

Abstract
The role of Notch signaling in melanoma drug resistance is not well understood. In this study, we show that although NOTCH proteins are upregulated in metastatic melanoma cell lines, Notch signaling inhibition had no effect on cell survival, growth, migration or the sensitivity of BRAFV600E-melanoma cells to MAPK inhibition (MAPKi). We found that NOTCH1 is downregulated in melanoma cell lines with intrinsic and acquired resistance to MAPKi. Forced expression of NICD1, the active form of Notch, caused apoptosis of the NOTCHlo, MAPKi-resistant cells, but not the NOTCHhi, MAPKi-sensitive melanoma cell lines. Whole transcriptome-sequencing analyses of NICD1-transduced MAPKi-sensitive and -resistant cells revealed differential regulation of endothelin1 (EDN1) by NICD1, i.e., downregulation in MAPKi-resistant cells and upregulation in MAPKi-sensitive cells. Knockdown of EDN1 partially mimicked the effect of NICD1 on the survival of MAPKi-resistant cells. We show that the opposite regulation of EDN1 by Notch signaling is mediated by the differential regulation of c-JUN by NICD1. Our data show that MAPKi-resistant melanoma cells acquire vulnerability to Notch signaling activation and suggest that Notch-cJUN-EDN1 axis is a potential therapeutic target in MAPKi-resistant melanoma.
Keywords: Notch, Cutaneous Melanoma, Drug resistance, BRAFV600E, Apoptosis, EDN1
INTRODUCTION
Notch signaling is required for the maintenance of melanoblasts and epidermal melanocyte stem cells (Aubin-Houzelstein et al., 2008; Moriyama et al., 2006). However, the role of Notch signaling in melanoma is less clear. While both oncogenic and tumor suppressor roles were assigned (Bonyadi Rad et al., 2016; Liu et al., 2006), clinical trials targeting Notch signaling for melanoma treatment proved unsuccessful (Lee et al., 2015).
MAPK pathway components BRAF and NRAS are commonly mutated in melanoma (Lo & Fisher, 2014). Despite the initial response to MAPK pathway targeted chemotherapy, most patients develop drug resistance (Shtivelman et al., 2014). A number of mechanisms, including Notch signaling activation, are known to contribute to MAPKi resistance (Hugo et al., 2015; Martz et al., 2014; Tsao, Chin, Garraway, & Fisher, 2012). In BRAFV600E melanoma cells, activation of Notch1 signaling was reported to enhance acquired resistance to MAPKi, and knockdown of Notch1 was able to resensitize resistant cells to MAPK inhibitors (Martz et al., 2014). In long-term cultures, Notch signaling inhibition was shown to prolong the sensitivity of melanoma cells to BRAFi (Zhu et al., 2016). Furthermore, in BRAFV600E mutant melanoma with PTEN loss, targeting Notch signaling enhances the efficacy of ERKi (Krepler et al., 2016). Therefore, in BRAFV600E melanoma, Notch signaling seems to play a role in the context of their dependence on MAPK signaling. However, the exact mechanism by which Notch signaling modifies sensitivity of melanoma to MAPKi is not clear.
Here, we show that although NOTCH proteins are upregulated in metastatic melanoma cell lines, pharmacological inhibition of Notch signaling had no effect on cell survival, growth, migration or the sensitivity of BRAFV600E-melanoma cells to MAPKi. However, NOTCH1 expression was inversely related to MAPKi-resistance. Forced expression of NICD1, the active form of Notch1, alone was sufficient to induce apoptosis, independent of MAPK pathway inhibition, in melanoma cells with both intrinsic and acquired resistance to MAPKi. RNAseq analysis of NICD1 expressing MAPKi-sensitive and -resistant cells identified differential regulation of EDN1 by Notch, through the transcription factor c-JUN, as the mechanism for selective killing of MAPKi-resistant cells. Our data suggest a novel vulnerability of MAPKi-resistant melanoma and highlight Notch-c-JUN-EDN1 axis as a potential target to bypass MAPKi resistance in melanoma.
MATERIALS AND METHODS
Chemicals and Antibodies
Gamma-secretase inhibitors [DAPT #S2215, RO4929097 #S1575, LY411575 #S2714 and Semagacestat (LY450139) #S1594], BRAF inhibitor (PLX4032 #S1267), MEK inhibitor (AZD6244 #S1008), caspase inhibitor (z-VAD-FMK #S7023), autophagy inhibitors (chloroquine phosphate # S4157and 3-Methyladenine (3-MA) #S2767), Endothelin A receptor (EDNRA) antagonist (BQ-123 #S7883), EDNRA and EDNRB antagonist (Bosentan #S4220) and monensin (#S2324) were from Selleckchem (Houston, TX). Recombinant Endothelin-1 (ET-1 #E7764) was from Sigma (St. Louis, MO).
Antibodies to Notch receptors (1-3) and ligands, LaminB, MEK1/2, cleaved caspase-3 and -9, PARP, ERK1/2, pERK1/2, pro- and anti-apoptotic markers, HES1, c-JUN, p c-JUN, CREB, and pCREB were from Cell Signaling Technology (Danvers, MA). Notch4 antibody was from Santa Cruz Biotechnology (Dallas, TX). β-actin and GAPDH antibodies were from Sigma and Proteintech (Rosemont, IL), respectively. (See Supplementary Materials)
Lentiviruses and plasmids
NICD1 expression plasmid (pHX-CMV-NIC-IRES-GFP), empty vector control plasmid and pCI-HES1 expression plasmid were described previously (Maddodi, Bhat, Devi, Zhang, & Setaluri, 2010). Lentiviral EDN1 shRNA-hCMV TurboRFP set (#V3SH11243-07EG1906) and SMARTvector Non-targeting hCMV-TurboRFP (#VSC11714) were from GE Dharmacon (Lafayette, CO). Cells were transduced [Multiplicity of Infection (MOI) 10-115] in the presence of 8μg/ml polybrene.
Cell Culture
Human metastatic melanoma cell lines MRA-2, MRA-4, MRA-5, MRA-6, and MRA-9 were provided by Dr. Mark Albertini (Department of Medicine, UW-Madison), genotyped for BRAF and NRAS mutations and confirmed with western blot with anti- BRAFV600E antibody and cultured as described (See Table S1) (Rodriguez et al., 2017). Human melanocytes were from UW Skin Disease Research Center. Cells were grown in Medium 254 supplemented with Human Melanocyte Growth Supplement-2, PMA-free (HMGS-2) (Thermo Fisher, Grand Island, NY). All cells were grown at 37°C and 5% CO2.
Cell survival and growth assay
Equal number of cells were plated in 96-well plates. Cells were treated in 5-6 replicates with the appropriate vehicle or different concentrations of the inhibitor ranging from 1nM to 10μM for 72h. MTT dye (Thiazolyl Blue Tetrazolium Bromide) (Sigma) was added, incubated for one hour, cells were lysed using DMSO and absorbance at 540nm was recorded.
Immunoblotting
Cultured cells were scraped and lysed in RIPA buffer, sonicated for 20s and centrifuged. For cell fractionation, cells were trypsinized, lysed and fractionated using the NE-PER™ Nuclear and Cytoplasmic Extraction kit (#78833, Life Technologies, CA). Protein was estimated using BCA Protein Assay Kit (Thermo Fisher). Equal amounts of protein (15-30μg) were loaded for SDS-PAGE (7.5% or 9% gels). Equivalent volumes of cytoplasmic and nuclear fractionations, were loaded and run on a 7.5% SDS-PAGE. Proteins were transferred to PVDF membrane, blocked with 5% non-fat milk in TBS/0.1% Tween and incubated with primary antibodies at appropriate dilutions (Table S2) overnight at 4°C followed by HRP conjugated secondary antibodies for 1h at room temperature. Blots were stripped and/or re-probed for housekeeping proteins actin or GAPDH. Alternatively, blots were incubated with primary antibodies to proteins of interest together with anti-actin or -GAPDH. Protein bands were visualized using ECL Start Western Blotting Detection Reagent (Amersham) and ImageQuant LAS 4000 (GE Healthcare, Marlborough, MA).
q-RT-PCR
Total RNA was isolated using the miRNeasy Mini Kit (#217004, Qiagen, Hilden, Germany) either from 70-90 % confluent plates or 30h after lentiviral transduction of cells plated in 6-well plates. Total RNA (0.5μg) was used to generate cDNA using the High-Capacity cDNA Reverse Transcription Kit (#4368814, Thermo Fisher). Transcript levels of the target genes were then quantified by qRT-PCR using TaqMan probes (Table S3). GAPDH and ACTB were used as controls.
RNAseq
Cells were plated in 6-well plates, and transduced with the NICD1 or empty vector lentiviruses. At 30h post-transduction, total RNA was isolated using the miRNeasy Mini Kit (#217004, Qiagen). RNAseq was performed at Sanford Burnham Prebys Medical Discovery Institute (Orlando, FL) by Dr. Ranjan Perera. Multiplexed libraries were pooled and single-end 50-bp sequencing was performed on Illumina Hiseq 2500. See Supplementary Methods for details.
RNAseq analysis
Quality control was carried out using FastQC; no samples were omitted from the analysis. Reads were mapped against the Hg19 Refseq reference via Bowtie 0.12.8 allowing up to two mismatches and up to 20 multiple hits. The expected counts and transcripts per million (TPMs) were estimated via RSEM 1.2.3. Differential expression analysis, clustering, and gene set analysis were performed in R. Differentially expressed genes (NICD1 vs. control) for different cell types were identified using EBSeq (see Supplementary Methods for details). The RNAseq data are available NCBI (Accession GSE110179).
RESULTS
NOTCH receptors and ligands are upregulated in metastatic melanoma cells
Western blot analysis of a panel of melanocytes and metastatic melanoma cells showed that NOTCH1, -2 and -3 proteins and Notch ligands (Jagged1, -2, DLL3, -4) are upregulated (Fig. 1A), and the nuclear NICD1 (Notch intracellular domain, the active form of NOTCH) levels were also higher in melanoma cells (Fig. 1B). Treatment of melanoma cells with γ-secretase inhibitors resulted in downregulation in NICD1 levels (Fig. S1A) and a decrease in HES1 mRNA, an indicator of Notch signaling activity (Fig. S1B). These data show that Notch signaling is constitutively active in melanoma cells and membrane NOTCH receptors are cleaved to produce active NICD1 by the activity of γ-secretase. However, treatment with γ-secretase inhibitor had no consistent effect on the survival of NOTCH1hi (MRA-2, -4, -6 and -9) or NOTCH1lo (MRA-5) melanoma cells (Fig. S1C), but a prolonged treatment with high concentration (3μM) of γ-secretase inhibitor RO4929097 caused a modest growth inhibition in both NOTCH1hi and NOTCH1lo melanoma cells (Fig. 1C).
Figure 1. Notch receptors and ligands are upregulated in metastatic melanoma cells, but sustained Notch signaling is not required for melanoma cells.
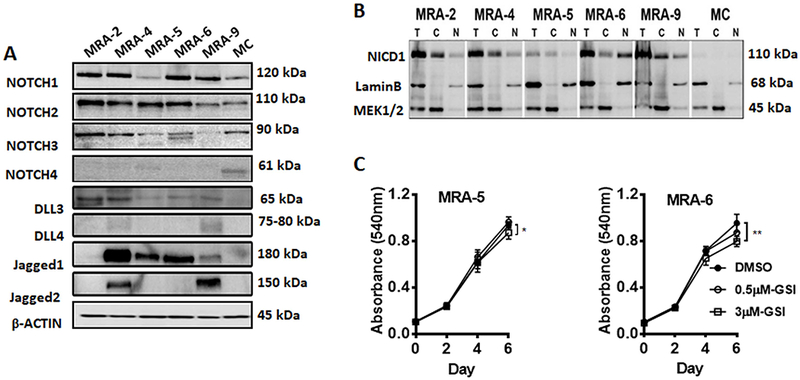
Western blot analysis of A. NOTCH receptors and ligands, DLL3-4, Jagged1 and -2 expression in human melanocytes (MC) and MRA series metastatic melanoma cell lines, and B. Cytoplasmic and nuclear NICD1 levels in melanocytes and metastatic melanoma cells (T: 4% total protein, C: cytoplasmic fraction, N: nuclear fraction). Blots were incubated with anti-NICD1 antibody together with antibodies to MEK1/2 and LaminB, which show relative enrichment of cytoplasmic and nuclear proteins. Numbers on the right indicate the molecular weight (kDa) of the respective proteins. Results are representative of at least two independent experiments. C. Effect of treatment with γ-secretase inhibitor on melanoma cell growth. MRA-5 and MRA-6 cells were plated in 96-well plates and treated with DMSO or 0.5μM or 3μM RO4929097. Cell survival was assessed up to 6 days. Data shown are mean ± SD of five to six replicates normalized to DMSO control.
NOTCH1 is downregulated in MAPKi-resistant BRAFV600E mutant melanoma cells
We noted that the two BRAFV600E melanoma cell lines MRA-5 and MRA-6 had a markedly different levels of NOTCH1 and NICD1 protein. NOTCH1lo MRA-5 cells are intrinsically resistant to both the BRAFV600E inhibitor, vemurafenib (PLX4032) and the MEK inhibitor, selumetinib (AZD6244), whereas NOTCH1hi MRA-6 cells are sensitive to both inhibitors (Fig. S2) (Rodriguez et al., 2017), suggesting a possible association between Notch signaling and MAPKi-sensitivity. Therefore, we asked whether NOTCH1 downregulation is also associated with acquired resistance to MAPKi. To test this, we employed vemurafenib resistant MRA-6BR and selumetinib-resistant MRA-6MR cells derived from the MAPKi-sensitive MRA-6 cell line (Fig. S2) (Rodriguez et al., 2017). Western blot analysis showed a marked downregulation in NOTCH1 in both MRA-6BR and -6MR cells compared to the parental MAPKi-sensitive MRA-6 cells (Fig. 2A).
Figure 2. Notch downregulation is associated with acquired resistance to MAPKi and forced expression of NICD1 inhibits survival of MAPKi-resistant cells.
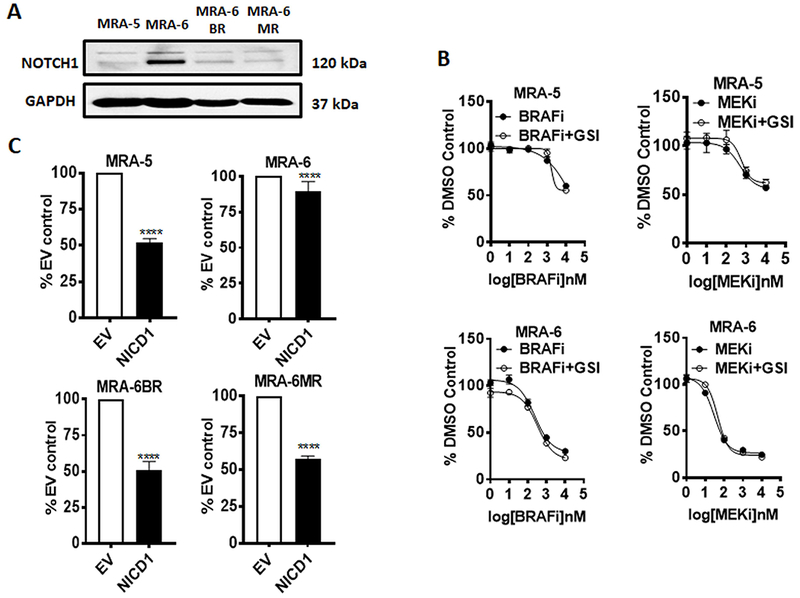
A. Western blot analysis of NOTCH1 protein expression in MAPKi-resistant (MRA-5, -6BR and -6MR) cells compared to MAPKi-sensitive (MRA-6) cells. Blots were incubated with anti-NOTCH1 antibody together with anti-GAPDH antibody. Numbers on the right indicate the molecular weight (kDa) of the respective proteins. Results shown represent at least two independent experiments. B. Dose response curve for MRA-5 and MRA-6 cells treated with either PLX4032 (BRAFi) or AZD6244 (MEKi) alone or in combination with RO4929097 (γ-secretase inhibitor). Cells were plated in 96-well plates and treated with DMSO or increasing concentrations of either PLX4032 or AZD6244 with or without 3μM RO4929097 for 72h and cell survival was assessed using MTT assay. Data shown are mean ± SD of five to six replicates normalized to DMSO control and represent two independent experiments. C. Cells were plated in 96-well plates and transduced with NICD1 or empty vector lentivirus and cell survival at 48h was assessed using MTT assay. Data shown are mean ± SD of five to six replicates, normalized to empty vector control, and represent three independent experiments. Unpaired Student t-test was used to analyze the data. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤0.0001.
To understand whether downregulation of Notch signaling contributes to MAPKi resistance, we tested the effect of inhibition of Notch signaling on the sensitivity of melanoma cells to MAPKi. We treated MRA-5 and MRA-6 cells with increasing concentrations of the BRAFV600E inhibitor PLX4032 or MEK inhibitor AZD6244 in combination with the γ-secretase inhibitor RO4929097. Intriguingly, Notch signaling inhibition did not affect the MAPKi sensitivity of BRAFV600E melanoma cell lines (Fig. 2B) suggesting that although downregulation of Notch is associated with MAPKi-resistance in BRAFV600E melanoma cells, it is not a mechanism for the resistance.
Forced expression of NICD1 induces cell death in MAPKi-resistant BRAFV600E melanoma cells
We then asked whether forced expression of NICD1 (active Notch1) sensitizes MAPKi-resistant cells to MAPKi. We transduced the MAPKi-sensitive MRA-6 and MAPKi-resistant MRA-5, MRA-6BR and -6MR cells with NICD1-GFP lentivirus. Transduction efficiency was monitored by GFP expression (Fig. S3A) and NICD1 expression was confirmed by western blotting (Fig. S3B). Interestingly, transduction with the NICD1 lentivirus alone (without MAPKi treatment) caused a marked decrease in the survival of the MAPKi resistant MRA-5, MRA-6BR and MRA-6MR cells but not MAPKi-sensitive MRA-6 cells (Figs. 2C, S3C, S3D and S3E) although MRA-6 cells had higher NICD1 expression (Fig. S3B). Furthermore, overexpression of NICD had no effect on the survival of NRAS mutant melanoma cell line MRA-9 (Fig. S3F).
To investigate the mechanism of NICD1-induced cell death in the MAPKi-resistant cells, we assessed biochemical markers of apoptosis. Figs. 3A, B show cleavage of caspase-3, caspase-9 and PARP in NICD1-transduced MRA-5, but not in MRA-6 cells. Addition of the pan-caspase inhibitor z-VAD, but not autophagy inhibitors (chloroquine or 3’-methyladenine), rescued MRA-5 cells from NICD1-induced cell death. This effect of caspase inhibitor was more prominent in the intrinsically resistant MRA-5 cells than in the MRA-6BR and -6MR cells with acquired resistance (Figs. 3C, S4A and S4B).
Figure 3. NICD1 expression induces apoptosis in BRAFV600E MAPKi-resistant melanoma cells.
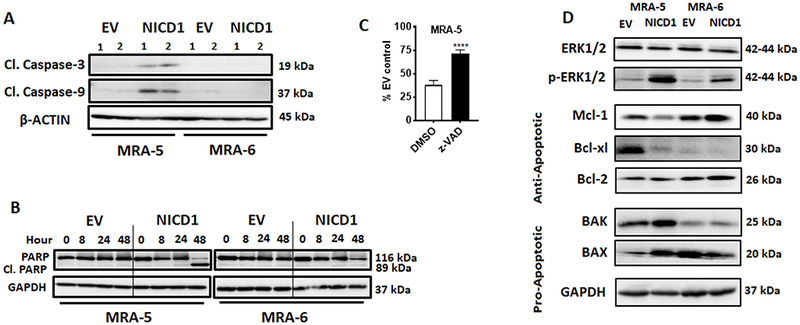
A. Cells were plated in 60mm plates and transduced with either NICD1 or empty vector lentivirus. Cells were harvested at 48h post-transduction and, lysed and analyzed for cleaved caspase-3 and -9. 1 and 2 indicate low (1) and high (2) MOI of lentivirus. Numbers on the right indicate the molecular weight (kDa) of the respective protein. B. Cells were plated in 60mm plates and transduced with either NICD1 or empty vector lentivirus. Cells were harvested at indicated times after transduction, lysed and analyzed for cleaved PARP. Numbers on the right indicate the molecular weight (kDa) of the respective proteins. C. Cells were plated in 96-well plates and transduced with either NICD1 or empty vector lentivirus. Twenty four hours after transduction cells were treated with DMSO or the apoptosis inhibitor z-VAD-FMK (5-10μM) for additional 48h. Cell survival was assessed by MTT assay. Data shown are mean ± SD of five to six replicates normalized to DMSO control and are representative of two independent experiments. Unpaired Student t-test was used to analyze the data. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤0.0001. D. Cells were plated in 60mm plates and transduced with either NICD1 or empty vector lentivirus. Cells were harvested at 48h post-transduction, lysed and analyzed for ERK1/2 and p-ERK1/2, anti-apoptotic (Mcl-1, Bcl-xl, Bcl-2), and pro-apoptotic (Bak, BAX). Numbers on the right indicate the molecular weight (kDa) of the respective proteins.
Western blot analysis showed a marked decrease in anti- apoptotic proteins Mcl-1 and Bcl-xl and increase in pro-apoptotic proteins BAK and BAX in NICD1-transduced MAPKi-resistant MRA-5 cells (Fig. 3D). Interestingly, NICD1 overexpression also caused a marked upregulation of phospho-ERK in both MAPKi-resistant and -sensitive cells (Fig. 3D) suggesting that upregulation of Notch signaling in MAPKi-resistant melanoma cells induces apoptosis independent of MAPK pathway inhibition.
Role of HES1 in NICD1-induced apoptosis of MAPKi-resistant cells
In NICD1-transduced cells, endogenous NOTCH1 expression was also upregulated consistent with the positive feedback regulation of Notch signaling (Chen et al., 2017) (Fig. S3B). Although upregulation of endogenous NOTCH1 protein is comparable in both the MAPKi-sensitive and MAPKi-resistant cells, there was no corresponding increase in NICD1 in the MAPKi-resistant cells (unlike in the MAPKi-sensitive cells), suggesting that additional mechanism(s) are involved in the regulation of NICD1 levels in MAPKi-resistant cells (Fig. S3B).
HES1 is a major downstream target and effector of Notch signaling (Jarriault et al., 1995). HES1 has been implicated in Notch-induced apoptosis (Nefedova, Sullivan, Bolick, Dalton, & Gabrilovich, 2008; Zweidler-McKay et al., 2005). NICD1 expression caused a marked upregulation of HES1 protein in both MAPKi-resistant MRA-5 cells and MAPKi-sensitive MAR-6 cells (Fig. 4A). However, overexpression of HES1 had no significant effect on the survival of MAPKi-resistant cells (Figs. 4B and 4C) suggesting that Notch target gene HES1 does not contribute to NICD1-induced apoptosis of MAPKi-resistant cells.
Figure 4. Role of HES1 in NICD1-induced apoptosis in MAPKi-resistant cells.
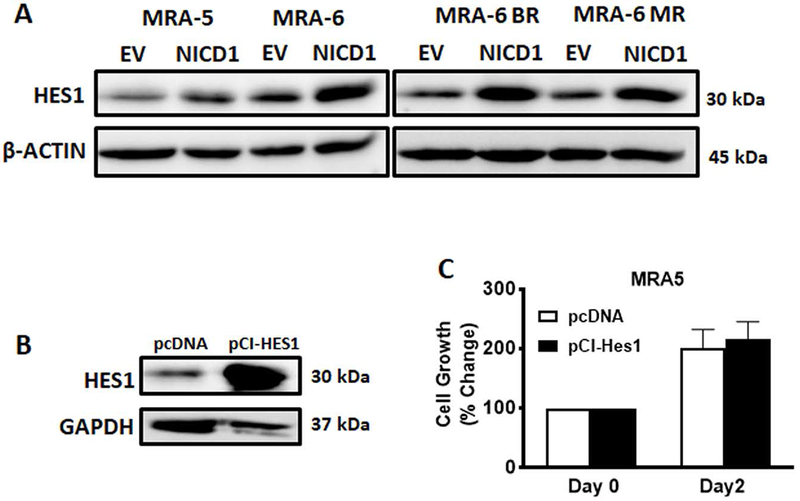
A. Cells were plated in 60mm plates and transduced with NICD1 or empty vector lentivirus and 48h after transduction, cells were harvested and lysed for western blot analysis of HES1 expression using anti-HES1 antibody. Numbers on the right indicate the molecular weight (kDa) of the respective proteins. B and C. Cells were plated in 6-well plates and transfected with 2μg pCI-HES1 expression plasmid or empty vector. B. Transfected Cells were trypsinized after 48h, plated in a 6-well plate, harvested after 24h and lysed for western blot analysis using anti-HES1 antibody. Numbers on the right indicate the molecular weight (kDa) of the respective protein. C. Transfected cells were plated after 24h in a 96-well plate and cell survival was assessed using MTT at days 0 and 2. Data shown are mean ± SD of five to six replicates and represent two independent experiments. Unpaired Student t-test was used to analyze the data. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤0.0001.
Differential effect of NICD1 on EDN1 in MAPKi- sensitive and -resistant melanoma cells
To identify NICD1-induced changes in gene expression that contribute to activation of apoptosis in MAPKi- resistant cells, we performed RNA-seq analysis of MRA-6, MAPKi-resistant MRA-5, MRA-6BR and MRA-6MR cells transduced with the empty vector or NICD1 lentivirus for 30h (prior to induction of apoptosis). We found that 452 genes consistently showed NICD1-associated differential expression compared to empty vector control (at a 1% false discovery rate) across all the three MAPKi-resistant cells (MRA-5, MRA-6BR, MRA-6MR) (Table S4), whereas NICD1-overexpression affected the expression of 1985 genes in MAPKi-sensitive MRA-6 cells. The top ten Gene Ontology (GO) categories (Figs. S5A, S5B and Tables S5 and S6) of the differentially expressed genes were associated with cell migration, cell differentiation, tissue morphogenesis, but not cell proliferation or cell death pathways.
Since NICD1 overexpression induced apoptosis selectively in the MAPKi-resistant cells, we then queried the above datasets for genes that show changes in expression in a) opposite direction consistently between the MAPKi-resistant vs. MAPKi-sensitive cells (gene set #1 consisting10 genes), b) only in MAPKi-resistant cells (gene set # 2 consisting 18 genes), or c) only in MAPKi-sensitive cells (gene set #3 consisting 38 genes) (Figs. 5A and S5C, S5D, respectively).
Figure 5. Whole transcriptome analysis of NICD1-transduced BRAFV600E mutant MAPKi-resistant and sensitive melanoma cells.
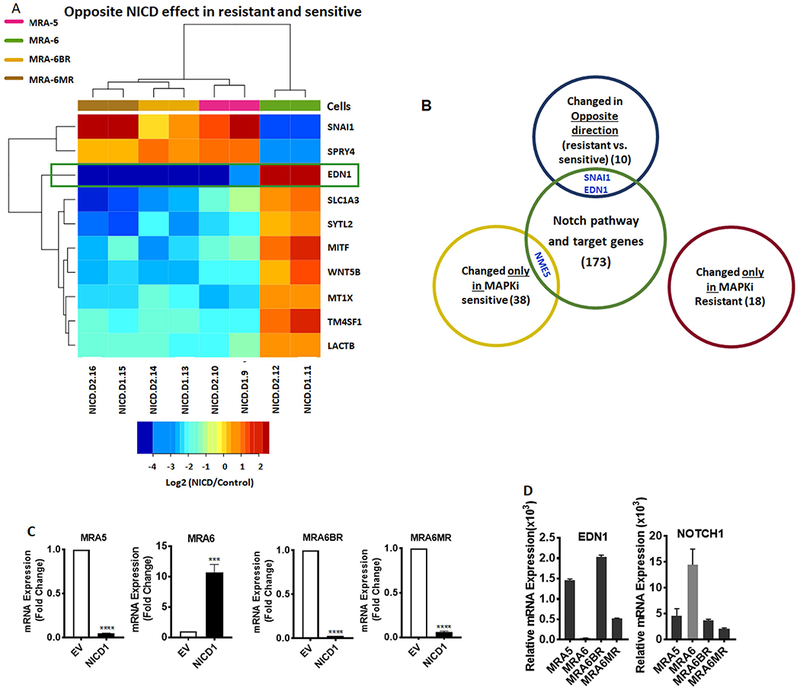
A. Gene expression in NICD1-transduced cells, relative to empty vector-transduced control cells. Expression of 10 genes showing significant differential expression by NICD1 that is directionally opposite in MAPKi-sensitive and MAPKi-resistant cells. Differential expression was scored by EBSeq posterior probability exceeding 0.99, mean fold exceeding 1.5, and directional consistency within resistant cells, as indicated in Methods. B. Venn diagram showing intersection of the three gene lists described in Fig.5A, Fig. S5C and Fig. S5D with Notch signaling pathway genes, Notch signaling target genes and apoptotic pathway genes. C. MAPKi-sensitive and MAPKi-resistant melanoma cells were plated in 6-well plates and transduced with either NICD1 or empty vector lentivirus. Total RNA was isolated 30h after transduction and qRT-PCR was performed for EDN1 mRNA expression using TaqMan probes. GAPDH mRNA expression was used for normalization. D. qRT-PCR analyses for EDN1 and NOTCH1 mRNA expression in MAPKi-sensitive and MAPKi-resistant melanoma cells using gene specific TaqMan probes. GAPDH and ACTB mRNA expression were used for normalization of EDN1 and NOTCH1, respectively. Data shown are mean ± SD of three replicates. Unpaired Student t-test was used to analyze the data. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤0.0001.
We intersected these gene sets with the Notch signaling pathway and target genes (Table S7) and identified three candidate genes; NME5, EDN1 (endothelin 1) and SNAI1 (Fig. 5B). Interestingly, NICD1 expression did not cause de novo activation of novel Notch target genes uniquely in the MAPKi-resistant cells suggesting that NICD1-induced cell death in MAPKi-resistant cells is due to differential regulation of Notch target genes between MAPKi-resistant and MAPKi-sensitive cell.
NME5/NM23-H5, a metastasis suppressor gene (Boissan & Lacombe, 2012; Steeg et al., 1988), was upregulated only in MAPKi-sensitive cells, whereas NME5 mRNA expression was not altered in MAPKi-resistant cells and, therefore, was not analyzed further.
Expression of EDN1 and SNAI1 changed in the opposite direction between MAPKi-sensitive and resistant cells. These genes were previously reported to be regulated by Notch signaling (Matsuno, Coelho, Jarai, Westwick, & Hogaboam, 2012; Meier-Stiegen et al., 2010). SNAI1 is known primarily for its role in melanoma tumor progression (Lin et al., 2010; Massoumi et al., 2009). EDN1, on the other hand, has been recently implicated in melanoma drug resistance (Smith et al., 2017).
We validated the effect of NICD1 on EDN1 mRNA expression by qRT-PCR (Fig. 5C). NICD1 overexpression resulted in downregulation of EDN1 in all three MAPKi-resistant cell lines, whereas EDN1 mRNA was upregulated in NICD1-transduced MAPKi-sensitive MRA-6 cells. Interestingly, basal expression of EDN1 mRNA was also higher in NOTCH1lo MAPKi-resistant cells compared to NOTCH1hi MAPKi-sensitive cells (Fig. 5D), suggesting an inverse relationship between NOTCH1 and EDN1 expression. A query of The Cancer Genome Atlas dataset (TCGA, PanCancer Atlas) (Gao et al., 2013) showed that in melanoma tumor samples NOTCH1 and EDN1 mRNAs show a tendency toward a mutually exclusive upregulation (Fig. S6A).
EDN1 knockdown partially mimics NICD1 overexpression and sensitizes BRAFV600E melanoma cells to MEKi
To test whether downregulation of EDN1 contributes to apoptosis activation, we performed EDN1 knockdown using shRNA lentivirus (Fig. S6B). EDN1 knockdown decreased the survival of both MAPKi-resistant MRA-5 and MAPKi-sensitive MRA-6 cells (Fig. 6A). Cell killing by EDN1 knockdown was less effective than NICD1 overexpression in MAPKi-resistant MRA-5 cells (Fig. 6A) suggesting that downregulation of EDN1 partly accounts for the NICD1-induced apoptosis of MAPKi-resistant melanoma cells.
Figure 6. EDN1 knockdown partially mimics the effect of NICD1 overexpression in MAPKi-resistant melanoma cells.
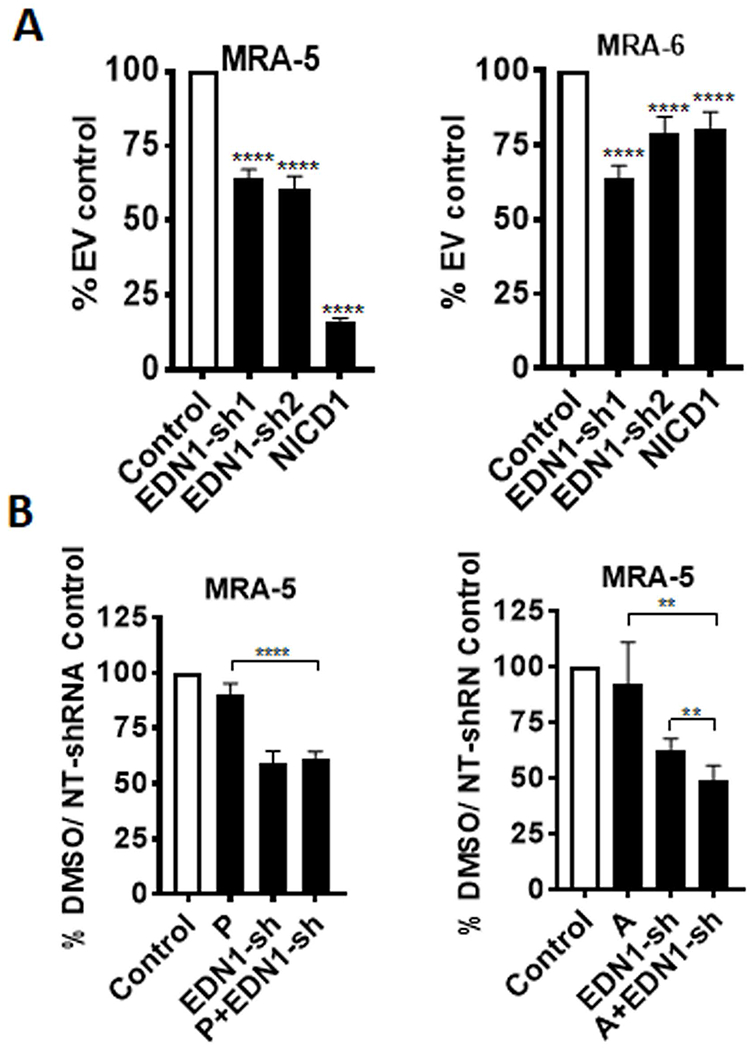
A. Cells were plated in 96-well plates and transduced with EDN1-shRNA lentivirus or non-targeting shRNA lentivirus, or NICD1 or empty vector control lentivirus. Cell survival was assessed using MTT assay after 72h. Data shown are mean ± SD of five to six replicates, normalized to empty vector control, and represent two independent experiments. Unpaired Student t-test was used to analyze the data. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤0.0001. B. Cells were plated in 96-well plates and transduced with EDN1-shRNA lentivirus or non-targeting shRNA lentivirus. Twenty four hours post-transduction cells were treated with 1μM PLX4032 (P) or 1μM AZD6244 (A). Cell survival was assessed after 72 h of treatment using MTT assay. Data shown are mean ± SD of five to six replicates, normalized to DMSO/non-targeting (NT)-shRNA control and are representative of two independent experiments. Unpaired Student t-test was used to analyze the data. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤0.0001.
To test whether downregulation of EDN1 also sensitizes BRAFV600E cells to MAPKi, we treated EDN shRNA lentivirus transduced MRA-5 cells with either the BRAFV600E inhibitor PLX4032 or the MEK inhibitor AZD6244. As shown in Fig. 6B, although compared to PLX4032 treatment alone the combination of PLX4032 treatment with EDN1-KD significantly reduced cell survival, compared to EDN1-KD alone, the combination of PLX4032 treatment with EDN1-KD did not have any additional effect. On the other hand, combination of AZD6244 with EDN1-KD was more effective than either treatment with PLX4032 or EDN1-KD alone. The reason for the selective sensitization to MEKi by EDN1 downregulation is not clear.
Intracrine effect of EDN1 on MAPKi-resistant cells
EDN1 is a secreted peptide that acts as autocrine and paracrine factor by binding to its receptor EDNRA and/or EDNRB (Nussdorfer, Rossi, Malendowicz, & Mazzocchi, 1999). Our RNAseq analysis showed that NICD1 expression also consistently upregulated (8-36 fold) mRNA for EDN1 receptor EDNRA (but not EDNRB) in both MAPKi -sensitive and -resistant cells. Since upregulation of EDN1 by NICD1 in MAPKi-sensitive MRA-6 cells seemed to provide a survival advantage, we asked whether secretion of EDN1 contributes to the survival of MRA-6 cells in response to NICD1 overexpression. Treatment of NICD1-transduced MRA-6 cells with monensin (an intracellular protein transport inhibitor), did not affect their survival (Fig. S6C). We asked whether exogenous addition of recombinant endothelin-1 (ET-1) protein can, conversely, rescue MAPKi-resistant MRA-5 cells form NICD1-induced cell death. Supplementing the culture medium with ET-1, however, had no effect on the survival of NICD1-transduced MRA-5 cells (Fig. 7A). Up-regulation of p-CREB in human melanocytes treated with ET-1 confirmed the activity of the exogenously added ET-1(Tada, Pereira, Beitner-Johnson, Kavanagh, & Abdel-Malek, 2002) (Fig. S6D).
Figure 7. Effect of NICD1 expression on EDN1 in MAPKi resistant cells is mediated by c-JUN.
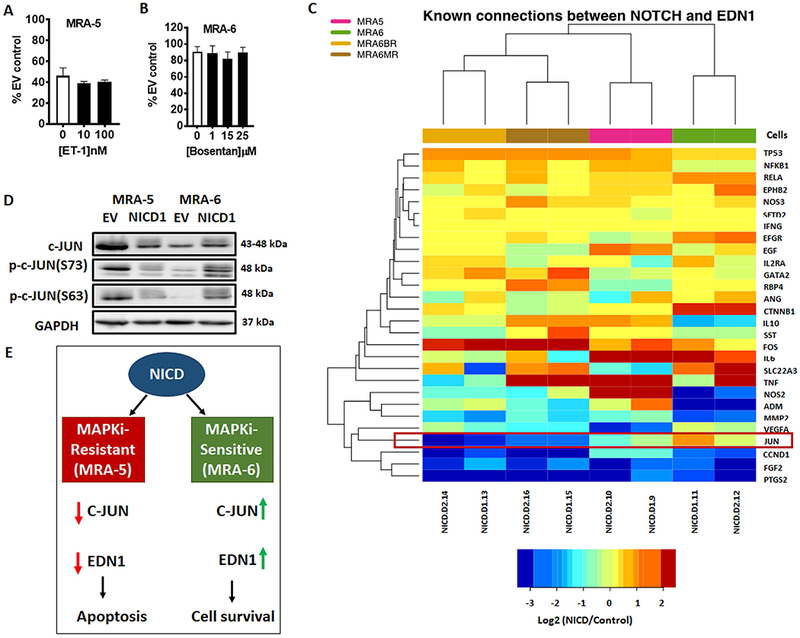
A. Cells were plated in 96-well plates and transduced with NICD1 or empty vector lentivirus for 8h, treated with recombinant ET-1 (10nM or 100nM) for 40h (i.e., 48h post-lentiviral transduction) and cell survival was assessed by MTT assay. Data shown are mean ± SD of five to six replicates, normalized to empty vector control, and are representative of two independent experiments. Unpaired Student t-test was used to analyze the data. B. Cells were plated in 96-well plates and transduced with NICD1 or empty vector lentivirus for 24h, treated with bosentan (1μM, 15 μM, or 25 μM) and incubated for additional 24h. Cell survival was assessed by MTT assay. Data shown are mean ± SD of five to six replicates normalized to empty vector control. C. Change in expression of genes linked to EDN1 regulation by Notch in NICD1-transduced cells relative to empty vector-transduced control cells. D. Cells were plated in 60mm plates and transduced with either NICD1 or empty vector lentivirus for 48h, harvested and lysed for western blot analyses of c-JUN and phospho-c-JUN. Numbers on the right indicate the molecular weight (kDa) of the respective proteins. E. Schematic model for the differential effect of Notch signaling in MAPKi-sensitive and MAPKi-resistant melanoma cells. NICD1 overexpression in MAPKi-sensitive cells causes upregulation of EDN1 through the upregulation of c-JUN.
Furthermore, treatment of NICD1-transduced MAPKi-sensitive cells with either the endothelin receptor A (EDNRA) antagonist, BQ-123, or the dual endothelin receptor (EDNRA and EDNRB) antagonist, bosentan, did not induce cell death (Figs. S6E and 7B). Taken together with the effect of shRNA-mediated knockdown of endogenous EDN1, these data suggest an intracrine mechanism of action for EDN1 in promoting the survival of melanoma cells treated with MAPKi.
Role of c-JUN in the mechanism of action of NICD1
To understand the mechanism of action of NICD1 in regulation of EDN1, we identified a list of genes reported to mediate the effect of Notch signaling and also regulate EDN1 based on a curated PubMed search (Poon, Quirk, DeZiel, & Heckerman, 2014). We queried RNAseq dataset for genes, specifically transcription factors that show change in opposite direction between MAPKi-resistant and MAPKi-sensitive cells in response to NICD1 overexpression. We found that only JUN mRNA levels showed change in the opposite direction similar to the change in EDN1 expression. NICD1 overexpression caused a downregulation of JUN mRNA in MAPKi-resistant cells and a modest upregulation of JUN mRNA levels in MAPKi-sensitive cells (Fig. 7C).
c-JUN is a component of AP-1, a heterodimer consisting of c-FOS and c-JUN subunits. Notch has been reported to downregulate AP-1 complex (Chu & Bresnick, 2004) and AP-1 has been shown to regulate EDN1 transcription (Kawana, Lee, Quertermous, & Quertermous, 1995; Kojima, Ohata, & Yasue, 1997; Stricklett, Strait, & Kohan, 2011). Western blot analysis confirmed that NICD1 overexpression caused a marked downregulation in c-JUN and phospho-c-JUN in MAPKi-resistant MRA-5 cells, whereas c-JUN and phospho-c-JUN levels showed a slight increase in NICD1-transduced MAPKi-sensitive MRA-6 cells (Fig. 7D). These data show that NICD1 downregulates EDN1 in MAPKi-resistant cells by downregulating c-JUN and suggest that in the context of MAPKi resistance, Notch signaling is downregulated resulting in upregulation of EDN1, which can act as an intracrine survival factor (Fig. 7E).
DISCUSSION
Here, we demonstrated that activation of Notch signaling induces cell death by selective downregulation of EDN1 in a subset of melanoma cells resistant to MAP kinase inhibition. Although Notch signaling has been implicated in melanoma, its specific role is still not fully understood. We found that although Notch receptors and some Notch ligands are upregulated in the melanoma cell lines tested, sustained activation of Notch signaling is not essential for melanoma cells in vitro. Previously, Notch inhibition was shown to reduce the growth of primary but not metastatic melanoma cells (Balint et al., 2005). Inhibition of Notch3 was also reported to decrease proliferation of primary, but not metastatic, melanoma cells (Howard et al., 2013).
Notch signaling has also been reported to contribute to resistance of melanoma cells to MAPK inhibitors (Krepler et al., 2016; Martz et al., 2014). Our data, on the contrary, show that downregulation of NOTCH1 is associated with resistance of BRAFV600E melanoma cells to MAPKi and forced expression of active Notch, NICD1, induces cell death preferentially in MAPKi-resistant, but not MAPKi-sensitive, BRAFV600E melanoma cells.
Activation or inhibition of apoptosis by Notch in different cancer cell types has been reported. For example, Notch activation was shown to induce apoptosis in B-cell lymphoblastic leukemia, but not in T-cell lymphoblastic leukemia (Kannan et al., 2011). Therefore, in the context of Notch-induced apoptosis, MAPKi-resistant melanoma cells appear to behave similar to B-ALL, whereas MAPKi-sensitive cells seem to mimic T-ALL cells. Additionally, in neural progenitor cells and hepatocellular carcinoma, Notch was shown to activate apoptosis (Qi et al., 2003; Yang et al., 2004), whereas in myeloma and breast epithelial cells Notch seems to inhibit apoptosis (Meurette et al., 2009; Nefedova et al., 2008).
Downstream of Notch, a divergent role for HES1 in Notch-induced apoptosis has been reported. For example, in myeloma and B-cell malignancies Notch-induced apoptosis was shown to be HES1-dependent (Nefedova et al., 2008; Zweidler-McKay et al., 2005), whereas in CD34+ hematopoietic progenitor cells, apoptosis appears to be HES1-independent (Chadwick et al., 2007). In our studies, although NICD1 upregulated HES1, as expected, HES1 overexpression alone was not sufficient to induce apoptosis.
Whole transcriptome sequencing and differential gene expression analysis identified EDN1 as a mediator of the differential action of NICD1 in MAPKi-resistant vs. MAPKi-sensitive cells. Notch signaling and EDN1 expression showed an inverse relationship in the melanoma cell lines we studied. Analysis of TCGA melanoma tumor data set also showed a tendency for mutually exclusive upregulation of EDN1 and NOTCH1 mRNA. However, most samples with EDN1 or NOTCH1 mRNA overexpression were from patients with either primary melanoma or lymph node metastases, but not on MAPKi treatment.
Although transcription of EDN1 has not been reported to be directly regulated by Notch, other transcription factors regulated by Notch, specifically the activator protein-1 (AP-1), are known to regulate EDN1 transcription (Kawana et al., 1995). AP-1 is a heterodimer composed of FOS and JUN proteins. AP-1, and specifically c-JUN, has been extensively described and implicated in melanoma and apoptosis (Kappelmann, Bosserhoff, & Kuphal, 2014). An AP-1 binding sequence is present upstream of EDN1 gene and this site was shown to be required for EDN1 promoter activity (Kawana et al., 1995). In cervical carcinoma cells, NOTCH1 upregulation was shown to decrease c-FOS expression and suppress AP-1 activity (Talora, Sgroi, Crum, & Dotto, 2002). Notch signaling has also been reported to inhibit AP-1 activity (Chu & Bresnick, 2004). NOTCH1 downregulation sensitized murine erythroleukemia cells to apoptosis and NOTCH1 activation abrogated phosphorylation of c-Jun N-terminal kinase (JNK) (Jang et al., 2004). In our study, we found differential regulation of c-JUN and EDN1 expression by NICD1 in MAPKi-sensitive and MAPKi-resistant cells. Our data suggest that NICD1 activates apoptosis in MAPKi-resistant cells by downregulation of AP-1 (c-JUN)-dependent EDN1 mRNA expression.
Endothelins (EDN1 and EDN3) are secreted peptide hormones and act as autocrine and paracrine factors by binding to their receptors EDNRA and EDNDB (Nussdorfer et al., 1999). Interestingly, neither exogenously added endothelin-1 nor blocking endothelin receptors had any effect on apoptosis of MAPKi- resistant cells or survival of MAPKi-sensitive cells, respectively. However, knockdown of EDN1 alone caused cell death in both MAPKi-sensitive and -resistant cells. These data suggest that EDN1 acts intracellularly rather than as a secreted autocrine/paracrine factor through binding to EDN1 receptors on the cell surface. Intracrine-like function of EDN1 was also proposed in rat cardiomyocytes that express endothelin receptors on the nuclear membranes (Merlen et al., 2013).
EDN1 has been implicated in drug resistance in melanoma (Smith et al., 2017). In melanoma tumors resected from patients on BRAFi treatment, high levels of EDN1 were shown to confer drug resistance. In the same study, upregulation of EDN1 was also found in short-term cultures of isolated tumor cells from melanoma patients on MAPKi treatment. In agreement with these observations, we found that MAPKi-resistant cells express higher levels of EDN1 compared to MAPKi-sensitive cells. Smith et al. (2017) proposed a paracrine role for EDN1 in BRAFi resistance since blocking EDN receptors in vitro with the EDNRA/B antagonist bosentan sensitized melanoma cells to BRAFi. However, in our study, conditioned media from NICD1-transduced MAPKi sensitive cells MRA-6 did not significantly affect the survival of NICD1-transduced MAPKi resistant MRA-5 cells (Fig. S6F). Interestingly, Jagged1/Notch1 signaling was reported to increase ETB(R) expression indirectly by raising the level of phosphorylated signal transducer and activator of transcription 3 (STAT3) in mouse astrocytes after brain injury (LeComte, Shimada, Sherwin, & Spees, 2015).
We queried a publically available RNAseq dataset (Hugo et al., 2015) for the levels of expression for EDN1 and NOTCH1 in melanoma tumors pre-MAPKi treatment and with acquired resistance to MAPKi, and in cell lines treated with MAPKi. Although EDN1 and NOTCH1 levels did not change significantly in patient samples with acquired MAPKi-resistance, EDN1 expression was upregulated in cell lines. However, NOTCH1 levels were not significantly changed, suggesting mechanisms other than Notch signaling also contribute to EDN1 upregulation.
In summary, our study shows that attenuation of Notch signaling is associated with resistance to MAPKi. Reactivation of Notch signaling in MAPKi-resistant cells preferntailly downregulates EDN1, a target of AP-1/c-JUN, and induces cell death. Our data also suggest that EDN1 functions as an intracrine factor unlike its widely known role as a paracrine/autocrine factor. These findings highlight the value of Notch and EDN1 expression levels as potential biomarkers for MAPKi-sensitivity and the potential of targeting intracellular EDN1 as a therapeutic option to overcome MAPKi-resistance. It is known that combination of BRAF and MEK inhibitors improve survival in BRAF-mutant metastatic melanoma. However, resistance develops in the majority of patients (Welsh, Rizos, Scolyer, & Long, 2016). Therefore, it is possible that additional modalities such as inhibition of EDN1 signaling could improve the efficacy of and overcome resistance to MAPKi therapy in a subset of such patients.
Supplementary Material
SIGNIFICANCE:
Resistance of melanoma to MAPK targeted therapy remains a major challenge. Several signaling pathways, including Notch signaling, that contribute to MAPKi resistance have been identified. However, the exact mechanism by which Notch signaling modifies sensitivity of melanoma to MAPKi is not clear. Here, we showed that Notch signaling downregulation is associated with resistance to MAPKi. Reactivation of Notch signaling preferentially downregulates EDN1, a target of AP-1/c-JUN, and induces cell death in MAPKi-resistant cells. These findings highlight the value of Notch and EDN1 expression levels as potential biomarkers for MAPKi-sensitivity and Notch-cJUN-EDN1 axis is a potential therapeutic target in MAPKi-resistant melanoma.
ACKNOWLEDGEMENTS
This work was supported by VA Merit Review Award (1ǀ01BX002623) to VS, in part by the UW Skin Diseases Research Center (NIH/NIAMS P30 AR066524), Data Center grant (U54AI117924) and the UWCCC Biostatistics Shared Resource (P30CA014520). We also would like to acknowledge Clara Nemr for technical help.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors do not have conflicts of interest to disclose.
REFERENCES
- Aubin-Houzelstein G, Djian-Zaouche J, Bernex F, Gadin S, Delmas V, Larue L, & Panthier JJ (2008). Melanoblasts’ proper location and timed differentiation depend on Notch/RBP-J signaling in postnatal hair follicles. J Invest Dermatol, 128(11), 2686–2695. doi: 10.1038/jid.2008.120 [DOI] [PubMed] [Google Scholar]
- Balint K, Xiao M, Pinnix CC, Soma A, Veres I, Juhasz I, Liu ZJ (2005). Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest, 115(11), 3166–3176. doi: 10.1172/jci25001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissan M, & Lacombe ML (2012). [NM23, an example of a metastasis suppressor gene]. Bull Cancer, 99(4), 431–440. doi: 10.1684/bdc.2012.1550 [DOI] [PubMed] [Google Scholar]
- Bonyadi Rad E, Hammerlindl H, Wels C, Popper U, Ravindran Menon D, Breiteneder H, Schaider H (2016). Notch4 Signaling Induces a Mesenchymal-Epithelial-like Transition in Melanoma Cells to Suppress Malignant Behaviors. Cancer Res, 76(7), 1690–1697. doi: 10.1158/0008-5472.can-15-1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick N, Nostro MC, Baron M, Mottram R, Brady G, & Buckle AM (2007). Notch signaling induces apoptosis in primary human CD34+ hematopoietic progenitor cells. Stem Cells, 25(1), 203–210. doi: 10.1634/stemcells.2005-0303 [DOI] [PubMed] [Google Scholar]
- Chen KY, Srinivasan T, Tung KL, Belmonte JM, Wang L, Murthy PKL, Shen X (2017). A Notch positive feedback in the intestinal stem cell niche is essential for stem cell self-renewal. Molecular Systems Biology, 13(4). doi: 10.15252/msb.20167324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, & Bresnick EH (2004). Evidence that C promoter-binding factor 1 binding is required for Notch-1-mediated repression of activator protein-1. J Biol Chem, 279(13), 12337–12345. doi: 10.1074/jbc.M311510200 [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Schultz N (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal, 6(269), pl1. doi: 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JD, Moriarty WF, Park J, Riedy K, Panova IP, Chung CH, Alani RM (2013). Notch signaling mediates melanoma-endothelial cell communication and melanoma cell migration. Pigment Cell Melanoma Res, 26(5), 697–707. doi: 10.1111/pcmr.12131 [DOI] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Lo RS (2015). Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell, 162(6), 1271–1285. doi: 10.1016/j.cell.2015.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MS, Miao H, Carlesso N, Shelly L, Zlobin A, Darack N, Miele L (2004). Notch-1 regulates cell death independently of differentiation in murine erythroleukemia cells through multiple apoptosis and cell cycle pathways. J Cell Physiol, 199(3), 418–433. doi: 10.1002/jcp.10467 [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, & Israel A (1995). Signalling downstream of activated mammalian Notch. Nature, 377(6547), 355–358. doi: 10.1038/377355a0 [DOI] [PubMed] [Google Scholar]
- Kannan S, Fang W, Song G, Mullighan CG, Hammitt R, McMurray J, & Zweidler-McKay PA (2011). Notch/HES1-mediated PARP1 activation: a cell type-specific mechanism for tumor suppression. Blood, 117(10), 2891–2900. doi: 10.1182/blood-2009-12-253419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappelmann M, Bosserhoff A, & Kuphal S (2014). AP-1/c-Jun transcription factors: Regulation and function in malignant melanoma. European Journal of Cell Biology, 93(1), 76–81. doi: 10.1016/j.ejcb.2013.10.003 [DOI] [PubMed] [Google Scholar]
- Kawana M, Lee ME, Quertermous EE, & Quertermous T (1995). Cooperative interaction of GATA-2 and AP1 regulates transcription of the endothelin-1 gene. Mol Cell Biol, 15(8), 4225–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M, Ohata K, & Yasue H (1997). Structural organization and chromosomal assignment of the swine endothelin-1 gene. J Vet Med Sci, 59(6), 431–435. [DOI] [PubMed] [Google Scholar]
- Krepler C, Xiao M, Samanta M, Vultur A, Chen HY, Brafford P, Villanueva J (2016). Targeting Notch enhances the efficacy of ERK inhibitors in BRAF-V600E melanoma. Oncotarget, 7(44), 71211–71222. doi: 10.18632/oncotarget.12078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeComte MD, Shimada IS, Sherwin C, & Spees JL (2015). Notch1-STAT3-ETBR signaling axis controls reactive astrocyte proliferation after brain injury. Proc Natl Acad Sci U S A, 112(28), 8726–8731. doi: 10.1073/pnas.1501029112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, Margolin KA (2015). Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer, 121(3), 432–440. doi: 10.1002/cncr.29055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Baritaki S, Militello L, Malaponte G, Bevelacqua Y, & Bonavida B (2010). The Role of B-RAF Mutations in Melanoma and the Induction of EMT via Dysregulation of the NF-kappaB/Snail/RKIP/PTEN Circuit. Genes Cancer, 1(5), 409–420. doi: 10.1177/1947601910373795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, Herlyn M (2006). Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res, 66(8), 4182–4190. doi: 10.1158/0008-5472.can-05-3589 [DOI] [PubMed] [Google Scholar]
- Maddodi N, Bhat KM, Devi S, Zhang SC, & Setaluri V (2010). Oncogenic BRAFV600E induces expression of neuronal differentiation marker MAP2 in melanoma cells by promoter demethylation and down-regulation of transcription repressor HES1. J Biol Chem, 285(1), 242–254. doi: 10.1074/jbc.M109.068668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Wood KC (2014). Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci Signal, 7(357), ra121. doi: 10.1126/scisignal.aaa1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T, Bosserhoff AK (2009). Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med, 206(1), 221–232. doi: 10.1084/jem.20082044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno Y, Coelho AL, Jarai G, Westwick J, & Hogaboam CM (2012). Notch signaling mediates TGF-beta1-induced epithelial-mesenchymal transition through the induction of Snai1. Int J Biochem Cell Biol, 44(5), 776–789. doi: 10.1016/j.biocel.2012.01.021 [DOI] [PubMed] [Google Scholar]
- Meier-Stiegen F, Schwanbeck R, Bernoth K, Martini S, Hieronymus T, Ruau D, Just U (2010). Activated Notch1 target genes during embryonic cell differentiation depend on the cellular context and include lineage determinants and inhibitors. PLoS One, 5(7), e11481. doi: 10.1371/journal.pone.0011481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlen C, Farhat N, Luo X, Chatenet D, Tadevosyan A, Villeneuve LR, Allen BG (2013). Intracrine endothelin signalling evokes IP3-dependent increases in nucleoplasmic Ca(2+) in adult cardiac myocytes(). Journal of molecular and cellular cardiology, 62, 189–202. doi: 10.1016/j.yjmcc.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, & Brennan K (2009). Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res, 69(12), 5015–5022. doi: 10.1158/0008-5472.can-08-3478 [DOI] [PubMed] [Google Scholar]
- Moriyama M, Osawa M, Mak SS, Ohtsuka T, Yamamoto N, Han H, Nishikawa S (2006). Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J Cell Biol, 173(3), 333–339. doi: 10.1083/jcb.200509084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, & Gabrilovich DI (2008). Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood, 111(4), 2220–2229. doi: 10.1182/blood-2007-07-102632 [DOI] [PubMed] [Google Scholar]
- Nussdorfer GG, Rossi GP, Malendowicz LK, & Mazzocchi G (1999). Autocrine-paracrine endothelin system in the physiology and pathology of steroid-secreting tissues. Pharmacol Rev, 51(3), 403–438. [PubMed] [Google Scholar]
- Poon H, Quirk C, DeZiel C, & Heckerman D (2014). Literome: PubMed-scale genomic knowledge base in the cloud. Bioinformatics, 30(19), 2840–2842. doi: 10.1093/bioinformatics/btu383 [DOI] [PubMed] [Google Scholar]
- Qi R, An H, Yu Y, Zhang M, Liu S, Xu H, Cao X (2003). Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res, 63(23), 8323–8329. [PubMed] [Google Scholar]
- Rodriguez CI, Castro-Perez E, Prabhakar K, Block L, Longley BJ, Wisinski JA, Setaluri V (2017). EPAC-RAP1 Axis-Mediated Switch in the Response of Primary and Metastatic Melanoma to Cyclic AMP. Mol Cancer Res, 15(12), 1792–1802. doi: 10.1158/1541-7786.mcr-17-0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, … Fisher DE (2014). Pathways and therapeutic targets in melanoma. Oncotarget, 5(7), 1701–1752. doi: 10.18632/oncotarget.1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MP, Rowling EJ, Miskolczi Z, Ferguson J, Spoerri L, Haass NK, Wellbrock C (2017). Targeting endothelin receptor signalling overcomes heterogeneity driven therapy failure. EMBO Mol Med, 9(8), 1011–1029. doi: 10.15252/emmm.201607156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA, & Sobel ME (1988). Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst, 80(3), 200–204. [DOI] [PubMed] [Google Scholar]
- Stricklett PK, Strait KA, & Kohan DE (2011). Novel regulation of endothelin-1 promoter activity by protein kinase C. Cell Biochem Biophys, 61(3), 643–650. doi: 10.1007/s12013-011-9250-0 [DOI] [PubMed] [Google Scholar]
- Tada A, Pereira E, Beitner-Johnson D, Kavanagh R, & Abdel-Malek ZA (2002). Mitogen- and Ultraviolet-B-Induced Signaling Pathways in Normal Human Melanocytes. Journal of Investigative Dermatology, 118(2), 316–322. doi: 10.1046/j.0022-202x.2001.01694.x [DOI] [PubMed] [Google Scholar]
- Talora C, Sgroi DC, Crum CP, & Dotto GP (2002). Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev, 16(17), 2252–2263. doi: 10.1101/gad.988902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Chin L, Garraway LA, & Fisher DE (2012). Melanoma: from mutations to medicine. Genes Dev, 26(11), 1131–1155. doi: 10.1101/gad.191999.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh SJ, Rizos H, Scolyer RA, & Long GV (2016). Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer, 62, 76–85. doi: 10.1016/j.ejca.2016.04.005 [DOI] [PubMed] [Google Scholar]
- Yang X, Klein R, Tian X, Cheng HT, Kopan R, & Shen J (2004). Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol, 269(1), 81–94. doi: 10.1016/j.ydbio.2004.01.014 [DOI] [PubMed] [Google Scholar]
- Zhu G, Yi X, Haferkamp S, Hesbacher S, Li C, Goebeler M, Schrama D (2016). Combination with gamma-secretase inhibitor prolongs treatment efficacy of BRAF inhibitor in BRAF-mutated melanoma cells. Cancer Lett, 376(1), 43–52. doi: 10.1016/j.canlet.2016.03.028 [DOI] [PubMed] [Google Scholar]
- Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, Pear WS (2005). Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood, 106(12), 3898–3906. doi: 10.1182/blood-2005-01-0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.