealed that they serve distinct, and non-redundant functions linking various sarcolemmal proteins to the spectrinactin network. Loss of ankyrin-B is well-established in causing defects in cardiac calcium handling and arrhythmias [47]. There is unequivocal evidence of a critical role for ankyrin-G in properly localizing Nav1.5 in cardiomyocytes [48]. Indeed, ankyrin-G is key to the organization of the entire signaling platform that regulates Nav1.5 activity at the ICD; although βIV-spectrin is necessary for the targeting of CamKII to the signaling platform, it in turn requires ankyrin-G to be present at the ICD [49].
There is a second family, the 4.1 proteins, that serves as a link between the spectrinactin cytoskeletal complexes and membrane proteins, and have also been increasingly implicated in the regulation of Na+-signaling [50]. The family consists of four proteins: 4.1R, 4.1N, 4.1G and 4.1B. Expression of all but 4.1B have been confirmed in cardiomyocytes [51], and each has been shown to localize to the lateral sarcolemma, T-tubules, sarcoplasmic reticulum, and ICDs [51,52]. Research specifically on 4.1R shows that it co-localizes with Na+/K+-ATPase and NCX1, and most noticeably, sorts away from components of cell adhesion complexes [52]. Consistent with these findings, analysis of 4.1R knockout hearts revealed a reduction in NCX1 current density, prolongation of action potential duration, reduced Nav1.5-α expression, and bradycardia, but no perturbation of cell-cell adhesions [53]. The current interpretation is that 4.1 proteins associate with and regulate the localization and function of the same proteins as do the ankyrins but localize these proteins to distinct membrane and signaling domains. The reasons for this sorting, and its consequences for cardiac function and pathologies, remain to be elucidated.
Non-muscle myosins also associate with cytoplasmic actin micro-filaments, and these interactions are necessary in order to produce the contractile forces needed for cell polarity, migration, and adhesion [54]. The myosin II family consists of the classical myosin motors that reside within the sarcomere as well as three non-muscle myosins (NMIIs). Two NMIIs reside in the cardiomyocyte: NMII-B at Z-disks and ICDs and NMII-C at only ICDs [55]. Only NMII-B seems essential for cardiac development as whole-body knockouts of the protein are embryonically lethal, and cardiac-specific conditional knockouts develop hypertrophy stemming from ICD disruption [56,57]. A study of the association of NMIIs with actin isoforms revealed that NMIIs have two-to fourfold greater coupling efficiencies with β- and γ-actin compared to α-actin. NMIIs also have faster sliding velocities with β- and γ-actin filaments. Moreover, NMII-C has preferential association for β-actin over the γ isoform, whereas NMII-B has no predilection for either [58]. The differences in NMII associations with cytoplasmic actin isoforms, as well as their differential localization, may then point to different functional roles for β- and γ-actin in cardiomyocytes. Recently, it has been shown that NMII is critical to the tension-sensing capabilities of cardiomyocytes. The contractions produced by NMII are layered on top of the more rapid sarcomeric myosin-based contractions. Under normal conditions, these contractions result in oscillatory stretching that is integrated by the mechanosensitive costameric protein talin [59].
Several non-conventional myosins (i.e. members of myosin classes outside of the myosin II family), may be critical to cardiac function, though information on their functionality is scarce in the cardiomyo-cyte, if they are at all present. Some of the non-conventional myosins are thought to function as organelle transporters, but there is also a body of evidence describing their roles as tension sensors [60], which supports the NMII data discussed above. Myosin VI is a motor that moves towards the minus end of actin filaments (in contrast to all others which are plus-end directed [61]) and has many functions including organelle transport and cytoskeletal-based motility [62]. In skeletal muscle, it has been shown to localize to the sarcoplasmic reticulum [63]. Myosin VI is also well known to associate with the adaptor protein disabled 2 [64], a promoter of cardiomyocyte differentiation [65]. Conversely, myosin XVIIIb has been established as important in the heart, as embryonic lethality occurs in the knockout mouse due to significant cardiac defects [66]. These mice display sarcomeric disarray, but little else has been determined about the cardiac-specific function of myosin XVIIIb and how it may interact with the cytoskeleton. Interestingly, it has been shown to translocate to the nucleus upon differentiation in skeletal muscle suggesting a possible role in muscle-specific gene transcription [67]. There also is a growing population of studies in non-cardiac cells reporting the ability of the non-conventional myosins to interact with microtubules [60]. Myosin V and VI depletion in neurons increases mitochondrial transport along microtubules [68], and myosin X binds microtubules and helps to position centrosomes during cell division [69]. These data reinforce the notion that there are many layers of interconnectedness between the different cytoskeletal systems.
2.3. Importance of cytoplasmic actin and associated proteins in disease processes
Because there has not been much research on the basic biology of cytoplasmic actin in cardiomyocytes, the data on how β- and γ-actin change during cardiovascular disease or even contribute to cardiomyopathies is extremely scarce. One study has shown that within feline cardiomyocytes, β-actin is upregulated in response to right ventricular pressure overload. Conversely, immuno-neutralization of this isoform reduces cardiac contractility [14]. Beyond this study, we do not know how cytoplasmic actin changes localization or function either in human heart disease or other experimental animal models of cardio-vascular disease. Evidence suggests that at least one NEPF, cofilin-2, is important in human dilated cardiomyopathy. Cofilin-2 localizes to the cardiac protein aggregates that develop in a subset of patients with dilated hearts. In the aggregates, cofilin-2 was phosphorylated, rendering it inactive. This was validated in vitro with the overexpression of a phosphomimetic version of cofilin-2 in NRVMs, which caused the development of actin-based stress fibers [70]. Stress fibers have similarly been observed in developing chick hearts [71]. Actin stress fibers are important in mechanotransduction and localize to focal adhesions [72], which supports the role of cytoplasmic actin in tension development and sensing in cardiomyocytes when stress fibers are present. As mentioned above, NMII actions on cytoplasmic actin are important for normal tension-sensing in cardiomyocytes. However, in diseased hearts, deposition of NMII increases at costameres and contributes to aberrant tension sensing and cardiac remodeling [59]. Additionally, mutations in non-conventional myosin VI are linked with familial hypertrophic cardiomyopathy [73].
The largest body of information on the roles of cytoplasmic actin or accessory proteins in heart disease has resulted from studies on spectrins and ankyrins. Many alterations in β-spectrin function have been outlined in human disease and failure. In Beckwith-Wiedmann syndrome, a stem cell disorder, reduced βII-spectrin levels are associated with congenital arteriovenous malformations [74,75]. Studies from mouse models have elucidated the mechanisms of such changes, finding that βII-spectrin expression is essential for normal embryonic development [76]. βII-spectrin deficiency in the heart also causes sinus node dysfunction, cytoskeletal remodeling, ryanodine receptor mislocalization, arrhythmias and more rapid progression to heart failure in response to increased afterload [40]. Beyond βII-spectrin, βIV-spectrin has been shown to regulate the targeting of STAT3 to the ICD in response to pressure overload [77] and has reduced expression during heart failure in humans [78].
Ankyrins also contribute to several forms of cardiovascular disease. Human Ank2 mutations have been associated with a spectrum of traits called “ankyrin-B syndrome,” which confers both arrhythmias and electrical conduction issues as well as a predisposition to aging-related disorders [79,80]. Ankyrin-B has been suggested to be cardioprotective [81,82] and is downregulated in a canine model of myocardial infarction [83] and in human atrial fibrillation [84]. Brugada syndrome, a potentially fatal arrhythmic condition caused by altered Nav1.5 function, has been linked to disruptions in the interaction between this sodium channel and ankyrin-G [85,86]. Ankyrin-G also plays a role in the broader maintenance of normal cardiomyocyte structure and adaptive remodeling in response to pressure overload [87].
3. Microtubules
Microtubules are hollow barrel-shaped structures that are comprised of heterodimers of two tubulin isoforms, α and β. These structures maintain polarity with β subunits oriented towards the plus end and α subunits directed towards the minus end. The loss and addition of tubulin subunits occurs more rapidly at the plus than the minus end. Polymerization is powered by the hydrolysis of GTP bound to β-tubulin that allows the addition of new subunits. When in the GDP-bound state, subunits can more easily fall away and the microtubule depolymerizes. This property of “dynamic instability” allows for rapid growing and shrinking of microtubules [88]. Along with this characteristic of microtubules, they can also be cut by severing enzymes such as katanin, spastin, and fidgetin [89]. Of note, α- or β- tubulin each have several isotypes, the numbers of which vary across mammals. The isotype that becomes incorporated in a heterodimer can change the stability and function of microtubules, and changes in expression of isotypes are associated with the development of cancer and developmental disorders [90,91]. Although nothing is known about the specificity of α-tubulin isotypes in the heart, the isotypes of β-tubulin have distinct localizations within adult rat cardiomyocytes: βI-tubulin has scattered cytoplasmic distribution, βII associates with the outer mitochondrial membrane, βIII localizes to Z-disks, and βIV comprises the cytoskeletal microtubule network [92].
A third isoform, γ-tubulin, is a centrosomal protein that localizes to microtubule organizing centers (MTOCs) during cell division [93]. Our knowledge of MTOCs and γ-tubulin functions is largely the result of studies in mitotic cells. However, cardiomyocytes lose their replicative capacity shortly after birth, begging the question of what happens to γ-tubulin in the postnatal heart. During the postnatal period, MTOCs in mouse cardiomyocytes move towards the perinuclear space as centrosomes dissolve, promoting cell cycle arrest. Cardiomyocytes of newts and zebrafish, both species with enhanced cardiac regenerative capacity, instead maintain their centrosome integrity into adulthood [94]. Despite loss of centrosome integrity shortly after birth, there still may be roles for these structures and their associated proteins in the adult mammalian cardiomyocyte.
As previously mentioned, most of what we know about micro-tubules stems from studies in mitotic cells, where the major function of these structures is to facilitate the segregation of sister chromatids. With microtubules freed from these duties in post-mitotic cardiomyocytes (and with their MTOCs moved to the nuclear envelope) they are likely to have evolved a whole host of novel functions and duties in these cells, many of which remain to be explored. In line with this, it has been posited that the disbanding of the centrosome in the postnatal mammalian cardiomyocyte is partially due to a need for microtubules to serve as support structures to handle the increased hemodynamic stress on the adult heart [94].
3.1. Function and post-translational modifications of microtubules
Indeed, microtubules are important regulators of a cardiomyocyte’s ability to respond to shear stress, providing resistance against compression [95]. The shock absorbing function of microtubules is important to crossbridge cycling, as microtubule depolymerization with colchicine increases myocyte shortening and relaxation [96]. The in-creases in cardiomyocyte shortening and relaxation that occur after depolymerizing microtubules with colchicine can be mimicked by blocking the detyrosination of microtubules [96], which occurs by cleavage of the C-terminal tyrosine on α-tubulin [97]. More heavily detyrosinated microtubules buckle and form sinusoidal patterns during contraction, whereas tyrosinated microtubules are straighter and more permissive to the sarcomere shortening. Detyrosinated microtubules also more readily associated with desmin intermediate filaments, suggesting that these two networks are interdependent in the mechanical modulation of cardiomyocyte stiffness [98].
Microtubules are subject to other post translational modifications such as phosphorylation, acetylation, and glutamylation. The majority of these occur on the C-terminal tails of α- and β-tubulin leading these regions to be considered “hotspots for chemical diversity” [99]. All of these modifications, in concert with detyrosination usually occur on fully polymerized microtubules. One exception is the phosphorylation of β-tubulin in an unincorporated dimer, which prevents its inclusion in microtubules. However, this phosphorylation occurs by cyclin-dependent kinase 1 during mitosis, so it is unknown if this phosphorylation event has a function in the non-dividing adult cardiomyocyte [100]. The phosphorylation of polymerized microtubules in cardiomyocytes is also not well understood, though it is known from other studies that microtubules become phosphorylated on the inside of their barrel-like structures [101].
Acetylation is unlike other tubulin modifications in that it occurs on the N-terminus of α-tubulin at lysine 40 [102]. Strikingly, the acetylation of microtubules can be controlled by the activity of formins, discussed above as NEPFs that enhance actin polymerization. Re-searchers found that expression of 13 of 15 mammalian formins were able to induce microtubule acetylation in HeLa and NIH3T3 cells [103]. This ability stems from the binding of G-actin monomers to the myocardin-related transcription factor (MRTF), which regulates the transcription of α-tubulin acetyltransferase 1 (αTAT1). Thus, when formins are abundant, the G-actin pool is small such that monomers are less likely to bind and inhibit MRTF. This results in the presence of more αTAT1 to enhance the acetylation of microtubules [104].
Glutamylation is another modification that can affect the functionality of microtubules, wherein a single molecule or chain of glutamate is added to the c-terminal tails of either α- or β-tubulin. The number of glutamates added to a tubulin subunit regulates the ability of spastin to sever the microtubule. The addition of chains of more than three to eight glutamates inhibits spastin activity, and can actually signal across adjacent microtubules to further inhibit severing [105]. To our knowledge, there is currently no literature on the function of spastin or microtubule glutamylation in cardiomyocytes. In addition to their ability to change the overall architecture of microtubules, many of the posttranslational modifications discussed above can disrupt or enhance the ability of motor proteins to move along microtubule, drastically changing the microtubule-based signaling network [101].
Microtubules also aid in tension sensing and contractility through their interaction with other components of cytoskeletal networks. Studies in recent years have begun to describe the interconnectedness between microtubules and intermediate filament and cytoplasmic actin networks in cardiomyocytes. However, research from the skeletal muscle field highlights just how many layers there can be to the crosstalk between microtubules and other cytoskeletal elements. In skeletal muscle, dystrophin can bind to and prevent the polymerization of microtubules, despite being known as a protein that classically tethers cytoplasmic actin filaments to costameres [106]. In this subcortical costameric space, microtubules also interact with ankyrin-B. Ankyrin-B is a nexus that connects the sarcomere to microtubules via its association with dynactin-4 (a cofactor for microtubule motor dynein 1) [107], in addition to its previously mentioned role of linking to cytoplasmic actin via βII-spectrin.
Ankyrin-B, is in turn, targeted to the M-line in myocytes by the giant protein obscurin [108]. In skeletal muscle of obscurin-null mice, the M-line localization of ankyrin-B was lost, and this was accompanied by impaired microtubule and dystrophin organization, suggesting that obscurin is a nodal protein for crosstalk between cytoplasmic actin and microtubule networks [109]. Another layer to this interconnectedness was found through studies on the striated muscle of Caenorhabditis elegans. In these cells, obscurin binds to a ubiquitin ligase adaptor protein, MEL-26. This interaction decreases degradation of the micro-tubule-severing protein, katanin, and leads to microtubule disarray and concomitant sarcomeric dysfunction [110]. In cardiomyocytes, the communication between microtubules and other cytoskeletal elements is likely to be no less complicated. Indeed, we know that obscurin is also critical in the heart, as several small isoforms of obscurin have been recently identified as mediators of cardiomyocyte adhesion and hypertrophy [111], and osbcurin mutations can lead to cardiomyopathies [112]. Furthermore, the body of data body of data in skeletal muscle discussed here suggests that it is important to look beyond the established functions of cytoskeletal proteins and consider the possibility of non-canonical cytoskeletal interactions and modifications in the heart.
3.2. Roles of microtubule-based transport in cardiac function
Proteins such as kinesins and dyneins serve as motors to transport cargo along microtubules. Kinesins are responsible for anterograde trafficking of cargoes, while dyneins move cargo in the retrograde direction. Segregation of separate pools of kinesins and dyneins occur with cyclical mechanical stress that aligns microtubules parallel to the axis of stretch in NRVMs. This is accompanied by the accumulation of dyneins near the nucleus and MTOC, while kinesins move towards the cell periphery [113]. Beyond this there is a paucity of cardiomyocyte-specific information on the function of these proteins, their interaction with various adaptors, and how they all work in concert to transport cargo. Inhibition of class I histone deacetylases (HDACs) was recently shown to stimulate the expression of Jun amino-terminal kinase (JNK)-interacting protein-1 in cardiomyocytes. This protein, JIP1, known to regulate the transport of autophagosomes and mitochondria, in turn stimulates expression of KIF5A, a kinesin heavy chain family member, and its association to polymerized microtubules [114]. On the minus end of microtubules, dynein directly interacts with Kv1.5 voltage-gated potassium channels, serving as an internalization mechanism for the channel [115].
The transport of mRNA throughout the cell occurs along the microtubule network. Microtubule associated protein 4 (MAP4) decreases the speed by which mRNAs are moved along microtubules in cardio-myocytes [1]. MAP4 may have a more general role in striated muscle cell differentiation as a novel isoform of MAP4, oMAP4, was found to be critical for the elongation of myotubes during skeletal muscle cell development [116]. Microtubules are also responsible for the transportation and alignment of mitochondria. Nearly 30 years ago, β-tubulin was shown to label the outer mitochondrial membrane in cardiomyocytes [117]. Much more recently, a 20 kilodalton isoform of connexin 43 (GJA1–20k), which localizes to the interface between mitochondria and microtubules, has been shown to facilitate transport of mitochondria along microtubules [118]. Although those experiments were conducted in various non-contractile cell types, in cardiomyocytes specifically, GJA1–20k also stabilizes and increases the length of actin microfilaments. This stabilization is, in turn, needed for the proper orientation of microtubules towards the ends of cardiomyocytes so that full-length connexin 43 may be delivered to ICDs [119]. This is the first evidence of the ability of cytoplasmic actin’s ability to provide a roadmap for microtubules in the heart, and further highlights the interdependence of the different cytoskeletal networks. Notably, it is not only polymerized microtubules that can affect mitochondrial dynamics. The voltage dependent anion channel on the outer mitochondrial membrane can be blocked by the binding of dimeric tubulin, leading to a decrease in the respiration rate of heart mitochondria [120].
3.3. Microtubule dysfunction in cardiovascular disease
It is well-documented the cardiomyocyte microtubules and their associated proteins become dysregulated during various forms of cardiomyopathy. Perhaps the best studied incidence of microtubule dysregulation in the heart is the proliferation and densification of microtubule networks during hypertrophic cardiomyopathy [121]. Microtubule networks have been examined during other cardiomyopathies, such as tachycardia-induced dilated cardiomyopathy, but no changes in overall microtubule network density occurred [122]. During hypertrophic cardiomyopathy, the massive upregulation in microtubule network density is accompanied by increased expression of the microtubule binding protein MAP4 [123]. It is the overexpression of MAP4 that causes the polymerization of microtubule dimers and not simply overexpression of tubulin isoforms βI or βIV [123]. Other groups have found that MAP4 itself becomes de-phosphorylated at serines 924 and 1056 during cardiac pathological hypertrophy, contributing to the densification of the microtubule network [124]. Contrastingly, another study reported increased phosphorylation of serines 737 and 760 during pressure overload surgery in mice [125]. Clearly the phosphorylation of MAP4 is a complicated picture, and more studies are needed to understand the relative contributions of these phosphorylation sites to MAP4 and microtubule biology.
The posttranslational modifications of microtubules are also implicated in the development of cardiovascular diseases. The higher the degree to which microtubules are detyrosinated leads to increased myocyte stiffness and dysfunction [98]. Indeed, heavily detyrosinated microtubules are found in myocytes from failing human hearts, and suppressing this detyrosination allows for the restoration of contractility [126]. Changes in the acetylation of tubulin occur during several other forms of cardiomyopathy. In a mouse model of desmin-related cardiomyopathy that features toxic protein aggregates, α-tu-bulin is hyperacetylated. Increasing expression of HDAC6, which deacetylates α-tubulin, enhanced aggregate formation, suggesting that higher levels of acetylated of α-tubulin are protective in desmin-related cardiomyopathy. Supporting this was the finding that HDAC6 inhibition enhanced autophagic flux in these hearts. The authors also observed increases in acetylated tubulin after voluntary exercise in mice, providing a physiological role for this modification in the heart [127]. Samples from human atrial fibrillation patients also displayed activation of HDAC6 as well as decreased acetylated and total tubulin levels [128]. The acetylation of tubulin is also thought to be a characteristic of diabetic cardiomyopathy. Cardiomyocyte microtubules from rats with streptozotocin-induced diabetes have enhanced stability and contractile dysfunction [129]. More recent data show that experimental induction of diabetes causes a downregulation of the deacetylase SIRT2, and an accompanying upregulation of acetylated α-tubulin, promoting the overall stability of cardiomyocyte microtubules [130].
Dysregulation of microtubules can lead to impaired trafficking of proteins that are essential for the proper function of cardiomyocyte structures and organelles. The remodeling of microtubules under heart failure conditions has been reported to cause mislocalization of mitochondria and concurrent aberrant intracellular calcium release [131]. Ischemia-reperfusion injury also disrupts the association of βII-tubulin and mitochondria, causing the βII isoform to colocalize with α-actinin at Z-disks [132]. Stabilization of microtubules via paclitaxel treatment can rescue the mislocalization of connexin 43 that occurs in a mouse model of laminopathy that develops dilated cardiomyopathy [133]. However, microtubule stabilization does not always improve function (as seen in diabetes-induced cardiac dysfunction)—treatment with taxol can cause t-tubule remodeling and sarcoplasmic reticulum dysfunction due to aberrant localization of junctophilin 2 [134]. This mislocalization, nevertheless, can be rescued by expression of a dominant negative form of kinesin 1, suggesting that both microtubules and their motor proteins are critical to calcium handling in cardiomyocytes [134].
Microtubule-associated proteins that are traditionally described as having mitotic functions may also play important roles in heart disease, despite the fact that adult cardiomyocytes are terminally differentiated. Centromere protein F (CENP-F) interacts with microtubules and is key in directing the alignment of chromosomes during cell division [135]. Nonetheless, cardiomyocyte-specific genetic ablation of CENP-F causes progressive dilated cardiomyopathy. Underlying the dilation are a reduction in the number of ICDs, resulting in reduced coupling of cardiomyocytes, as well as an overall lack of proper microtubule architecture [136]. The same group has also identified a single nucleotide polymorphism in CENP-F that is associated with the development of systolic heart failure [137].
4. Intermediate filaments
This third group of cytoskeletal proteins is also critical to the maintenance of proper function in cardiomyocytes. Lamin and desmin intermediate filaments are by far the most well-studied of these proteins due to their roles in desminopathies and laminopathies in humans. Recently though, research in the field has expanded to show important roles in the cardiomyocyte for lesser known intermediate filament proteins such as synemin and syncoilin [138,139]. We have highlighted some of the important interactions between intermediate filaments and the microtubule and cytoplasmic actin networks in the above sections; however, we have chosen not to include an in-depth exploration of intermediate filaments here due to the recent publication of an excellent and thorough review by the Capetanaki group on the cardiac-specific functions of these cytoskeletal proteins [140].
5. Conclusions
Due to the complex nature of the cytoskeleton, it is difficult to cover every aspect of these structures, their function, and their associated proteins and motors in one review. However, we hope that we have highlighted some of the exciting new research emerging from the field of cardiomyocyte cytoskeletal dynamics. We have also attempted to underscore just how little is understood about cytoplasmic actin and microtubules in cardiomyocytes in comparison to other fields. This review hopefully has shed light on the abundant opportunities for further exploration that can lead to a more complete understanding of the likely specialized function of cytoskeletal networks in the unique cardiomyocyte.
Acknowledgements
We’d like to thank Demetria Fischesser for her expert editorial advice.
Funding
KMG is supported by a National Institutes of Health grant (F32HL138747) and JWM is supported by an American Heart Association postdoctoral fellowship (17POST33630095).
Footnotes
Disclosures
There are no conflicts of interest to disclose.
References
- [1].Scholz D, McDermott P, Garnovskaya M, Gallien TN, Huettelmaier S, DeRienzo C, Cooper G.t., Microtubule-associated protein-4 (MAP-4) inhibits microtubule-dependent distribution of mRNA in isolated neonatal cardiocytes, Cardiovasc. Res 71 (3) (2006) 506–516. [DOI] [PubMed] [Google Scholar]
- [2].Vasiliev JM, Gelfand IM, Domnina LV, Ivanova OY, Komm SG, Olshevskaja LV, Effect of colcemid on the locomotory behaviour of fibroblasts, J. Embryol. Exp. Morpholog 24 (3) (1970) 625–640. [PubMed] [Google Scholar]
- [3].Perrin BJ, Ervasti JM, The actin gene family: function follows isoform, Cytoskeleton (Hoboken) 67 (10) (2010) 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bergeron SE, Zhu M, Thiem SM, Friderici KH, Rubenstein PA, Ion-dependent polymerization differences between mammalian beta- and gamma-nonmuscle actin isoforms, J. Biol. Chem 285 (21) (2010) 16087–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Belyantseva IA, Perrin BJ, Sonnemann KJ, Zhu M, Stepanyan R, McGee J, Frolenkov GI, Walsh EJ, Friderici KH, Friedman TB, Ervasti JM, Gammaactin is required for cytoskeletal maintenance but not development, Proc. Natl. Acad. Sci. U. S. A 106 (24) (2009) 9703–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bunnell TM, Ervasti JM, Delayed embryonic development and impaired cell growth and survival in Actg1 null mice, Cytoskeleton 67 (9) (2010) 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jaeger MA, Sonnemann KJ, Fitzsimons DP, Prins KW, Ervasti JM, Context-dependent functional substitution of alpha-skeletal actin by gamma-cytoplasmic actin, FASEB J 23 (7) (2009) 2205–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shawlot W, Deng JM, Fohn LE, Behringer RR, Restricted beta-galactosidase expression of a hygromycin-lacZ gene targeted to the beta-actin locus and embryonic lethality of beta-actin mutant mice, Transgenic Res 7 (2) (1998) 95–103. [DOI] [PubMed] [Google Scholar]
- [9].Shmerling D, Danzer CP, Mao X, Boisclair J, Haffner M, Lemaistre M, Schuler V, Kaeslin E, Korn R, Burki K, Ledermann B, Kinzel B, Muller M, Strong and ubiquitous expression of transgenes targeted into the beta-actin locus by Cre/lox cassette replacement, Genesis 42 (4) (2005) 229–235. [DOI] [PubMed] [Google Scholar]
- [10].Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM, Beta-actin specifically controls cell growth, migration, and the G-actin pool, Mol. Biol. Cell 22 (21) (2011) 4047–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rybakova IN, Patel JR, Ervasti JM, The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin, J. Cell Biol 150 (5) (2000) 1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ervasti JM, Costameres: the Achilles’ heel of herculean muscle, J. Biol. Chem 278 (16) (2003) 13591–13594. [DOI] [PubMed] [Google Scholar]
- [13].Pardo JV, Pittenger MF, Craig SW, Subcellular sorting of Isoactins - selective association of gamma-actin with skeletal-muscle mitochondria, Cell 32 (4) (1983) 1093–1103. [DOI] [PubMed] [Google Scholar]
- [14].Balasubramanian S, Mani SK, Kasiganesan H, Baicu CC, Kuppuswamy D, Hypertrophic stimulation increases beta-actin dynamics in adult feline cardio-myocytes, PLoS One 5 (7) (2010) e11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bennett PM, Riding the waves of the intercalated disc of the heart, Biophys. Rev 10 (4) (2018) 955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Skwarek-Maruszewska A, Hotulainen P, Mattila PK, Lappalainen P, Contractility-dependent actin dynamics in cardiomyocyte sarcomeres, J. Cell Sci 122 (2009) 2119–2126 Pt 12. [DOI] [PubMed] [Google Scholar]
- [17].vonArx P, Bantle S, Soldati T, Perriard JC, Dominant negative effect of cytoplasmic actin isoproteins on cardiomyocyte cytoarchitecture and function, J. Cell Biol 131 (6) (1995) 1759–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Morikawa Y, Zhang M, Heallen T, Leach J, Tao G, Xiao Y, Bai Y, Li W, Willerson JT, Martin JF, Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in hippo-deficient mice, Sci. Signal 8 (375) (2015) ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Carlier MF, Pantaloni D, Control of actin dynamics in cell motility, J. Mol. Biol 269 (4) (1997) 459–467. [DOI] [PubMed] [Google Scholar]
- [20].Wegner A, Head to tail polymerization of actin, J. Mol. Biol 108 (1) (1976) 139–150. [DOI] [PubMed] [Google Scholar]
- [21].Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P, Mena, a relative of VASP and Drosophila enabled, is implicated in the control of microfilament dynamics, Cell 87 (2) (1996) 227–239. [DOI] [PubMed] [Google Scholar]
- [22].Aguilar F, Belmonte SL, Ram R, Noujaim SF, Dunaevsky O, Protack TL, Jalife J, Todd Massey H, Gertler FB, Blaxall BC, Mammalian enabled (Mena) is a critical regulator of cardiac function, Am. J. Physiol. Heart Circ. Physiol 300 (5) (2011) H1841–H1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Belmonte SL, Ram R, Mickelsen DM, Gertler FB, Blaxall BC, Cardiac over-expression of Mammalian enabled (Mena) exacerbates heart failure in mice, Am. J. Physiol. Heart Circ. Physiol 305 (6) (2013) H875–H884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Benz PM, Merkel CJ, Offner K, Abesser M, Ullrich M, Fischer T, Bayer B, Wagner H, Gambaryan S, Ursitti JA, Adham IM, Linke WA, Feller SM, Fleming I, Renne T, Frantz S, Unger A, Schuh K, Mena/VASP and alphaII-Spectrin complexes regulate cytoplasmic actin networks in cardiomyocytes and protect from conduction abnormalities and dilated cardiomyopathy, Cell Commun. Signal 11 (2013) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, Carlier MF, Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis, Cell 119 (3) (2004) 419–429. [DOI] [PubMed] [Google Scholar]
- [26].Al Haj A, Mazur AJ, Radaszkiewicz K, Radaszkiewicz T, Makowiecka A, Stopschinski BE, Schonichen A, Geyer M, Mannherz HG, Distribution of for-mins in cardiac muscle: FHOD1 is a component of intercalated discs and costa-meres, Eur. J. Cell Biol 94 (2) (2015) 101–113. [DOI] [PubMed] [Google Scholar]
- [27].Bartolini F, Gundersen GG, Formins and microtubules, Biochim. Biophys. Acta 1803 (2) (2010) 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S, Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1, Nat. Cell Biol 3 (1) (2001) 8–14. [DOI] [PubMed] [Google Scholar]
- [29].Nejedla M, Sadi S, Sulimenko V, de Almeida FN, Blom H, Draber P, Aspenstrom P, Karlsson R, Profilin connects actin assembly with microtubule dynamics, Mol. Biol. Cell 27 (15) (2016) 2381–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bamburg JR, Bernstein BW, Roles of ADF/cofilin in actin polymerization and beyond, F1000 Biol Rep 2 (2010) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Craig SW, Pardo JV, Gamma actin, spectrin, and intermediate filament proteins colocalize with vinculin at costameres, myofibril-to-sarcolemma attachment sites, Cell Motil. Cytoskeleton 3 (5–6) (1983) 449–462. [DOI] [PubMed] [Google Scholar]
- [32].Langley RC, Cohen CM, Association of Spectrin with Desmin intermediate filaments, J. Cell. Biochem 30 (2) (1986) 101–109. [DOI] [PubMed] [Google Scholar]
- [33].Cianci CD, Zhang Z, Pradhan D, Morrow JS, Brain and muscle express a unique alternative transcript of alphaII spectrin, Biochemistry 38 (48) (1999) 15721–15730. [DOI] [PubMed] [Google Scholar]
- [34].Baines AJ, Pinder JC, The spectrin-associated cytoskeleton in mammalian heart, Front. Biosci 10 (2005) 3020–3033. [DOI] [PubMed] [Google Scholar]
- [35].Zhang Y, Resneck WG, Lee PC, Randall WR, Bloch RJ, Ursitti JA, Characterization and expression of a heart-selective alternatively spliced variant of alpha II-spectrin, cardi+, during development in the rat, J. Mol. Cell. Cardiol 48 (6) (2010) 1050–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bennett V, Healy J, Membrane domains based on ankyrin and spectrin associated with cell-cell interactions, Cold Spring Harb. Perspect. Biol 1 (6) (2009) a003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ, A. beta (IV)-spectrin/CaMKII signaling complex is essential for membrane excit-ability in mice, J. Clin. Invest 120 (10) (2010) 3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bennett PM, Baines AJ, Lecomte MC, Maggs AM, Pinder JC, Not just a plasma membrane protein: in cardiac muscle cells alpha-II spectrin also shows a close association with myofibrils, J Muscle Res. Cel. M 25 (2) (2004) 119–126. [DOI] [PubMed] [Google Scholar]
- [39].Stankewich MC, Cianci CD, Stabach PR, Ji L, Nath A, Morrow JS, Cell organization, growth, and neural and cardiac development require alphaII-spectrin, J. Cell Sci 124 (2011) 3956–3966 Pt 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Smith SA, Sturm AC, Curran J, Kline CF, Little SC, Bonilla IM, Long VP, Makara M, Polina I, Hughes LD, Webb TR, Wei Z, Wright P, Voigt N, Bhakta D, Spoonamore KG, Zhang C, Weiss R, Binkley PF, Janssen PM, Kilic A, Higgins RS, Sun M, Ma J, Dobrev D, Zhang M, Carnes CA, Vatta M, Rasband MN, Hund TJ, Mohler PJ, Dysfunction in the betaII spectrin-dependent cytoskeleton underlies human arrhythmia, Circulation 131 (8) (2015) 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bennett V, Stenbuck PJ, Identification and partial purification of ankyrin, the high affinity membrane attachment site for human erythrocyte spectrin, J. Biol. Chem 254 (7) (1979) 2533–2541. [PubMed] [Google Scholar]
- [42].Davis JQ, Bennett V, Brain ankyrin. A membrane-associated protein with binding sites for spectrin, tubulin, and the cytoplasmic domain of the erythrocyte anion channel, J. Biol. Chem 259 (21) (1984) 13550–13559. [PubMed] [Google Scholar]
- [43].Kordeli E, Lambert S, Bennett V, AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier, J. Biol. Chem 270 (5) (1995) 2352–2359. [DOI] [PubMed] [Google Scholar]
- [44].Li ZP, Burke EP, Frank JS, Bennett V, Philipson KD, The cardiac Na+-Ca2+ exchanger binds to the cytoskeletal protein ankyrin, J. Biol. Chem 268 (16) (1993) 11489–11491. [PubMed] [Google Scholar]
- [45].Yoshida K, Harada K, Proteolysis of erythrocyte-type and brain-type ankyrins in rat heart after postischemic reperfusion, J. Biochem 122 (2) (1997) 279–285. [DOI] [PubMed] [Google Scholar]
- [46].Peters LL, John KM, Lu FM, Eicher EM, Higgins A, Yialamas M, Turtzo LC, Otsuka AJ, Lux SE, Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain, J. Cell Biol 130 (2) (1995) 313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT, Bennett V, A cardiac arrhythmia syndrome caused by loss of ankyrin-B function, P Natl Acad Sci USA 101 (24) (2004) 9137–9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA, Shibata E, Anderson ME, Mohler PJ, Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway, J. Cell Biol 180 (1) (2008) 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Makara MA, Curran J, Little SC, Musa H, Polina I, Smith SA, Wright PJ, Unudurthi SD, Snyder J, Bennett V, Hund TJ, Mohler PJ, Ankyrin-G co-ordinates intercalated disc signaling platform to regulate cardiac excitability in vivo, Circ. Res 115 (11) (2014) 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Baines AJ, Bennett PM, Carter EW, Terracciano C, Protein 4.1 and the control of ion channels, Blood Cells Mol. Dis 42 (3) (2009) 211–215. [DOI] [PubMed] [Google Scholar]
- [51].Taylor-Harris PM, Keating LA, Maggs AM, Phillips GW, Birks EJ, Franklin RC, Yacoub MH, Baines AJ, Pinder JC, Cardiac muscle cell cytoskeletal protein 4.1: analysis of transcripts and subcellular location–relevance to membrane integrity, microstructure, and possible role in heart failure, Mamm. Genome 16 (3) (2005) 137–151. [DOI] [PubMed] [Google Scholar]
- [52].Pinder JC, Taylor-Harris PM, Bennett PM, Carter E, Hayes NV, King MD, Holt MR, Maggs AM, Gascard P, Baines AJ, Isoforms of protein 4.1 are differentially distributed in heart muscle cells: relation of 4.1R and 4.1G to components of the Ca2+ homeostasis system, Exp. Cell Res 318 (13) (2012) 1467–1479. [DOI] [PubMed] [Google Scholar]
- [53].Stagg MA, Carter E, Sohrabi N, Siedlecka U, Soppa GK, Mead F, Mohandas N, Taylor-Harris P, Baines A, Bennett P, Yacoub MH, Pinder JC, Terracciano CM, Cytoskeletal protein 4.1R affects repolarization and regulates calcium handling in the heart, Circ. Res 103 (8) (2008) 855–863. [DOI] [PubMed] [Google Scholar]
- [54].Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR, Non-muscle myosin II takes centre stage in cell adhesion and migration, Nat. Rev. Mol. Cell Biol 10 (11) (2009) 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Takeda K, Yu ZX, Qian S, Chin TK, Adelstein RS, Ferrans VJ, Nonmuscle myosin II localizes to the Z-lines and intercalated discs of cardiac muscle and to the Z-lines of skeletal muscle, Cell Motil. Cytoskeleton 46 (1) (2000) 59–68. [DOI] [PubMed] [Google Scholar]
- [56].Ma X, Adelstein RS, In vivo studies on nonmuscle myosin II expression and function in heart development, Front. Biosci. (Landmark Ed) 17 (2012) 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ma X, Takeda K, Singh A, Yu ZX, Zerfas P, Blount A, Liu C, Towbin JA, Schneider MD, Adelstein RS, Wei Q, Conditional ablation of nonmuscle myosin II-B delineates heart defects in adult mice, Circ. Res 105 (11) (2009) 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Muller M, Diensthuber RP, Chizhov I, Claus P, Heissler SM, Preller M, Taft MH, Manstein DJ, Distinct functional interactions between actin isoforms and nonsarcomeric myosins, PLoS One 8 (7) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pandey P, Hawkes W, Hu J, Megone WV, Gautrot J, Anilkumar N, Zhang M, Hirvonen L, Cox S, Ehler E, Hone J, Sheetz M, Iskratsch T, Cardiomyocytes sense matrix rigidity through a combination of muscle and non-muscle myosin contractions, Dev. Cell 44 (3) (2018) 326–336 (e3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Woolner S, Bement WM, Unconventional myosins acting unconventionally, Trends Cell Biol 19 (6) (2009) 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, Carragher BI, Milligan RA, Sweeney HL, Myosin VI is an actin-based motor that moves backwards, Nature 401 (6752) (1999) 505–508. [DOI] [PubMed] [Google Scholar]
- [62].Buss F, Kendrick-Jones J, Multifunctional myosin VI has a multitude of cargoes, Proc. Natl. Acad. Sci. U. S. A 108 (15) (2011) 5927–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Karolczak J, Sobczak M, Majewski L, Yeghiazaryan M, Jakubiec-Puka A, Ehler E, Slawinska U, Wilczynski GM, Redowicz MJ, Myosin VI in skeletal muscle: its localization in the sarcoplasmic reticulum, neuromuscular junction and muscle nuclei, Histochem. Cell Biol 139 (6) (2013) 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Morris SM, Arden SD, Roberts RC, Kendrick-Jones J, Cooper JA, Luzio JP, Buss F, Myosin VI binds to and localises with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton, Traffic 3 (5) (2002) 331–341. [DOI] [PubMed] [Google Scholar]
- [65].Hofsteen P, Robitaille AM, Chapman DP, Moon RT, Murry CE, Quantitative proteomics identify DAB2 as a cardiac developmental regulator that inhibits WNT/beta-catenin signaling, P Natl Acad Sci USA 113 (4) (2016) 1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ajima R, Akazawa H, Kodama M, Takeshita F, Otsuka A, Kohno T, Komuro I, Ochiya T, Yokota J, Deficiency of Myo18B in mice results in embryonic lethality with cardiac myofibrillar aberrations, Genes Cells 13 (10) (2008) 987–999. [DOI] [PubMed] [Google Scholar]
- [67].Salamon M, Millino C, Raffaello A, Mongillo M, Sandri C, Bean C, Negrisolo E, Pallavicini A, Valle G, Zaccolo M, Schiaffino S, Lanfranchi G, Human MYO18B, a novel unconventional myosin heavy chain expressed in striated muscles moves into the myonuclei upon differentiation, J. Mol. Biol 326 (1) (2003) 137–149. [DOI] [PubMed] [Google Scholar]
- [68].Pathak D, Sepp KJ, Hollenbeck PJ, Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria, J. Neurosci 30 (26) (2010) 8984–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kwon M, Bagonis M, Danuser G, Pellman D, Direct microtubule-binding by Myosin-10 orients centrosomes toward retraction Fibers and subcortical actin clouds, Dev. Cell 34 (3) (2015) 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Subramanian K, Gianni D, Balla C, Assenza GE, Joshi M, Semigran MJ, Macgillivray TE, Van Eyk JE, Agnetti G, Paolocci N, Bamburg JR, Agrawal PB, Del Monte F, Cofilin-2 phosphorylation and sequestration in myocardial aggregates: novel pathogenetic mechanisms for idiopathic dilated cardio-myopathy, J. Am. Coll. Cardiol 65 (12) (2015) 1199–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sumida H, Ashcraft RA Jr., Thompson RP, Cytoplasmic stress fibers in the developing heart, Anat. Rec 223 (1) (1989) 82–89. [DOI] [PubMed] [Google Scholar]
- [72].Livne A, Geiger B, The inner workings of stress fibers - from contractile machinery to focal adhesions and back, J. Cell Sci 129 (7) (2016) 1293–1304. [DOI] [PubMed] [Google Scholar]
- [73].Mohiddin SA, Ahmed ZM, Griffith AJ, Tripodi D, Friedman TB, Fananapazir L, Morell RJ, Novel association of hypertrophic cardiomyopathy, sensorineural deafness, and a mutation in unconventional myosin VI (MYO6), J. Med. Genet 41 (4) (2004) 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yao ZX, Jogunoori W, Choufani S, Rashid A, Blake T, Yao W, Kreishman P, Amin R, Sidawy AA, Evans SR, Finegold M, Reddy EP, Mishra B, Weksberg R, Kumar R, Mishra L, Epigenetic silencing of beta-spectrin, a TGF-beta signaling/scaffolding protein in a human cancer stem cell disorder: Beckwith-Wiedemann syndrome, J. Biol. Chem 285 (46) (2010) 36112–36120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Drut R, Quijano G, Altamirano ME, Jones MC, Maffessoli OB, Vascular mal-formation and choroid plexus adrenal heterotopia: new findings in Beckwith-Wiedemann syndrome? Fetal. Pediatr. Pathol 25 (4) (2006) 191–197. [DOI] [PubMed] [Google Scholar]
- [76].Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L, Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice, Science 299 (5606) (2003) 574–577. [DOI] [PubMed] [Google Scholar]
- [77].Unudurthi SD, Nassal D, Greer-Short A, Patel N, Howard T, Xu X, Onal B, Satroplus T, Hong D, Lane C, Dalic A, Koenig SN, Lehnig AC, Baer LA, Musa H, Stanford KI, Smith S, Mohler PJ, Hund TJ, BetaIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload, J. Clin. Invest 128 (12). (2018) 5561–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hund TJ, Snyder JS, Wu XQ, Glynn P, Koval OM, Onal B, Leymaster ND, Unudurthi SD, Curran J, Camardo C, Wright PJ, Binkley PF, Anderson ME, Mohler PJ, Beta(IV)-Spectrin regulates TREK-1 membrane targeting in the heart, Cardiovasc. Res 102 (1) (2014) 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mohler PJ, Ankyrins and human disease: what the electrophysiologist should know, J. Cardiovasc. Electrophysiol 17 (10) (2006) 1153–1159. [DOI] [PubMed] [Google Scholar]
- [80].Mohler PJ, Healy JA, Xue H, Puca AA, Kline CF, Allingham RR, Kranias EG, Rockman HA, Bennett V, Ankyrin-B syndrome: enhanced cardiac function balanced by risk of cardiac death and premature senescence, PLoS One 2 (10). (2007) e1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li J, Kline CF, Hund TJ, Anderson ME, Mohler PJ, Ankyrin-B regulates Kir6.2 membrane expression and function in heart, J. Biol. Chem 285 (37) (2010) 28723–28730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kashef F, Li J, Wright P, Snyder J, Suliman F, Kilic A, Higgins RS, Anderson ME, Binkley PF, Hund TJ, Mohler PJ, Ankyrin-B protein in heart failure: identification of a new component of metazoan cardioprotection, J. Biol. Chem 287 (36) (2012) 30268–30281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hund TJ, Wright PJ, Dun W, Snyder JS, Boyden PA, Mohler PJ, Regulation of the ankyrin-B-based targeting pathway following myocardial infarction, Cardiovasc. Res 81 (4) (2009) 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, Koval O, Li J, Gudmundsson H, Gumina RJ, Karck M, Schott JJ, Probst V, Le Marec H, Anderson ME, Dobrev D, Wehrens XH, Mohler PJ, Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation, Circulation 124 (11). (2011) 1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ, Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families, Circulation 102 (20) (2000) 2509–2515. [DOI] [PubMed] [Google Scholar]
- [86].Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V, Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes, Proc. Natl. Acad. Sci. U. S. A 101 (50) (2004) 17533–17538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Makara MA, Curran J, Lubbers ER, Murphy NP, Little SC, Musa H, Smith SA, Unudurthi SD, Rajaram MVS, Janssen PML, Boyden PA, Bradley EA, Hund TJ, Mohler PJ, Novel mechanistic roles for Ankyrin-G in cardiac Remodeling and heart failure, JACC Basic Trans. Sci 3 (5) (2018) 675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Walker RA, O’Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP, Salmon ED, Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies, J. Cell Biol 107 (4) (1988) 1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].McNally FJ, Roll-Mecak A, Microtubule-severing enzymes: from cellular functions to molecular mechanism, J. Cell Biol 217 (12) (2018) 4057–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Belvindrah R, Natarajan K, Shabajee P, Bruel-Jungerman E, Bernard J, Goutierre M, Moutkine I, Jaglin XH, Savariradjane M, Irinopoulou T, Poncer JC, Janke C, Francis F, Mutation of the alpha-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration, J. Cell Biol 216 (8) (2017) 2443–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Parker AL, Teo WS, McCarroll JA, Kavallaris M, An emerging role for tubulin Isotypes in modulating Cancer biology and chemotherapy resistance, Int. J. Mol. Sci 18 (7) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Guzun R, Karu-Varikmaa M, Gonzalez-Granillo M, Kuznetsov AV, Michel L, Cottet-Rousselle C, Saaremae M, Kaambre T, Metsis M, Grimm M, Auffray C, Saks V, Mitochondria-cytoskeleton interaction: distribution of beta-tubulins in cardiomyocytes and HL-1 cells, Biochim. Biophys. Acta 1807 (4) (2011) 458–469. [DOI] [PubMed] [Google Scholar]
- [93].Joshi HC, Gamma-tubulin: the hub of cellular microtubule assemblies, Bioessays 15 (10) (1993) 637–643. [DOI] [PubMed] [Google Scholar]
- [94].Zebrowski DC, Vergarajauregui S, Wu CC, Piatkowski T, Becker R, Leone M, Hirth S, Ricciardi F, Falk N, Giessl A, Just S, Braun T, Weidinger G, Engel FB, Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nishimura S, Nagai S, Katoh M, Yamashita H, Saeki Y, Okada J, Hisada T, Nagai R, Sugiura S, Microtubules modulate the stiffness of cardiomyocytes against shear stress, Circ. Res 98 (1) (2006) 81–87. [DOI] [PubMed] [Google Scholar]
- [96].Caporizzo MA, Chen CY, Salomon AK, Margulies KB, Prosser BL, Microtubules provide a viscoelastic resistance to Myocyte motion, Biophys. J 115 (9) (2018) 1796–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hallak ME, Rodriguez JA, Barra HS, Caputto R, Release of tyrosine from tyrosinated tubulin. Some common factors that affect this process and the assembly of tubulin, FEBS Lett 73 (2) (1977) 147–150. [DOI] [PubMed] [Google Scholar]
- [98].Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, Prosser BL, Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes, Science 352 (6284) (2016) aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Garnham CP, Roll-Mecak A, The chemical complexity of cellular microtubules: tubulin post-translational modification enzymes and their roles in tuning micro-tubule functions, Cytoskeleton 69 (7) (2012) 442–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Fourest-Lieuvin A, Peris L, Gache V, Garcia-Saez I, Juillan-Binard C, Lantez V, Job D, Microtubule regulation in mitosis: tubulin phosphorylation by the cyclin-dependent kinase Cdk1, Mol. Biol. Cell 17 (3) (2006) 1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wloga D, Gaertig J, Post-translational modifications of microtubules, J. Cell Sci 123 (2010) 3447–3455 Pt 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].L’Hernault SW, Rosenbaum JL, Chlamydomonas alpha-tubulin is post-translationally modified by acetylation on the epsilon-amino group of a lysine, Biochemistry 24 (2) (1985) 473–478. [DOI] [PubMed] [Google Scholar]
- [103].Thurston SF, Kulacz WA, Shaikh S, Lee JM, Copeland JW, The ability to induce microtubule acetylation is a general feature of formin proteins, PLoS One 7 (10) (2012) e48041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Fernandez-Barrera J, Alonso MA, Coordination of microtubule acetylation and the actin cytoskeleton by formins, Cell. Mol. Life Sci 75 (17) (2018) 3181–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Valenstein ML, Roll-Mecak A, Graded control of microtubule severing by tubulin Glutamylation, Cell 164 (5) (2016) 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Belanto JJ, Mader TL, Eckhoff MD, Strandjord DM, Banks GB, Gardner MK, Lowe DA, Ervasti JM, Microtubule binding distinguishes dystrophin from utrophin, Proc. Natl. Acad. Sci. U. S. A 111 (15) (2014) 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ayalon G, Hostettler JD, Hoffman J, Kizhatil K, Davis JQ, Bennett V, Ankyrin-B. interactions with spectrin and dynactin-4 are required for dystrophin-based protection of skeletal muscle from exercise injury, J. Biol. Chem 286 (9) (2011) 7370–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Cunha SR, Mohler PJ, Obscurin targets Ankyrin-B and protein phosphatase 2A to the cardiac M-line, J. Biol. Chem 283 (46) (2008) 31968–31980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Randazzo D, Giacomello E, Lorenzini S, Rossi D, Pierantozzi E, Blaauw B, Reggiani C, Lange S, Peter AK, Chen J, Sorrentino V, Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity, J. Cell Biol 200 (4) (2013) 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wilson KJ, Qadota H, Mains PE, Benian GM, UNC-89 (obscurin) binds to MEL-26, a BTB-domain protein, and affects the function of MEI-1 (katanin) in striated muscle of Caenorhabditis elegans, Mol. Biol. Cell 23 (14) (2012) 2623–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ackermann MA, King B, Lieberman NAP, Bobbili PJ, Rudloff M, Berndsen CE, Wright NT, Hecker PA, Kontrogianni-Konstantopoulos A, Novel obscurins mediate cardiomyocyte adhesion and size via the PI3K/AKT/mTOR signaling pathway, J. Mol. Cell. Cardiol 111 (2017) 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hu LYR, Ackermann MA, Hecker PA, Prosser BL, King B, O’Connell KA, Grogan A, Meyer LC, Berndsen CE, Wright NT, Lederer WJ, Kontrogianni-Konstantopoulos A, Deregulated Ca2+ cycling underlies the development of arrhythmia and heart disease due to mutant obscurin, Sci. Adv 3 (6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dhein S, Schreiber A, Steinbach S, Apel D, Salameh A, Schlegel F, Kostelka M, Dohmen PM, Mohr FW, Mechanical control of cell biology. Effects of cyclic mechanical stretch on cardiomyocyte cellular organization, Prog. Biophys. Mol. Biol 115 (2–3) (2014) 93–102. [DOI] [PubMed] [Google Scholar]
- [114].Blakeslee WW, Lin YH, Stratton MS, Tatman PD, Hu T, Ferguson BS, McKinsey TA, Class I HDACs control a JIP1-dependent pathway for kinesin-microtubule binding in cardiomyocytes, J. Mol. Cell. Cardiol 112 (2017) 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Loewen ME, Wang ZR, Eldstrom J, Zadeh AD, Khurana A, Steele DF, Fedida D, Shared requirement for dynein function and intact microtubule cytoskeleton for normal surface expression of cardiac potassium channels, Am. J. Physiol-Heart C 296 (1) (2009) H71–H83. [DOI] [PubMed] [Google Scholar]
- [116].Mogessie B, Roth D, Rahil Z, Straube A, A novel isoform of MAP4 organises the paraxial microtubule array required for muscle cell differentiation, Elife 4 (2015) e05697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Saetersdal T, Greve G, Dalen H, Associations between Beta-tubulin and mitochondria in adult isolated heart Myocytes as shown by immunofluorescence and Immunoelectron microscopy, Histochemistry 95 (1) (1990) 1–10. [DOI] [PubMed] [Google Scholar]
- [118].Fu Y, Zhang SS, Xiao SH, Basheer WA, Baum R, Epifantseva I, Hong TT, Shaw RM, Cx43 isoform GJA1–20k promotes microtubule dependent mitochondrial transport, Front. Physiol 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Basheer WA, Xiao S, Epifantseva I, Fu Y, Kleber AG, Hong T, Shaw RM, GJA1–20k arranges actin to guide Cx43 delivery to cardiac intercalated discs, Circ. Res 121 (9) (2017) 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL, Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration, P Natl Acad Sci USA 105 (48) (2008) 18746–18751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sato H, Nagai T, Kuppuswamy D, Narishige T, Koide M, Menick DR, Cooper G.t., Microtubule stabilization in pressure overload cardiac hypertrophy, J. Cell Biol 139 (4) (1997) 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Takahashi M, Tsutsui H, Kinugawa S, Igarashi-Saito K, Yamamoto S, Yamamoto M, Tagawa H, Imanaka-Yoshida K, Egashira K, Takeshita A, Role of microtubules in the contractile dysfunction of myocytes from tachycardia-induced dilated cardiomyopathy, J. Mol. Cell. Cardiol 30 (5) (1998) 1047–1057. [DOI] [PubMed] [Google Scholar]
- [123].Takahashi M, Shiraishi H, Ishibashi Y, Blade KL, McDermott PJ, Menick DR, Kuppuswamy D, Cooper G.t., Phenotypic consequences of beta1-tubulin expression and MAP4 decoration of microtubules in adult cardiocytes, Am. J. Physiol. Heart Circ. Physiol 285 (5) (2003) H2072–H2083. [DOI] [PubMed] [Google Scholar]
- [124].Cheng G, Takahashi M, Shunmugavel A, Wallenborn JG, DePaoli-Roach AA, Gergs U, Neumann J, Kuppuswamy D, Menick DR, Cooper G.t., Basis for MAP4 dephosphorylation-related microtubule network densification in pressure overload cardiac hypertrophy, J. Biol. Chem 285 (49) (2010) 38125–38140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Li L, Zhang Q, Zhang X, Zhang J, Wang X, Ren J, Jia J, Zhang D, Jiang X, Zhang J, Mei H, Chen B, Hu J, Huang Y, Microtubule associated protein 4 phosphorylation leads to pathological cardiac remodeling in mice, EBioMedicine 37 (2018) 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Chen CY, Caporizzo MA, Bedi K, Vite A, Bogush AI, Robison P, Heffler JG, Salomon AK, Kelly NA, Babu A, Morley MP, Margulies KB, Prosser BL, Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure, Nat. Med 24 (8) (2018) 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].McLendon PM, Ferguson BS, Osinska H, Bhuiyan MS, James J, McKinsey TA, Robbins J, Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy, Proc. Natl. Acad. Sci. U. S. A 111 (48) (2014) E5178–E5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ, Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation, Circulation 129 (3) (2014) 346–358. [DOI] [PubMed] [Google Scholar]
- [129].Howarth FC, Qureshi MA, White E, Calaghan SC, Cardiac microtubules are more resistant to chemical depolymerisation in streptozotocin-induced diabetes in the rat, Pflugers Arch 444 (3) (2002) 432–437. [DOI] [PubMed] [Google Scholar]
- [130].Yuan Q, Zhan L, Zhou QY, Zhang LL, Chen XM, Hu XM, Yuan XC, SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy, Eur. J. Pharmacol 764 (2015) 554–561. [DOI] [PubMed] [Google Scholar]
- [131].Miragoli M, Sanchez-Alonso JL, Bhargava A, Wright PT, Sikkel M, Schobesberger S, Diakonov I, Novak P, Castaldi A, Cattaneo P, Lyon AR, Lab MJ, Gorelik J, Microtubule-dependent mitochondria alignment regulates calcium release in response to Nanomechanical stimulus in heart Myocytes, Cell Rep 14 (1) (2016) 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Bagur R, Tanguy S, Foriel S, Grichine A, Sanchez C, Pernet-Gallay K, Kaambre T, Kuznetsov AV, Usson Y, Boucher F, Guzun R, The impact of cardiac ischemia/reperfusion on the mitochondria-cytoskeleton interactions, Biochim. Biophys. Acta 1862 (6) (2016) 1159–1171. [DOI] [PubMed] [Google Scholar]
- [133].Macquart C, Juttner R, Le Dour C, Chatzifrangkeskou M, Schmitt A, Gotthardt M, Bonne G, Muchir A, Microtubule cytoskeleton regulates connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene, Hum. Mol. Genet (2018) 1–10. [DOI] [PubMed] [Google Scholar]
- [134].Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, Yuan C, Kutschke W, Zimmerman K, Weiss RM, Wehrens XH, Hong J, Johnson FL, Santana LF, Anderson ME, Song LS, Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure, Circulation 129 (17) (2014) 1742–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Holt SV, Vergnolle MA, Hussein D, Wozniak MJ, Allan VJ, Taylor SS, Silencing Cenp-F weakens centromeric cohesion, prevents chromosome alignment and activates the spindle checkpoint, J. Cell Sci 118 (2005) 4889–4900 Pt 20. [DOI] [PubMed] [Google Scholar]
- [136].Dees E, Miller PM, Moynihan KL, Pooley RD, Hunt RP, Galindo CL, Rottman JN, Bader DM, Cardiac-specific deletion of the microtubule-binding protein CENP-F causes dilated cardiomyopathy, Dis. Model. Mech 5 (4) (2012) 468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Manalo A, Schroer AK, Fenix AM, Shancer Z, Coogan J, Brolsma T, Burnette DT, Merryman WD, Bader DM, Loss of CENP-F results in dilated cardiomyopathy with severe disruption of cardiac Myocyte architecture, Sci. Rep 8 (1) (2018) 7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Garcia-Pelagio KP, Chen L, Joca HC, Ward C, Jonathan Lederer W, Bloch RJ, Absence of synemin in mice causes structural and functional abnormalities in heart, J. Mol. Cell. Cardiol 114 (2018) 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Howman EV, Sullivan N, Poon EP, Britton JE, Hilton-Jones D, Davies KE, Syncoilin accumulation in two patients with desmin-related myopathy, Neuromuscul. Disord 13 (1) (2003) 42–48. [DOI] [PubMed] [Google Scholar]
- [140].Tsikitis M, Galata Z, Mavroidis M, Psarras S, Capetanaki Y, Intermediate filaments in cardiomyopathy, Biophys. Rev 10 (4) (2018) 1007–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]