

Summary
Whether cell forces or extracellular matrix (ECM) can impact genome integrity is largely unclear. Here, acute perturbations (~1 hr) to actomyosin stress or ECM elasticity cause rapid and reversible changes in lamin-A, DNA damage, and cell cycle. The findings are especially relevant to organs such as the heart because DNA damage permanently arrests cardiomyocyte proliferation shortly after birth and thereby eliminates regeneration after injury including heart attack. Embryonic hearts, differentiated iPS-cells, and various nonmuscle cell types all show that actomyosin-driven nuclear rupture causes cytoplasmic mis-localization of DNA repair factors and excess DNA damage. Binucleation and micronuclei increase as telomeres shorten, which all favor cell cycle arrest. Deficiencies in lamin-A and repair factors exacerbate these effects, but lamin-A-associated defects are rescued by repair factor overexpression and by contractility modulators in clinical trials. Contractile cells on stiff ECM normally exhibit low phosphorylation and slow degradation of lamin-A by matrix-metalloprotease-2 (MMP2), and inhibition of this lamin-A turnover and also actomyosin contractility is seen to minimize DNA damage. Lamin-A is thus stress-stabilized to mechano-protect the genome.
Keywords: mechanobiology, lamin-A, collagen-I, matrix, contractility, heart, development
Graphical Abstract
Introduction
Proliferation of many cell types slows dramatically shortly after birth and is absent in key adult tissues (Li, et al., 1996), which presents a major challenge to regeneration. Cell growth in culture is modulated by the stiffness of extracellular matrix (ECM) which generally promotes actomyosin stress (Engler, et al., 2006; Paszek, et al., 2005) and affects the conformation (Sawada, et al., 2006), post-translational modification (PTM) (Guilluy, et al., 2014), localization (Dupont, et al., 2011), and degradation (Dingal and Discher, 2014) of mechanosensitive proteins. Actomyosin links to ECM and to nuclei, such that rapid changes to ECM and tissue mechanics that result from chronic or acute injury or even drug therapies (e.g. cardiac arrest or cardiomyopathy treatment (Green, et al., 2016)) can in principle affect the nucleus (Takaki, et al., 2017; Wiggan, et al., 2017; Kanellos, et al., 2015) and perhaps the DNA within. DNA damage and telomere shortening are indeed well-documented in injuries that affect heart (Chang, et al., 2018; Higo, et al., 2017; Sharifi-Sanjani, et al., 2017; Oh, et al., 2003) as well as nonmuscle tissue – but DNA damage and repair are rarely studied in developing organs, and relationships to proliferation, ECM stiffness, and actomyosin stress are understudied.
DNA damage and senescence increase with many disease-linked mutations, including those in the nucleoskeletal protein lamin-A (LMNA) that forms a structural meshwork around chromatin (Turgay, et al., 2017; Shimi, et al., 2015; Gruenbaum, et al., 2005). LMNA deficiencies associate with elevated DNA damage (Graziano, et al., 2018; Liu, et al., 2005) and result in accelerated aging of stiff tissues similar to deficiencies in DNA repair factors (e.g. KU80) (Li, et al., 2007). Moreover, progeroid syndromes are caused only by mutations in LMNA and DNA repair factors, but LMNA’s primary function in development remains hotly debated (Burke and Stewart, 2013), with suggested roles in gene positioning and regulation (Harr, et al., 2015) seeming at odds with largely normal development of human and mouse mutants until weeks after birth. Surprisingly, senescence or apoptosis of cells with LMNA defects is rescued by culturing cells on almost any ECM (versus rigid plastic (de La Rosa, et al., 2013; Hernandez, et al., 2010)) and by treatment with at least one drug affecting both cytoskeleton and nucleo-cytoplasmic trafficking (Larrieu, et al., 2018; Larrieu, et al., 2014). Relationships between lamins, actomyosin stress, ECM mechanics, and DNA damage are nonetheless obscure – especially in tissues.
Embryonic hearts beat spontaneously for days after isolation from early chick embryos, and beating is acutely sensitive to myosin-II inhibition (Fig.1A) as well as enzymatic stiffening or softening of ECM (Majkut, et al., 2013). The latter studies reveal an optimal stiffness for beating that is likewise evident for cardiomyocytes (CMs) cultured on gels (Majkut, et al., 2013; Engler, et al., 2008; Jacot, et al., 2008). DNA damage is conceivably optimized in heart as it triggers a switch from proliferation to senescence in post-natal hearts (Puente, et al., 2014). DNA damage is also implicated in telomere attrition and binucleation of CMs that signal irreversible exit from cell cycle (Aix, et al., 2016). We postulated embryonic hearts with rapidly tunable mechanics could prove useful as a tissue model for clarifying protein-level mechanosensing mechanisms in vivo that could be studied thoroughly in vitro with many cell types.
Figure 1. Contractility or collagen perturbations result in rapid ~1h changes in LMNA, DNA damage, and cell cycle.
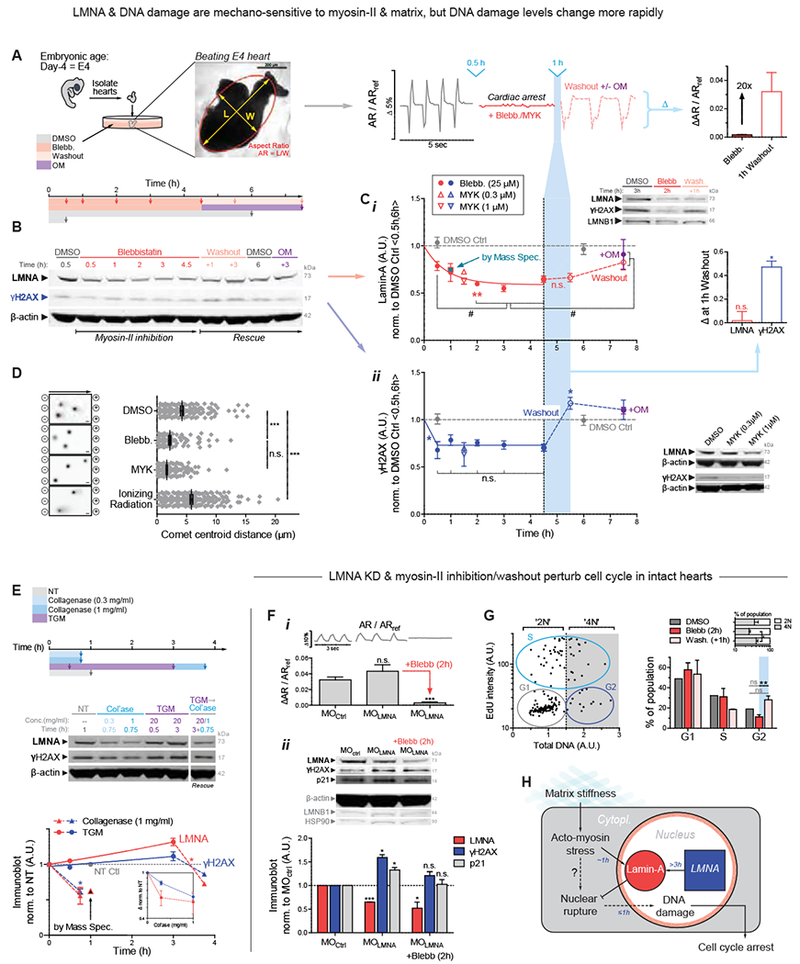
(A) Chick hearts from day 4 (E4) beat at 1-2 Hz for up to 5 d. Middle: Aspect ratio (AR) beating strain is arrested by myosin-II inhibition, but recovers with drug washout ± myosin-II activator, OM.
(B) Immunoblot of hearts inhibited for varying durations, followed by washout ± OM (8 hearts per lysate).
(c) (i) Immunoblot densitometry reveals LMNA decreases in ~45 min upon myosin-II inhibition, but LMNA recovery takes >3h upon washout. MS confirms 1h data with 29 LMNA-specific peptides, and inset shows LMNB1 remains unchanged. (ii) DNA damage γH2AX also decreases, but spikes upon blebb washout (5.5h, light blue). Bar graph of LMNA and γH2AX changes 1h post-washout. Bottom right immunoblot shows MYK treatment for 1.5 h also affects LMNA and γH2AX. One-way ANOVA (Dunnett’s test vs DMSO Ctrl avg of 0.5h,6h): *p<0.05, **<0.01; t-test (vs plateau average of 2, 3, 4.5h time points): #p<0.05. All error bars indicate ±SEM.
(D) Electrophoretic Comet assay of DNA damage validates γH2AX quantitation, with γ-irradiation control.
(E) Immunoblot of hearts treated with collagenase and/or transglutaminase (TGM) (8 hearts per lysate). Matrix digestion rapidly decreases LMNA and γH2AX, consistent with low stress; cross-linking has opposite and reversible effect by 3h (t-test: *p<0.05; superfluous blot lanes have been removed and indicated by a blank space).
(F) (i) Morpholino KD of LMNA (MOLMNA) does not affect beating versus MOCtrl, but (ii) MOLMNA increases DNA damage (γH2AX) and cell cycle inhibitor p21, unless rescued by myosin-II inhibition (i,ii). Two immunoblots (anti-LMNA, p21, β-actin) & (anti-LMNB1, γH2AX, HSP90) display proteins of interest above loading controls. (8 hearts per lysate). One-way ANOVA: *p<0.05, ***<0.001.
(G) (i) Representative plot of EdU vs DNA intensity for cell cycle phase (G1 vs S vs G2; or ‘2N’ vs ‘4N’). (ii) Blebb washout increases G2 and 4N cells (vs 2N). n>77 cells per cond.; t-test: **p<0.01.
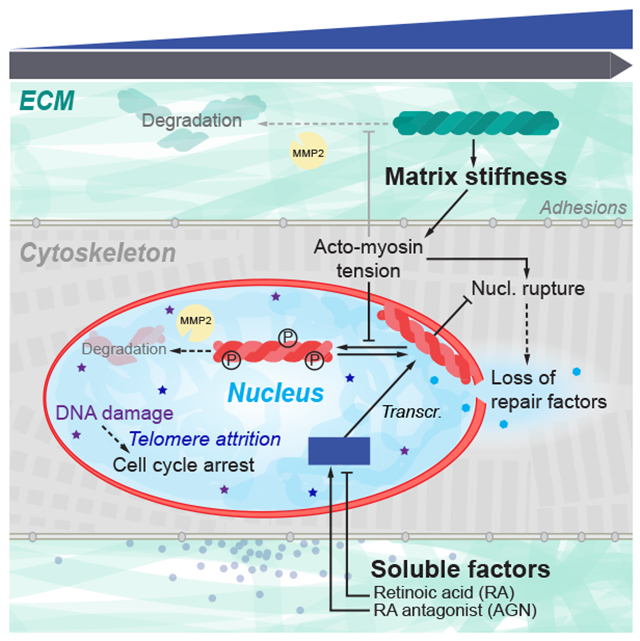
(H)Proposed mechanosensing pathway.
Although LMNA is reportedly ‘undetectable’ in early embryos (Stewart and Burke, 1987), it progressively accumulates in tissues such as heart, bone, and lung (Solovei, et al., 2013; Rober, et al., 1989; Lehner, et al., 1987) and is highest in adults within these mechanically stressed, stiff tissues that are collagen-rich (Swift, et al., 2013). LMNA deficiencies accordingly produce measurable defects weeks after birth in such tissues, including heart (Kubben, et al., 2011; Worman and Bonne, 2007). We therefore hypothesized that LMNA in normal embryos mechanosenses the earliest microenvironment in the first organ and increases not only to stiffen the nucleus (Osmanagic-Myers, et al., 2015; Dahl, et al., 2008; Pajerowski, et al., 2007) but also to regulate repair factors that confer resistance to both DNA damage and cell cycle arrest.
Results
Contractility and collagen perturbations rapidly impact LMNA and DNA damage in hearts
To first assess the consequences of cytoskeletal stress on nuclei in a tissue, we arrested embryonic hearts (day-4: E4) with reversible inhibitors of myosin-II (Fig.1A). Spontaneous beating of hearts stopped in minutes with blebbistatin (‘blebb’), but the heart re-started just as quickly upon washout of drug (Fig.1A right). An activator of cardiac myosin-II omecamtiv mecarbil (‘OM’) – in clinical trials for heart failure ( NCT02929329) – re-started the hearts reliably. Quantitative immunoblots and mass spectrometry (MS) measurements (Fig.1B, 1C-i, S1A&B) revealed a rapid decrease in LMNA (~45 min decay constant) while LMNB remained unchanged (Fig.1C-i, upper right inset, Fig.S1C), and similar results were evident for a cardiac myosin-II-specific inhibitor ‘MYK’ (mavacamten analog) – in clinical trials for hypertrophic cardiomyopathy ( NCT02842242). Despite rapid recovery of beating after drug washout (Fig.S1D), LMNA recovered more slowly (>3h), indicating slow synthesis.
DNA damage in post-natal development with LMNA deficiencies (Liu, et al., 2005) led us to hypothesize acute, embryonic changes in phospho-histone γH2AX, as a primary marker of DNA damage. A rapid decrease in DNA damage was surprising with myosin-II inhibition (Fig.1C-ii) given the decrease LMNA, but electrophoretic comet assay confirmed the γH2AX results (Fig.1D). It is useful to keep in mind that the heart beats reasonably well with LMNA deficiencies and mutations. Because blebbistatin washout recovers beating while LMNA remains low, we anticipated a large spike in DNA damage shortly after washout (Fig.1C-ii, right inset). LMNA and DNA damage eventually reached control levels (in ~hrs), but the spike highlights the disruptive effects of actomyosin stress on genome integrity.
Actomyosin contractility is generally downstream of ECM stiffness (Ulrich, et al., 2009; Engler, et al., 2006), including for immature cardiomyocytes (CMs) (Engler, et al., 2008; Jacot, et al., 2008). Acute perturbations of collagen matrix might therefore be expected to affect DNA damage. Collagenase treatment for 45 min indeed resulted in rapid decreases in DNA damage and LMNA (Fig.1E), consistent with rapid softening of E4 hearts (~50%) and weaker beating (Majkut, et al., 2013). Treatment with tissue transglutaminase (TGM), a cross-linker of ECM that stiffens heart and thereby increases basal tension (>2-fold in 2h (Majkut, et al., 2013)), increased γH2AX and LMNA (only after 3h) except when collagenase was subsequently added (Fig.1E). LMNA thus decreases quickly or increases slowly in response to changes in ECM stiffness or actomyosin tension, both of which appear to also affect DNA damage. Effects are also generally reversible.
DNA damage in LMNA-deficient hearts perturbs cell cycle and causes aberrant beating
Excess DNA damage has been shown to impact cell cycle in post-natal CMs (Puente, et al., 2014), and so we next sought to assess the biological consequences of DNA damage in LMNA-suppressed embryonic hearts. Morpholino-mediated knockdown of LMNA (‘MOLMNA’; ~40% KD in 24h) was achieved with no significant effect on contractile beating (Fig.1F-i, S1E). LMNA is thus not primarily upstream of beating, consistent with knockout mice (Singh, et al., 2013). Although past studies also suggest LMNA is not detectable in early embryonic hearts and is therefore dispensable (Solovei, et al., 2013; Stewart and Burke, 1987), morpholino-knockdown increased γH2AX in E4 hearts (Fig.1F-ii). Similar effects were seen by modest repression with retinoic acid (RA) of LMNA (−20% only after 3d) that acts transcriptionally (Swift, et al., 2013), while RA-antagonist (AGN) upregulated LMNA and suppressed DNA damage (Fig.S1F,G). Blebbistatin was sufficient to rescue the DNA damage in MOLMNA knockdown hearts, supporting our hypothesis that high contractility coupled to low LMNA causes excess damage.
Bona fide DNA damage can perturb cell cycle (Puente, et al., 2014); the cell cycle inhibitor p21 indeed trended with γH2AX in MO-treated hearts (Fig.1F-ii). EdU integration before and after blebbistatin/washout (per Fig.1A–C) further revealed an increase in cells in G2, with fewer cells in S, and an increase in 4N cells (Fig.1G). Given that acute DNA damage can inhibit cell cycle in developing heart and is somehow coupled to matrix-myosin-lamins (Fig.1H), we assessed DNA damage that was directly and abruptly increased with etoposide (4-fold, 1h) (Fig.2A). This inhibitor of replication/transcription quickly caused aberrant beating that could be reversed upon drug washout (Fig.2B); many cardiac laminopathies exhibit arrhythmias and broader conduction defects (Fatkin, et al., 1999). No such effects were seen with the same dose of H2O2 (for oxidative stress) relative to DMSO control hearts, unless H2O2 exposure was sustained for days. DNA damage can have multiple causes, ranging from drugs and oxidative stress to nucleases, but reversal of damage from natural sources or etoposide in hours (Fig.1F,G,2A) suggest an important role for DNA repair factors (Fig.2C).
Figure 2. DNA damage in embryonic hearts causes aberrant beating.
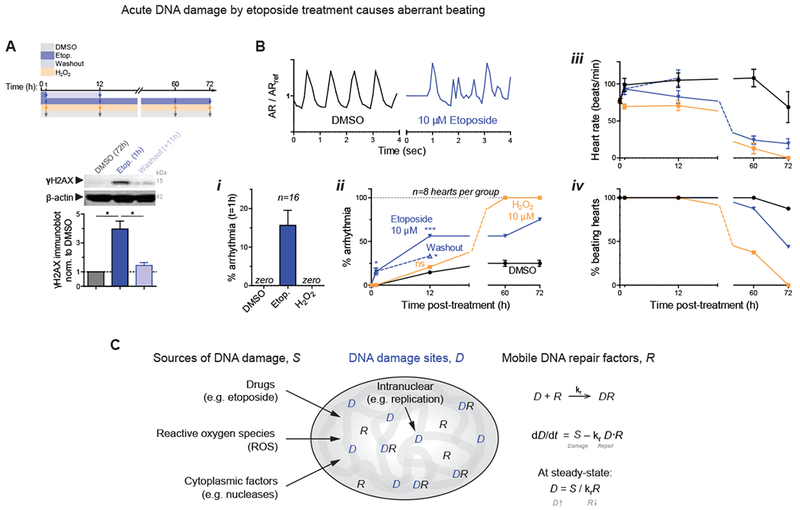
(A) 1h etoposide reversibly increases DNA damage 4-fold (8 hearts per grp). All error bars indicate ±SEM.
(B) Beating (top) after 1h of treatment is only affected by etoposide. (i,ii) %-hearts exhibiting arrhythmia. Oxidant H2O2 shows distinct kinetics. (iii,iv) Beating decreases and arrests compared to DMSO. 8~16 hearts per grp; t-test *p<0.05, **<0.01, ***<0.001.
(C) Schematic: the level of DNA damage that results from various sources depends also on levels of repair factors in the nucleus.
LMNA knockdown in intact hearts human iPS-CMs: nuclear rupture & loss of DNA repair
To address how increased mechanical stress causes DNA damage, the integrity of the nucleus was scrutinized in intact E5 hearts doubly transfected with DNA repair protein GFP-KU80 (XRCC5) and with a cytoplasmic protein that can bind nuclear DNA, mCherry-cGAS (cyclic GMP-AMP synthase (Raab, et al., 2016)) (Fig.3A-i). Cytoplasmic mis-localization of KU80 and concomitant formation of cGAS puncta at the nuclear periphery provided evidence of nuclear rupture in transfected hearts (Fig.3A-i, yellow arrow), and the fraction of such double-positive cells increased with MOLMNA knockdown unless blebbistatin was added (Fig.3A-ii). Myosin-II inhibition disrupted sarcomeres, which rapidly recovered upon drug washout (Fig.3A-iii), consistent with beating (Fig.1A–C); and mis-localization of KU80 and cGAS showed the same trends (Fig.3A-iV), without any apparent changes in cell viability (Fig.S2A-i). DNA damage again spiked upon blebbistatin washout and remained excessively high with addition of a nuclear import inhibitor (‘Imp-i’, ivermectin) that keeps KU80 mis-localized to cytoplasm (Fig.3B-i,ii, S2A-ii). LMNA immunofluorescence (IF) for MOLMNA and blebbistatin/washout hearts (Fig.3B-iii & C) confirmed immunoblot trends (Fig. 1C-i, F) and showed Imp-i kept LMNA levels low upon washout.
Figure 3. LMNA knockdown in intact hearts and in hiPS-CMs: nuclear envelope rupture and loss of DNA repair factors.
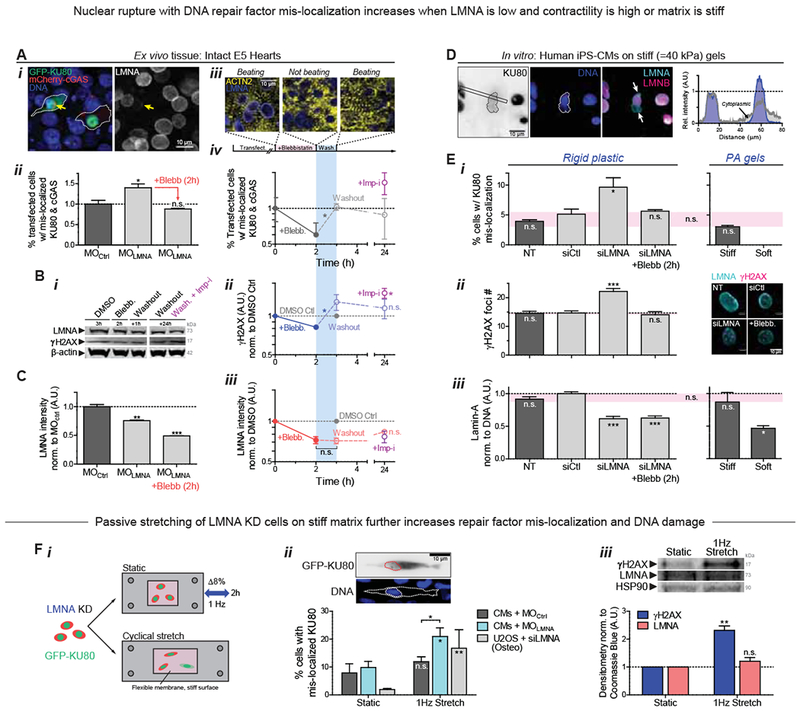
(A) (i) Images of E4 hearts transfected with GFP-KU80 and mCherry-cGAS. Arrow: cell with low LMNA and ruptured nucleus, with cytoplasmic mis-localization of GFP-KU80 and mCherry-cGAS puncta at nuclear envelope. (ii) LMNA KD increases mis-localized KU80 and cGAS but is rescued by myosin-II inhibition. (iii) Images of striation (α-actinin-2) before blebb, 2h after, and upon washout (1h). (iv) %-Ruptured nuclei (double +’s) decreases with myosin-II inhibition but increases upon washout. Mis-localized KU80 increases further with import inhibitor ivermectin, Imp-i. (n>111 double-transfected cells per cond’n; t-test: *p<0.05). All error bars indicate ±SEM.
(B) (i,ii) anti-γH2AX and (i,iii) anti-LMNA immunoblot of blebb-treated E4 hearts after washout ± Imp-i (8 hearts per lysate; t-test: *p<0.05; superfluous blot lanes have been removed and is indicated by a blank space).
(C) LMNA immunofluorescence with MOLMNA ± blebb. (n>139 cells per cond.).
(D) Human iPS-derived cardiomyocytes (hiPS-CMs) on stiff 40 kPa gels for 24h, showing cytoplasmic KU80 in cells with nuclei having LMNA-rich, LMNB-depleted blebs at high curvature sites (arrows).
(E) (i,ii) hiPS-CM on soft 0.3 kPa gels minimizes rupture. On rigid plastic, siLMNA KD (~40%, n>119 cells per cond. (iii)) increases mis-localized KU80 (i) and DNA damage by γH2AX foci (right images). (n>80 cells per cond.; Dunnett’s test vs NT Ctrl)
(F) (i) Passive cyclic stretch of LMNA-knockdown cells expressing GFP-KU80 (8% strain, 1Hz) increases (ii) %-rupture with cytoplasmic KU80 (upper inset) and (iii) γH2AX. (n>55 cells per cond.; t-test: #p<0.05, ##<0.01). (Unless noted, ; one-way ANOVA: *p<0.05, **<0.01, ***<0.001)
Human induced pluripotent stem cell-derived CMs (hiPS-CMs) cultured on soft or stiff gels with collagen-ligand constitute another model for clarifying mechanisms at the single cell level. Nuclei in hiPS-CMs ‘beat’ (Fig.S2B) and express low LMNA relative to some well-studied human cell lines (e.g. lung-derived A549 cells) and relative to mature heart which is much stiffer than embryos (Swift, et al., 2013). Cell morphologies and sarcomere striations resemble those of embryonic/fetal CMs. On very stiff gels (40 kPa) but not on gels as soft as embryos (0.3 kPa), hiPS-CMs exhibited cytoplasmic, mis-localized KU80 (Fig.3D), consistent with envelope rupture under high nuclear stress (Fig.1H). Nuclear blebs typical of ruptured nuclei formed at points of high curvature in such cells (Xia, et al., 2018), appearing rich in LMNA but depleted of LMNBs (Fig.3D). Stress-induced nuclear rupture was also imaged as rapid and stable accumulation of mCherry-cGAS upon nuclear probing with a high curvature Atomic Force Microscopy (AFM) tip (<1μm), using nano-Newton forces similar to those exerted by a cell’s actomyosin (Fig.S2C).
KD of LMNA (‘siLMNA’) doubled the fraction of cells with cytoplasmic KU80 (Fig.3E-i, S2D-i) and increased γH2AX foci (Fig.3E-ii & S2D-ii). These observations seem consistent with iPS-CM results for LMNA defects, which show nuclear blebbing (Lee, et al., 2017), focal loss of nuclear membrane (Siu, et al., 2012), cytoskeletal defects (Bollen, et al., 2017), and fibrosis, all of which increase with electrical stimulation of contraction. We found LMNA levels in hiPS-CMs exhibited a sensitivity to substrate stiffness similar to that in embryonic hearts (Fig.3E-iii), and we saw blebbistatin treated siLMNA cells on rigid plastic rescued nuclear rupture (Fig.3E-i) as well as DNA damage (Fig.3E-ii). Relaxation of rigidity-driven actomyosin stress can thus limit nuclear rupture.
Cytoplasmic mis-localization of KU80 is merely representative: repair factors 53BP1 and RPA2 also simultaneously mis-localized to the cytoplasm upon LMNA KD (Fig.S2E-i). Time-lapse images of GFP-53BP1 expressing cells further revealed mis-localization persists for hours (Fig.S2E-ii). However, total KU80 levels did not vary (Fig.S2F), suggesting nuclear retention is key.
To assess whether mis-localization of repair factors is a general consequence of increased mechanical stress, LMNA KD of both CM’s and U2OS osteosarcoma cells was followed by cyclic stretch at a frequency and magnitude similar to that of the intact heart (8% uniaxial strain at 1 Hz frequency) (Fig.3F-i). Mis-localized GFP-KU80 increased >2-fold with imposed stretching of MOLMNA knockdown E4 CMs but not MOCtrl (Fig.3F-ii), and the fold-increase was even more dramatic with the non-beating U2OS cells because baseline mis-localization was very low. DNA damage also increased with stretching of MOLMNA CMs while LMNA levels showed an insignificant increase (Fig.3F-iii). An acute increase in stress without a proportionate increase in LMNA can thus cause mis-localization of repair factors and DNA damage, which could affect tissue-level function (Fig.2A,B). Etoposide treated hiPS-CM organoids indeed show aberrant beating (Fig.S2G).
Loss of Repair Factors or LMNA: DNA damage, binucleation micronuclei, cell cycle arrest
To assess the impact of a limited capacity for DNA repair in cells, key individual DNA repair factors were partially depleted individually and in combination (‘si-Combo’). BRCA1 is implicated in myocardial infarction and ischemia (Shukla, et al., 2011), and RPA1 appeared abundant and constant in level in our MS of drug/enzyme-perturbed hearts (Fig.1C,E). DNA damage increased in hiPS-CMs for all repair factor KDs, and the combination proved additive (Fig.4A-i).
Figure 4. Knockdown of LMNA or DNA repair factors increases DNA damage and perturbs cell cycle.
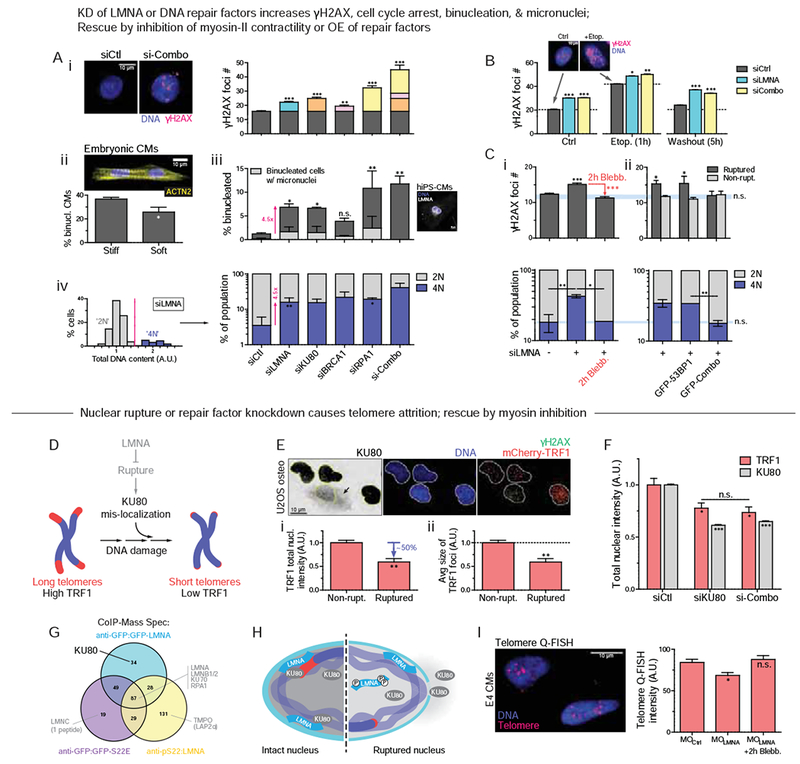
(A) KD of LMNA or repair factors (KU80, BRCA1, RPA1, or ‘si-Combo’) in hiPS-CMs increases: (i) γH2AX (ii,iii) binucleated CMs (suppressed on soft matrix, inset) and micronuclei, and (iv) 4N cells (n>77 cells per cond.; all error bars indicate ±SEM)
(B) KDs increase γH2AX before/during/after 10 μM etoposide (n>39 cells per cond).
(C) (i) Blebb rescues siLMNA-induced DNA damage and 4N cells (n>400 cells per cond.). (ii) Overexpression of DNA repair factors (GFP-KU70 + KU80 + BRCA1 = ‘GFP-Combo’) rescues excess DNA damage (top) and 4N (bottom) in siLMNA cells only in ruptured nuclei (n>296 cells per cond.; t-test: **p<0.01, ***<0.001).
(D) Schematic: effect of repair factor mis-localization on telomeres.
(E) U2OS expressing mCherry-TRF1. Ruptured nuclei (n=7) with cytoplasmic KU80 exhibit (i) lower TRF1 and (ii) smaller TRF1 foci (t-test: *p<0.05).
(F) siKU80 and si-Combo decrease TRF1 (n>89 cells per cond.).
(G) ColP-MS of WT LMNA, phosphorylated LMNA (anti-S22), and phospho-mimetic LMNA (GFP-S22E).
(H) Cartoon: LMNA-KU80-telomere interactions at the lamina.
(I) Telomere Q-FISH in embryonic CMs shows MOLMNA shortens telomere unless rescued by blebb (n>81 nuclei per cond.). (Unless noted, one-way ANOVA: *p<0.05, ***<0.001)
Excess DNA damage in post-natal hearts has been associated with CM binucleation (Aix, et al., 2016) – a hallmark of irreversible cell cycle exit. Soft ECM suppressed binucleation of embryonic CMs relative to stiff ECM (Fig.4A-ii) while suppressing mis-localization of repair factors (Fig.3E-i). Partial KD of DNA repair factors directly increased the fraction of binucleated hiPS-CMs as well as those with micronuclei (Fig.4A-iii), both of which correlated strongly with γH2AX. Repair factor KD further increased the fraction of ‘4N’ cells (vs ‘2N’; Fig.4A-iV, S3A), in agreement with trends for blebbistatin-treated hearts (Fig.1G) and with mitotic arrest. LMNA KD had all of the same effects, consistent with a role in repair factor retention in nuclei, and consistent also with a late checkpoint, KD showed more DNA damage per DNA in ‘4N’ cells relative to siCtrl cells (Fig.S3B). Importantly, with siLMNA or siCombo KD, increased γH2AX and cell cycle perturbations were evident not only at steady state but also within 1hr of damaging DNA with etoposide (per Fig.2) – which partially recovered after drug washout (Fig.4B).
Myosin-II inhibition once again rescued the DNA damage as well as cell cycle effects in siLMNA KD cells (Fig.4C-i), whereas siCombo cells maintained high DNA damage (Fig.S3C). Furthermore, over-expression of relevant DNA repair factors (with GFP-KU70, KU80, BRCA1 = ‘GFP-Combo’) rescued only the excess DNA damage in nuclear-ruptured cells compared to non-ruptured (Fig.4C-ii). A baseline level of damage in the latter cells was not affected, likely because repair factors become limiting only upon nuclear rupture or depletion. A combination of factors (‘GFP-Combo’) rescued the excess damage while one repair factor had no effect on its own (53BP1) even on the ruptured nuclei. Rescue of DNA damage again suppressed the fraction of ‘4N’ cells (Fig.4B lower) and cells in G2 (Fig.S3D).
Since cardiac telomere attrition has been implicated in CM binucleation (Aix, et al., 2016) as well as in cardiomyopathies and heart failure (Sharifi-Sanjani, et al., 2017), we assessed rupture-associated telomere shortening as a more localized form of DNA damage (Fig.4D). U2OS osteosarcoma cells with ruptured nuclei (cytoplasmic anti-KU80) showed lower total intensity of TRF1 (telomeric repeat-binding factor-1 as an mCherry fusion; Fig.4E-i) and smaller TRF1 foci size (Fig.4E-ii), as well as higher γH2AX in these LMNA KD cells. Importantly, partial KD of either KU80 or multiple repair factors also caused telomere attrition in cells with normal LMNA levels (Fig.4F). Proper retention of DNA repair factors within stressed nuclei thus requires sufficient LMNA, but LMNA also interacts with key repair factors (including KU70/80, RPA1) as confirmed by immunoprecipitation-MS (IP-MS) (Fig. S3E). IP-MS further indicates KU80 interacts preferentially with non-phosphorylated LMNA (‘GFP-WT’) compared to its phospho-solubilized nucleoplasmic form (per IP by anti-pSer22 or phosphomimetic mutant ‘GFP-S22E’) (Fig.4G), suggesting that LMNA scaffolds the repair of DNA at the periphery – which often includes telomeres (Fig.4H). Importantly, analysis of telomeres in embryonic CMs by quantitative FISH (Q-FISH) revealed LMNA KD causes telomere shortening, unless actomyosin is inhibited (Fig.4I). The findings motivate deeper mechanistic insights into how LMNA adjusts to actomyosin stress (Fig.1A-E & 2C, 2E-iii) to thereby regulate DNA damage and cell cycle.
LMNA effectively stiffens nuclei during development and increases with collagen-I ECM
To begin to assess in normal beating hearts how LMNA senses stress, nuclear deformations were imaged at embryonic day-4 (E4) – just 1~2d after the first heartbeat (Fig.5A). Transfection of a minor fraction of CMs in hearts with mCherry-Histone-H2B or GFP-LMNA facilitated imaging, and each ~1Hz contraction of heart tissue was seen to strain individual CM nuclei by up to ~5-8%. Recall that 8% stretch at 1-Hz was sufficient to rupture CM nuclei (Fig.3F). While transfection did not affect overall tissue-level beating, GFP-LMNA nuclei deformed much less than neighboring control nuclei (Fig.5A-ii), consistent with in vitro studies showing LMNA stiffens the nucleus (Pajerowski, et al., 2007; Lammerding, et al., 2006). MS of lysates quantified 29 unique LMNA peptides at E4 (Fig.S1C), confirming our early detection of functional LMNA (Fig.1B) despite past reports (Solovei, et al., 2013; Stewart and Burke, 1987), and showed a rapid increase in LMNA expression by ~3-fold from soft E4 hearts to stiffer E10 hearts (Fig.5B,C). B-type lamins remained relatively constant, in agreement with studies of diverse soft and stiff adult tissues that suggest LMNA-specific mechanosensitivity (Fig.S4A–C) (Swift, et al., 2013). Trends are confirmed by confocal IF and immunoblots of embryonic heart (Fig.5C-i,ii insets, S4D).
Figure 5. Early embryonic LMNA increases with collagen-I and tissue stiffness Et.
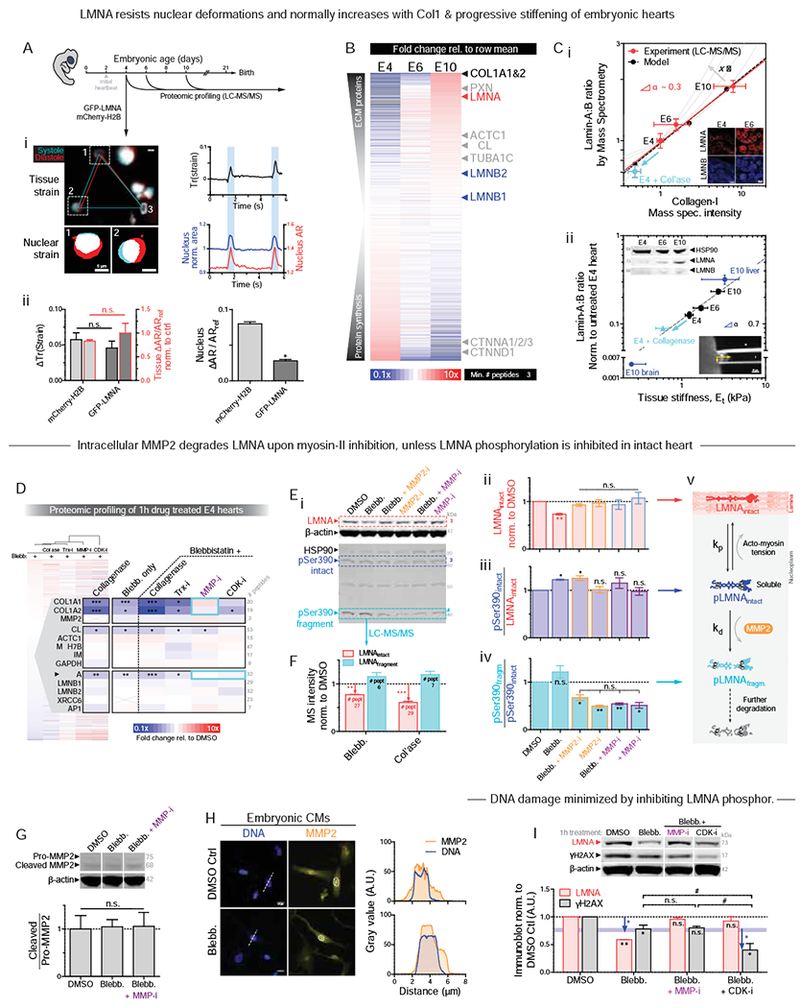
(A) (i) E4 heart beating with GFP-LMNA transfection shows nuclear ‘beating’. Right: Trace of tissue strain tensor quantified using fiduciary transfected nuclei per (Taber, et al., 1994), or nuclear strain from nuclear area and aspect ratio (AR). (ii) Overexpression does not affect local or global tissue strain (left), but GFP-LMNA nuclei deform less than mCherry-H2B nuclei (n>8 nuclei per cond.; t-test: *p<0.05). All error bars indicate ±SEM.
(B) Proteomic heatmap of E4, 6, and 10 hearts (n=8,2,1 hearts per lysate, resp.), ranked by fold-change from time-average: collagen-I and LMNA top the list.
(C) (i) Lamin-A:B ratio from MS scales with collagen-I and fits ‘lose it or use it’ model of tension-inhibited turnover of structural proteins, with stress-sensitivity denoted as x. Inset: Confocal images of LMNA & B in hearts taken at same settings suggest LMNA increases while LMNB remains constant. (ii) Lamin-A:B scaling vs Et from micropipette aspiration (lower inset), with exponent α similar for adult tissue (α ~ 0.6) (Swift, et al., 2013). Cyan: softening by collagenase decreases LMNA. Upper immunoblot (norm. to HSP90): LMNB varies by <20% from E4-E10 as LMNA increases (n>8 hearts per lysate).
(D) MS intensity fold-changes relative to DMSO for ‘Trx-i’ inhibitor of translation (cycloheximide, 1μM); ‘MMP-i’ pan-MMP inhibitor (GM6001, 10μM); ‘CDK-i’ (RO3306, at >3.5 μM doses to inhibit many CDKs) to suppress LMNA phosphorylation. Cyan boxes indicate rescues (n=8 hearts per lysate; t-test vs DMSO: *p<0.05, **<0.01, ***<0.001).
(E) (i) E4 hearts treated with blebb ± MMP-i’s (2h; n=6 hearts per lysate). pSer390 bands at intact MW (73 kDa) and 42 kDa, consistent with fragments (Baghirova, et al., 2016; Prudova, et al., 2010) predicted in silico (Song, et al., 2012). (ii) Decreased LMNA with blebb is rescued by MMP-i’s, including MMP2-specific ARP-100. (iii) Blebb increases pSer390 of 73 kDa band. (iv) Blebb does not affect pSer390 ratio of 42 kDa and 73 kDa bands, except with MMP-i. (n=7 hearts per lysate; one-way ANOVA: *p<0.05, ** p<0.01) (v) Turnover schematic.
(F) MS validation of LMNA degradation.
(G) Neither blebb nor MMP-i affect catalytic activation of MMP2 (n=8 hearts per lysate).
(H) IF shows nucleoplasmic MMP2 in embryonic CMs is unaffected by blebb.
(I) MMP-i rescues blebb-induced decrease in LMNA, but does not further suppress γH2AX (n=6 hearts per lysate). γH2AX is further decreased with CDK-i to inhibit LMNA phosphorylation (n=8 hearts per lysate; one-way ANOVA: *p<0.05, **<0.01; t-test: #p<0.05).
Collagen-I (α1, α2) exhibited the largest fold-change in MS analyses of heart development (Fig.5B), with calibrated spike-ins of purified collagen-I matching past measurements of collagen in developing chick hearts (Woessner, et al., 1967) (Fig.S4E). Collagen-I becomes the most abundant protein in developed animals and is a major determinant of tissue stiffness (Shoulders and Raines, 2009), and so its increase together with many other structural proteins (Fig.S4C) is consistent with heart stiffening (Fig.S2F) (Majkut, et al., 2013). Mechanosensitive proteins at the interface between adhesions and actomyosin cytoskeleton were also upregulated, including paxillin (Zaidel-Bar, et al., 2007) and vinculin (Huang, et al., 2017), consistent with increased adhesion and contractile stress (Geiger, et al., 2009). Down-regulation of protein synthesis and catenins suggest a transition from a rapidly growing, epithelial-like embryo to a more specialized, terminally differentiated state.
Lamin-A:B ratio increased with collagen-I (Fig.5C-i, S4F) and with tissue stiffness Et (Fig.5-ii), with a power law exponent α=0.3 (vs collagen-I) similar to that for diverse tissue proteomes (Cho, et al., 2017). Experiments fit a ‘lose it or use it’ mathematical model (Fig.5C-i) wherein mechanical tension on filamentous collagen-I and LMNA (Dingal and Discher, 2014) suppresses protease-driven degradation (Fig.5C-i). The plot versus Et yielded α~0.7 that is similar for adult tissues (α~0.6 (Swift, et al., 2013)), with soft E10 brain and stiff E10 liver fitting well (Fig.S4F,G). Importantly, 45min collagenase softened E4 hearts by ~50% and decreased LMNA (Fig.5C), per Fig.1A–E.
Different combinations of drugs were used to identify mechanisms for how LMNA levels change in tight coordination with collagen-I and tissue stiffness. MS-derived heatmaps of drug-treated E4 hearts (e.g. collagenase, blebb) reveal rapid and large decreases in collagen-I, vinculin, and LMNA (Fig.5D; confirmed by immunoblot, Fig.S5A). Little to no change was observed across most of the detected proteome including DNA repair factors (KU70, RPA1 (Fig.S5B-i)), matrix-metalloproteinase MMP2 (pro- and cleaved forms, Fig.S5B-ii,iii), cardiac myosin-II (MYH7B), vimentin (VIM), and B-type lamins. Similar results were obtained even in the presence of the translation inhibitor cycloheximide (‘Trx-i’), except for decreases in a few cytoskeletal proteins that indicate rapid transcription/translation (e.g. actin (Katz, et al., 2012)). Decreased translation thus does not explain the heart’s decreases in collagen-I and LMNA with blebbistatin and/or collagenase.
To clarify how myosin-II inhibition (intracellular) leads to a rapid decrease in collagen-I (ECM) in intact hearts, a pan-inhibitor of matrix-metalloproteinases (‘MMP-i’) was used with blebbistatin. The combination surprisingly prevented collagen-I degradation. In contrast, a CDK inhibitor (‘CDK-i’, likely affecting CDK4/6) that limits interphase phosphorylation of LMNA (e.g. pSer22 normalized to total LMNA: ‘pSer22/LMNA’ in Fig.S5A) rescued blebbistatin-induced decreases in LMNA levels without affecting the collagen-I decrease. Vinculin’s decrease concurs with decreased actomyosin tension regardless of MMP-i (Fig.5D), but MMP-i surprisingly rescued blebbistatin-induced decreases in LMNA similar to CDK-i (Fig.5D, S5A).
Mechano-protection of the genome is limited by LMNA phospho-solubilization
We hypothesized that one or more MMPs directly degrade LMNA. Nuclear MMP2 is well-documented for many cell types including CMs (Xie, et al., 2017), and LMNA (but not B-type lamins) has been speculated to be a proteolytic target (Baghirova, et al., 2016). Beating E4 hearts treated with MMP-i or an MMP2-specific inhibitor, MMP2-i, rescued the blebbistatin-induced decrease in LMNA in immunoblots that were also probed with a previously untested anti-pSer390 (Fig.5E-i,ii). For other phosphosites, interphase phosphorylation, solubilization (Kochin, et al., 2014), and subsequent degradation of LMNA (Naeem, et al., 2015; Bertacchini, et al., 2013) are already known to be suppressed by actomyosin tension on nuclei (Buxboim, et al., 2014; Swift, et al., 2013). Intact phospho-LMNA (73 kDa) increased upon blebbistatin treatment (Fig.5E-iii, ‘pSer390intact/LMNAintact’), but even more dramatic was the suppression by MMP2-i and MMP-i (regardless of blebbistatin) of a 42 kDa pSer390 fragment (‘pSer390fragm/pSer390intact’) (Fig.5E-iv). Fragment size matches predictions for MMP2 digestion (Song, et al., 2012), but is likely a transient intermediate that is further degraded (Buxboim, et al., 2014) (Fig.5E-v). MS of hearts treated with blebbistatin or collagenase not only confirmed the trend for the fragment (>6 LMNA peptides) (Fig.5F) but also showed MMP2 levels remain constant, as supported by immunoblots for MMP2’s activation (‘cleaved/pro-MMP2’ ratio, Fig.5G, S5B). Anti-MMP2 (for both pro- and active forms) was mostly nucleoplasmic in isolated CMs with localization unaffected by actomyosin inhibition (Fig.5H). Nucleoplasmic MMP2 in neonatal CMs (Baghirova, et al., 2016; Kwan, et al., 2004) suggests that, independent of maturation stage, active MMP2 is localized appropriately for degradation of phospho-LMNA downstream of actomyosin tension (Fig.5E-v).
DNA damage was assessed after acute 2h treatments of CDK-i/MMP-i plus blebbistatin. Whereas MMP-i had no effect on γH2AX relative to blebbistatin alone, CDK-i plus blebbistatin greatly suppressed DNA damage (Fig.5I). Since CDK-i maintains LMNA at the lamina (unlike MMP-i) and thereby stiffens and stabilizes the nucleus (per Fig.5A and single cell measurements (Buxboim, et al., 2014)), the results suggest that even low levels of nuclear stress are resisted to minimize both rupture and DNA damage in the embryonic heart.
Isolated CMs establish ECM elasticity and contractility in LMNA turnover by MMP2
Intact heart presents many complications to understanding LMNA regulation and its function. As the heart stiffens in development, for example, collagen ligand also increases greatly for adhesion (Fig.5B,C), sarcomere assembly becomes very dense (Fig.6A, S6A-i), cells and nuclei elongate (Fig.S6A-ii, S6B) with stabilization by microtubules (Robison, et al., 2016), cells align (Fig.S6C), and nuclear volumes decrease (Fig.S6D). Gels with constant collagen-I ligand and of controlled stiffness (0.3 – 40 kPa) (Fig.6B) showed – after just 24h – the same stiffness-correlated morphology and cytoskeleton trends for isolated E4 CMs as the intact heart from E4 to ~E11 (Fig.6C). Although matrix elasticity had large effects on size, shape, sarcomere assembly, and contractility of early CMs (Fig.S6A-i,ii, S6E) similar to later stage CMs (Ribeiro, et al., 2015; Engler, et al., 2008; Jacot, et al., 2008), blebbistatin quickly disrupted cell spreading, striation, and elongation on stiff gels (10 kPa) and on collagen-coated rigid plastic (Fig.6C). Gels that mimic the stiffness of E4 heart (1-2 kPa) were optimal for cell beating (Fig.S6F), consistent with past studies (Majkut, et al., 2013; Engler, et al., 2008), and nuclear ‘beating strain’ in isolated CMs (Fig.S6F-i, S6G-i) was similar in magnitude to that in intact heart (5-8%) (Fig.5A). Surprisingly, lamin-A:B intensity increased monotonically with gel stiffness rather than exhibiting an optimum (Fig.6D-i, S6G-ii). LMNA’s increase depended on myosin-II, as did cell/nuclear aspect ratio (AR), spreading, and sarcomere assembly (Fig.6C, 5D-ii, S6H). Monotonic increases with stiffness of ECM agrees with elevated isometric tension as documented for other cell types (e.g. (Engler, et al., 2006)). Remarkably, MMP-i or MMP2-i rescued the blebbistatin-induced decrease in lamin-A:B at the single cell level (Fig.5D-i) as seen for intact embryonic hearts (Fig.5D-i), despite no significant effects on cell morphology or striation (Fig.6C,D-ii).
Figure 6. LMNA in isolated embryonic CMs is sensitive to substrate stiffness and actomyosin stress.
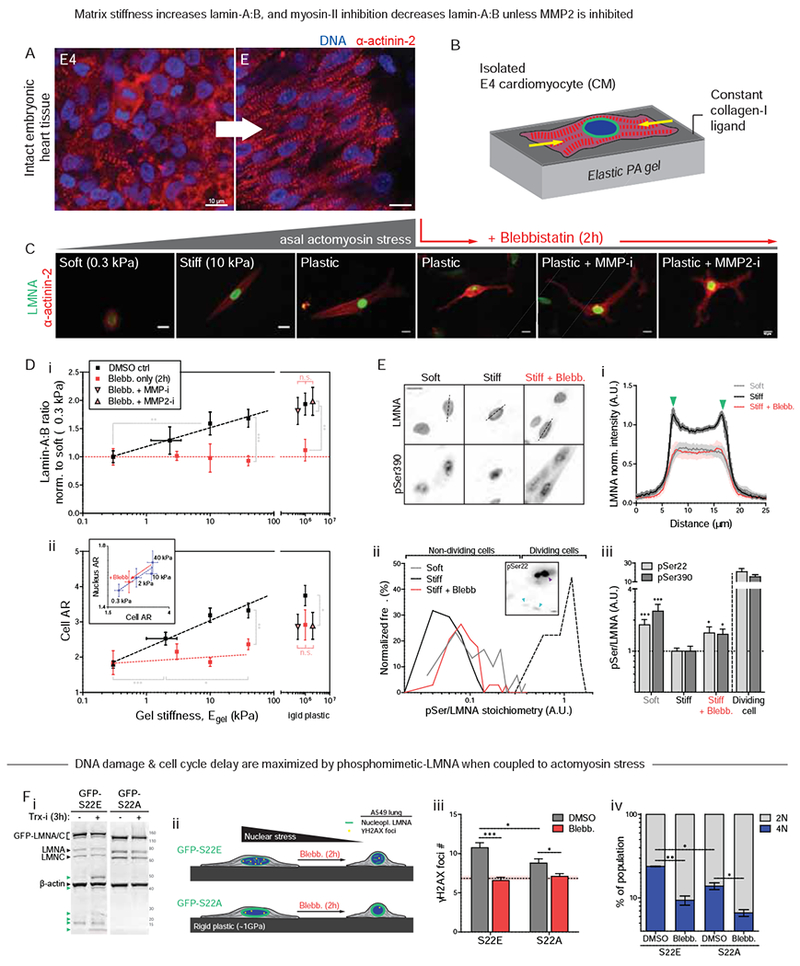
(A) Airyscan images of embryonic heart.
(B) CM cultures on PA gels of controlled stiffness (0.3 −40 kPa) and constant collagen-I ligand.
(C) Well-separated E4 CMs at 24h on gels. As with tissue (A), isolated CMs spread, elongate, and striate more with stiffness. Blebb on rigid plastic causes rounded or dendritic CM, with loss of striation. MMP inhibitors have no effect.
(D) (i) Lamin-A:B increases with gel stiffness except with blebb (n>50 cells per grp). Effects are rescued by MMP-i’s on rigid plastic. (ii) Cell and nuclear AR (inset) increase as CMs polarize on stiffer gels. Blebb reduces cell/nuclear AR, but MMP-i’s show not affect (n>47 cells per cond.; all error bars indicate ±SEM).
(E) (i) LMNA is more nucleoplasmic in CMs on soft gels (0.3 kPa) than stiff (40 kPa). Blebb gives higher nucleoplasmic signal. (ii) Histograms of phosphorylation (‘pSer/LMNA’) for pSer22 and pSer390. Signal for both sites are higher for soft gels and stiff gels + blebb. pSer/LMNA for rare mitotic CMs (yellow) are typically 10~20-fold higher than interphase CMs (cyan arrows). (iii) pSer/LMNA ratios for pSer22 and 390 (n>11 nuclei per cond.).
(F) (i) Phospho-mimetic mutant GFP-S22E (± Trx-i, 3h) probed with anti-LMNA shows low LMNA versus GFP-S22A cells. Stable lines were made within a stable shLMNA line to minimize both endogenous LMNA and overexpression artifacts. Low-MW fragments (green arrows) are not detected in GFP-S22A cells. (ii,iii) Basal DNA damage and (iv) %4N cells are highest in GFP-S22E cells, but blebb minimizes differences (n>93 nuclei per cond.; t-test *p<0.05, **<0.01, ***<0.001; irrelevant blot lanes have been removed and is indicated by a blank space).
Since myosin-II-dependent increases in lamin-A:B versus gel stiffness for isolated CMs (Fig.6D-i) align with lamin-A:B’s increase with heart stiffness and contractility during development (Fig.5B,C), LMNA phosphorylation was re-examined (per Fig.5D, S5A). IF with anti-pSer22 & pSer390 proved consistent with tension-suppressed LMNA phosphorylation and turnover: cells on soft gels showed high nucleoplasmic phospho-signal as did cells on stiff gels treated with blebbistatin (Fig.6E). The latter also showed cytoplasmic phospho-signal, consistent with multi-site phosphorylation in interphase (Kochin, et al., 2014). Regardless, LMNA was similarly low and decreased at the nuclear envelope (Fig.6E-i). Phospho-signal normalized to total LMNA (‘pSer/LMNA’) was also 2~3-fold higher in CMs on soft gels compared to stiff gels (Fig.6E-ii,iii). Blebbistatin-treated cells on stiff gels again showed increased pSer/LMNA for both phospho-sites. Signal in interphase cells was, however, >10-fold lower than in rare mitotic CMs (Fig.6E-ii, inset: purple arrow). Studies of stable phospho-mimetic mutants (GFP-S22A & GFP-S22E) conformed to the model, with higher nucleoplasmic signal but lower overall levels for S22E (Fig.6F-i, S7A). Inhibition of translation for 3 hrs (Trx-i) showed more degradation only for S22E (Fig.6F-i & S7B), and MMP2-i treatment increased S22E intensity (Fig.S7C) consistent with inhibition of phospho-LMNA turnover. S22E-phosphomimetic cells also exhibited higher DNA damage (Fig.6F-ii,iii) and more cells in late cell cycle (‘4N’, Fig.6F-iv) that were more suppressed by acute blebbistatin than S22A cells. Reduction of nuclear stress by soft matrix or actomyosin inhibition at the single cell level thus favors LMNA interphase phosphorylation, nucleoplasmic solubilization, and subsequent degradation – while also suppressing DNA damage and cell cycle arrest.
Discussion
LMNA defects cause disease through “cell-extrinsic mechanisms” that likely include ECM and/or cytoskeletal stress. Mosaic mice in which 50% of the cells express defective LMNA maintain a normal lifespan, whereas mice with 100% defective cells die within weeks of birth (de La Rosa, et al., 2013). Cultures on rigid plastic of the same cells (and similar cell types (Hernandez, et al., 2010)) exhibit premature senescence/apoptosis, as is common with excess DNA damage, but growth and viability are surprisingly rescued upon culture on almost any type of ECM. Soft matrix certainly reduces cytoskeletal stress and suppresses nuclear rupture and DNA damage in CMs with low LMNA (Fig.3E). The findings are further consistent with the observation that laminopathies spare soft tissues such as brain, independent of lineage or developmental origin, but generally affect stiff and mechanically stressed adult tissues including muscle or bone (Cho, et al., 2018).
Early embryos are soft, with minimal ECM, but as tissues form and sustain higher physical stresses, collagen-I accumulates and stiffening of the developing tissue minimizes the strain on resident cells. However, increasing stiffness and stress necessitate an adaptive mechanism to protect against nuclear stress, and accumulation of mechanosensitive LMNA fulfills this role – unless actomyosin stress is inhibited (Fig.1A–C). LMNA thus mechano-protects DNA by retaining repair factors in the nucleus (Fig.3A–D), which thereby prevents excessive DNA damage in stiff microenvironments and/or under high actomyosin stress (Fig.7A). Rapid change in LMNA protein independent of any transcription/translation (Trx-i) rules out many possible contributing pathways to the equally rapid DNA damage response. Such conclusions about causality are difficult to otherwise achieve with experiments conducted over many hrs/days such as with mouse models. Results with the cardiac myosin-II specific inhibitor (‘MYK’) in clinical trials for some cardiomyopathies ( NCT02842242) further suggest an unconventional method to attenuate DNA damage in heart (Fig.1C,D).
Figure 7. LMNA mechanosensing based on a ‘lose it or use it’ mechanism for tension-inhibited turnover that also applies to collagen-I.
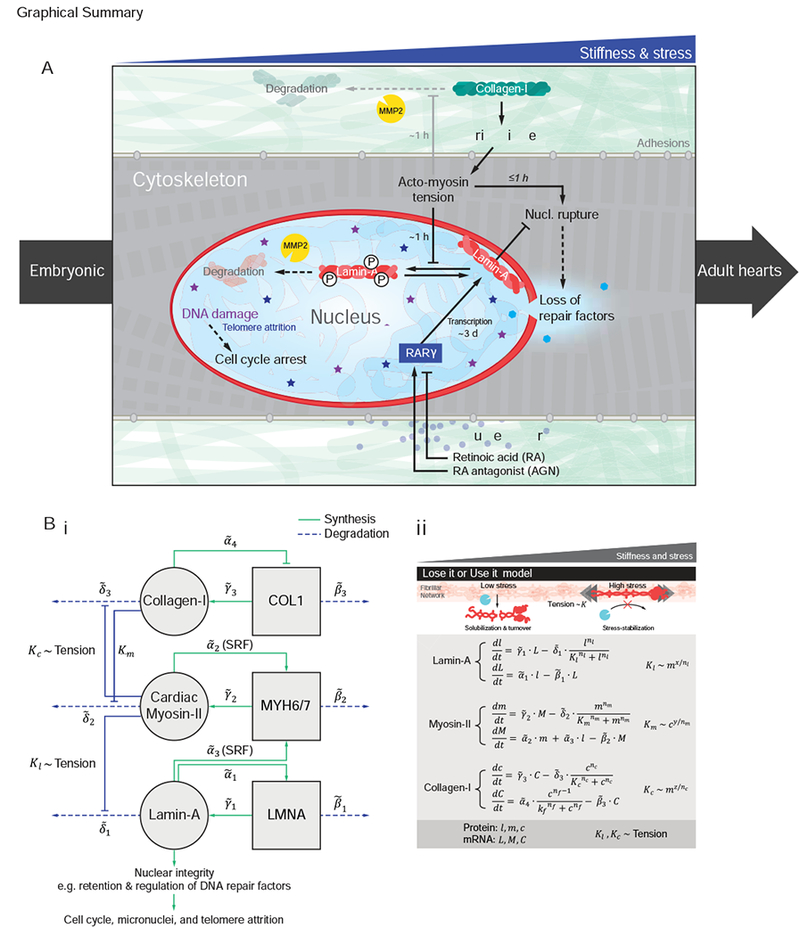
(A) Summary sketch.
(B) (i) Gene circuit model adapted from Dingal et al. (Dingal and Discher, 2014), with (ii) governing equations. Squares = genes, circles = protein. LMNA and myosin-II protein (l and m) are weak regulators of the Serum Response Factor (SRF) pathway. LMNA upregulates its own transcription (via RARγ). LMNA and collagen-I protein turnover with x and z sensitivity of degradation to myosin tension. Myosin-II protein turnover depends on matrix elasticity E which scales with collagen-I (Fig.S4E).
Loss of DNA repair factors could compromise genome integrity, but enhanced entry of cytoplasmic nucleases (Maciejowski, et al., 2015) would seem unlikely to generate γH2AX foci distributed throughout the nucleoplasm (Fig.3E-ii, 4D). Indeed, cytoplasmic proteins that bind DNA strongly (e.g. cGAS) are restricted to rupture sites. Rupture results in ‘global’ mis-localization of multiple DNA repair factors in several pathways (Fig.S2E-i) (Xia, et al., 2018; Irianto, et al., 2017), and repair factor depletion causes excess DNA damage before, during, and after transiently induced damage (by 1h etoposide, Fig.4B).
Remarkably, MMPs that degrade and remodel collagen-I matrix within hours or less are found here to directly regulate LMNA of the ‘nuclear matrix’ (Fig.5D–I, 5A,B), which indicates a surprising inside-outside symmetry (Fig.7) to the ‘lose it or use it’ mechanism (Fig.5C, 7B) of stress-stabilizing fibers. While both phospho-LMNA and MMP2 are found mostly in the nucleoplasm of CMs, the specific functions of MMP2 in the nucleus, the location of LMNA degradation, and its activation/localization mechanisms remain to be elucidated. Nuclear MMPs are not new (Xie, et al., 2017), but tension-regulation of their substrates has not been described. Our studies of isolated CMs on soft/stiff gels further demonstrate steady-state LMNA levels are determined primarily by a basal tone related to average morphologies of cells and nuclei, as opposed to dynamic contractions that intermittently strain the nucleus (Fig.3F, 5D, S6G). Basal and dynamic strain might contribute to a ‘tension-time integral’ model that seems predictive for hypertrophic versus dilated cardiomyopathy disease fates (Davis, et al., 2016). Roles for MMPs in such diseases and compensatory changes in LMNA could emerge.
Reasons why cells do not simply maintain high LMNA levels at all times in all tissues could be physical as well as biochemical. An overly rigid nucleus might disrupt cytoskeletal assembly and function. Also, LMNA fine-tunes differentiation and maturation by regulating many cytoskeletal genes via the MKL1/SRF pathway and by interacting with regulatory factors at the nuclear periphery. Nonetheless, accumulation of LMNA in stiff tissues of the embryo begins earlier than reported (Stewart and Burke, 1987) and tracks non-linearly what eventually becomes the most abundant protein in adult animals, collagen-I (Fig.5C). Collagenase softens tissue and decreases LMNA consistent with scaling trends, and the scaling LMNA ~ collagen-I0.3 in embryos matches meta-analyses of all available transcriptomes analyzed for diverse normal and diseased adult and developing hearts across species (Cho, et al., 2017) (Fig.S7A). The scaling is consistent with that across proteomes of diverse chick embryo tissues at E18 (Fig.S7B) as well as adult mouse tissues (α=0.4) (Swift, et al., 2013), and underscores a universality of normal regulation. Even the tissue-dependent timing of LMNA detection (Solovei, et al., 2013; Rober, et al., 1989; Lehner, et al., 1987) correlates well with stiffness of a tissue in adults (Fig.S7C; adapted from (Swift, et al., 2013; Rober, et al., 1989)). Mechano-coupled accumulation of LMNA and collagens that begin in early development could thus persist well into maturation and perhaps even to disease and aging of adult tissues (Fig.7G). Thus, mechano-protection of the genome by LMNA likely plays a critical role not only in embryonic development, but also in a broad range of adult diseases.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dennis E. Discher (discher@seas.upenn.edu).
Experimental model and subject details
Embryonic chick hearts and cardiomyocytes
White Leghorn chicken eggs (Charles River Laboratories; Specific Pathogen Free (SPF) Fertilized eggs, Premium #10100326) were used to extract embryonic hearts. SPF chicken eggs from Charles River are produced using filtered-air positive-pressure (FAPP) poultry housing and careful selection of layer flocks. Every flock’s SPF status is validated in compliance with USDA memorandum 800.65 and European Pharmacopoeia 5.2.2 guidelines. SPF eggs were incubated at 37°C with 5% CO2 and rotated once per day until the desired developmental stage (e.g. four days for E4; Hamburger-Hamilton stage 23-24 (HH23-24)). Embryos were extracted at room temperature (RT) by windowing eggs, carefully removing extra-embryonic membranes with sterile sonicated forceps, and cutting major blood vessels to the embryonic disc tissue to free the embryo. The extracted embryo was then placed in a dish containing PBS on a 37°C-heated plate, and quickly decapitated. For early E2-E5 embryos, whole heart tubes were extracted by severing the conotruncus and sino venosus. For more mature (>E5) embryos, embryonic discs were extracted by windowing the egg, cutting out the embryo with the overlying vitelline membrane intact, lifting out the embryo adherent to the vitelline membrane and placing in a dish of pre-warmed PBS. Extra-embryonic tissue was carefully cut away using dissection scissors and the embryo was teased away from the vitelline membrane using forceps. Whole hearts (>E5) were extracted by severing the aortic and pulmonary vessels. The pericardium was carefully sliced and teased away from the ventricle using extra-fine forceps. E10 brain and liver tissue were collected from the presumptive midbrain and hepatic diverticulum, respectively. All tissues were incubated at 37°C in pre-warmed chick heart media (α-MEM supplemented with l0 % FBS and 1% penn-strep, Gibco, #12571) for at least 1 h for stabilization, until ready for use.
To isolate primary embryonic CMs from tissue, whole hearts were diced to sub-millimeter size and digested with Trypsin/EDTA (Gibco, #25200-072). Approximately 1 ml of Trypsin per E4 heart was added and incubated for 13 min at 37°C with gentle shaking, then placed upright for 2 min to let large pieces of tissue settle to the bottom of the dish. The supernatant was carefully removed and replaced with an equal volume of fresh Trypsin for a second round of 15 min incubation at 37°C. Digestion was blocked by adding an equal volume (1:1) of chick heart media. Isolated cells were plated onto collagen-I-coated polyacrylamide (PA) gels and were allowed to adhere for >4h. Spontaneously beating CMs were imaged using an Olympus I81 microscope with a 40x air objective configured for phase contrast after 24 hrs in culture. As with intact embryonic hearts, cell area and AR were measured with time to quantify beating.
Induced pluripotent stem cell-derived cardiomyocytes (iPS-CMs)
Normal human iPS cells (obtained from Dr. Joseph Wu, Stanford Cardiovascular Institute (CVI) BioBank) were cultured following the protocol provided: Matrigel (BD Matrigel, hESC qualified: #354277) was suspended in cold 4°C DMEM/F12 medium 1:200 dilution (DMEM/F12 medium #10-092-CM-Fisher), mixed gently, and 1ml of this suspension was added to one 6-well plate (Corning Catalog #353046) and incubated for 1hr at RT to allow Matrigel to coat the surface. The solution was gently aspirated and small aggregates of human iPS cells were added to each well in mTesr1 medium containing 10 μM of ROCK inhibitor (Y27632, 2HCl – 50 mg: #50-863-7-Fisher). Culture medium was replaced daily (no ROCK inhibitor) until the cells reached ~85% confluency.
Human induced pluripotent stem cells (hiPSCs) were differentiated into cardiomyocytes (hiPS-CMs) using the “Cardiomyocytes differentiation and maintenance kit” from Stem Cell technologies (#05010 & #05020). The differentiation process was followed as described by the manufacturer’s protocol. Briefly, the mTesr1 medium with ROCK inhibitor (10 μM) was replaced with differentiation medium A, cultured for 2 days (Day 0), then subsequently to medium B for 2 days (Day 2) and again switched to medium C twice (Day 4 & 6). From Day 8 onwards, maintenance medium (Day 8) was added/refreshed every 2 days until spontaneous CM beating was observed through imaging.
Cell lines
A549 (human lung adenocarcinoma) cell lines were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA) and U2OS cell lines were obtained from the laboratory of Roger Greenberg, University of Pennsylvania. ATCC provides cell line authentication test recommendations per Tech Bulletin number 8 (TB-0111-00-02; 2010). These include: microscopy-based cell morphology check, growth curve analysis, and mycoplasma detection (by DNA staining) which were conducted on all cell lines used in these studies. All cell lines maintained the expected morphology and standard growth rates with no mycoplasma detected. U2OS and A549 cell lines were cultured in DMEM high-glucose media and Ham’s F12 nutrient mixture (Gibco, Life Technologies), respectively, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (Sigma-Aldrich).
Method details
Mass spectrometry (LC-MS/MS) of whole heart lysates
Mass spectrometry (LC-MS/MS, or ‘MS’) samples were prepared using procedures outlined in Swift et al.. Briefly, ~1 mm3 gel sections were carefully excised from SDS-PAGE gels and were washed in 50% 0.2 M ammonium bicarbonate (AB), 50% acetonitrile (ACN) solution for 30 min at 37°C. The washed slices were lyophilized for >15 min, incubated with a reducing agent (20 mM TCEP in 25 mM AB solution), and alkylated (40 mM iodoacetamide (IAM) in 25 mM AB solution). The gel sections were lyophilized again before in-gel trypsinization (20 mg/mL sequencing grade modified trypsin, Promega) overnight at 37Ό with gentle shaking. The resulting tryptic peptides were extracted by adding 50% digest dilution buffer (60 mM AB solution with 3% formic acid) and injected into a high-pressure liquid chromatography (HPLC) system coupled to a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) via a nano-electrospray ion source.
Raw data from each MS sample was processed using MaxQuant (version 1.5.3.8, Max Planck Institute of Biochemistry). MaxQuant’s built-in Label-Free Quantification (LFQ) algorithm was employed with full tryptic digestion and up to 2 missed cleavage sites. Peptides were searched against a FASTA database compiled from UniRef100 gallus gallus (chicken; downloaded from UniProt), plus contaminants and a reverse decoy database. The software’s decoy search mode was set as ‘revert’ and a MS/MS tolerance limit of 20 ppm was used, along with a false discovery rate (FDR) of 1%. The minimum number of amino acid residues per tryptic peptide was set to 7, and MaxQuant’s ‘match between runs’ feature was used for transfer of peak identifications across samples. All other parameters were run under default settings. The output tables from MaxQuant were fed into its bioinformatics suite, Perseus (version 1.5.2.4), for protein annotation and sorting.
Immunoblotting
Hearts isolated from chick embryos (E4, E6, and E10; n > 8, 4, and 2 per sample, respectively) were rinsed with pre-warmed PBS, excised into sub-millimeter pieces, quickly suspended in ice-cold 1x NuPAGE LDS buffer (Invitrogen; diluted 1:4 in 1x RIPA buffer, plus 1% protease inhibitor cocktail, 1% β-mercaptoethanol), and lysed by probe-sonication on ice (10 × 3s pulses, intermediate power setting). Samples were then heated to 80Ό for 10 min and centrifuged at maximum speed for 30 min at 4C. SDS-PAGE gels were loaded with 5 – 15 μL of lysate per lane (NuPAGE 4-12% Bis-Tris; Invitrogen). Lysates were diluted with additional 1x NuPAGE LDS buffer if necessary. Gel electrophoresis was run for 10 min at 100 V and 1 h at 160 V. Separated samples were then transferred to a polyvinylidene fluoride membrane using an iBlot Gel Transfer Device (Invitrogen). The membrane was blocked with 5% non-fat dry milk in TTBS buffer (Tris-buffered saline, BioRad; with 0.1% Tween-20), washed x3 in TTBS, and incubated with primary antibodies against: LMNA (1:500, CST, #4777), γH2AX (1:1000, EMD Millipore, #05-636), HSP90 (1:1000, Abcam, #ab13495), β-actin (1:200, Santa Cruz, #sc-47778), LMNB (1:200, Santa Cruz, #sc-6217), LMNB1 (1:1000, Abcam, ab16048), LMNA pSer22 (1:500, CST, #2026), and/or LMNA pSer390 (1:500, EMD Millipore, custom-made Ab) diluted in TBS to final concentrations generally on the order of ~1 μg/ml at 4 °C overnight. After washing x3 with TTBS, the membrane was incubated with 1:2000 diluted secondary Ab: anti-mouse HRP-conjugated IgG (GE Healthcare), at RT for >1.5 hrs. The membrane was washed x3 again with TTBS and developed with ChromoSensor (GenScript) for ~3 min at RT. Immunoblot images were obtained using a HP Scanjet 4850. Densitometry was performed using ImageJ (NIH).
Ex vivo drug perturbations of embryonic hearts
For actomyosin perturbation experiments, intact E4 hearts were incubated in 25 μM blebbistatin (EMD Millipore, #203390, stock solution 50 mg/ml in DMSO), 1 μM Omecamtiv Mecarbil (OM, Cytokinetics), or 0.3 – 1 μM MYK-581 (gift from MyoKardia, analog of MYK-461 (Green, et al., 2016)) in heart medium at 37°C, for the desired duration. Drug solutions were kept away from light to minimize photo-deactivation. Treated hearts were compared to a control sample treated with an equal concentration of vehicle solvent DMSO in heart culture media. Drug solutions were gently washed out with pre-warmed chick heart media x3 for recovery experiments. For collagen matrix perturbations, E4 heart tissue was incubated in 0.3 – 1 mg/ml concentrations of collagenase (Sigma, #C7657) for ~45 min, or 20 mg/mL transglutaminase (Sigma, T5398) for up to 3 hrs at 37°C. Enzyme activity was blocked by replacing with chick heart media containing 5% BSA. Nuclear import/export of KU80 was inhibited using 5 μM ivermectin (Sigma I8898), 10 μM mifepristone (Sigma M8046), or45 nM leptomycin B (Sigma L2913) for 24h. For transcriptional modulation of LMNA, hearts were incubated with 1 μM retinoic acid (RA, Fisher Scientific) or antagonist to retinoic acid (AGN-193109, Santa Cruz), for short (3 hrs) or long (72 hrs) treatment times. For MMP inhibition during blebbistatin treatment, hearts were incubated with 10 μM GM6001 (‘MMP-i’, EMD Millipore, #CC1010) or with 30 μM of ARP-100 (‘MMP2-i’,Santa Cruz, CAS 704888-90-4). 1 μM cycloheximide (‘Trx-I’, Sigma, C7698-1G) was added to block protein synthesis during actomyosin and/or matrix perturbations. 5 μM ivermectin (Sigma, I8898) was used to inhibit nuclear import. A minimum of 8 hearts were treated and pooled per lysate/experimental condition.
Morpholino knockdown in intact hearts
Vivo-Morpholinos (MO) against LMNA mRNA and the corresponding 5’-mispair control were designed according to the manufacturer’s guidelines (Gene Tools, Inc.): 5’-TTGGCATTCCTGCAACGCCGC-3’ (MOLMNA) and 5’-TTCGCATTGCTGGAACGGCCC-3’ (MOCtrl). Desired concentrations (1, 2, 5, 10, 30 μM) were prepared by dilution in heart growth medium at 37°C, and the resulting solutions were added to E4 hearts for 24h before analysis.
Alkaline Comet assay
Alkaline Comet assays was performed according to manufacturer’s protocol (Cell Biolabs). Briefly, cells were trypsinized, mixed with liquefied agarose at 37°C, placed drop-wise onto the supplied glass slide , and incubated for 15 min at 4°C for the agarose to gel. Lysis buffer from the kit was then added to the solidified gel and incubated for 45 min, followed by an additional 30 min incubation with alkaline solution. Electrophoresis was performed at 300 mA for 30 min followed by a 70% ethanol wash, before samples were air dried overnight. Finally, DNA dye in the kit was added to each sample for 15 minutes, followed by epifluorescence imaging as described above.
Immunofluorescence imaging
Cells were rinsed with pre-warmed PBS, fixed with 4% paraformaldehyde (PFA, Fisher) for 15 min, washed x3 with PBS and permeabilized with 0.5% Triton-X (Fisher) in PBS for 10 min. Permeabilized cells were then blocked with 5% BSA in PBS for >1.5 hrs. Samples were incubated overnight with primary antibodies in 0.5% BSA solution, with gentle agitation at 4 °C. The primary antibodies used were: LMNA (1:500, CST, #4777), LMNB (1:500, Santa Cruz, #sc-6217), LMNA pSer22 (1:500, CST, #2026), LMNA pSer390 (1:500, EMD Millipore, new custom-made Ab), α-actinin-2 (1:500, Abcam, #ab9465), γH2AX (EMD Millipore, #05-636), KU80 (1:500, Cell Signaling), 53BP1 (1:300, Novus, #NB100-304), RPA2 (1:500, Abcam). Samples were washed x3 in 0.1% BSA in PBS and incubated with the corresponding secondary antibodies at 1:500 dilution for 1.5 hrs at RT (Alexa Fluor 488, 546 and 647 nm; Invitrogen). Cells on gels or glass coverslips were mounted with mounting media (Invitrogen ProLong Gold Antifade Reagent). Images of adherent cells were taken with an Olympus IX81. All images in a given experiment were taken under the same imaging conditions and were analyzed using ImageJ (NIH).
For confocal imaging of embryonic heart tissue, heart samples were rinsed with PBS, fixed with 4% paraformaldehyde (PFA, Fisher) for 45 min, washed x3 with PBS, and permeabilized with 0.5% Triton-X (Fisher) in PBS for 3 hrs. Samples were then incubated overnight with primary antibodies in 0.5% BSA solution with gentle agitation at 4 °C (as describe d above), and washed x3 in 0.1% BSA and 0.05% Triton-X in PBS. Corresponding secondary antibodies were added at 1:500 dilution for 1.5 hrs at RT. Immunostained hearts were then mounted between glass coverslips with mounting media (Invitrogen ProLong Gold Antifade Reagent). Images were taken with Leica TCS SP8 system with either a 63×/1.4 NA oil-immersion or 40×/1.2 NA water-immersion objective.
siRNA knockdown and GFP-repair factor rescue
All siRNAs used in this study were purchased from Dharmacon (ON-TARGETplus SMARTpool; siBRCA1, L-003461-00; siBRCA2, L-003462-00; siKu80, L-010491-00; siRPA1, L-015749-01; siLMNA, L-004978-00 and non-targeting siRNA, D-001810-10). GFP-KU70 and GFP-KU80 were gifts from Dr. Stuart L Rulten from University of Sussex, Brighton, UK, and GFP-53BP1 was a gift from Dr. Roger Greenberg from University of Pennsylvania. Cells were plated 24 hours prior to transfection. Lipofectamine/nucleic acid complexes were prepared according to the manufacturer’s instructions (Lipofectamine 2000, Invitrogen), by mixing siRNA (25 nM) or GFPs (0.2-0.5ng/ml) with 1 μg/ml Lipofectamine 2000. Final solutions were added to cells and incubated for 3 days (for siRNAs) or 24 hours (for GFPs) in corresponding media containing 10% FBS.
mCherry D450A FOK1 TRF1 expression
Death domain (DD)- Oestrogen receptor (ER)-mCherry-TRF1-FokI constructs were cloned and concentrated TRF1-FokI lentivirus with polybrene (8 mg/ml) diluted in media was added to U2OS cells at a minimum titer. Doxycycline-inducible TRF1-FokI lines were generated using the Tet-On 3G system. Doxycycline was used at a concentration of 40 ng/ml for 16–24 h to induce expression of TRF1- D450A FokI. Shield-1 (Cheminpharma LLC) and 4-hydroxytamoxifen (4-OHT) (Sigma) were both used at a concentration of 1 μM for 2 h, to allow for TRF1-D450A FokI stabilization and translocation into the nucleus.
Quantitative Fluorescence In Situ Hybridization and Telomere Analysis
Isolated cells were attached on slides, fixed and stained for PNA telomeric probe as we previously described in (Tichy, et al., 2017). Images of mMuSCs were taken using a Nikon eclipse 90i wide-field epifluorescence microscope equipped with a Prior Proscan III motorized stage, a Photometrics Coolsnap HQ2 14-bit digital camera, and a Nikon 100×/1.40 Plan Apo VC objective. Telomeres were analyzed with the investigators blinded to genotypes and/or conditions using open-source software Telometer, as previously described. The program generates statistics on the entire region of the nucleus. Analysis includes the intensity sum of all Cy5 telomere pixels for a given nucleus (proportional to the cell’s total telomere length) and the intensity sum of all DAPI pixels for the nucleus (proportional to total cellular nuclear DNA content).
Quantification and statistical analysis
Quantification of beating strain
Transfected E4 chick hearts were video-imaged using an Olympus I81 at 4x magnification, with a CCD camera. Procedures described by Taber et al. (Taber, et al., 1994) were followed to calculate 2D tissue beating strain. Briefly, >3 groups of 3 cells located within 20 microns of each other were selected as fiduciary markers along the outer walls of ventricular tissue. A custom Matlab program was written to track their displacements and compute trace of the tissue strain tensor (Taber, et al., 1994). Alternatively, for higher throughput quantification of contractility changes (n>10 hearts per sample, e.g. following blebbistatin treatment), morphological measurements (projected 2D area, aspect ratio (AR), circularity, perimeter, etc.) of beating whole-hearts were traced with time. Morphology measurements were normalized to baseline reference values, and peak-to-valley ‘amplitudes’ were averaged over >5 beats to quantify tissue beating strain (e.g. ΔAR/ARref). A similar approach was taken to quantify nuclear deformations: the projected 2D area and aspect ratio (AR) of transfected nuclei (e.g. expressing GFP-LMNA) were traced with time, and the normalized amplitudes were measured and compared across experimental conditions.
Statistics
All statistical analyses were performed and graphs were generated using GraphPad Prism 5. All error bars reflect ± SEM. Unless stated otherwise, all comparisons for groups of three or more were analyzed by one-way ANOVA followed by a Dunnett’s multiple comparison test. Pairwise sample comparisons were performed using student’s t-test. Population distribution analyses were performed using the Kolmogorov-Smirnov test, with α = 0.05. Information regarding statistical analyses are included in the figure legends. For all figures, the p-values for statistical tests are as follows: n.s. = not significant, *p<0.05, **<0.01, ***<0.001 (or #p<0.05, ##<0.01 for multiple tests within the same dataset).
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-lamin A/C (4C11) | Cell Signaling Technology | #4777s |
| Mouse monoclonal anti-phospho-Histone H2A.X (Ser139), clone JBW301 | EMD Millipore | #05-636 |
| Rabbit polyclonal anti-HSP90 | Abcam | ab13495 |
| Rabbit polyclonal anti-lamin B1 | Abcam | ab16048 |
| Mouse monoclonal anti-β-actin (C4) | Santa Cruz Biotechnology | sc-47778 |
| Goat polyclonal anti-lamin-B (M-20) | Santa Cruz Biotechnology | sc-6217 |
| Rabbit polyclonal anti-phospho-lamin A/C (Ser22) | Cell Signaling Technology | #2026 |
| Rabbit polyclonal anti-phospho-lamin A/C (Ser390) | EMD Millipore | N/A (custom) |
| Biological Samples | ||
| Human induced pluripotent stem cell-derived cardiomyocytes (iPS-CMs) | Stanford Cardiovascular Institute (SCVI) Biobank | http://med.stanford.edu/scvibiobank.html |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Blebbistatin | EMD Millipore | #203390 |
| Omecamtiv mecarbil | Selleckchem | S2623 |
| MYK (MYK-581) | MyoKardia | N/A (gift) |
| Collagenase | Sigma-Aldrich | C7657 |
| Transglutaminase | Sigma-Aldrich | T5398 |
| Ivermectin (Imp-i’) | Sigma-Aldrich | I8898 |
| All-trans-retinoic acid (RA) | Fisher Scientific | AC207341000 |
| Retinoic acid receptor antagonist (AGN193109) | Santa Cruz Biotechnology | sc-217589 |
| GM6001 (pan-MMP inhibitor, ‘MMP-i’) | EMD Millipore | CC1010 |
| ARP-100 (MMP2 inhibitor, ‘MMP2-i’) | Santa Cruz Biotechnology | sc-203522 |
| Cycloheximide (CHX, ‘Trx-i’) | Sigma-Aldrich | C7698 |
| PNA Probe (TelC-Cy5) | PNA bio | # F1003 |
| Critical Commercial Assays | ||
| Comet Assay Kit | Cell Biolabs | STA-350 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| A549 lung carcinoma cell line | ATCC | CCL-185 |
| U2OS osteosarcoma cell line expressing mCherry–TRF1 | Laboratory of Roger Greenberg, Univ. of Pennsylvania | N/A |
| Experimental Models: Organisms/Strains | ||
| White Leghorn chicken eggs: Specific Pathogen Free (SPF) fertilized | Charles River Laboratories | Premium #10100326 |
| Oligonucleotides | ||
| Vivo-Morpholino against chick LMNA (MOLMNA): 5’-TTGGCATTCCTGCAACGCCGC-3’ | Gene Tools | N/A |
| 5’ mismatch control Vivo-Morpholino (MOCtrl): 5’-TTCGCATTGCTGGAACGGCCC-3’ | Gene Tools | N/A |
| Software and Algorithms | ||
| MaxQuant Version 1.5.3.8 | Max Planck Institute of Biochemistry | http://www.coxdocs.org/doku.php?id=maxquant:common:download_and_installation |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Telometer | Johns Hopkins School of Medicine | http://demarzolab.pathology.jhmi.edu/telometer/index.html |
| Other | ||
Acknowledgements
This work was supported by: National Institutes of Health/National Heart Lung and Blood Institute Awards R01 HL124106 and R21 HL128187, National Cancer Institute PSOC Award U54 CA193417, the US-Israel Binational Science Foundation, Charles Kaufman Foundation Award KA2015-79197, and National Science Foundation grant agreement CMMI 15-48571.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests The authors declare no competing interests.
References
- Aix E, Gutierrez-Gutierrez O, Sanchez-Ferrer C, Aguado T, and Flores I (2016). Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation. The Journal of cell biology 213, 571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghirova S, Hughes BG, Poirier M, Kondo MY, and Schulz R (2016). Nuclear matrix metalloproteinase-2 in the cardiomyocyte and the ischemic-reperfused heart. Journal of molecular and cellular cardiology 94, 153–161. [DOI] [PubMed] [Google Scholar]
- Bertacchini J, Beretti F, Cenni V, Guida M, Gibellini F, Mediani L, Marin O, Maraldi NM, de Pol A, Lattanzi G, et al. (2013). The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 27, 2145–55. [DOI] [PubMed] [Google Scholar]
- Bollen IAE, Schuldt M, Harakalova M, Vink A, Asselbergs FW, Pinto JR, Kruger M, Kuster DWD, and van der Velden J (2017). Genotype-specific pathogenic effects in human dilated cardiomyopathy. The Journal of physiology 595, 4677–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B, and Stewart CL (2013). The nuclear lamins: flexibility in function. Nature reviews. Molecular cell biology 14, 13–24. [DOI] [PubMed] [Google Scholar]
- Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PC, Athirasala A, Kao YR, Cho S, Harada T, Shin JW, et al. (2014). Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Current biology : CB 24, 1909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang ACY, Chang ACH, Kirillova A, Sasagawa K, Su W, Weber G, Lin J, Termglinchan V, Karakikes I, Seeger T, et al. (2018). Telomere shortening is a hallmark of genetic cardiomyopathies. Proceedings of the National Academy of Sciences of the United States of America 115, 9276–9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Abbas A, Irianto J, Ivanovska IL, Xia Y, Tewari M, and Discher DE (2018). Progerin phosphorylation in interphase is lower and less mechanosensitive than lamin-A,C in iPS-derived mesenchymal stem cells. Nucleus 9, 230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Irianto J, and Discher DE (2017). Mechanosensing by the nucleus: From pathways to scaling relationships. The Journal of cell biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl KN, Ribeiro AJ, and Lammerding J (2008). Nuclear shape, mechanics, and mechanotransduction. Circulation research 102, 1307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR, et al. (2016). A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 165, 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Rosa J, Freije JM, Cabanillas R, Osorio FG, Fraga MF, Fernandez-Garcia MS, Rad R, Fanjul V, Ugalde AP, and Liang Q (2013). Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nature communications 4, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingal PC, and Discher DE (2014). Systems mechanobiology: tension-inhibited protein turnover is sufficient to physically control gene circuits. Biophysical journal 107, 2734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–83. [DOI] [PubMed] [Google Scholar]
- Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, and Discher DE (2008). Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. Journal of cell science 121, 3794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, and Discher DE (2006). Matrix elasticity directs stem cell lineage specification. Cell 126, 677–89. [DOI] [PubMed] [Google Scholar]
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr., Spudich S, De Girolami U, et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. The New England journal of medicine 341, 1715–24. [DOI] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, and Bershadsky AD (2009). Environmental sensing through focal adhesions. Nature reviews. Molecular cell biology 10, 21–33. [DOI] [PubMed] [Google Scholar]
- Graziano S, Kreienkamp R, Coll-Bonfill N, and Gonzalo S (2018). Causes and consequences of genomic instability in laminopathies: Replication stress and interferon response. Nucleus 9, 258–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, et al. (2016). A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, and Wilson KL (2005). The nuclear lamina comes of age. Nature reviews. Molecular cell biology 6, 21–31. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Osborne LD, Van Landeghem L, Sharek L, Superfine R, Garcia-Mata R, and Burridge K (2014). Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature cell biology 16, 376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr JC, Luperchio TR, Wong X, Cohen E, Wheelan SJ, and Reddy KL (2015). Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. The Journal of cell biology 208, 33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, et al. (2010). Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Developmental cell 19, 413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo T, Naito AT, Sumida T, Shibamoto M, Okada K, Nomura S, Nakagawa A, Yamaguchi T, Sakai T, Hashimoto A, et al. (2017). DNA single-strand break-induced DNA damage response causes heart failure. Nature communications 8, 15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DL, Bax NA, Buckley CD, Weis WI, and Dunn AR (2017). Vinculin forms a directionally asymmetric catch bond with F-actin. Science 357, 703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, et al. (2017). DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration. Current biology : CB 27, 210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacot JG, McCulloch AD, and Omens JH (2008). Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophysical journal 95, 3479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellos G, Zhou J, Patel H, Ridgway RA, Huels D, Gurniak CB, Sandilands E, Carragher NO, Sansom OJ, Witke W, et al. (2015). ADF and Cofilin 1 Control Actin Stress Fibers, Nuclear Integrity, and Cell Survival. Cell reports 13, 1949–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ZB, Wells AL, Park HY, Wu B, Shenoy SM, and Singer RH (2012). beta-Actin mRNA compartmentalization enhances focal adhesion stability and directs cell migration. Genes & development 26, 1885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Min J, Recknagel AK, Riccio M, and Butcher JT (2011). Quantitative three-dimensional analysis of embryonic chick morphogenesis via microcomputed tomography. Anatomical record 294, 1–10. [DOI] [PubMed] [Google Scholar]
- Kochin V, Shimi T, Torvaldson E, Adam SA, Goldman A, Pack CG, Melo-Cardenas J, Imanishi SY, Goldman RD, and Eriksson JE (2014). Interphase phosphorylation of lamin A. Journal of cell science 127, 2683–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Voncken JW, Konings G, van Weeghel M, van den Hoogenhof MM, Gijbels M, van Erk A, Schoonderwoerd K, van den Bosch B, Dahlmans V, et al. (2011). Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus 2, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JA, Schulze CJ, Wang W, Leon H, Sariahmetoglu M, Sung M, Sawicka J, Sims DE, Sawicki G, and Schulz R (2004). Matrix metalloproteinase-2 (MMP-2) is present in the nucleus of cardiac myocytes and is capable of cleaving poly (ADP-ribose) polymerase (PARP) in vitro. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 18, 690–2. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, and Lee RT (2006). Lamins A and C but not lamin B1 regulate nuclear mechanics. The Journal of biological chemistry 281, 25768–80. [DOI] [PubMed] [Google Scholar]
- Larrieu D, Britton S, Demir M, Rodriguez R, and Jackson SP (2014). Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344, 527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu D, Vire E, Robson S, Breusegem SY, Kouzarides T, and Jackson SP (2018). Inhibition of the acetyltransferase NAT10 normalizes progeric and aging cells by rebalancing the Transportin-1 nuclear import pathway. Science signaling 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Lau YM, Cai ZJ, Lai WH, Wong LY, Tse HF, Ng KM, and Siu CW (2017). Modeling Treatment Response for Lamin A/C Related Dilated Cardiomyopathy in Human Induced Pluripotent Stem Cells. Journal of the American Heart Association 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CF, Stick R, Eppenberger HM, and Nigg EA (1987). Differential expression of nuclear lamin proteins during chicken development. The Journal of cell biology 105, 577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, and Gerdes AM (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. Journal of molecular and cellular cardiology 28, 1737–46. [DOI] [PubMed] [Google Scholar]
- Li H, Vogel H, Holcomb VB, Gu Y, and Hasty P (2007). Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Molecular and cellular biology 27, 8205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, et al. (2005). Genomic instability in laminopathy-based premature aging. Nature medicine 11, 780–5. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majkut S, Idema T, Swift J, Krieger C, Liu A, and Discher DE (2013). Heart-specific stiffening in early embryos parallels matrix and myosin expression to optimize beating. Current biology : CB 23, 2434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeem AS, Zhu Y, Di WL, Marmiroli S, and O’Shaughnessy RF (2015). AKT1-mediated Lamin A/C degradation is required for nuclear degradation and normal epidermal terminal differentiation. Cell death and differentiation 22, 2123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT02842242 A Phase 2 Open-label Pilot Study Evaluating MYK-461 in Subjects With Symptomatic Hypertrophic Cardiomyopathy and Left Ventricular Outflow Tract Obstruction. In https://ClinicalTrials.gov/show/NCT02842242).
- NCT02929329 Registrational Study With Omecamtiv Mecarbil/AMG 423 to Treat Chronic Heart Failure With Reduced Ejection Fraction. In https://ClinicalTrials.gov/show/NCT02929329).
- Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, and Schneider MD (2003). Telomere attrition and Chk2 activation in human heart failure. Proceedings of the National Academy of Sciences of the United States of America 100, 5378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmanagic-Myers S, Dechat T, and Foisner R (2015). Lamins at the crossroads of mechanosignaling. Genes & development 29, 225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajerowski JD, Dahl KN, Zhong FL, Sammak PJ, and Discher DE (2007). Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences of the United States of America 104, 15619–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer cell 8, 241–54. [DOI] [PubMed] [Google Scholar]
- Prudova A, auf dem Keller U, Butler GS, and Overall CM (2010). Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Molecular & cellular proteomics : MCP 9, 894–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, et al. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Dumenil AM, Manel N, et al. (2016). ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–62. [DOI] [PubMed] [Google Scholar]
- Ribeiro AJ, Ang YS, Fu JD, Rivas RN, Mohamed TM, Higgs GC, Srivastava D, and Pruitt BL (2015). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proceedings of the National Academy of Sciences of the United States of America 112, 12705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rober RA, Weber K, and Osborn M (1989). Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105, 365–78. [DOI] [PubMed] [Google Scholar]
- Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, and Prosser BL (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, and Sheetz MP (2006). Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 127, 1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi-Sanjani M, Oyster NM, Tichy ED, Bedi KC Jr., Harel O, Margulies KB, and Mourkioti F (2017). Cardiomyocyte-Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. Journal of the American Heart Association 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T, Kittisopikul M, Tran J, Goldman AE, Adam SA, Zheng Y, Jaqaman K, and Goldman RD (2015). Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Molecular biology of the cell 26, 4075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, and Raines RT (2009). Collagen structure and stability. Annual review of biochemistry 78, 929–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla PC, Singh KK, Quan A, Al-Omran M, Teoh H, Lovren F, Cao L, Rovira II, Pan Y, Brezden-Masley C, et al. (2011). BRCA1 is an essential regulator of heart function and survival following myocardial infarction. Nature communications 2, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Hunt CR, Pandita RK, Kumar R, Yang CR, Horikoshi N, Bachoo R, Serag S, Story MD, Shay JW, et al. (2013). Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Molecular and cellular biology 33, 1210–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu CW, Lee YK, Ho JC, Lai WH, Chan YC, Ng KM, Wong LY, Au KW, Lau YM, Zhang J, et al. (2012). Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 4, 803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–98. [DOI] [PubMed] [Google Scholar]
- Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, and Pike RN (2012). PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PloS one 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C, and Burke B (1987). Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 51, 383–92. [DOI] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber LA, Sun H, Clark EB, and Keller BB (1994). Epicardial strains in embryonic chick ventricle at stages 16 through 24. Circulation research 75, 896–903. [DOI] [PubMed] [Google Scholar]
- Takaki T, Montagner M, Serres MP, Le Berre M, Russell M, Collinson L, Szuhai K, Howell M, Boulton SJ, Sahai E, et al. (2017). Actomyosin drives cancer cell nuclear dysmorphia and threatens genome stability. Nature communications 8, 16013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichy ED, Sidibe DK, Tierney MT, Stec MJ, Sharifi-Sanjani M, Hosalkar H, Mubarak S, Johnson FB, Sacco A, and Mourkioti F (2017). Single Stem Cell Imaging and Analysis Reveals Telomere Length Differences in Diseased Human and Mouse Skeletal Muscles. Stem cell reports 9, 1328–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, Ben-Harush K, Dubrovsky-Gaupp A, Sapra KT, Goldman RD, and Medalia O (2017). The molecular architecture of lamins in somatic cells. Nature 543, 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich TA, de Juan Pardo EM, and Kumar S (2009). The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer research 69, 4167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggan O, Schroder B, Krapf D, Bamburg JR, and DeLuca JG (2017). Cofilin Regulates Nuclear Architecture through a Myosin-II Dependent Mechanotransduction Module. Scientific reports 7, 40953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woessner JF Jr., Bashey RI, and Boucek RJ (1967). Collagen development in heart and skin of the chick embryo. Biochimica et biophysica acta 140, 329–38. [DOI] [PubMed] [Google Scholar]
- Worman HJ, and Bonne G (2007). “Laminopathies”: a wide spectrum of human diseases. Experimental cell research 313, 2121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, Pfeifer CR, Alvey CM, Ji J, Liu D, Cho S, et al. (2018). Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. The Journal of cell biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Mustafa A, Yerzhan A, Merzhakupova D, Yerlan P, A NO, Wang X, Huang Y, and Miao L (2017). Nuclear matrix metalloproteinases: functions resemble the evolution from the intracellular to the extracellular compartment. Cell death discovery 3, 17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Leone LM, and Kaufman LJ (2009). Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophysical journal 97, 2051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Milo R, Kam Z, and Geiger B (2007). A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. Journal of cell science 120, 137–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.