

Abstract
Background & Aims:
Mechanical forces contribute to portal hypertension (PHTN) and fibrogenesis. We investigated the mechanisms by which forces are transduced by liver sinusoidal endothelial cells (LSECs) into pressure and matrix changes.
Methods:
We isolated primary LSECs from mice and induced mechanical stretch with a Flexcell device, to recapitulate the pulsatile forces induced by congestion, and performed microarray and RNA-sequencing analyses to identify gene expression patterns associated with stretch. We also performed studies with C57BL/6 mice (controls), mice with deletion of neutrophil elastase (NE–/–) or PAD4 (Pad4–/–) (enzymes that formation of neutrophil extracellular traps [NETs]), and mice with LSEC-specific deletion of Notch1 (Notch1iΔEC). We performed partial ligation of the suprahepatic inferior vena cava (pIVCL) to simulate congestive hepatopathy-induced portal hypertension in mice; some mice were given subcutaneous injections of sivelestat or underwent bile-duct ligation. Portal pressure was measured using a digital blood pressure analyzer and we performed intravital imaging of livers of mice.
Results:
Expression of the neutrophil chemoattractant CXCL1 was upregulated in primary LSECs exposed to mechanical stretch, compared to unexposed cells. Intravital imaging of livers in control mice revealed sinusoidal complexes of neutrophils and platelets and formation of NETs after pIVCL. NE–/– and Pad4–/– mice had lower portal pressure and livers had less fibrin compared to control mice after pIVCL and bile-duct ligation; neutrophil recruitment into sinusoidal lumen of liver might increase portal pressure by promoting sinusoid microthrombi. RNA sequencing of LSECs identified proteins in mechanosensitive signaling pathways that are altered in response to mechanical stretch, including integrins, Notch1, and calcium signaling pathways. Mechanical stretch of LSECs increased expression of CXCL1 via integrin-dependent activation of transcription factors regulated by Notch and its interaction with the mechanosensitive piezo calcium channel.
Conclusions:
In studies of LSECs and knockout mice, we identified mechanosensitive angiocrine signals released by LSECs which promote PHTN by recruiting sinusoidal neutrophils and promoting formation of NETs and microthrombi. Strategies to target these pathways might be developed for treatment of PHTN.
Keywords: Congestive hepatopathy, mouse model, chemokine, extracellular matrix
Graphical Abstract
Introduction
Portal hypertension (PHTN) is a common sequelae of chronic liver disease which constitutes the principle driver of mortality and liver transplantation in patients with cirrhosis. The pathophysiology of PHTN is complex and is regulated at multiple levels, including paracrine signaling within sinusoids, alterations in vasoconstrictive tone, and more grossly, by angio-architectural distortion of the liver1. Chronic hepatic congestion, or congestive hepatopathy (CH), is a cause of PHTN that occurs in conditions such as congestive heart failure and Budd-Chiari syndrome which perturb efficient blood flow in the liver. Despite the increasing prevalence of CH in an aging population, the mechanism by which chronic hepatic congestion promotes PHTN and fibrosis has been poorly understood. Our partial inferior vena cava ligation (pIVCL) murine model of CH revealed that chronic congestion drives fibrosis through sinusoidal thrombosis and mechanical forces2. However, the molecular pathways that mediate and integrate these processes remain incompletely defined. Furthermore, sinusoidal thrombosis has been implicated in chronic liver disease progression in human studies3, 4, suggesting that sinusoidal thrombogenesis may be broadly applicable to other etiologies of fibrosis-related PHTN.
Recent studies implicate neutrophils in the formation and propagation of thrombosis5, 6. Neutrophils accumulate early in the formation of thrombosis7, promote propagation of the coagulation cascade, and aggregate with other thrombogenic mediators, most notably, platelets5, 8. Neutrophil interaction with platelets can contribute to thrombosis through the formation of neutrophil extracellular traps, or NETs9, 10. NETs are composed of a backbone of extracellular DNA fibers bound to histones and granular proteins such as myeloperoxidase and neutrophil elastase (NE)11. Various cellular components of neutrophils and NETs, including histone and granular proteins, can initiate or propagate coagulation, and NETs have been identified as pro-thrombotic structures in the setting of sepsis and deep vein thrombosis9, 10, 12–14. The role of NETs in the development of PHTN has not been mechanistically explored.
Tissue-specific endothelial cells, including LSECs, secrete “angiocrine” factors which serve as critical regulators of metabolism, organ homeostasis and regeneration15. Mechanosensitive proteins contribute to a paradigm of “mechanocrine” signaling where changes in mechanical forces and the physical environment are transduced into secretion of angiocrine signals which impact neighboring cells. Integrins are transmembrane proteins that link extracellular matrix molecules with the cellular cytoskeleton and thereby sense and respond to a spectrum of biochemical and mechanical signals16. Studies have shown reciprocal interactions between integrins and the Notch pathway17, a mechanosensitive signaling pathway which is critical to endothelial function and hepatic vasculature18, 19. Mice with selective deletion of the Notch1 receptor in LSECs have distorted vascular architecture, dilated sinusoids, and increased portal pressures, suggesting that the Notch1 receptor regulates sinusoidal structure and tone18. Piezo proteins are components of mechanosensitive nonselective ion channels which have also been implicated in modulation of vascular tone20, 21. However comprehensive RNA-sequencing based analysis of pathways activated by mechanical stretch in LSECs has not been previously explored.
In this study, we first examined the impact of pathologic levels of mechanical stretch on LSEC signaling. We used non-biased high-throughput screening with microarray and RNA-sequencing technology to identify novel mechanocrine signals generated by LSECs. These signals were further tested in hypothesis-driven in vivo and in vitro studies to elucidate events in the sinusoidal microenvironment which contribute to the pathophysiology of PHTN. We demonstrate that mechanical stretch of LSECs induces Notch-dependent upregulation and secretion of the neutrophil chemotactic chemokine CXCL1. CXCL1-mediated neutrophil chemotaxis propagates PHTN by interacting with platelets to promote extrusion of NETs, and inhibition of NET formation attenuates PHTN. We show that integrins and piezo channels serve as mechanosensors which activate the Notch signaling pathway to upregulate CXCL1. These results have the potential to augment the spectrum of therapeutic targets to treat PHTN and its complications in CH and other forms of chronic liver injury.
Materials and Methods
More detailed Materials and Methods are included in the Supplementary Materials.
Animal experiments
Partial inferior vena cava ligation
C57BL/6 mice (8–10 weeks) were purchased from Envigo Laboratories, and NE−/− and Pad4−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). Notch1flox/flox mice were crossed with Cdh5(PAC)-CreERT mice to generate mice with LSEC-specific deletion of Notch1 (Notch1iΔEC). Mice were subjected to pIVCL for 4 or 6 weeks to induce PHTN and fibrosis as previously described2, 22. In other protocols, after pIVCL, C57BL/6 mice were injected subcutaneously with sivelestat (30 mg/kg) or equal volume of dimethyl sulfoxide (DMSO) control. All animal work was performed under Mayo Institutional Animal Care and Use Committee oversight.
Bile duct ligation
C57BL/6 mice (8–10 weeks) were subjected to bile duct ligation (BDL) for 4 weeks as previously described22.
Portal pressure measurements
Portal pressure was measured using a digital blood pressure analyzer (Digi-Med)23. After calibration of the analyzer, a 16-guage catheter attached to a pressure transducer was inserted into the portal vein. The average portal pressure (mm Hg) was then recorded.
Intravital imaging
Intravital imaging of the liver was done using an inverted spinning-disk confocal microscopy system (Olympus IX81)24. Please refer to the Supplementary Materials and Methods for more detail.
Statistical analysis
Means are expressed as means ± standard error. Significance was established using the Student’s t-test and analysis of variance when appropriate.
Results
Liver sinusoidal endothelial cells (LSECs) subjected to cyclic stretch secrete the neutrophil chemoattractant CXCL1
CH is characterized by passive hepatic congestion and sinusoidal dilatation which imparts mechanical stretch on LSECs. Given the critical role of LSECs in mechanical sensing of sinusoidal forces as well as recent studies implicating angiocrine signaling in diverse liver functions and diseases25, 26, we aimed to elucidate the role of “mechanocrine” signaling mechanisms in the pathogenesis of CH. We isolated primary murine LSECs and subjected them to cyclic biaxial stretch with a Flexcell device. Cyclic stretch was imposed at an intensity and frequency (20% strain, 1 Hz) intended to recapitulate the cardiac cycle and therefore mimic the forces experienced by LSECs during CH27. We then performed microarray screening of genes related to endothelial cell function. Microarray screening of LSECs subjected to cyclic stretch showed transcriptional upregulation of a number of cytokines which impact inflammatory cell chemotaxis, including CXCL1, CXCL2, and Ccl2 (Figure 1a). We pursued neutrophil chemotactic signals given the prominent role that neutrophils play in formation of thromboses, which we hypothesize are integral to the pathophysiology of CH-induced PHTN. We confirmed an increase in CXCL1 in LSECs subjected to cyclic biaxial stretch by quantitative PCR (Figure 1b, upper panel), and ELISA analysis (Figure 1b, lower panel). These findings suggest that LSECs subjected to cyclic stretch generate angiocrine signals which have the potential to recruit neutrophils and possibly propagate microthrombus formation, fibrosis, and PHTN. Prior studies suggest that CXCL1 induces neutrophil chemotaxis28, 29. To confirm a functional role of CXCL1 as a neutrophil chemoattractant, a microfluidic gradient generator was utilized to create a gradient of CXCL130 (Supplementary Figure 1). Neutrophils were plated on a surface coated with fetal bovine serum, and their migration was studied over the ensuing hour. Neutrophils migrated towards higher concentrations of CXCL1 (Figure 1c), confirming the ability of CXCL1 to promote neutrophil chemotaxis. We next tested this proposed pathway in vivo.
Figure 1. Cyclic stretch upregulates CXCL1.

(A) Primary murine LSECs were subjected to cyclic stretch with a Flexcell device Microarray analysis of genes relevant to endothelial cell biology reveals upregulation of a number of genes impacting inflammatory cell chemotaxis, including CXCL1 (fold change 6.92). The top 15 genes are shown (THBD: thrombomodulin, SELE: selectin, endothelial cell, CXCL1: C-X-C motif chemokine ligand 1, CXCL2: C-X-C motif chemokine ligand 2, SELL: selectin, lymphocyte, CCL2: chemokine (C-C motif) ligand 2, PTGS2: prostaglandin-endoperoxide synthase 2, TYMP: thymidine phosphorylase, PLG: plasminogen, PROCR: protein C receptor, endothelial, CCL5: chemokine (C-C motif) ligand 5, SELPLG: selectin, platelet (p-selectin) ligand, TGFB1: transforming growth factor, beta 1, PTGIS: prostaglandin I2 (prostacyclin), IL6: interleukin 6). (B) CXCL1 upregulation by cyclic stretch was demonstrated with quantitative PCR (upper panel), and ELISA (lower panel). (C) Neutrophils plated in a fibronectin-coated microfluidic device migrate toward a CXCL1 chemotactic gradient (n=3–5; *P≤0.05 for all panels).
Pro-thrombotic NETs form in the sinusoids in congestive hepatopathy
Our in vitro results thus far indicate that LSEC responses to mechanical force leads to angiocrine signals that can recruit neutrophils. To directly examine the role of neutrophils in CH in vivo, we performed intravital imaging 24 hours after pIVCL and sham procedures. Intravital microscopy of livers 24 hours after sham procedures revealed normal sinusoidal architecture with sparse neutrophils which traverse through the sinusoids and rapidly exit via the hepatic vein (Figure 2a, left panel; Supplementary Movie 1, left panel). Visualization of livers 24 hours after pIVCL revealed significant sinusoidal dilatation and vascular accumulation of neutrophils which aggregate with platelets (Figure 2a, right panel; Supplementary Movie 1, right panel). We next performed transmission electron microscopy which confirmed early recruitment of neutrophils within the liver sinusoids (Figure 2b) and spatial association with erythrocytes and platelets after pIVCL (Figures 2b and 2c). These findings suggest that neutrophil stasis within the sinusoids may serve as a nidus of platelet interaction and thrombosis in the setting of CH.
Figure 2. Neutrophils and platelets infiltrate liver sinusoids early after pIVCL.

(A) Livers of mice were subjected to in vivo intravital imaging 24 hours after sham (left panel) and pIVCL (right panel) procedures. Mice were injected with fluorescently-conjugated anti-Ly6G and anti-CD49b antibodies and Sytox Green. Neutrophils are shown in red, platelets in blue, and extracellular DNA with Sytox Green. After the sham procedure, scant neutrophils are seen in the sinusoids. In contrast, IVC ligation induces sinusoidal dilation and accumulation of neutrophils which aggregate with platelets. All images were taken at the same fluoresence intensity. (B) Transmission electron microscopy shows infiltration of neutrophils within liver sinusoids 24 hours after pIVCL which associate with erythrocytes (right panel). In contrast, sinusoids are non-dilated with scant erythrocytes after sham operation (left panel) (EC, endothelial cell; N, neutrophil; E, erythrocyte; scale bars, 10 μm). (C) Transmission electron microscopy reveals a neutrophil during late-stage NETosis and its aggregation with platelets 24 hours after pIVCL (N, neutrophil; P, platelets; scale bars, 10 μm). (D)Images of NETs were acquired 24 hours after sham (upper panels) and pIVCL (lower panels) procedures. Extracellular DNA was stained with Sytox Green, histone with red-conjugated antibody, and NE with blue-conjugated antibody. Images from each channel were overlaid to visualize colocalization of NET components. (E) Expression of citrullinated histone 3, a byproduct of NET formation, is increased in liver lysates of mice six weeks after pIVCL (quantification in the adjacent graph). Samples are shown in triplicate. (F) Immunofluorescent staining of liver sections shows increased deposition of fibrin (red) and MPO (green) after pIVCL. Intensity of fibrin and MPO and their colocalization were quantified by ImageJ and displayed in the panel below the images. (G) Serum from patients with cardiac cirrhosis have significantly increased levels of circulating dsDNA-cit-Histone (left panel) and dsDNA-MPO complexes (right panel) compared with healthy controls (n=4–5; *P≤0.05 for all panels).
Platelet interaction with activated neutrophils is a potent inducer of NET formation13, 31, and recent studies suggest that endothelial cells also play a key role in neutrophil chemotaxis and induction of NET formation32, 33. NET components, including histones and granular proteins, have the capability to then initiate or propagate thrombosis and coagulation9, 34. Having visualized extensive neutrophil-platelet aggregation lining liver sinusoids 24 hours after pIVCL, we hypothesized that this interaction may promote the formation of NETs which then propagate microvascular thrombosis, fibrosis, and PHTN in CH. We utilized combination staining with intravital microscopy to visualize colocalization of three key NET components: extracellular DNA (extDNA), neutrophil elastase (NE), and histones. Visualization of livers 24 hours after pIVCL revealed colocalization of these structures within the lining of the liver sinusoids which was absent after the sham procedure (Figure 2d). Furthermore, neutrophils were visualized during the process of NET formation (Figure 2c) aggregating with platelets on TEM. This morphological data confirms that neutrophils are recruited into the sinusoids early after pIVCL and rapidly form NETs after engaging with LSECs and platelets. Consistent with prior histologic observations of CH, few neutrophils were observed within the parenchyma after pIVCL35, 36.
Given the role of sinusoidal thrombosis in portal hypertension and CH, we next used complementary microscopy techniques to ascertain the relationship of NETs with sinusoidal thrombosis. Six weeks after surgery, livers of mice who had undergone pIVCL had significantly increased levels of citrullinated histone 3, the protein byproduct of the peptidyl arginine deiminase 4 (PAD4) enzyme which is a critical mediator of NET formation37 (Figure 2e). The formation of NETs results in the release of double-stranded DNA (dsDNA) which is commonly measured as a marker of NET formation38. Indeed, serum levels of circulating dsDNA were significantly increased in mice which had undergone pIVCL when compared with sham-operated animals (Supplementary Figure 2a). Finally, we then performed additional immunostaining which revealed increased peri-sinusoidal deposition of myeloperoxidase (MPO), a neutrophil granular protein, after pIVCL (Figure 2f). Importantly, MPO was spatially associated with fibrin, suggesting that NET formation serves as a nidus of sinusoidal thrombosis in our pIVCL model of CH. These findings confirm the formation of NETs after the pIVCL procedure and their contribution to thrombosis. To further test our hypothesis, liver samples were obtained from patients who had undergone corrective surgery (Fontan procedure) for congenital heart disease in addition to healthy controls. The Fontan procedure creates an anastomosis between the vena cava or right atrium with the pulmonary arteries and imposes chronic venous passive congestion on the liver as a result39. We performed immunostaining which confirmed that livers from patients with Fontan physiology had increased sinusoidal fibrin which was spatially associated with myeloperoxidase (Supplementary Figure 2b). Serum obtained from patients with cardiac cirrhosis contained higher levels of circulating dsDNA-MPO and dsDNA-cit-Histone complexes compared with healthy controls (Figure 2g), confirming the relevance of our findings in patients with CH. This in total suggests that NET formation contributes to thrombogenesis in patients with CH.
Genetic deletion of neutrophil elastase impairs thrombosis, fibrosis, and PHTN in CH.
Sinusoidal microvascular thromboses are critical mediators of PHTN and fibrosis in CH4. Our studies thus far indicate that pIVCL induces neutrophil accumulation within sinusoids and the formation of NETs. Furthermore, spatial association of NET components with fibrin suggest that NETs contribute to the formation of microvascular thrombosis after the pIVCL procedure14. To determine the impact of NET formation on PHTN and fibrosis, neutrophil elastase (NE−/−) deficient mice were subjected to pIVCL and sham procedures and then compared to wild type mice. NE is a granular protein which is a key component of and driver of NET formation40. Six weeks after pIVCL, NE−/− mice had significantly lower portal pressures (Figure 3a) when compared with wild type mice undergoing pIVCL. Expression of hepatic fibrosis markers α-SMA, collagen 1, and fibronectin were also significantly decreased in NE−/− mice compared to wild type controls as demonstrated by PCR and immunoblotting (Figure 3b and 3c). Hydroxyproline assay confirmed decreased liver collagen content in NE−/− mice (Figure 3d). Next, we utilized immunofluorescent studies to confirm that NE deficiency attenuates intrahepatic thrombosis in the setting of CH. These studies revealed decreased fibrin immunostaining as well as decreased spatial association with collagen (Figure 3e). These results suggest that NE deficiency attenuates PHTN and fibrosis in the setting of CH by preventing thrombosis.
Figure 3. NE−/− mice have attenuated increase in portal pressure and fibrosis after pIVCL.

(A) NE−/− mice have significantly lower portal pressures 6 weeks after pIVCL compared to WT mice (ANOVA P≤0.05). (B) Quantitative reverse transcription polymerase chain reaction from whole liver mRNA shows lower mRNA levels of α-SMA (ANOVA P≤0.05) and collagen 1 (ANOVA P≤0.05) in NE−/− mice after pIVCL compared to WT controls. (C) Western blot analysis reveals decreased fibronectin (ANOVA P≤0.05) and α-SMA (ANOVA P≤0.05) protein levels in whole liver of NE−/− mice after pIVCL compared to WT controls. Hsc70 is a loading control. Quantification is shown in the adjacent panel. (D) Hydroxyproline assay shows lower collagen content in livers of NE−/− mice after pIVCL compared to WT mice (ANOVAP ≤0.05). (E) Collagen (red, ANOVA P≤0.05) and fibrin (green, ANOVA P≤0.05) immunofluorescence was significantly lower in NE−/− mice after pIVCL compared to WT controls. Quantification was performed with ImageJ and displayed in the adjacent graphs (n=5–7; *P≤0.05 for all panels). (F) Pad4−/− mice have significantly lower portal pressures after pIVCL when compared with WT controls (n=4–6; ANOVA P≤0.05).
NET formation has been identified despite inhibition of NE in certain sterile forms of injury41. Citrullination of histones by Pad4 is a critical step in NET formation, and genetic inhibition of Pad4 prevents NET formation42. To verify the role of NETs in thrombosis and PHTN in CH, peptidyl arginine deaminase 4 (Pad4−/−) deficient mice or wild type controls were subjected to pIVCL or sham. Pad4−/− mice had significantly attenuated portal pressure increases when compared with wild type mice after pIVCL (Figure 3f). Immunofluorescent studies confirmed that fibrin formation and sinusoidal MPO deposition was also attenuated after pIVCL in Pad4−/− mice (Supplementary Figure 3a), indicating that Pad4 deficiency prevents NET formation and fibrin clot. These results corroborate the critical role of NETs in driving thrombosis and PHTN in CH.
NE deficiency decreases portal pressure after BDL
To further test our hypothesis that NETs promote PHTN in CH through thrombogenesis, we studied the impact of NE deficiency after BDL, a second model of PHTN. BDL is a cholestatic model of liver cirrhosis which induces hepatocellular injury, cholangiocyte proliferation, and parenchymal infiltration of inflammatory cells, including neutrophils and Kuppfer cells, which culminates in PHTN43, 44. To test our hypothesis that sinusoidal neutrophil recruitment impacts portal pressures, BDL was performed on wild-type (WT) and NE−/− mice. Four weeks after BDL, NE−/− mice had attenuated portal pressure increase compared with WT controls (Figure 4a). NE deficiency also attenuated fibrin formation after BDL, as demonstrated by immunofluorescence (Figure 4b). In contrast to CH, deficiency of NE and NETosis did not interrupt fibrogenesis after BDL, as assessed by PCR, biochemical, and microscopic techniques (Supplementary Figures 4a-4c). We concluded that NE inhibition attenuates portal pressure increases in two model of liver disease by impairing thrombogenesis.
Figure 4. NE−/− mice have decreased PHTN after BDL.

(A) NE−/− mice have lower portal pressures after BDL when compared with WT mice (ANOVA P≤0.05). (B) Immunofluorescent staining shows decreased fibrin content in NE−/− mice after BDL compared to WT mice. Quantification was performed with ImageJ and displayed in the adjacent graph. (C) Sivelestat was administered subcutaneously three times a week for six weeks following pIVCL. Mice treated with sivelestat had lower portal pressures after pIVCL compared with mice treated with DMSO (ANOVA P≤0.05). (D) Fibrin (red) and myeloperoxidase (green, ANOVA P≤0.05) immunofluorescence was significantly lower after sivelestat treatment compared to DMSO treatment. Quantification was performed with ImageJ and displayed in the adjacent graphs (n=5–7; *P≤0.05 for all panels).
To corroborate our genetically derived findings, we next employed pharmacologic NE inhibition after pIVCL. Sivelestat is an inhibitor of NE which has been safely employed in specific clinical scenarios, including interstitial pneumonia45 and acute lung injury46. Sivelestat was administered via subcutaneous injection three times a week for six weeks following pIVCL. Sivelestat-treated mice had significantly lower portal pressures 6 weeks after pIVCL when compared with vehicle-treated mice (Figure 4c). Consistent with our other models of PHTN, livers from sivelestat-treated mice demonstrated less fibrin formation compared with DMSO-treated mice (Figure 4d). Sivelestat treatment also decreased sinusoidal myeloperoxidase (MPO) deposition, suggesting that it disrupts NET formation (Figure 4D).
Cyclic stretch activates integrin-dependent Notch signaling
Given the aforementioned evidence that LSEC responses to mechanical stretch and sinusoidal thrombosis contribute to the pathophysiology of PHTN, we next returned to our in vitro models to explore the mechanosensitive pathways which transduce mechanical stretch in LSECs. For this purpose, we performed RNA-sequencing (RNA-seq) to compare gene expression profiles of stretched and unstretched control primary murine LSECs (Supplementary Figure 5a). Based on two selection criteria (fold change ≥ 2 & FDR < 0.05), 561 genes were identified as transcriptionally regulated by cyclic stretch (Supplementary Figure 5b). This analysis confirmed upregulation of CXCL1 observed from the earlier targeted microarray analysis (logFc 0.981). While CXCL1 is produced by multiple liver cell types in a species and stimulus dependent manner47, 48, RNAseq from cell lines generated from resident liver cell populations demonstrated that LSEC are a major source of CXCL1 under basal conditions (Supplementary Figure 6a) and that CXCL1 production predominates over CXCL2 and CCL2 in LSEC (Supplementary Figure 6b). This was confirmed from in silico analysis of publically accessible RNAseq data from FANTOM as well (Supplementary Figure 6c). Among the larger network of genes impacted by cyclic stretch in LSECs were those related to cellular morphology and function. Network analysis of genes related to cellular morphology and function revealed a network of mechanosensitive cell signaling pathways and molecules, including integrin subunits, the Notch pathway, and molecules related to calcium signaling such as calcium/calmodulin-dependent protein kinase II (CAMKII) and sarco/endoplasmic reticulum calcium-ATPase (SERCA) (Supplementary Figure 7a). Ingenuity Pathway Analysis confirmed that genes related to integrin-linked kinase (ILK) and integrin signaling were significantly impacted by cyclic stretch, including actin subunits, vinculin, and the integrin subunits (Figure 5a). This analysis also confirmed upregulation of the transcriptional target of the Notch pathway, Hairy and Enhancer of Split 1 (Hes1, logFC 1.213), which was verified at the protein level via Western Blot (Figure 5c). Prior studies demonstrate that interactions between the Notch pathway and integrins regulate development and carcinogenesis17, 49, 50, and our network analysis of genes related to morphology and function suggested a potential interaction with calcium signaling (Supplementary Figure 7a). We therefore hypothesized that the integrin-Notch interaction may intersect with calcium-based signaling to mediate CXCL1-dependent angiocrine signaling.
Figure 5. Cyclic stretch utilizes integrins to activate the Notch pathway.

(A) RNA was isolated from primary murine LSECs that were subjected to cyclic stretch. RNA-seq revealed significant upregulation of genes related to integrin signaling. (B) RGD-peptide prevents stretch-induced upregulation of CXCL1 (ANOVA P≤0.05), Hes1 (ANOVA P≤0.05) and Hey1 (ANOVA P≤0.05). (C) Protein expression of Hes1 is higher in HUVEC subjected to cyclic stretch compared with unstretched controls. Quantification was performed using ImageJ with fold change represented in the adjacent graph. (D) Notch1 agonist Jagged-1 upregulates CXCL1 mRNA levels. (E) HUVEC transfected with an siRNA against the Notch1 receptor have lower mRNA levels of CXCL1 after cyclic stretch compared with HUVEC transfected with siControl (ANOVA P≤0.05). siRNA knockdown of Notch is shown in adjacent panel. (F) Treatment of HUVEC with the Notch inhibitor DAPT decreases mRNA expression of CXCL1 (ANOVA P≤0.05) (n=3–5; *P≤0.05 for all panels).
Studies suggest that integrins enhance Notch signaling by regulating intracellular processing and endosomal trafficking of the Notch receptor51. We hypothesized that the Notch signaling pathway serves as a mechanotransducer downstream of stretch-induced integrin activation to culminate in CXCL1 release. To test this hypothesis, we treated HUVEC with arginine-glycine-aspartate (RGD) peptide which inhibits integrin-mediated signaling52. Inhibition of integrin signaling prevented stretch-induced upregulation of the Notch transcriptional targets Hes1 and Hey1 (Figure 5b). Integrin inhibition also prevented upregulation of CXCL1 by stretch, suggesting that integrins activate the Notch pathway to promote CXCL1 expression. Indeed, upregulation of Hes1 was verified at the protein level by Western Blot (Figure 5c), and incubation of HUVEC with the Notch agonist Jagged-1 increased mRNA levels of CXCL1 (Figure 5d). Conversely, transfection of pooled siRNA to Notch1 (siNotch1) decreased CXCL1 upregulation in the setting of stretch (Figure 5e), and primary LSEC isolated from mice with LSEC-specific deletion of Notch1 (Notch1iΔEC) have decreased expression of CXCL1 (Supplementary Figure 8a). Finally, pharmacologic inhibition of Notch signaling with the gamma-secretase inhibitor DAPT also abrogated the stretch-induced stimulation of CXCL1 secretion (Figure 5f). These data suggest that initial integrin mechanosensation activates CXCL1 mechanocrine signaling through the Notch pathway in LSECs.
The Notch pathway interacts with piezo channels to mediate stretch-induced CXCL1 secretion
Our RNA-sequencing suggests interaction of integrins with calcium metabolism in response to mechanical stretch (Supplementary Figure 7a). Integrin activation has been shown to act upstream of calcium channel activation to modulate cell formation and function53, and recent studies suggest that integrins regulate activity of calcium-permeable mechanosensitive ion channels through traction imposed by myosin54, 55. We found that our network analysis of genes related to cell morphology and function impacted by cyclic stretch includes multiple molecules which have been linked to calcium signaling through piezo ion channels, including ERK1/256, SERCA57, Akt58, and CAMKII59. Given the aforementioned evidence of integrin interaction with the Notch pathway, we hypothesized that the Notch pathway may link integrins to piezo channels to impact downstream mechanocrine signaling. To test this hypothesis, we treated HUVEC with the specific piezo1 activator Yoda160 which increased mRNA expression of the Notch transcriptional targets Hes1 and Hey1 as well as CXCL1 (Figure 6a). Conversely, inhibition of piezo1 activity with the pharmacologic inhibitor ruthenium red prevented stretch-induced Notch activation and CXCL1 upregulation (Figure 6b). Transfection of HUVEC with siRNA to piezo1 similarly abrogated this response (Figure 6c). To verify the role of the Notch signaling pathway in piezo1-induced CXCL1 upregulation, HUVEC were transfected with pooled siRNA to Notch1 and then treated with Yoda1. Transfection with siNotch attenuated CXCL1 upregulation in response to Yoda1 (Figure 6d). To ascertain for a biochemical interaction between piezo channels and the Notch pathway, immunoprecipitation assays were performed on protein lysates from primary murine ECs. The cleaved Notch1 receptor was immunoprecipitated and prepared for western blot for piezo1. Indeed, Notch1 receptor and piezo1 protein co-precipitated suggesting a physical interaction between these two proteins (Supplementary Figure 9a). In summary, our in vitro studies construct a multifaceted pathway of mechanocrine signaling which engages integrins, mechanosensitive piezo ion channels, and the Notch pathway in a functional engagement that induces CXCL1 secretion.
Figure 6. Notch pathway interacts with piezo1 channels to upregulate CXCL1.

(A) Stimulation of HUVEC with the piezo1 activator Yoda1 increases mRNA levels of Hes1, Hey1, and CXCL1. (B) Inhibition of piezo1 channels with ruthenium red decreases mRNA levels of CXCL1, Hes1, and Hey1 in HUVEC (CXCL1 ANOVA P≤0.05; Hes1 ANOVA P≤0.05; Hey1 ANOVA P≤0.05). (C) Transfection of HUVEC with siRNA pool to piezo1 decreases upregulation of CXCL1 as well as the Notch targets, Hes1 and Hey1, by cyclic stretch (Hey1 ANOVA P≤0.05; Hes1 ANOVA P≤0.05; CXCL1 ANOVA P≤0.05). siRNA knockdown of piezo1 is shown. (D) Transfection of HUVEC with siRNA pool to Notch1 attenuates the upregulation of CXCL1 by Yoda1 (P≤0.05). siRNA knockdown of Notch1 is shown. (E) Primary LSECs were isolated from mice 48 hours after pIVCL and sham procedures. Quantitative reverse transcription PCR showed increased mRNA levels of Hes1, Hey1, and CXCL1in primary LSECs after IVC ligation compared to sham controls (n=3–5, *P≤0.05 for all panels). (F) Mice with LSEC-specific deletion of Notch1 (Notch1iΔEC) have lower portal pressures when compared with Notchfl/fl mice 4 weeks after IVC ligation (n=7–11; ANOVA P≤0.05).
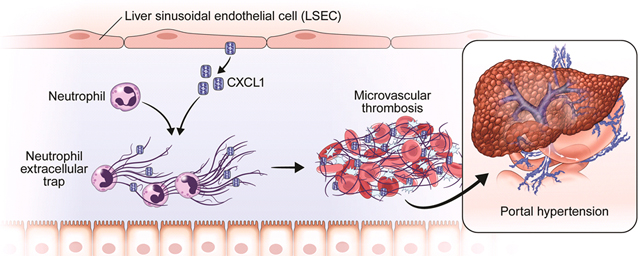
To verify the relevance of our in vitro model of mechanical stretch to the pIVCL model of CH, primary murine ECs were isolated from mice 48 hours after pIVCL. Primary murine ECs isolated 48 hours after pIVCL also demonstrate transcriptional upregulation of the Notch pathway and CXCL1, confirming the relevance of Notch signaling and downstream CXCL1 upregulation in the setting of CH (Figure 6e). Finally, to test the in vivo role of Notch pathway, we performed IVC ligation and sham procedures in mice with LSEC-specific deletion of Notch1 (Notch1iΔEC). Four weeks after pIVCL, Notch1iΔEC mice had attenuated portal pressure increases compared with WT controls (Figure 6f). Endothelial deletion of Notch1 also blunted fibrin formation after pIVCL, as demonstrated by immunofluorescence microscopy (Supplementary Figure 9b). These results elucidate a novel pathway of endothelial mechanocrine signaling which drives PHTN and fibrosis in CH (Figure 7).
Figure 7. Proposed model of mechanocrine signaling and PHTN.

LSECs sense cyclic stretch through integrins. The insert box shows proposed molecular interactions between integrins, piezo1 channels, and the Notch1 receptor. Integrin activated piezo channels bind to the Notch1 receptor which leads to production of downstream transcription factors, Hes1 and Hey1 to promote CXCL1 generation. Integrins are thought to transmit mechanical forces to piezo channels through myosin54, 55. CXCL1 attracts neutrophils which induce sinusoidal thromboses through formation of NETs. Sinusoidal thromboses are pivotal mediators of PHTN through volume-pressure effects within the sinusoidal lumen.
Discussion
PHTN constitutes a common final pathway of chronic liver diseases and is a significant driver of morbidity and mortality. While significant progress has been made in identifying and treating the etiologies of specific liver diseases, few therapies exist to ameliorate their ultimate consequence of PTHN. In the current study, we identify pathways of mechanocrine signaling which are instigated by mechanical stretch and which culminate in microvascular thrombosis, fibrosis, and PHTN (Figure 7). This study contains the following novel findings: 1) mechanical stretch induces Notch-dependent upregulation and secretion of the CXCL1 chemokine by LSECs, 2) CH instigates early vascular recruitment of neutrophils and aggregation with platelets and LSECs via sinusoidal CXCL1 secretion 3) neutrophils/NETs promote microvascular thrombosis and fibrosis in CH, 4) microvascular thrombosis drives PHTN through volume effect in the sinusoids, and 5) interaction of integrins with piezo channels activates the Notch signaling pathway, upregulates CXCL1, and drives CH-induced PHTN. Together, our results identify a novel pathway of endothelial mechanocrine signaling and demonstrate the downstream functional impact of this signaling mechanism on the pathophysiology of PHTN.
CH is characterized by scant parenchymal infiltration of neutrophils, contributing to the traditional description of CH as a non-inflammatory form of fibrosis36, 61. However, our results elucidate a previously unrecognized but critical role that neutrophils within the hepatic sinusoids play in the pathophysiology of CH. We observed that deficiency of NE abrogates the development of fibrosis and microvascular thrombosis in our murine model of CH, suggesting that sinusoidal recruitment of neutrophils is in fact a key component of the pathophysiology of CH. Although neutrophil infiltration into liver tissue is more prominent histologically in cholestatic liver diseases than in CH, we found that NE deficiency decreased portal pressure but did not impact fibrosis after BDL. This observation suggests that thrombosis may not be as critical to the development of fibrosis in cholestatic liver diseases. In CH, fibrosis may result from ischemia and thrombosis induced PHTN which is not the case in cholestatic fibrosis. Thus, neutrophil recruitment to the sinusoids after pIVCL, as opposed to neutrophil recruitment to the parenchyma after BDL, may account for the differences we observed62, 63. Importantly, genetic and pharmacologic inhibition of NE impacts portal pressures in different models of chronic liver injury, suggesting that neutrophil recruitment and NE may constitute a broad novel therapeutic target in PHTN.
NETs are thrombogenic structures implicated in vascular thrombosis. Recent studies suggest that endothelial-derived factors promote neutrophil chemotaxis and NET formation32, 33. Platelets have also been well-described as mediators of NET formation via interaction of platelet toll-like receptor 4 (TLR4) with neutrophils12,64, 65. We postulate that interactions between neutrophils, platelets, and LSECs that we visualize in the liver sinusoids early after IVC ligation instigate NET formation in CH. Hemodynamic forces such as shear stress modulate NET formation in the setting of thrombosis66, suggesting that sinusoidal mechanical forces may further impact NETosis in CH. NETs capture red blood cells and promote clot maturation and stability through interactions with fibronectin, fibrinogen, and von Willebrand Factor14, 67. The release of histones and proteases from NETs further propagates endothelial injury and thrombin generation68. Neutrophil serine proteases such as NE are particularly thrombogenic components of NETs given their ability to promote degradation of tissue factor pathway inhibitor9. We found that genetic and pharmacologic inhibition of NE mitigates portal pressure increases in two models of chronic liver injury by decreasing fibrin formation. We postulate that fibrin physically modulates portal pressures through volume and pressure impact within sinusoids and thereby highlight a critical role of NETs-induced fibrin thrombi in PHTN pathogenesis.
Dilatation of the hepatic sinusoids as occurs in CH confers biaxial stretch on cells lining the sinusoids, notably LSECs and hepatic stellate cells (HSCs). LSECs are the primary sensors of disturbances in portal and arterial circulation. Despite their susceptibility to hemodynamic changes, the mechanisms of mechano-sensation and -transduction in LSECs have not been well-studied. Our RNA-seq analysis of LSECs subjected to cyclic stretch revealed upregulation of genes related to integrin signaling. Integrins interact with cytoskeletal proteins and transmit signals to the cell interior which impact cell differentiation, homeostasis, morphology, and survival69. Integrin subunits have also been implicated in collagen remodeling, suggesting that integrins both sense and modulate mechanical forces70, 71. Our study suggests that endothelial integrins may contribute to pressure changes in CH by transducing changes in the sinusoidal environment to generate inflammatory and fibrotic mechanocrine signals. Recent studies of LSEC gene expression in a fibrotic matrix also revealed changes of genes related to chemokine signaling and cytokine receptor interaction70. Generation of chemokines such as CXCL1 occurs in multiple liver cell types in addition to LSEC including hepatocytes and inflammatory cells72. This is likely dependent upon species as well as the physiologic or pathophysiologic stimulus. CXCL1 production may also occur as part of a larger chemokine program given redundancy of function and signal activation of multiple CXC chemokines73. Nonetheless, our study provides evidence that mechanical sensation by LSEC can drive chemokine dependent changes in vascular structure and function with CXCL1 serving as a prototype.
We found that integrin activation contributes to a mechanosensitive pathway which entails functional engagement of the Notch pathway with piezo proteins to generate CXCL1 release in response to mechanical stretch. Studies suggest that the Notch1 receptor is a pivotal mediator of liver sinusoidal structure and portal pressure18, 74, 75. Prior studies report that mice with disruption of Notch1 signaling in LSECs and hepatocytes develop a phenotype resembling nodular regenerative hyperplasia (NRH) with prominent angio-architectural distortions18,76. Our data suggest that the Notch1 receptor modulates sinusoidal tone and portal pressure in response to mechanical forces by regulating formation of fibrin within liver sinusoids demonstrating the multifaceted role of Notch signaling in modulation of portal pressure. We also found that Notch activation by mechanical stretch relies on integrin mechanosensation. Integrins exert traction forces on piezo channels likely through myosin which enhance their activation. Piezo1 channels then physically engage with the Notch1 receptor to modulate CXCL1 expression. While piezo1 expression has been described in human liver endothelium77, its role as a calcium channel in the development of liver disease has not been explored. Calcium influx impacts endothelial vasodilation and vascular tone through several mechanisms78, including calcium-dependent modulation of vasodilatory molecules such as eNOS and endothelium-derived hyperpolarization (factor) (EDH(F))20 and through activation of tissue transglutaminases and endothelial remodeling79. Recent studies implicate piezo channels in the upregulation of blood pressure during physical activity20. Our results suggest that piezo channels also contribute to vascular tone by participating in mechanocrine signaling pathways whose downstream targets impact portal pressure.
In total our work identifies a series of new pathophysiologic pathways and associated potential therapeutic targets in PHTN and other diseases within hepatic sinusoids which are attributed to mechanical forces.
Supplementary Material
Acknowledgements:
Mayo Center for Cell Signaling in Gastroenterology (NIDDK P30DK084567)
RNA sequencing accession number: GSE119547
Grant support: Supported by National Institutes of Health (NIH), USA R01 DK59615 and R01 AA21171 (VHS)
Abbreviations:
- PHTN
portal hypertension
- LSEC
liver sinusoidal endothelial cell
- pIVCL
partial inferior vena cava ligation
- NET
neutrophil extracellular trap
- NE
neutrophil elastase
- BDL
bile duct ligation
- CH
congestive hepatopathy
- DMSO
dimethyl sulfoxide
- HUVEC
human umbilical vein endothelial cells
- extDNA
extracellular DNA
- PAD4
protein arginine deiminase 4
- dsDNA
double-stranded DNA
- cit-Histone
citrullinated histone
- MPO
myeloperoxidase
- α-SMA
alpha smooth muscle actin
- CAMKII
calcium/calmodulin-dependent protein kinase II
- Hes1
Hairy and Enhancer of Split 1
- RGD peptide
arginine-glycine-aspartate peptide
- EC
endothelial cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have declared that no conflicts of interest exist.
Please see supplement for additional references.
References
- 1.Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J Hepatol 2015;62:S121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonetto DA, Yang HY, Yin M, et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology 2015;61:648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villa E, Camma C, Marietta M, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012;143:1253–60e1–4. [DOI] [PubMed] [Google Scholar]
- 4.Cerini F, Vilaseca M, Lafoz E, et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J Hepatol 2016;64:834–42. [DOI] [PubMed] [Google Scholar]
- 5.Sreeramkumar V, Adrover JM, Ballesteros I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014;346:1234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Wang Q, Wang J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) Deficiency is Protective Against Venous Thrombosis in Mice. Sci Rep 2017;7:14360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 2012;209:819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pak S, Kondo T, Nakano Y, et al. Platelet adhesion in the sinusoid caused hepatic injury by neutrophils after hepatic ischemia reperfusion. Platelets 2010;21:282–8. [DOI] [PubMed] [Google Scholar]
- 9.Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 2010;16:887–96. [DOI] [PubMed] [Google Scholar]
- 10.Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 2013;110:8674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532–5. [DOI] [PubMed] [Google Scholar]
- 12.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007;13:463–9. [DOI] [PubMed] [Google Scholar]
- 13.McDonald B, Urrutia R, Yipp BG, et al. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012;12:324–33. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 2010;107:15880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature 2016;529:316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gauthier NC, Roca-Cusachs P. Mechanosensing at integrin-mediated cell-matrix adhesions: from molecular to integrated mechanisms. Curr Opin Cell Biol 2018;50:20–26. [DOI] [PubMed] [Google Scholar]
- 17.LaFoya B, Munroe JA, Mia MM, et al. Notch: A multi-functional integrating system of microenvironmental signals. Dev Biol 2016;418:227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuervo H, Nielsen CM, Simonetto DA, et al. Endothelial notch signaling is essential to prevent hepatic vascular malformations in mice. Hepatology 2016;64:1302–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res 2007;100:1556–68. [DOI] [PubMed] [Google Scholar]
- 20.Rode B, Shi J, Endesh N, et al. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat Commun 2017;8:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coste B, Xiao B, Santos JS, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 2012;483:176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao S, Yaqoob U, Das A, et al. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest 2010;120:2379–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semela D, Das A, Langer D, et al. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology 2008;135:671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng Z, Surewaard BG, Wong CH, et al. CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe 2016;20:99–106. [DOI] [PubMed] [Google Scholar]
- 25.Ding BS, Nolan DJ, Butler JM, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010;468:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sackey-Aboagye B, Olsen AL, Mukherjee SM, et al. Fibronectin Extra Domain A Promotes Liver Sinusoid Repair following Hepatectomy. PLoS One 2016;11:e0163737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anwar MA, Shalhoub J, Lim CS, et al. The effect of pressure-induced mechanical stretch on vascular wall differential gene expression. J Vasc Res 2012;49:463–78. [DOI] [PubMed] [Google Scholar]
- 28.Cummings CJ, Martin TR, Frevert CW, et al. Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J Immunol 1999;162:2341–6. [PubMed] [Google Scholar]
- 29.Sawant KV, Poluri KM, Dutta AK, et al. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci Rep 2016;6:33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y, Sun J, Lin WH, et al. A compact microfluidic gradient generator using passive pumping. Microfluid Nanofluidics 2012;12:887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SJ, Jenne CN. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin Immunol 2016;28:546–554. [DOI] [PubMed] [Google Scholar]
- 32.Gupta AK, Joshi MB, Philippova M, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett 2010;584:3193–7. [DOI] [PubMed] [Google Scholar]
- 33.Gollomp K, Kim M, Johnston I, et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akiyoshi H, Terada T. Centrilobular and perisinusoidal fibrosis in experimental congestive liver in the rat. J Hepatol 1999;30:433–9. [DOI] [PubMed] [Google Scholar]
- 36.Arcidi JM Jr., Moore GW, Hutchins GM. Hepatic morphology in cardiac dysfunction: a clinicopathologic study of 1000 subjects at autopsy. Am J Pathol 1981;104:159–66. [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009;184:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Margraf S, Logters T, Reipen J, et al. Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock 2008;30:352–8. [DOI] [PubMed] [Google Scholar]
- 39.Asrani SK, Asrani NS, Freese DK, et al. Congenital heart disease and the liver. Hepatology 2012;56:1160–9. [DOI] [PubMed] [Google Scholar]
- 40.Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 2010;191:677–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.