

Abstract
Background & Aims:
The peroxisome proliferator activated receptor delta (PPARD) regulates cell metabolism, proliferation, and inflammation and has been associated with gastric and other cancers. Villin-positive epithelial cells are a small population of quiescent gastric progenitor cells. We expressed PPARD from a villin promoter to investigate the role of these cells and PPARD in development of gastric cancer.
Methods:
We analyzed gastric tissues from mice that express the Ppard (PPARD1 and PPARD2 mice) from a villin promoter, and mice that did not carry this transgene (controls), by histology and immunohistochemistry. We performed cell lineage tracing experiments and analyzed the microbiomes, chemokine and cytokine production, and immune cells and transcriptomes of stomachs of these mice. We also performed immunohistochemical analysis of PPARD levels in in 2 sets of human gastric tissue microarrays.
Results:
Thirty-eight percent of PPARD mice developed spontaneous, invasive gastric adenocarcinomas, with severe chronic inflammation. Levels of PPARD were increased in human gastric cancer tissues, compared with non-tumor tissues, and associated with gastric cancer stage and grade. We found an inverse correlation between level of PPARD in tumor tissue and patients survival time. Gastric microbiomes from PPARD and control mice did not differ significantly. Lineage-tracing experiments identified villin-expressing gastric progenitor cells (VGPCs) as the origin of gastric tumors in PPARD mice. In these mice, PPARD upregulated CCL20 and CXCL1, which increased infiltration of the gastric mucosa by immune cells. Immune cell production of inflammatory cytokines promoted chronic gastric inflammation and expansion and transformation of VGPCs, leading to tumorigenesis. We identified a positive-feedback loop between PPARD and interferon gamma signaling that sustained gastric inflammation to induce VGPC transformation and gastric carcinogenesis.
Conclusions:
We found PPARD overexpression in VPGCs to result in inflammation, dysplasia, and tumor formation. PPARD and VGPCs might be therapeutic targets for stomach cancer.
Keywords: tumor stem cell, IFNG, mouse model, nuclear factor
INTRODUCTION
Gastric cancer is the third leading cause of cancer mortality worldwide, with a 5-year survival rate of less than 25%1 The critical molecular factors that drive gastric tumor initiation and progression need to be identified to develop effective targeted strategies for gastric cancer prevention and treatment.
Gastric carcinogenesis is strongly associated with chronic inflammation2, 3. Although gastric cancer has marked heterogeneity, the two most common subtypes are intestinal-type gastric adenocarcinoma (GAC) and diffuse gastric cancer, each with different epidemiologic and pathophysiologic features. GAC is the more common type and typically emerges following chronic inflammation, intestinal metaplasia (IM), dysplasia, and finally invasive adenocarcinoma.
Intensive research efforts have been undertaken to define the cell types responsible for gastric cancer initiation. Accumulating evidence from animal model studies supports that gastric cancer originates from gastric stem cells4–10. Specifically, villin-positive epithelial cells that have been identified as a small population of quiescent gastric progenitor cells, called villin-expressing gastric progenitor cells (VGPCs)8, have been proposed as the cell of origin of gastric cancer8, 9. At present, the critical molecular changes that drive the transformation of these normal gastric progenitors/stem cells to promote gastric tumorigenesis remain poorly defined.
Peroxisome proliferator-activated receptor delta (PPARD) is a ligand-dependent nuclear receptor that functions as a transcription factor to regulate physiologic processes involved in cell metabolism, proliferation, and inflammation11, 12. PPARD expression is upregulated in many cancers, such as breast13, colon14, 15 and lung16 cancer. However, the functional role of PPARD in tumorigenesis has remained controversial. In particular, studies of intestinal tumorigenesis in Apcmin mice have reported conflicting results on the effect of germline PPARD knockout on intestinal tumorigenesis17, 18. For gastric cancer, limited studies have investigated the potential role of PPARD in gastric tumorigenesis. Helicobacter pylori (H. pylori), a class I carcinogen associated with human gastric cancer3, upregulates PPARD expression to increase gastric epithelial cell proliferation in humans and mice19. Genetic variants of PPARD might alter the risk of gastric cancer in humans20. GW501516, a synthetic selective PPARD agonist, promoted carcinogen 7,12-dimethylbenzanthracene–induced squamous gastric tumor of the forestomach, a rare form of human gastric cancer21. Together, these reports suggest a link between PPARD and gastric cancer; however, evidence to support this link remains insufficient, especially regarding the most common gastric cancer type, GAC.
Here, we provide direct evidence that PPARD can act as a potent driver of gastric cancer. We found that mice with villin promoter–driven PPARD overexpression in VGPCs spontaneously developed gastric tumors that progressed into large, invasive GAC. Our in-depth mechanistic studies uncovered a positive feedback loop between PPARD and interferon gamma as a novel mechanism by which PPARD drives gastric tumorigenesis.
MATERIALS AND METHODS
Please refer to the online Supplementary Material for detailed additional Methods.
Animals
PPARD mice were produced from two independent founders (PPARD1 and PPARD2) that were generated at The University of Texas MD Anderson Cancer Center Genetically Engineered Mouse Facility by pronuclear injection of mouse PPARD expression construct under the control of a villin promoter (p12.4Kvill–PPARD) into fertilized FVB oocytes (Supplementary Figure 1A)22. B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (LSL–tdTomato) mice were purchased from The Jackson Laboratory (#007914), and villin-cre mice were generated as previously described23. The two PPARD lines were found to exhibit similar PPARD expression levels and display similar phenotypes (Supplementary Figure 1B and C)22. Therefore, all subsequent experiments were performed using randomly selected PPARD1 and PPARD2 mice, designated as PPARD hereafter unless otherwise specified.
Statistical analysis
Statistical significance was determined by the unpaired Student t-test or analysis of variance (one-way or two-way) with Bonferroni adjustments for all multiple comparisons. The statistical significance of the correlation of two factors was determined by Spearman correlation analysis. Kaplan-Meier survival analysis and the log-rank test were used to compare survival outcomes. All tests were two-sided, and significance was defined at P < 0.05. Data were analyzed using SAS software, version 9.4 (SAS Institute, Cary, NC) or GraphPad Prism 7.01 (GraphPad Software, La Jolla, CA). Values presented are mean ± standard error of the mean (SEM) (*P < .05, **P < .01, ***P < .001, and ****p < .0001).
RESULTS
PPARD overexpression in villin-expressing cells drives GAC development
Longitudinal follow-up of two PPARD mouse lines (PPARD1 and PPARD2 mice) unexpectedly revealed that mice from both lines spontaneously developed large, invasive GAC (Figure 1A-D). Gastric tumorigenesis progressed in an age-dependent fashion from normal-appearing mucosa at 10 weeks to hyperplasia and low-grade dysplasia at 25 weeks, high-grade dysplasia at 35 weeks, and finally large, invasive GAC at 55 weeks (Figure 1 C, E-l). Gross lesions were initially found in the lesser curvature of the gastric corpus at 25 weeks and eventually expanded to occupy the whole gastric corpus at 55 weeks (Figure 1A). All PPARD1 and PPARD2 mice examined at ≥25 weeks spontaneously developed at least gastric hyperplasia. At 35 weeks, 59 of 97 mice (60.8%) developed low-grade or high-grade gastric dysplasia; and at 55 weeks, 87 of 120 mice (72.5%) developed GACs, including 45 mice (37.5%) with large, invasive GACs (Figure 1C). None of the wild-type (WT) littermates that were followed concomitantly developed gastric tumors.
Figure 1. Villin promoter–driven PPARD overexpression induced gastric adenocarcinoma in mice.

(A-C) Gross and histologic examination of the stomachs in PPARD mice. Stomach photographs of PPARD1 and PPARD2 mouse lines and their wild-type (WT) littermates (A), with black arrows indicating tumors, and mouse stomach to body weight ratios (B) and incidence of gastric lesions(C) for mouse lines shown in panel A at indicated ages (LGD: low-grade dysplasia, HGD: high-grade dysplasia).
(D) Representative hematoxylin and eosin (H&E) staining microphotographs of entire gastric mucosa sections (diagram, top) of 35-week-old PPARD and WT littermate mice. Red dotted circle indicates gastric tumor in the corpus.
(E-I) H&E staining of representative histologic features of gastric mucosa from PPARD mice showing a sequence of morphologic changes of the stomach: normal mucosa (E), t marked hyperplasia of glandular epithelium (F), LGD (G) and HGD (H) of glandular epithelium, and invasive adenocarcinoma into the gastric muscle layers (I).
(J) Representative images of PPARD immunohistochemical analysis (IHC) of human gastric tissue microarrays (#ST781, US Biomax).
(K) Total nuclear and cytoplasm PPARD IHC combined expression scores (CES) for the human gastric tissue microarrays in panel J.
Scale bars represent 1 cm (A), 1 mm (D, E-I [top]), 100 μm (E-I [bottom]), and 50 μm (J).
PPARD expression is upregulated in human GAC and negatively associated with the survival time of GAC patients
Examination of PPARD expression by immunohistochemistry in a human gastric tissue array showed that PPARD was weakly expressed in the nucleus of normal gastric mucosal cells but was upregulated in the paired GAC-adjacent tissues, which had chronic gastritis with IM, and was further increased in the nucleus and cytoplasm of the GAC cells (Figure 1J and K, Supplementary Figure 2A). Higher PPARD expression in GAC than in paired normal tissues was confirmed in a second set of 90 human GAC cases, in which elevated nuclear and cytoplasmic PPARD expression in GAC tissues was associated with higher clinical stage (I-IV), pathologic grade (G1-G3), and primary tumor category (T1-T4) as well as with metastasis to lymph nodes or distant organs (Supplementary Figure 2A-E). Interrogation of several public databases of gastric cancer patients showed that PPARD upregulation in gastric cancer was negatively correlated with overall survival, disease/progression-free survival, and post-progression survival (Supplementary Figure 2F-H).
Villin promoter–driven PPARD expression in the mouse stomach induces chronic inflammation, parietal cell loss, spasmolytic polypeptide-expressing metaplasia, and IM
Chronic inflammation scores of gastric mucosa were significantly higher in PPARD mice than in their WT littermates, and these scores increased with mouse age and with the progression of the lesions from hyperplasia to invasive adenocarcinoma in the PPARD mice (Figure 2A-C, Supplementary Figure 3A). Parietal cells were gradually lost as the mice aged (Figure 2D and E). While the majority of GACs were well differentiated, foci of moderately to poorly differentiated adenocarcinoma were noted in mice at 55 weeks old (Figure 2F and G).
Figure 2. Villin promoter–driven PPARD overexpression induced chronic inflammation and premalignant changes in mice.
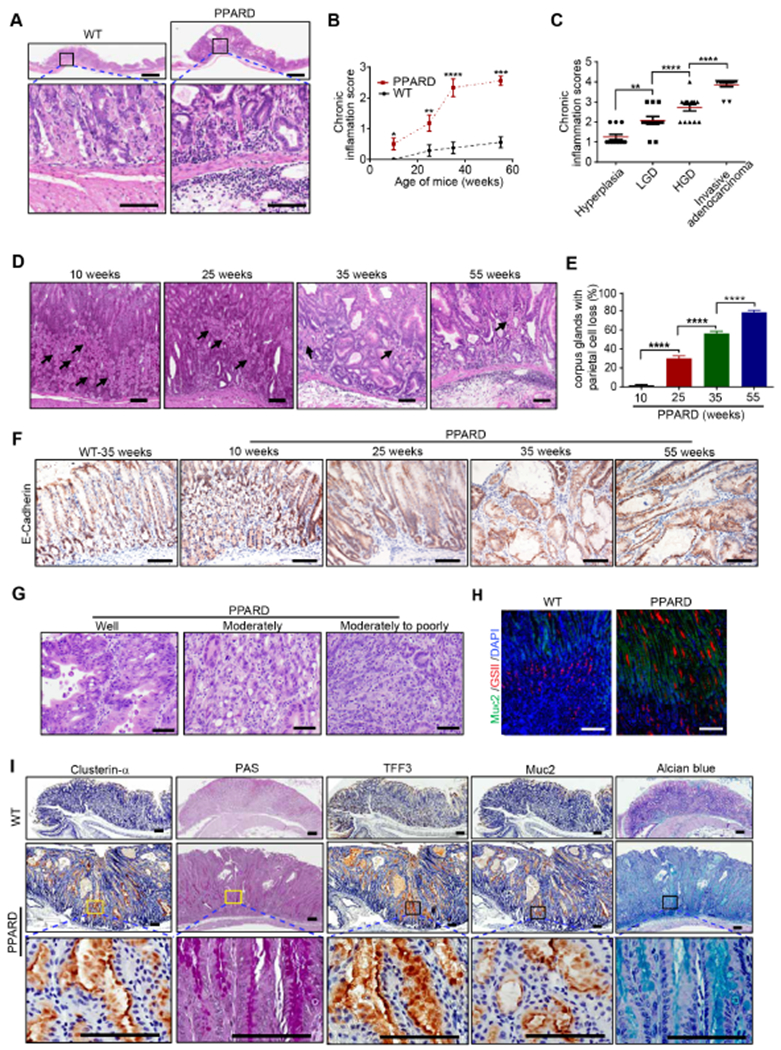
(A) Representative H&E-stained images of chronic inflammation induced in gastric corpus of PPARD mice in contrast to their WT littermates at 35 weeks.
(B, C) Gastric corpus inflammation scores of PPARD and WT mice by age (B) and morphology (C).
(D, E) Representative H&E images of gastric corpus from PPARD mice showing parietal cells indicated by black arrows (D) and their quantitation, analyzed for 50 glands per mouse (E). n=15-20 per group.
(F) Representative E-cadherin IHC microphotographs in gastric corpus of PPARD mice and WT mice at the indicated ages.
(G) Representative images of PPARD–induced gastric adenocarcinoma differentiation in PPARD mice at 55 weeks.
(H, I) Representative microphotographs of GSII and Muc2 immunofluorescence (H) and clusterin-α, TFF3, and Muc2 IHC and of PAS and alcian blue staining (I) in the gastric corpus of PPARD mice and WT littermates at 35 weeks.
Scale bars, 1 mm (A [top]) and 100 μm (A [bottom], D, F, G-I).
PPARD mice also developed two gastric metaplasia stages24: spasmolytic polypeptide-expressing metaplasia (SPEM) and IM, which were distinguished by expression markers (clusterin-α and GSII for SPEM and TFF3 and Muc2 for IM) and were stained using periodic acid–Schiff (PAS) for SPEM and alcian blue for IM24. At 10 weeks, PAS- and alcian blue–positive cells could be observed at the mucosal luminal surface and GSII-positive cells at the gland necks of the PPARD mice. As the mice aged, the cells stained with PAS and alcian blue gradually moved deeper to the base of the glands, while GSII-positive cells moved bi-directionally to the base and the lumen of the glands (Supplementary Figure 3B-D). At 35 weeks, PPARD mice had more SPEM and IM cells than WT littermates did (Figure 2H and I). Together, these data indicate that PPARD mice developed gastric parietal cell loss and SPEM and IM lesions as pre-malignant steps of GAC.
GAC development in PPARD mice was unrelated to stomach microbiota differences
Alteration of stomach microbiota, most notably H. pylori infection, can cause chronic inflammation to promote gastric cancer 3. Evaluation of the microbial composition of the stomach contents of PPARD mice and their WT littermates at 10 weeks showed that the stomach microbial communities of WT and PPARD mice were indistinguishable, as evidenced by a similar community richness and composition of the stomach microbiomes (Supplementary Figure 3E and F; 11,216 16Sv4 reads per sample).
PPARD overexpression in VGPCs expands VGPC population in PPARD mice
VGPCs are long-lived progenitor cells that can generate multi-lineage cell populations to reconstitute entire gastric glands8. To characterize the distributions and stem-like properties of VGPCs in the mouse stomach, we performed fate-mapping experiments using LSL-tdTomato;villin-cre mice generated by breeding LSL-tdTomato mice with villin-cre mice, in which the progeny of VGPCs are marked by tdTomato red fluorescent protein expression (tdTomato). Analyses of frozen tissue sections and isolated glands of the gastric corpus and antrum showed that only small portions of glands had tdTomato-marked VGPCs, which were in the isthmus of the gastric corpus glands and in the base of the gastric antrum glands (Supplementary Figure 4A-C). More importantly, VGPCs bi-directionally expanded upward into the lumen and deeper into the base of the glands to generate entire glands in the corpus, but not in the antrum, suggesting that corpus VGPCs are more stem-like than antrum VGPCs (Supplementary Figure 4A-C). tdTomato-expressing cells coexpressed villin in the gastric corpus (Supplementary Figure 4D), thus confirming that VGPCs retain villin expression.
To examine whether PPARD expression in VGPCs enhances the stem-like properties of VGPCs in the gastric corpus to drive gastric tumorigenesis, we traced VGPCs by villin immunofluorescence staining in the stomach. PPARD–induced gastric tumors were highly enriched with VGPCs, even at an early stage in tumor development in the lesser curvature of the gastric corpus (Figure 3A, Supplementary Figure 4E and F). The number of villin-marked VGPCs markedly increased in PPARD mice as they aged (Figure 3B and C). At 10 weeks, before gastric tumor development, VPGCs were confined to the isthmus of the glands, while at 25 weeks and 35 weeks, VPGCs expanded bi-directionally toward the lumen and base of the glands (Figure 3C). The VGPCs approaching the lumen developed tumor-associated morphologic changes earlier than did the VGPCs approaching the base (Figure 3C). WT littermates exhibited markedly less villin expression in the stomach, which was limited to the isthmus of corpus glands and the base of antrum glands (Supplementary Figure 4F-H). Villin antibody specificity was confirmed by strong staining of duodenal epithelial cells, in which villin is known to be highly expressed (Supplementary Figure 4F and I). Also, villin was expressed at the boundary of the corpus and forestomach (Supplementary Figure 4F and J), consistent with a previous report8. Numbers of PPARD–positive and villin-positive gastric epithelial cells increased in PPARD mice from ages 10 to 35 weeks (Figure 3D and E). Double immunostaining of villin and the cell proliferation marker Ki67 showed that the Ki67-positive VGPC population markedly increased in PPARD mice as the mice aged (Figure 3F [top] and G). These findings demonstrate that PPARD overexpression in VGPCs markedly expands the VGPC population in PPARD mice.
Figure 3. PPARD expanded villin-positive gastric progenitor cell population in mice.
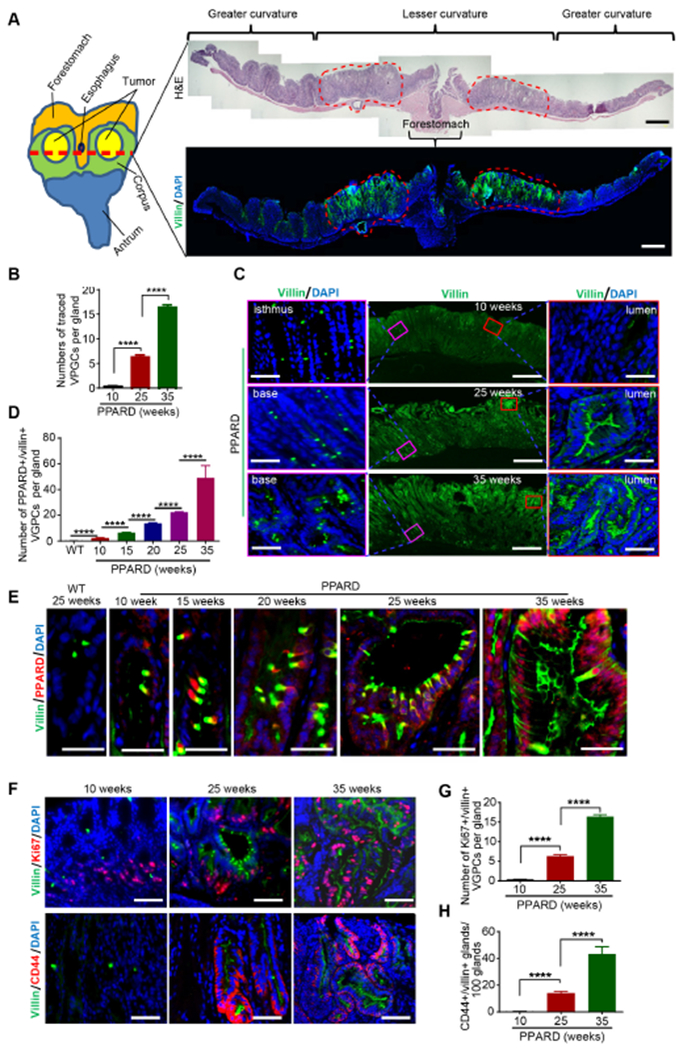
(A) Representative images for a PPARD mouse gastric corpus at age 25 weeks. Red dotted circles indicate gastric tumors. The red dotted line (left) in the diagram shows the cutting direction for H&E staining and villin immunostaining; the stomach was opened from the greater curvature.
(B) Numbers of villin-positive gastric progenitor cells (VGPCs) per gastric corpus gland in PPARD mice.
(C) Representative immunostaining images for villin in PPARD mouse gastric corpus, corresponding to panel B.
(D) Numbers of PPARD–positive and villin-positive cells in the corpus of PPARD mice and WT mice.
(E) Representative corpus images of villin and PPARD immunostaining, corresponding to panel D.
(F-H) Representative immunofluorescence images of villin and Ki67 (F, top) and of villin and CD44 (F, bottom) in the corpus of PPARD mice with corresponding quantitative results of Ki67- and villin-positive cells (G) and villin- and CD44-positive cells (H).
Fifty glands per mouse were analyzed (n=5 per group) for panel B, D, G and H. Scale bars, 1 mm (A[right], and C [middle]), 50 μm (C [left and right], E and F).
PPARD enhances stemness and tumorigenicity of VGPCs in PPARD mice
We next examined the effects of PPARD overexpression on VGPCs’ stemness properties. CD44, an established gastric cancer stem cell marker25, is a sensor of inflammatory microenvironments that activates stem cell proliferation26, 27. CD44 expression increased along with the villin upregulation and VGPC expansion as the PPARD mice aged (Figure 3F [bottom] and H). Consistent with this finding, mRNA and protein expression levels of PPARD, villin, and CD44 were also significantly higher in the corpus mucosa from PPARD mice than in that from WT littermates starting at 10 weeks of age and markedly increased further in PPARD mice at 35 weeks (Supplementary Figure 4K-N). This CD44 upregulation was accompanied by increased PPARD transcriptional activity, as indicated by upregulation of mRNA for PPARD target gene angiopoietin-like 4 (ANGPTL4), and by increased proliferation of gastric mucosa in PPARD mice compared with WT mice (Supplementary Figure 40 and P). Co-staining human corpus GAC glands for villin and CD44 expression, to determine the relevance of mouse findings to human GAC, showed strong co-expression of villin and CD44 (Supplementary Figure 4Q). Thus, PPARD overexpression in VGPCs upregulated the GAC cell stemness enhancer CD4425 in VGPCs.
We examined the mechanistic significance of PPARD in the self-renewal capacity and transformation of VGPCs using three-dimensional organoid culture28. Gastric organoids derived from PPARD mice were more numerous per gastric crypt and larger than the WT littermate–derived organoids (Supplementary Figure 5A-C). PPARD agonist GW501516 increased the number and size of both PPARD mouse–derived and WT mouse–derived organoids (Figure 4A [top] and B). Furthermore, GW501516 treatment altered the cell features and arrangement of WT spheroids (immature organoids), producing irregularly shaped spheroids with pseudo-stratification, lumen-like spaces, and apoptotic debris. In contrast, untreated WT mouse–derived spheroids exhibited well-organized circular or oval spheroids lined by a single layer of cuboidal or low columnar epithelium (Figure 4A [bottom]). PPARD mouse–derived spheroids had multilayered, disorganized, irregular epithelial cells that failed to form a lumen (Figure 4A [bottom]), and GW501516 treatment of these spheroids induced more such malignant features: multilayered, irregular cells that appeared to be attempting to form multiple small lumen-like spaces, which are typically seen in human adenocarcinomas. PPARD mouse–derived organoids had higher proportions of CD44 and villin-expressing cells than WT mouse–derived organoids did (Figure 4C and D). Organoids that were derived from triple-transgenic LSL-tdTomato; villin-cre; PPARD mice (td–PPARD) had a higher percentage of tdTomato–marked organoids than did organoids derived from their WT LSL-tdTomato; villin-cre control littermates (td-WT) (Figure 4E).
Figure 4. PPARD induced malignant transformation of VGPCs.
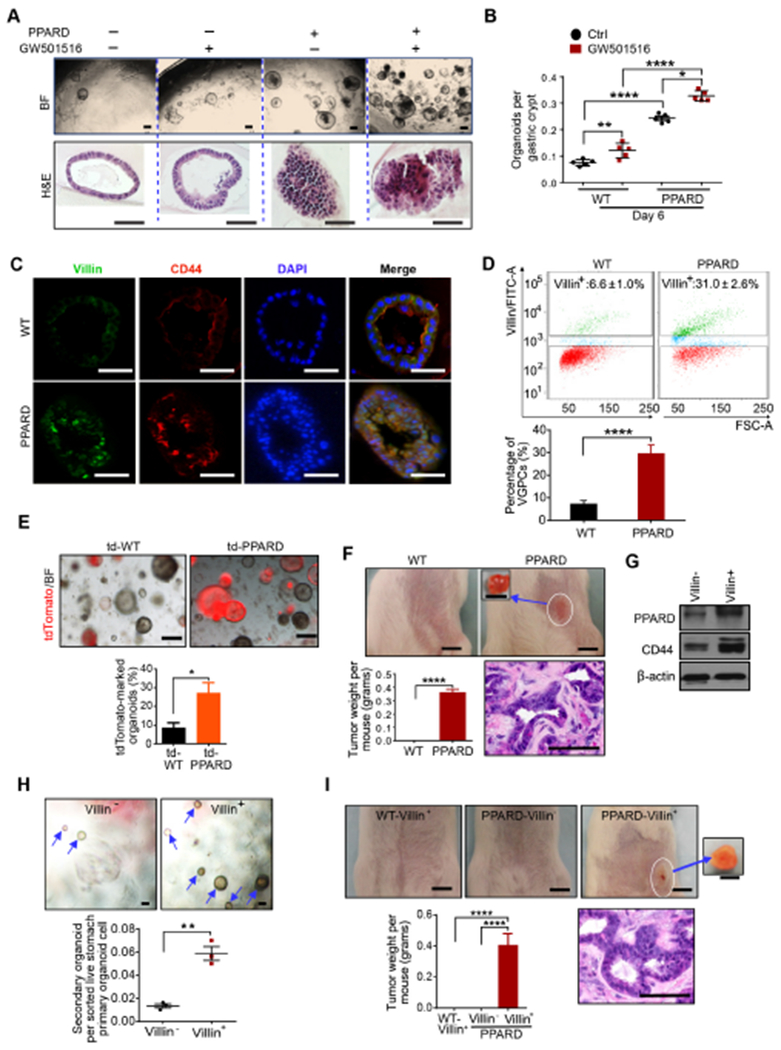
(A, B) Representative images (A) and numbers of primary organoids per gastric crypt (B) of PPARD and WT mice with and without GW501516 treatment (n=5 per group). BF indicates bright field light.
(C) Representative villin and CD44 immunostaining images of organoids described in panel A and B.
(D) Villin-positive cells (villin+) sorted by flow cytometry from gastric organoids of PPARD and WT mice.
(E) Representative primary gastric organoid images and precentage of tdTomato-marked gastric organoids at day 10 of culture for gastric organoids that are derived from LSL-tdTomato; villin-cre; PPARD (td–PPARD) mice and their WT LSL-tdTomato; villin-cre (td-WT) littermates (n=3 per group).
(F) Tumor formation by subcutaneous injection of primary gastric organoids, derived from PPARD mice and WT littermates (14 days after injection). Representative tumor images, tumor weights, and H&E-stained sections are shown.
(G-I) Flow cytometry–sorted villin-positive (villin+) and villin-negative (villin−) cells of primary organoids, derived from PPARD mice, were examined for PPARD and CD44 protein expression (G) , and secondary organoid formation (H), and tumor formation by subcutaneous injection of secondary organoid cells derived from sorted villin+ and villin− cells from PPARD mice, and sorted villin+ cells from control WT mice into syngeneic mice was assessed at 14 days after injection (I).
Scale bars, 200 μm (A [top]), 100 μm (A [bottom], C, E, F [bottom], H, and I [bottom]), and 5 mm (top for both F and I).
Next, we examined whether PPARD overexpression in VGPCs could enhance tumorigenicity in an organoid transplantation assay. Indeed, organoid cells derived from PPARD mice, but not those from their WT littermates, formed tumors when subcutaneously injected into immunocompetent syngeneic mice (Figure 4F). To better define the VGPC population within the heterogeneous organoids, we used FACS to sort organoid-derived cells into villin-positive (VGPC) and villin-negative sub-populations. Villin-positive cells had higher PPARD and CD44 expression and formed more secondary organoids in subsequent three-dimensional organoid culture compared with villin-negative cells in PPARD mice (Figure 4G and H). More importantly, only PPARD mouse–derived VGPCs, but not the WT mouse–derived VGPCs or PPARD mouse–derived villin-negative cells, formed tumors when injected into immunocompetent syngeneic mice (Figure 4I). Together, our findings provide strong evidence that PPARD overexpression in VGPCs not only increases their self-renewal capacity (Figure 4A-E, G and H, Supplementary Figure 5A-C) but also transforms these cells and confers on them a tumor-initiating capacity (Figure 4F and I).
To evaluate the mechanistic significance of PPARD in driving gastric tumorigenesis, we generated mouse gastric cancer cell lines using gastric tumor tissues from three PPARD mice at age 55 weeks. These cell lines formed secondary tumors when injected subcutaneously into syngeneic mice (Supplementary Figure 5D-F). These cell lines and the secondary tumors expressed the gastric epithelial marker keratin 19, confirming their gastric epithelial origin (Supplementary Figure 5G). Furthermore, PPARD downregulation by lentiviral shRNAs in these PPARD mouse gastric tumor–derived cell lines markedly inhibited tumorigenicity when the cells were injected subcutaneously into syngeneic mice (Supplementary Figure 5H-J), indicating that PPARD contributes substantially to the tumorigenicity of these cells. Downregulation of PPARD expression also significantly decreased CD44 expression and attenuated the spheroid-forming ability of these gastric cancer cells (Supplementary Figure 5H, K, and L), whereas PPARD agonist GW501516 promoted their spheroid-forming ability (Supplementary Figure 5M and N), further substantiating the mechanistic significance of PPARD in promoting stemness and tumorigenicity.
PPARD upregulation of CCL20 and CXCL1 chemo-attracts immune cell infiltration of gastric lesions to promote gastric chronic inflammation
Because gastric tumor development was strongly associated with chronic inflammation in PPARD mice (Figure 2A-C, Supplementary Figure 3A), we investigated the mechanisms by which PPARD overexpression increases recruitment of pro-inflammatory immune cells into gastric mucosa. We used the LEGENDplex Mouse Proinflammatory Chemokine Panel to screen for chemokine proteins in the culture medium from primary organoids derived from mouse gastric corpus crypts (Figure 4E) and secondary organoids derived from tdTomato sorted–VGPCs (Supplementary Figure 6A) from td–PPARD mice and td-WT littermates. Organoids derived from td–PPARD mice had significantly higher levels of secreted CCL20 and CXCL1 than did those from their td-WT littermates (Figure 5A-C). Consistent with this finding, CCL20 and CXCL1 mRNA expression levels in gastric digested glands, gastric crypt–derived primary organoid cells, and tdTomato-sorted VGPCs–derived secondary organoid cells were higher in td-PPARD mice than in their td-WT littermates (Figure 5D and E). Chemokine protein levels in sera from PPARD mice and their WT littermates were similar (Supplementary Figure 6B and C), suggesting that upregulation of CCL20 and CXCL1 expression due to PPARD overexpression in VGPCs was restricted to gastric mucosa. In addition, CCL20 and CXCL1 were upregulated in gastric mucosa of PPARD mice compared with WT mice at ages 10, 25, and 55 weeks (Supplementary Figure 6D and E), which further supports our chemokine screening and validation results (Figure 5A-E). To investigate whether chronic gastric inflammation also affects PPARD expression, we treated germ-free C57BL/6J mice with H. felis, a relative of H. pylori commonly used to study gastric inflammation and tumorigenesis in animal models29. H. felis cultures were characterized by Gram staining and PCR using Helicobacter-specific primers for helicobacter 16S rRNA amplicons30 (Supplementary Figure 6F and G). Successful H. felis colonization into gastric epithelial cells was confirmed by PCR testing30 (Figure 5F). Severe gastric inflammation was observed in H. felis–treated mice, but not in control mice (Supplementary Figure 6H). H. felis–treated mice had significantly higher Ppard, Ccl20, and Cxcl1 mRNA expression in gastric epithelial cells than did untreated C57BL/6J mice (Figure 5G-I).
Figure 5. PPARD in VGPCs induced CCL20 and CXCL1 secretion to promote chronic gastric inflammation.

(A) Heatmap of secreted chemokines in the culture media of primary organoids derived from gastric corpus crypts from td-PPARD and td-WT mice (Primary), and secondary organoids derived from tdTomato-marked VGPCs, sorted from primary organoid cells by flow cytometry at day 10 or 14 of culture, respectively.
(B, C) Quantitation of secreted CCL20 (C) and CXCL1 (D) protein levels in organoid culture medium described in panel A.
(D, E) CCL20 (D) and CXCL1 (E) mRNA expression levels in digested gastric glands, and primary and secondary organoid cells from mice as described in panel A.
(F) PCR gel image of Helicobacter-specific 16S rRNA amplicons (1.2 kb) for gastric epithelial cells from control and H. felis–infected germ-free C57BL/6J mice.
(G-I) PPARD (G), CCL20 (H), and CXCL1 (I) expression levels of gastric epithelial cells from the mice as described in panel F.
(J-L) Profile characterization of stomach-infiltrating inflammatory immune cells by flow cytometry in gastric tissues of PPARD mice. (J) Immune cells (CD45+), (K) CD3-positive cells (T cells) and B220-positive cells (B cells), and (L, left) CD11b-positive/Gr1-positive cells (granulocytes) and CD11b-positive/Gr1-negative cells (myeloid cells) among CD45-positive living immune cells from PPARD mice as described in panel J. (L, right) Expression of CD11c (dendritic cells) and F4/80 (macrophages) markers on CD45-positive/CD11b-positive/Gr1-negative (myeloid cells). WT mice had extremely low abundance of CD45-positive immune cells in gastric tissues (J).
(M) Pro-inflammatory cytokine mRNA expression levels in gastric corpus tissues from PPARD mice and WT littermates at 25 weeks.
We subsequently investigated the profiling of stomach-infiltrating immune cells by flow cytometry analyses of stomach corpuses of WT and PPARD mice. We found that PPARD mice had far more CD45-positive hematopoietic cells than WT mice did (Figure 5J). Moreover, among CD45-positive cells from PPARD mice, 23.8% were CD3 positive (T cells), 0.55% were B220 positive (B cells) (Figure 5K), 41% were CD11b and Gr1 positive (granulocytes/neutrophils), and 9.45% were CD11b positive and Gr1 negative (myeloid cells) (Figure 5L, left). Among the myeloid cells, 35.4% were CD11c positive and F4/80 negative (dendritic cells), and 6.47% were CD11c negative and F4/80 positive (macrophages) (Figure 5L, right). Thus, the majority of stomach-infiltrating immune cells in PPARD mice were T lymphocytes, granulocytes/neutrophils, and dendritic cells. Furthermore, we found that PPARD overexpression in VGPCs in PPARD mice was associated with upregulated expression of pro-inflammatory cytokines interferon gamma (IFNG), interleukin 1α (IL1 α), IL1β, IL6, and TNFα in the whole-stomach tissues but not in the isolated corpus epithelial glands of PPARD mice at age 25 weeks (Figure 5M, Supplementary Figure 6I, and data not shown), indicating that these upregulated cytokines were from non-epithelial cells and likely from the increased stomach-infiltrating immune cells.
IFNG signaling activation is the top canonical pathway regulated by PPARD during gastric tumorigenesis
To characterize the transcriptomic alterations accompanying PPARD overexpression in VGPCs, we performed RNA sequencing of corpus mucosa epithelial cells harvested from PPARD mice and WT littermates at ages 10, 25, and 55 weeks. Bioinformatics analyses identified 255 genes differentially expressed between PPARD and WT mice at these three ages using a cutoff of a log2 fold-change of PPARD over WT of <−1 or >1 and a false discovery rate of < .01 (Figure 6A and B, Supplementary Table 1). Pathway enrichment analysis of these 255 genes identified IFNG signaling as the top canonical pathway that PPARD activated, with an overlap ratio of 36.1% of the known genes involved in this pathway (Figure 6C). The next two top canonical pathways, with overlap ratios of 23.7% and 15.9% (Figure 6C, Supplementary Table 2), were the antigen presentation pathway and the pathway for activation of IFN regulatory factor by cytosolic pattern recognition receptors, respectively, both of which are related to IFNG signaling pathway activation and inflammatory immune response31, 32. Furthermore, Gene-Set Enrichment Analysis33 revealed that 62 of the 255 differentially expressed genes were IFNG response related (Figure 6D, Supplementary Figure 7A and B). The upregulation of the IFNG signaling genes as well as activation of the IFNG signaling pathway was confirmed by analyses at the mRNA and protein levels (Figure 6E-G, Supplementary Figure 7C-E). We hypothesized that this robust activation of the interferon response due to PPARD overexpression in VGPCs was induced by the stomach-infiltrating immune cells. Indeed, profiling results from whole-stomach tissues (but not the isolated epithelial cells) revealed substantially increased IFNG levels in the PPARD mice compared with the WT mice (Figure 5M).
Figure 6. PPARD activated the IFNG signaling pathway in the corpus of PPARD mice.

(A) Heatmap of RNA sequencing transcriptome analysis for the corpus mucosa of the PPARD mice and WT littermates.
(B) Venn diagram of total differentially expressed genes (DEGs) for the mice described in panel A.
(C) Ingenuity Pathway Analysis for the 255 shared DEGs. The top three pathways were identified. IRF, interferon regulatory factor.
(D) Gene Set Enrichment Analysis plot of the IFNG activation signature based on 255 shared DEGs described in panel B.
(E-G) Validation results of IFNG signaling pathway–related gene measurements by qRT-PCR (E), immunostaining (F), and Western blot (G) in corpus mucosa of indicated mice at 25 weeks.
Scale bars, 100 μm (F).
Crosstalk between PPARD and IFNG signaling accelerates VGPC expansion and gastric lesion development in PPARD mice
To further examine the interaction between IFNG and PPARD during PPARD–driven GAC tumorigenesis, we treated 15-week-old PPARD mice and their WT littermates with IFNG by intraperitoneal injection for 21 consecutive days. IFNG administration decreased the number of parietal cells and increased 5-bromo-2’-deoxyuridine incorporation and the proliferative (Ki67 positive) VGPC population in WT littermates, and these IFNG effects were markedly enhanced in PPARD mice (Figure 7A-D). IFNG also markedly increased gastric hyperplasia in PPARD mice (Figure 7A) and upregulated PPARD protein expression in WT mice and, more strikingly, in PPARD mice (Figure 7E). In two PPARD mouse–derived gastric cancer cell lines, IFNG also increased PPARD expression, which was accompanied by upregulation of genes responsive to IFNG signaling (Figure 7F-I). These results demonstrate a positive feedback loop between PPARD and IFNG signaling.
Figure 7. IFNG upregulated gastric PPARD expression and increased VGPC cell population, parietal cell loss, and gastric epithelial cell proliferation.

(A) Representative gastric corpus microphotographs of indicated mice groups. Scale bars, 50 μm.
(B-D) Numbers of parietal cells (B), 5-bromo-2’-deoxyuridine (BrdU)-positive cells (C), and activated VGPCs (Ki67+) (D) per gland in the gastric corpus of the indicated mice, corresponding to panel A. Fifty glands per mouse group were analyzed (n=4 per group).
(E) PPARD expression in corpus mucosa of the indicated mice, corresponding to panel A.
(F-I) IFNG effects on expression of PPARD and IFNG signaling downstream genes in PPARD mouse–derived gastric cancer cell lines. Mouse gastric cancer cells (mGC-1 and mGC-2) were treated with 30 ng/mL IFNG for 16 hours and then processed for mRNA (F-H) and protein (I) expression levels of indicated genes.
(J) Proposed conceptual model of the role of PPARD overexpression in promoting gastric tumorigenesis.
DISCUSSION
Our findings demonstrate profound effects of PPARD overexpression on GAC tumorigenesis. Unlike other mouse models of gastric cancer, which rarely spontaneously develop invasive GAC through the alteration of a single gene34, targeted PPARD overexpression in VGPCs was sufficient to induce invasive GAC. PPARD and WT mice harbored indistinguishable stomach microbiomes, suggesting microbe differences were unlikely the cause for the development of GAC in this mouse model. The clinical relevance of PPARD to human GAC is supported by our findings of PPARD upregulation in human GAC, which negatively impacted gastric cancer patients’ clinical outcomes.
PPARD overexpression in VGPCs specifically expanded a small population of stem-like VGPCs in the lesser curvature of the gastric corpus where gastric tumorigenesis was initiated. Our cell lineage tracing study showed that VGPCs in the gastric lesser curvature of the corpus possess the properties of stem cells and can serve as a cell of origin of GAC. PPARD upregulated CD44 and villin expression in VGPCs of gastric corpus tumor glands in PPARD mice, similar to concomitant villin and CD44 co-expression observed in human corpus GAC. More importantly, our data show that in gastric organoid cultures, PPARD strongly enhanced stem cell renewal, induced malignant morphological changes, and endowed organoids–derived VGPCs with tumorigenic capacity in vivo. A recent report showed that PPARD activation also enhances the stemness and tumorigenicity of intestinal progenitor cells35 in the presence of an APC mutation, a potent driver of intestinal tumorigenesis. Our findings demonstrate for the first time, in an in vivo setting, that PPARD overexpression is sufficient to transform gastric epithelial progenitor cells and to drive gastric tumorigenesis.
PPARD overexpression in VGPCs induced the expression of immune-attractive chemokines, thereby recruiting a variety of immune cells to the tumor microenvironment. These cells promoted chronic inflammation and the production of a repertoire of inflammatory cytokines, including IFNG, IL6, TNFα, and IL1β, which are well known to strongly promote carcinogenesis36, 37. Through chemokine panel screening and subsequent validation experiments, we found that PPARD overexpression strongly upregulated transcription and protein expression of the chemoattractants CCL20 and CXCL1 in VGPCS. CXCL1 mediates neutrophil recruitment38, and CCL20 is upregulated in H. pylori–infected gastric mucosa and attracts immune cells such as lymphocytes and dendritic cells toward epithelial cells39–41. Chronic gastric inflammation, as in the cases of H. pylori infection in humans and its relative H. felis in mice, plays a critical role in gastric tumorigenesis19, 29. These microbial agents seem to employ PPARD to promote gastritis and gastric tumorigenesis. Infection of gastric epithelial cells and gastric progenitor cells with H. pylori isolated from human gastric tumor tissues has been found to upregulate PPARD, CXCL1, and CCL2042. H. pylori infection has been shown to upregulate gastric PPARD expression, which returned to normal levels after eradication of H. pylori in rodent and human gastric mucosa19. We found similar results showing that H. felis infection of germ-free C57BL/6J mice induced severe gastric chronic inflammation and increased PPARD, CCL20, and CXCL1 expression in gastric epithelial cells. These findings support the role of PPARD in perpetuating chronic gastric inflammation to create a tumor-promoting microenvironment.
A positive feedback loop between PPARD and IFNG signaling perpetuates gastric cancer driving inflammatory microenvironment. Unbiased RNA transcriptome profile sequencing analyses revealed that IFNG signaling and IFNG–related signaling pathways were the top canonical pathways drastically activated by PPARD in our PPARD mice. Several lines of evidence support the role of aberrant IFNG pathway activation in promoting gastric tumorigenesis: (1) Gastric IFNG is upregulated in humans and mice by chronic H. pylori infection43, 44 and promotes H. pylori–induced gastric inflammation45. (2) IFNG intraperitoneal injection into mice expanded VGPCs and induced lesser curvature hypertrophy8. (3) Transgenic mice with IFNG overexpression in gastric parietal cells spontaneously developed inflammation, metaplasia, dysplasia, and eventually GAC in the gastric corpus46. (4) IFNG deficiency inhibits spontaneous development of gastric tumors in Huntingtin-interacting protein 1-related (Hiplr)–deficient mice47. More recently, sustained IFNG signaling was implicated in tumor immune evasion48 and resistance to checkpoint blockade49. Our findings demonstrate not only that PPARD upregulates IFNG signaling in gastric mucosa to promote tumorigenesis but also that IFNG in turn upregulates PPARD expression in gastric epithelial cells in vitro and in vivo. This paracrine signaling feedback loop between IFNG and PPARD represents a novel mechanism of gastric cancer promotion by chronic inflammation.
In summary, our results provide the first evidence that PPARD overexpression in VGPCs is sufficient to drive gastric tumorigenesis to invasive GAC. Our results also uncover a novel positive feedback loop between PPARD and IFNG signaling that creates an inflammatory tumor-promoting microenvironment enabling VGPC transformation and gastric tumorigenesis (Figure 7J). These findings provide new insights into the molecular pathogenesis of GAC that can form the basis for developing interventional strategies to target PPARD for the chemoprevention/treatment of gastric cancer.
Supplementary Material
Acknowledgements
This study made use of the MD Anderson Cancer Center Genetically Engineered Mouse Facility, Functional Genomics Core, Flow Cytometry and Cellular Imaging Facility, Sequencing and Microarray Facility, and Research Animal Support Facility—Smithville Laboratory Animal Genetic Services, supported by Cancer Center Support Grant CA016672. The study also used the Baylor College of Medicine Gnotobiotics Core, supported by P30-DK056338.
We thank Ms. Sarah J Bronson at the Department of Scientific Publications at MD Anderson Cancer Center for editing the manuscript.
Funding
This work was supported by the National Cancer Institute (R01-CA142969, R01-CA195686, and R01-CA206539 to I.S.), the Cancer Prevention and Research Institute of Texas (RP140224 to I.S.), and MD Anderson Institutional Research Seed Fund (to X. Zuo).
Abbreviations used in this paper:
- GAC
gastric adenocarcinoma
- VGPC
villin-expressing gastric progenitor cells
- PPARD
peroxisome proliferator-activated receptor delta
- WT
wild-type
- SPEM
spasmolytic polypeptide-expressing metaplasia
- IM
intestinal metaplasia
- GSII
(Griffonia simplicifolia lectin II)
- Muc
(Mucin)
- PAS
periodic acid–Schiff
- ANGPTL4
angiopoietin-like 4
- IFN
interferon
- IRF
interferon regulatory factor
- IL
interleukin
- H. felis
Helicobacter felis
- H. pylori
Helicobacter pylori
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors declare no conflicts.
Supplementary Material
Supplemental material includes supplementary methods, seven supplementary figures and three supplementary tables and can be found with this article online.
REFERENCES
- 1.Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2.Qadri Q, Rasool R, Gulzar GM, et al. H. pylori infection, inflammation and gastric cancer. J Gastrointest Cancer 2014;45:126–32. [DOI] [PubMed] [Google Scholar]
- 3.Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007;133:659–72. [DOI] [PubMed] [Google Scholar]
- 4.Hayakawa Y, Ariyama H, Stancikova J, et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015;28:800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayakawa Y, Jin G, Wang H, et al. CCK2R identifies and regulates gastric antral stem cell states and carcinogenesis. Gut 2015;64:544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demitrack ES, Gifford GB, Keeley TM, et al. Notch signaling regulates gastric antral LGR5 stem cell function. Embo j 2015;34:2522–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayakawa Y, Fox JG, Wang TC. The Origins of Gastric Cancer From Gastric Stem Cells: Lessons From Mouse Models. Cell Mol Gastroenterol Hepatol 2017;3:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiao XT, Ziel JW, McKimpson W, et al. Prospective identification of a multilineage progenitor in murine stomach epithelium. Gastroenterology 2007;133:1989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q, Jia Z, Wang L, et al. Disruption of Klf4 in villin-positive gastric progenitor cells promotes formation and progression of tumors of the antrum in mice. Gastroenterology 2012;142:531–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li XB, Yang G, Zhu L, et al. Gastric Lgr5(+) stem cells are the cellular origin of invasive intestinal-type gastric cancer in mice. Cell Res 2016;26:838–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neels JG, Grimaldi PA. Physiological functions of peroxisome proliferator-activated receptor beta. Physiol Rev 2014;94:795–858. [DOI] [PubMed] [Google Scholar]
- 12.Manickam R, Wahli W. Roles of Peroxisome Proliferator-Activated Receptor beta/delta in skeletal muscle physiology. Biochimie 2017;136:42–48. [DOI] [PubMed] [Google Scholar]
- 13.Yuan H, Lu J, Xiao J, et al. PPARdelta induces estrogen receptor-positive mammary neoplasia through an inflammatory and metabolic phenotype linked to mTOR activation. Cancer Res 2013;73:4349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta RA, Tan J, Krause WF, et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc Natl Acad Sci U S A 2000;97:13275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuo X, Xu W, Xu M, et al. Metastasis regulation by PPARD expression in cancer cells. JCI Insight 2017;2:e91419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedchenko TV, Gonzalez AL, Wang D, et al. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am J Respir Cell Mol Biol 2008;39:689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu M, Zuo X, Shureiqi I. Targeting peroxisome proliferator-activated receptor-beta/delta in colon cancer: how to aim? Biochem Pharmacol 2013;85:607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters JM, Gonzalez FJ, Muller R. Establishing the Role of PPARbeta/delta in Carcinogenesis. Trends Endocrinol Metab 2015;26:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagy TA, Wroblewski LE, Wang D, et al. beta-Catenin and p120 mediate PPARdelta-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology 2011;141:553–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeon C, Chang SC, Mu L, et al. Genetic variants of peroxisome proliferator-activated receptor delta are associated with gastric cancer. Dig Dis Sci 2013;58:2881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollock CB, Rodriguez O, Martin PL, et al. Induction of metastatic gastric cancer by peroxisome proliferator-activated receptordelta activation. PPAR Res 2010;2010:571783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo X, Xu M, Yu J, et al. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR-delta/beta overexpression. J Natl Cancer Inst 2014;106:dju052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuo X, Peng Z, Moussalli MJ, et al. Targeted genetic disruption of peroxisome proliferator-activated receptor-delta and colonic tumorigenesis. J Natl Cancer Inst 2009;101:762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldenring JR, Nam KT, Wang TC, et al. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology 2010;138:2207–10, 2210.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takaishi S, Okumura T, Tu S, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009;27:1006–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertaux-Skeirik N, Feng R, Schumacher MA, et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog 2015;11:e1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan Y, Zuo X, Wei D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med 2015;4:1033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flanagan DJ, Schwab RH, Tran BM, et al. Isolation and Culture of Adult Intestinal, Gastric, and Liver Organoids for Cre-recombinase-Mediated Gene Deletion. Methods Mol Biol 2016. [DOI] [PubMed] [Google Scholar]
- 29.Ericksen RE, Rose S, Westphalen CB, et al. Obesity accelerates Helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut 2014;63:385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Z, Feng Y, Fox JG. Identification of enterohepatic Helicobacter species by restriction fragment-length polymorphism analysis of the 16S rRNA gene. Helicobacter 2000;5:121–8. [DOI] [PubMed] [Google Scholar]
- 31.Mandai M, Hamanishi J, Abiko K, et al. Dual Faces of IFNgamma in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res 2016;22:2329–34. [DOI] [PubMed] [Google Scholar]
- 32.Schroder K, Hertzog PJ, Ravasi T, et al. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004;75:163–89. [DOI] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayakawa Y, Fox JG, Gonda T, et al. Mouse models of gastric cancer. Cancers (Basel) 2013;5:92–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beyaz S, Mana MD, Roper J, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016;531:53–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14:408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Fu L, Ning W, et al. Peroxisome proliferator-activated receptor delta promotes colonic inflammation and tumor growth. Proc Natl Acad Sci U S A 2014;111:7084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sawant KV, Poluri KM, Dutta AK, et al. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci Rep 2016;6:33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook KW, Letley DP, Ingram RJ, et al. CCL20/CCR6-mediated migration of regulatory T cells to the Helicobacter pylori-infected human gastric mucosa. Gut 2014;63:1550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu YY, Tsai HF, Lin WC, et al. Upregulation of CCL20 and recruitment of CCR6+ gastric infiltrating lymphocytes in Helicobacter pylori gastritis. Infect Immun 2007;75:4357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Borgne M, Etchart N, Goubier A, et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity 2006;24:191–201. [DOI] [PubMed] [Google Scholar]
- 42.Giannakis M, Chen SL, Karam SM, et al. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc Natl Acad Sci U S A 2008;105:4358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen S, Felley CP, Bouzourene H, et al. Inflammatory gene profiles in gastric mucosa during Helicobacter pylori infection in humans. J Immunol 2004;172:2595–606. [DOI] [PubMed] [Google Scholar]
- 44.Sawai N, Kita M, Kodama T, et al. Role of gamma interferon in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Infect Immun 1999;67:279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allison CC, Ferrand J, McLeod L, et al. Nucleotide oligomerization domain 1 enhances IFN-gamma signaling in gastric epithelial cells during Helicobacter pylori infection and exacerbates disease severity. J Immunol 2013;190:3706–15. [DOI] [PubMed] [Google Scholar]
- 46.Syu LJ, El-Zaatari M, Eaton KA, et al. Transgenic expression of interferon-gamma in mouse stomach leads to inflammation, metaplasia, and dysplasia. Am J Pathol 2012;181:2114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z, Demitrack ES, Keeley TM, et al. IFNgamma contributes to the development of gastric epithelial cell metaplasia in Huntingtin interacting protein 1 related (Hip1r)-deficient mice. Lab Invest 2012;92:1045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mojic M, Takeda K, Hayakawa Y. The Dark Side of IFN-gamma: Its Role in Promoting Cancer Immunoevasion. Int J Mol Sci 2017;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benci JL, Xu B, Qiu Y, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016;167:1540–1554.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.