

Abstract
The collapsin response mediator protein 2 (CRMP2) has emerged as a central node in assembling nociceptive signaling complexes involving voltage-gated ion channels. Concerted actions of posttranslational modifications, phosphorylation and SUMOylation, of CRMP2 contribute to regulation of pathological pain states. In the present study, we demonstrate a novel role for CRMP2 in spinal nociceptive transmission. We found that, of six possible post-translational modifications, three phosphorylation sites on CRMP2 were critical for regulating calcium influx in dorsal root ganglion sensory neurons. Of these, only CRMP2 phosphorylated at serine 522 by cyclin dependent kinase 5 (Cdk5) contributed to spinal neurotransmission in a bidirectional manner. Accordingly, expression of a non-phosphorylatable CRMP2 (S522A) decreased the frequency of spontaneous excitatory postsynaptic currents (sEPSCs), whereas expression of a constitutively phosphorylated CRMP2 (S522D) increased the frequency of sEPSCs. The presynaptic nature of CRMP2’s actions were further confirmed by pharmacological antagonism of Cdk5-mediated CRMP2 phosphorylation with S-N-benzy-2-acetamido3-methoxypropionamide ((S)-Lacosamide; (S)-LCM) which (i) decreased sEPSC frequency, (ii) increased paired pulse ratio, and (iii) reduced the presynaptic distribution of CaV2.2 and NaV1.7, two voltage-gated ion channels implicated in nociceptive signaling. (S)-LCM also inhibited depolarization-evoked release of the pro-nociceptive neurotransmitter calcitonin gene related peptide (CGRP) in the spinal cord. Increased CRMP2 phosphorylation in rats with spared nerve injury (SNI) was decreased by intrathecal administration of (S)-LCM resulting in a loss of pre-synaptic localization of CaV2.2 and NaV1.7. Together, these findings indicate that CRMP2 regulates presynaptic excitatory neurotransmission in spinal cord and may play an important role in regulating pathological pain. Novel targeting strategies to inhibit CRMP2 phosphorylation by Cdk5 may have great potential for the treatment of chronic pain.
Keywords: CRMP2, NaV1.7, CaV2.2, (S)-Lacosamide, spontaneous excitatory postsynaptic currents, CGRP
Introduction
Pain serves a crucial protective role by alerting the body to potentially harmful stimuli. However, pain that persists months or years after an injury has healed is viewed as pathological chronic pain [1–3]. In this state, pain occurs spontaneously and responses to noxious and innocuous stimuli are pathologically magnified. Ineffective treatment of chronic pain results from our incomplete understanding of mechanisms underlying abnormal neuronal circuits along nociceptive pathways.
Sensory signals are transmitted by primary afferent fibers into the superficial layers of the dorsal horn of the spinal cord, where the first sensory synapse formed by central terminals of primary afferent neurons and spinal second order neurons is critically involved in nociceptive transmission and regulation [4,5]. An increase of synaptic transmission, efficacy, and plasticity are key components in central sensitization, which is needed for pain chronification [6]. Throughout the central nervous circuits, high voltage-gated calcium channels, including the N-type (CaV2.2) channel, are dominant in controlling release of neurotransmitters release such as glutamate [7,8].
In studying regulators of CaV2.2 channels, a proteomic approach identified the axonally expressed collapsin response mediator protein 2 (CRMP2) as a biochemical and functional regulator of CaV2.2 channels in hippocampal and sensory neurons [9–11]. Uncoupling CRMP2 from CaV2.2 complex suppressed inflammatory and neuropathic pain [12–16]. Although CRMP2 was initially identified as a protein involved in axon specification/guidance within the developing central nervous system [17,18], it is now known that CRMP2’s cellular functions include ion channel trafficking [12,19–21]. These functions appear to be under the control of SUMOylation [22,23,20,24–26] or a multiplicity of phosphorylation events triggered by kinases including cyclin-dependent kinase 5 (Cdk5) [27], glycogen synthase kinase 3β (GSK3β) [28], Rho-associated protein kinase (ROCK) [29], and the Src-family kinases Yes [30] and Fyn [31].
It is now recognized that protein kinases are involved in the physiopathology of acute and chronic pain [32]. Phosphorylation of CRMP2 by kinases is balanced by its dephosphorylation by phosphatases. Cdk5 is the “priming” kinase responsible for first phosphorylating CRMP2 at Serine 522 [33], which then makes CRMP2-T514 (as well as of CRMP2-T518 and CRMP2-T509) available for phosphorylation by GSK3β [27]. We reported enhanced interaction of cyclin-dependent kinase 5 (Cdk5)-phosphorylated CRMP2 with presynaptic CaV2.2 which lead to enhanced calcium entry following membrane depolarization in DRG neurons [34]. Further, we demonstrated that CRMP2 phosphorylation by Cdk5 appears to be necessary and sufficient for peripheral neuropathic pain of varying etiologies [35,36,26]. Increased circulating CRMP2 autoantibodies were found in the acute stage of spinal cord injury, which predicts the subsequent development of neuropathic pain [37]. Hijacking CRMP2 phosphorylation resulted in normalization of ion channel current densities and excitability of dorsal root ganglion (DRG) neurons [22], as well as of hyperalgesia in a gene editing model of Neurofibromatosis type 1 related pain [38]. Pharmacological antagonism of Cdk5-mediated CRMP2 phosphorylation by S-N-benzy-2acetamido-3-methoxypropionamide ((S)-Lacosamide; (S)-LCM) inhibited calcium influx in sensory neurons [39] and reversed post-operative surgical and neuropathic pain behaviors [39].
Out studies have thus far established CRMP2 phosphorylation by Cdk5 to be an intrinsic pathological event participating in the establishment of chronic neuropathic pain. However, the precise role of CRMP2 (and its phosphorylated forms) within the sensory nervous circuitry, especially in the nociceptive central nervous system, remain unclear. Therefore, here we investigated the potential role of phosphorylation of CRMP2 on the interaction with presynaptic CaV2.2, and how that contributes to spinal nociceptive synaptic transmission.
Materials and Methods
Animals
Pathogen-free, male Sprague-Dawley rat pups (12–21 days old; Envigo) were used for electrophysiological experiments and adult male Sprague-Dawley rats (100g; Envigo) were used for all other experiments. All animals were housed in a temperature (23±3 °C) and light-controlled (12-h light/12-h dark cycle; lights on 08:00–20:00) rooms with chow and water available ad libitum. The Institutional Animal Care and Use Committee of the College of Medicine at the University of Arizona approved all experiments. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain. All electrophysiology and calcium imaging experiments were performed by experimenters who were blinded to the experimental groups and treatments.
Preparation of spinal cord slices
As described previously [40], young (postnatal 12–21 days) rats were deeply anesthetized with isoflurane (4% for induction and 2% for maintaining). For spinal nerve block, 0.3 mL of 2% lidocaine was injected to both sides of L4 to 5 lumbar vertebrae. Laminectomy was performed from mid-thoracic to low lumbar levels, and the spinal cord was quickly removed to cold modified ACSF oxygenated with 95% O2 and 5% CO2. The ACSF for dissection contained (in millimolar): 80 NaCl, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3.5 MgCl2, 25 NaHCO3, 75 sucrose, 1.3 ascorbate, 3.0 sodium pyruvate, with pH at 7.4 and osmolarity at 310 mOsm. Transverse 350- mm thick slices were obtained by a vibratome (VT1200S; Leica, Nussloch, Germany). Slices were then incubated for at least 1 hour at RT in an oxygenated recording solution containing (in millimolar): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 25 D-glucose, 1.3 ascorbate, 3.0 sodium pyruvate, with pH at 7.4 and osmolarity at 320 mOsm. The slices were then positioned in a recording chamber and continuously perfused with oxygenated recording solution at a rate of 3 to 4 mL/min before electrophysiological recordings at RT.
Electrophysiological recordings in spinal cord slices by whole-cell patch clamp
Whole cell recording experiments were performed as described previously [41]. Substantia gelatinosa neurons (lamina I/II) were visualized and identified in the slices by means of infrared differential interference contrast video microscopy on an upright microscope (FN1; Nikon, Tokyo, Japan) equipped with a 3.40/0.80 water-immersion objective and a charge-coupled device camera. Patch pipettes with resistance at 6 to 10 MΩ were made from borosilicate glass (Sutter Instruments, Novato, CA) on a 4-steps micropipette puller (P-90; Sutter Instruments, Novato, CA). The pipette solution contained the following (in millimolar): 120 potassium gluconate, 20 KCl, 2 MgCl2, 2 Na2-ATP, 0.5 Na-GTP, 20 HEPES, 0.5 EGTA, with pH at 7.28 and osmolarity at 310 mOsm. The membrane potential was held at 60 mV using a PATCHMASTER software in combination with a patch clamp amplifier (EPC10; HEKA Elektronik, Lambrecht, Germany).
The whole-cell configuration was obtained in voltage-clamp mode. To record spontaneous excitatory postsynaptic currents (sEPSCs), bicuculline methiodide (10 μM) and strychnine (2 μM) were added to the recording solution to block γ-aminobutyric acid-activated (GABA) and glycine-activated currents. Tetrodotoxin (TTX; 1 μM, Cat# ab120054, Abcam) was added to block action potentials when we were recording miniature excitatory postsynaptic currents (mEPSCs). Hyperpolarizing step pulses (5 mV in intensity, 50 milliseconds in duration) were periodically delivered to monitor the access resistance (15–25 MΩ), and recordings were discontinued if the access resistance changed by more than 20%. For each neuron, sEPSCs or mEPSCs were recorded for a total duration of 2 minutes. Currents were filtered at 3 kHz and digitized at 5 kHz. Data were further analyzed by the Mini-Analysis Program (Synatosoft Inc, NJ) to provide spreadsheets for the generation of cumulative probability plots. The amplitude and frequency of sEPSCs were compared between neurons from animals in control and the indicated groups.
Confocal imaging of spinal cord slices after electrophysiological recording
Following electrophysiological recordings and filling of recorded neurons with Biocytin (Millipore-Sigma, B4261), slices were fixed in 4% PFA for 20 minutes at room temperature. Non-specific staining was blocked in 3% BSA, 0.1% Triton X-100 in PBS for 1 hour at room temperature and slices were incubated with Streptavidin-AlexaFluor488 (Cat# S32354, Invitrogen,) and a primary antibody antidsRed (Table 1) overnight at +4°C. After thorough washing, slices were incubated with a secondary antibody (Goat anti-Mouse AlexaFluor 660, Cat# A21054 Invitrogen, or Goat anti-Mouse AlexaFluor 594, Cat# A11032, Invitrogen) for 4 hours at room temperature, washed and counterstained with DAPI. Images were obtained using a Zeiss LSM880 confocal microscope equipped with a EC Plan-Apochromat 40x (NA=1.3) lens.
Table 1 :
Antibodies used in this study
| Antibody | species | Catalog number | Company |
|---|---|---|---|
| CRMP2 | Rabbit | C2993 | Sigma, St. Louis, MO |
| CRMP2 pS522 | Rabbit | CP2191 | ECM Biosciences, Versailles, KY |
| CaV2.2 | Rabbit | TA308673 | Origene, Rockville, MD |
| NaV1.7 | Mouse | 75–103 | NeuroMab, Davis, CA |
| Synaptophysin | Mouse | MAB5258 | Thermofisher scientific, San Diego, CA |
| PSD95 | Mouse | MA1–045 | Thermofisher scientific, San Diego, CA |
| CaV2.3 | Rabbit | ACC-006 | Alomone, Jerusalem, Israel |
| CB1R | Rabbit | abl37410 | Abeam, Cambridge, UK |
| Actin | Rabbit | A2066 | Sigma, St. Louis, MO |
| Flotilin | Rabbit | F1180 | Sigma, St. Louis, MO |
| DsRed | Mouse | 51–8115GR | BD Pharmingen, San Jose, CA |
Preparation of acutely dissociated dorsal root ganglion neurons
Dorsal root ganglia from all levels were acutely dissociated from 100 g Sprague-Dawley rats and DRG neurons were isolated as we described previously [13,39,24,42,35,38,43–45,22,46]. In brief, removing dorsal skin and muscle and cutting the vertebral bone processes parallel to the dissection stage-exposed DRG. Dorsal root ganglia were then collected, trimmed at their roots, and enzymatically digested in 3 mL bicarbonate-free, serum-free, sterile DMEM (Cat# 11965, Thermo Fisher Scientific) solution containing neutral protease (3.125 mg.ml−1, Cat# LS02104; Worthington, Lakewood, NJ) and collagenase type I (5 mg.ml−1, Cat# LS004194, Worthington, Lakewood, NJ) and incubated for 60 minutes at 37°C under gentle agitation. Dissociated DRG neurons (~1.5 × 106) were then gently centrifuged to collect cells and washed with DRG media DMEM containing 1% penicillin/streptomycin sulfate from 10,000 μg/mL stock, 30 ng/mL nerve growth factor, and 10% fetal bovine serum (Hyclone) before plating onto poly-D-lysine- and laminin-coated glass 12- or 15-mm coverslips. All cultures were used within 48 hours.
Dorsal root ganglia neuron transfection
Collected cells were re-suspended in Nucleofector transfection reagent containing 4 μg of the indicated pdsRed-N2 plasmids described earlier [22]. Then, cells were subjected to electroporation protocol O-003 in an Amaxa Biosystem (Lonza, Basel, Switzerland) and plated onto poly-d-lysine-coated glass 15 mm glass coverslips. Transfection efficiencies were routinely between 20% and 30% with about ~10% cell death. Successfully transfected cells were identified by dsRed fluorescence.
Calcium imaging in acutely dissociated dorsal root ganglion (DRG) neurons
Dorsal root ganglion neurons were loaded for 30 minutes at 37°C with 3 μM Fura-2AM (Cat# F1221, Thermo Fisher, stock solution prepared at 1mM in DMSO, 0.02% pluronic acid, Cat#P-3000MP, Life technologies) to follow changes in intracellular calcium([Ca2+]c) in a standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM Na HEPES, pH 7.4, 5 mM glucose exactly as previously described [19,13,47–49,43,45]. Fluorescence imaging was performed with an inverted microscope, NikonEclipseTi-U (Nikon Instruments Inc., Melville, NY), using objective Nikon Fluor 4X and a Photometrics cooled CCD camera Cool SNAP ES2 (Roper Scientific, Tucson, AZ) controlled by Nis Elements software (version 4.20, Nikon Instruments). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters (340 ± 5 and 380 ± 7) were controlled by a Lambda 10 to 2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505-nm dichroic mirror at 535 ± 25 nm. To minimize photobleaching and phototoxicity, the images were taken every ~10 seconds during the time-course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c was monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels. For analysis, only transfected DRG neurons, identified by dsRed florescence, were used. Our analysis included all transfected DRGs based on our previous reports demonstrating that inhibition of CRMP2 with (i) either a knockdown strategy (CRMP2-siRNA), (ii) peptide-based interference strategies (TAT-CBD3, TAT-CBD3-A6K, TAT-CBD3-L5M, TAT-CNRP1), or (iii) pharmacological antagonism ((S)-LCM) blunted depolarization evoked calcium influx in all DRG neurons tested independently of their size [50,12,51,43,45,39].
Calcitonin gene–related peptide (CGRP) release from lumbar slices
Rats were deeply anesthetized with 5% isofluorane and then decapitated. Two vertebral incisions (cervical and lumbar) were made in order to expose the spinal cord. Pressure was applied to a saline-filled syringe inserted into the lumbar vertebral foramen, and the spinal cord was extracted. Only the lumbar region of the spinal cord was used for the CGRP release assay. Baseline treatments (#1 and #2) involved bathing the spinal cord in Tyrode’s solution. The excitatory solution consisting of 90 mM KCl was paired with the treatment for fraction #4. These fractions (10 minutes, 400 μL each) were collected for measurement of CGRP release. Samples were immediately flash frozen and stored in a −20°C freezer. (S)-LCM (10 μM) or vehicle (0.9% saline) was added to the pretreatment and co-treatment fractions (#3 and 4). The concentration of CGRP released into the buffer was measured by enzyme-linked immunospecific assay (Cat# 589001, Cayman Chemical, Ann Arbor, MI).
Synapse enrichment and fractionation
Adult rats were killed by isofluorane overdose and decapitation, the spinal cords dissected and the lumbar dorsal horn collected. Only the dorsal horn of the spinal cord was used as this structure contains the synapses arising from the DRG. Synaptosomes were isolated as described previously [52]. Fresh tissues were homogenized in ice-cold Sucrose 0.32M, HEPES 10 mM, pH 7.4 buffer. The homogenates were centrifuged at 1000xg for 10 min at 4°C to pellet the insoluble material. The supernatant was harvested and centrifuged at 12000xg for 20 min at 4°C to pellet a crude membrane fraction. The pellet was then re-suspended in a hypotonic buffer (4 mM HEPES, 1 mM EDTA, pH 7.4) and the resulting synaptosomes pelleted by centrifugation at 12000xg for 20 min at 4°C. The synaptosomes were then incubated in 20 mM HEPES, 100 mM NaCl, 0.5% triton X, pH= 7.2) for 15 min on ice and centrifuged at 12000xg for 20 min at 4°C. The supernatant was considered as the non-postsynaptic density (nonPSD) membrane fraction, sometimes referred to as the triton soluble fraction. The pellet containing the postsynaptic density fraction (PSD) was then solubilized (20 mM HEPES, 0.15 mM NaCl, 1% triton X100, 1% deoxycholic acid, 1% SDS, pH= 7.5). The integrity of non-PSD and PSD fractions was verified by immunoblotting for PSD95, which was enriched in PSD fraction, and synaptophysin which was enriched in non-PSD fraction (see Figure 3A). All buffers were supplemented with protease and phosphatase inhibitor cocktails. Protein concentrations were determined using the BCA protein assay.
Figure 3. CRMP2 phosphorylation alters sEPSC frequency in lumbar dorsal horn.

(A) Representative recording traces of sEPSC recordings from SG neurons from the indicated groups. (B) A cumulative distribution of sEPSCs frequency recorded from SG neurons transfected with CRMP2 S522A revealed a rightward shift toward longer inter-event interval, while transfected with CRMP2 S522D showed a leftward shift toward shorter inter-event interval. (C) A cumulative distribution of sEPSCs amplitude. No significant change was observed in this parameter. Summary of amplitudes (D) and frequencies (E) of sEPSCs for different groups are shown. Overexpression of CRMP2 S522A resulted in a decrease in the frequency of sEPSCs (D), while neurons transfected with CRMP2 S522D showed an increase in the frequency of sEPSCs. There is no difference in the amplitude (E) of sEPSCs between groups. Data are expressed as means ± SEM. *p < 0.05 (versus baseline); #p < 0.01 (versus as indicated), one-way ANOVA followed by Tukey’s post hoc test.
Immunoblot preparation and analysis
Tissue lysates prepared from adult Sprague-Dawley rats were generated by homogenization and sonication in RIPA buffer (50mM Tris-HCl, pH 7.4, 50 mM NaCl, 2 mM MgCl2, 1% [vol/vol] NP40, 0.5% [mass/vol] sodium deoxycholate, 0.1% [mass/vol] SDS) as described previously [13]. Protease inhibitors (Cat# B14002; Bimake, Houston, TX), phosphatase inhibitors (Cat# B15002, Bimake), and benzonase (Cat#71206, Millipore, Billerica, MA). Protein concentrations were determined using the BCA protein assay (Cat# PI23225, Thermo Fisher Scientific, Waltham, MA). Indicated samples were loaded on 4–20% Novex® gels (Cat# EC60285BOX, Thermo Fisher Scientific, Waltham, MA). Proteins were transferred for 1h at 120 V using TGS (25mM Tris pH=8.5, 192mM glycine, 0.1% (mass/vol) SDS), 20% (vol/vol) methanol as transfer buffer to polyvinylidene difluoride (PVDF) membranes 0.45μm (Cat# IPVH00010, Millipore, Billerica, MA), pre-activated in pure methanol. After transfer, the membranes were blocked at room temperature for 1 hour with TBST (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1 % Tween 20), 5% (mass/vol) non-fat dry milk, then incubated separately with the indicated primary antibodies (Table 1) in TBST, 5% (mass/vol) BSA, overnight at 4°C. Following incubation in horseradish peroxidase-conjugated secondary antibodies from Jackson immunoresearch, blots were revealed by enhanced luminescence (WBKLS0500, Millipore, Billerica, MA) before exposure to photographic film. Films were scanned, digitized, and quantified using Un-Scan-It gel version 6.1 scanning software by Silk Scientific Inc. For all experiments, CRMP2 phosphorylation levels were always normalized to total CRMP2 levels in the same sample.
Statistics
Statistical analyses were performed using GraphPad Prism 7 (GraphPad, La Jolla, CA). Data were sourced from a minimum of three independent biological replicates unless indicated otherwise. All data represent the mean ± S.E.M. All data was first tested for a Gaussian distribution using a D’agostino-Pearson test (GraphPad Prism 7 Software). The statistical significance of differences between means was determined by either parametric or non-parametric Student’s t-test, analysis of variance (ANOVA) followed by post hoc comparisons (Tukey) using Prism 7. Statistical significance was set at α < 0.05.
Results
CRMP2 phosphorylation status controls depolarization-evoked calcium influx
CRMP2 expression levels and phosphorylation by Cdk5 alter depolarization-evoked calcium influx and calcium currents in cortical neurons [34,11]. As a first step to study the function of CRMP2 in spinal synaptic transmission from DRG sensory neurons, we explored how CRMP2 post-translational modifications could regulate the function of sensory neuronal voltage-gated calcium channels. DRG neurons were transfected with plasmids expressing wildtype CRMP2 with a dsRed tag (to detect the transfected neurons with red fluorescence, Figure 1A) or CRMP2 harboring inactivating mutations (change of the phosphorylated amino acid to an alanine) of known CRMP2 phosphorylation sites [36]. N-type (CaV2.2) channels have been reported to account for ~30–50% of the total calcium current given that there is considerable heterogeneity of the neuronal population comprising the DRG [53,54]. The transfected DRGs were challenged with 90mM KCl (a concentration known to activate mostly CaV2.x channels [55]) and measured the evoked calcium influx in the transfected neurons (Figure 1A). Expressing wildtype CRMP2 in DRG neurons did not affect depolarization-evoked calcium influx (peak or area under the curve, AUC) compared to dsRed-transfected controls DRGs (Figure 1B–D). Depolarization-evoked calcium influx was also unaffected in DRGs expressing CRMP2 mutations of the Fyn (Y32F) or Rho (T555A) kinase sites but expressing a CRMP2 deficient for the Cdk5 (S522A)kinase site resulted in decreased evoked calcium influx (Figure 1B–D). To confirm that this phosphorylation site was important for CRMP2 regulation of depolarization-evoked calcium influx, we used CRMP2 mutations that mimic the presence of a phosphorylated residue (mutation from serine/threonine to an aspartate). With the phospho-mimetic CRMP2 mutants, we observed that the depolarization-evoked calcium influx in DRGs expressing S522D (Cdk5) CRMP2 mutant was statistically similar to control DRGs, whereas DRGs expressing the T555D (RhoK) CRMP2 mutant exhibited decreased depolarization evoked calcium influx compared to control DRGs (Figure 1E–G). Finally, we also tested the effect of the loss of CRMP2 SUMOylation on depolarization evoked calcium influx and found that preventing CRMP2 SUMOylation (K374A) had no effect (Figure 1E–G).
Figure 1: CRMP2 post-translational modifications affect depolarization-evoked calcium influx.

Dorsal root ganglion neurons were transfected during plating with a dsRed fused CRMP2 plasmids. (A) representative experiment (dsRed fluorescence and pseudocolored fluorescent images visualized for Fura2-AM before (middle panel) and after stimulations with 90 mM KCl (right panel). In this experiment, transfected neurons responded to KCl. (B), (E) and (H) Traces of response average for the indicated transfections. Arrows indicate the initiation of a 15 second stimulation period of the DRG sensory neurons with 90 mM KCl as indicated. (C), (F) and (I) Bar graph shows peak calcium response averages ± S.E.M. of DRG sensory neurons transfected with the indicated plasmid. (D), (G) and (J) Bar graph showing the area under the curve (Averages ± S.E.M.) of DRG sensory neurons transfected with the indicated plasmid. Statistical significance compared to dsRed-CRMP2 wildtype transfected cells is indicated (*p<0.05, one-way ANOVA with Dunnett’s post-hoc test, n=12–82 cells per condition).
CRMP2 controls calcium influx by regulating CaV2.2 function [12,11]. So, we next tested whether the inhibition of calcium influx observed with the CRMP2 mutants S522A and T555D was due to CaV2.2 loss of function. Using ω-conotoxin GVIA to block all CaV2.2 channels [56], we asked if CRMP2 mutations could further decrease the depolarization-evoked calcium influx. Treatment with 500 nM ω-conotoxin GVIA efficiently inhibited calcium influx in DRG neurons (Figure 1H–J). Expressing CRMP2 mutations did not result in further inhibition of the KCl-evoked calcium influx (Figure 1H–J). This suggests that the inhibition of the calcium influx observed with the CRMP2 mutants S522A and T555D (Figure 1A–G) is entirely due to inhibition of CaV2.2. These results highlight 2 phosphorylation sites on CRMP2 that are important for depolarization evoked calcium influx. However, CRMP2 phosphorylation by RhoK was not detected in rat dorsal horn of the spinal cord [36] and is therefore unlikely to participate in spinal neurotransmission. Cdk5 phosphorylation is a priming event for subsequent GSK3β mediated phosphorylation of CRMP2 [27]. Thus, we hypothesized that CRMP2 phosphorylation by Cdk5 underlies CRMP2’s function in spinal neurotransmission.
Phosphorylation of CRMP2 changes the frequency of spontaneous excitatory postsynaptic currentin lumbar dorsal horn
To determine whether the function of neurons transfected with CRMP2 and its phosphorylation by Cdk5 are changed, we performed electrophysiological analyses in whole-cell configuration to measure spontaneous excitatory postsynaptic currents (sEPSCs) of neurons in the Substantia Gelatinosa (SG) region of the lumbar dorsal horn. We used in vivo transfection of the plasmids to interrogate the role of CRMP2 expression and phosphorylation by Cdk5 in spinal neurotransmission. At 24 hours after in vivo transfection, dsRed fluorescence could be visualized in the dorsal horn of the spinal cord (Figure 2A–C). Only post-synaptic cells adjacent to dsRed fluorescence (i.e. pre-synaptic, white spots in Figure 2C) were selected for analysis (Figure 3A). Inter-event interval and amplitude cumulative distribution curves for sEPSCs are shown in Figure 3B, C. We found that both the amplitude and frequency of sEPSCs were not different between dsRed- (control) and wildtype-CRMP2 transfected neurons (Figure 3D, E). In contrast, overexpression of the CRMP2 S522A mutant resulted in a decrease in the frequency of sEPSCs compared with dsRed- or wildtype-CRMP2-transfeced cells (Figure 3D, E). Moreover, neurons transfected with the constitutively active (i.e. phospho-mimetic) CRMP2 S522D mutant showed an increase in the frequency of sEPSCs in comparison with dsRed- or wildtype-CRMP2-transfeced cells (Figure 3D, E). Collectively, these data demonstrate that dephosphorylated CRMP2 suppresses, while phosphorylated CRMP2 increases, spinal excitatory synaptic transmission.
Figure 2. Location of biocytin-filled recorded neurons.

(A, left) Photomicrograph of the slice preparation showing that the substantia gelatinosa (SG) can be identified as a translucent pale band in the superficial dorsal horn (lamina I/II) enabling positioning of the recording electrode to this region. (A, middle) infrared differential interference contrast image, and (A, right) image of the same cell (indicated by a dashed red box in middle panel) with part of the recording electrode after whole-cell configuration. (B) Schematic diagram of the intrathecal (i.t.) transfected experiment schedule. (C) Identification of SG neurons (green, biocytin) recorded and the transduced presynapses (red spots, dsRed) in the transverse spinal cord slices.
CRMP2 phosphorylation by Cdk5 impacts the presynaptic localization of voltage gated ion channels
The findings that CRMP2 phosphorylation by Cdk5 regulates the frequency but not the amplitude of spinal sEPSCs suggests a presynaptic origin of CRMP2’s effect on spinal neurotransmission. Along these lines, it is well known that frequency of sEPSC is governed by DRG neuron action potential firing which is dependent on voltage gated Na+ channels function and on neurotransmitter release which is dependent on voltage gated calcium channel activation. We previously demonstrated that CRMP2 can regulate the membrane localization and function of two voltage-gated ion channels important in nociceptive signaling, CaV2.2 and NaV1.7 [12,22,20]. Thus, here we asked if inhibiting CRMP2 phosphorylation affects its presynaptic provenance and causes a resulting change in localization of CaV2.2 and NaV1.7. We have been characterizing a small molecule (S)-lacosamide ((S)-LCM) as a specific antagonist of CRMP2 phosphorylation (S522). (S)-LCM does not affect slow inactivation of the sodium channels, thus differentiating from its (R) enantiomer, sold clinically as Vimpat ® [57]. We first extracted synaptosomes from the dorsal horn of the spinal cord of rats one hour after injection with (S)LCM (20μg in 5μl, i.th.) and isolated pre- and post-synaptic fractions (Figure 4A). Synaptosomes had higher levels of the marker synaptophysin while the post-synaptic density (PSD) was enriched in the prototypical protein post synaptic density 95 (PSD95) (Figure 4A). We next focused on the pre-synaptic fraction because CRMP2 expression is mostly pre-synaptic in the dorsal horn of the spinal cord [36]. By western blot analysis we found that (S)-LCM decreased the level of CRMP2 phosphorylation in the presynaptic fraction (Figure 4B–C). Notably, there was a concomitant decrease in the levels of CaV2.2 and NaV1.7 in the presynaptic fraction (Figure 4B–C). These results demonstrate that CRMP2 phosphorylation by Cdk5 controls the presynaptic localization of both CaV2.2 and Nav1.7 in the dorsal horn of the spinal cord.
Figure 4: Inhibition of CRMP2 phosphorylation with (S)-lacosamide decreases CaV2.2 and NaV1.7 presynaptic localization in the dorsal horn.

(A) Immunoblots showing the integrity of the synaptic fractionation from lumbar dorsal horn of the spinal cord. The non-post synaptic density (PSD) fraction was enriched in the pre-synaptic marker Synaptophysin and the PSD fraction was enriched in the post-synaptic marker PSD95. The membrane-associated protein flotillin was used as a loading control. (B) Immunoblots showing the presynaptic CRMP2 expression, CRMP2 p522, CaV2.2 and NaV1.7 levels in the lumbar dorsal horn of the spinal cord of animals having received (S)-Lacosamide (20μg in 5 μl, i.th.) compared to vehicle (0.1% DMSO in saline). Spinal cords were harvested from rats 1 hour following treatment. Flotilin is used as a loading control. (C) Bar graph showing decreased CRMP2 p522 concomitant with decreased CaV2.2 and NaV1.7 levels at the presynaptic sites of lumbar dorsal horn of the spinal cord in (S)-lacosamide treated animals. Mean ± SEM, *p<0.05, MannWhitney compared to the contralateral side.
(S)-LCM decreased the frequency of spontaneous, but not miniature, excitatory postsynaptic current in lumbar dorsal horn
So far, our results have established that pharmacological antagonism of Cdk5-mediated CRMP2 phosphorylation can control the presynaptic localization of key nociceptive ion channels. Whether this antagonism has a presynaptic or postsynaptic effect is not known. Therefore, we recorded, in the wholecell configuration, sEPSCs and mEPSCs of neurons in the SG region of lumbar dorsal horn. No change was observed in the amplitude of both sEPSCs (Figure 5A–C) and mEPSCs (Figure 5F–H) between slices treated with 10 μM (S)-Lacosamide versus control. There was no change in the frequency of mEPSCs (Figure 5I, J), sIPSC or mIPSC (data not shown), however, the frequency of sEPSCs was significantly decreased with perfusion of 10 μM (S)-Lacosamide (1.96 ± 0.21 Hz vs 1.26 ± 0.17 Hz, P<0.01, vs control) (Figure 5D, E). These observations indicate that CRMP2 is involved in glutamatergic transmission in the spinal cord and are suggestive of presynaptic suppression of (S)-LCM in lumbar dorsal horn glutamatergic transmission.
Figure 5. sEPSC frequency is reduced by pharmacological antagonism of CRMP2 phosphorylation by (S)-Lacosamide.

Perfusion of 10 μM (S)-Lacosamide decreased spontaneous excitatory synaptic transmission (A-E) in lumbar dorsal horn neurons. (A) Representative sEPSCs recording traces of cells from control (0.1% DMSO) and (S)-Lacosamide groups. (B, D) Cumulative distribution of the sEPSCs amplitude and the inter-event interval recorded from control and (S)-LCM treated slices. (S)-LCM did not affect the distribution of the amplitude but shifted the distribution to a longer inter-event interval. (C, E) Summary of amplitudes and frequencies of sEPSCs for both groups are shown. Perfusion of 10 μM (S)-Lacosamide did not change the frequency and amplitude of miniature excitatory synaptic transmission (mEPSC)(H, J). (F) Representative mEPSCs recording traces of cells from both groups. (G, I) Cumulative distribution of the mEPSCs amplitude and the inter-event interval. (H, J) Summary of amplitudes and frequencies of mEPSCs for both groups are shown.. Data are expressed as means ± SEM. *p < 0.05 (versus control group); unpaired t-test with Welch’s correction.
(S)-LCM increased the paired pulse ratio of excitatory post synaptic current in lumbar dorsal horn
To determine whether (S)-LCM alters glutamate release, we measured paired-pulse ratios (PPRs) of eEPSCs in SG neurons in response to two consecutive stimulations (with a 50 ms interval). PPR is a measure related to synaptic neurotransmitter release that is commonly used to assess changes in presynaptic function [58,59]. At excitatory synapses, the second stimulation generates a larger eEPSC than the first, because of high residual calcium concentrations in the neuron induced by the first stimulation (Figure 6A). PPRs were increased by prefusion with (S)-LCM compared with control slices (Figure 6A, B). From this data, we infer that (S)-LCM modulates glutamatergic transmission by a presynaptic mechanism. Notably, (S)-LCM also decreased the first eEPSC amplitude, further corroborating its role on of calcium channels by modulating CRMP2 phosphorylation.
Figure 6. Paired pulse ratio is increased by pharmacological antagonism of CRMP2 phosphorylation by (S)-Lacosamide.

(A, B) Paired pulse ratios of eEPSCs are shown. (S)-LCM increased the PPR in SG neurons. Example traces show the relative change in the second eEPSC before (black trace) and during perfusion of drug (red trace). PPR was calculated by dividing the second pulse by the first (PSC2/PSC1). Data are expressed as means ± SEM. *p < 0.05 (versus control (0.1% DMSO) group); unpaired t-test with Welch’s correction.
CRMP2 phosphorylation regulates nociceptive neurotransmitter release from spinal cord.
Presynaptic CGRP-release from sensory neurons is a known mediator of pro-nociceptive neuronal signaling [60–62]. Therefore, we tested if the decrease sEPSC frequency caused by (S)-LCM could in turn inhibit depolarization-evoked CGRP-release from rat spinal cord. To test this, we used an ex-vivo method for evoked CGRP release from the lumbar region of the rat spinal cord. An enzyme-linked immunosorbent assay (ELISA) was used to measure CGRP content; samples were collected every 10 min. Basal CGRP levels were 0.64 ± 0.13 pg/ml/mg of tissue (Figure 7, fractions #1 & 2). Vehicle (0.1% DMSO) or a 10 μM concentration of (S)-LCM was added (Figure 7, fraction #3) 10 min prior to stimulation with 90 mM KCl (Figure 7, fraction #4). Treatment with (S)-LCM did not elicit any CGRP release from the spinal cords (Figure 7, fraction #3). Under depolarization, treatment with (S)-Lacosamide resulted in a ~56% decrease (CGRP level for vehicle was 10.26 ± 0.71 and for (S)-LCM 4.47 ± 0.54 pg/ml/mg of tissue) of the depolarization-evoked increase in CGRP release seen in vehicle-treated tissue (Figure 7, fraction #4). These results show that inhibiting CRMP2 phosphorylation by Cdk5 with (S)-LCM results in decreased depolarization-evoked CGRP release.
Figure 7. CGRP release from spinal cord is inhibited by (S)-Lacosamide.

KCl depolarization-evoked CGRP release was measured from spinal cord tissue isolated from naïve adult rats as a result of pre- and co-incubation with 0.1% DMSO or a 10μM concentration of (S)-Lacosamide as indicated. Bar graph shows immunoreactive CGRP levels observed in bath solution normalized to the weight of each spinal cord tissue. Statistical significance is indicated by asterisks for fraction 4 (*p<0.05; 2-way ANOVA post hoc Sidak test, n=6) in comparison with control tissue.
CRMP2 phosphorylation controls nociceptive ion channels pre-synaptic localization in neuropathic pain
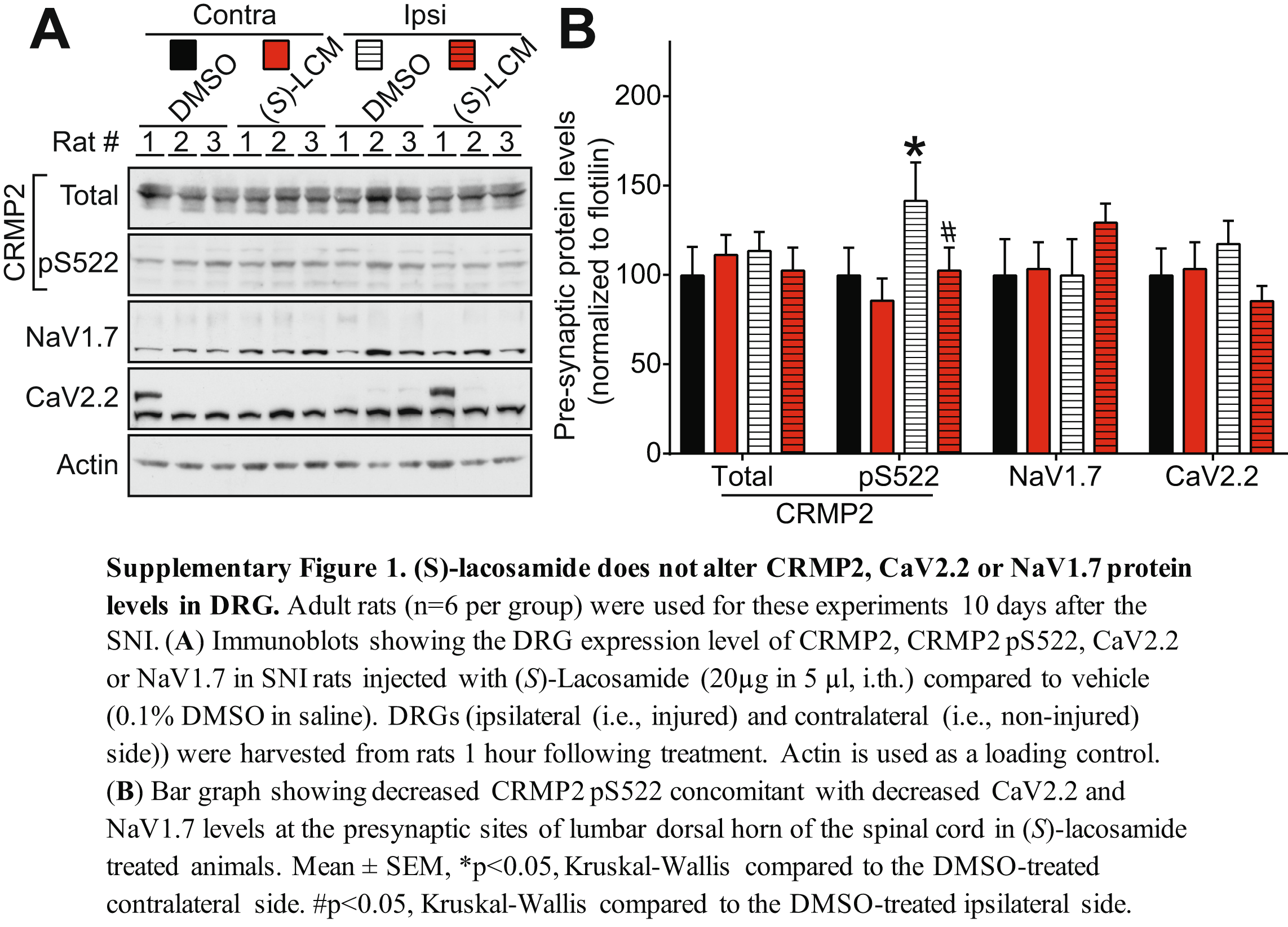
Thus far, the results show that CRMP2 phosphorylation on S522 controls depolarization-evoked calcium influx and sEPSC frequency through a pre-synaptic mechanism. We previously reported that in a model of neuropathic pain – spared nerve injury (SNI) [63] –, CRMP2 phosphorylation level on S522 is increased [36]. Inhibiting CRMP2 phosphorylation in vivo using (S)-LCM reversed allodynia in rats with SNI [39]. Thus, we asked if this (S)-LCM mediated inhibition of sEPSC, neurotransmitter release and allodynia could be related to decreased pre-synaptic localization of the nociceptive ion channels CaV2.2 and NaV1.7. Adult rats were injured following the SNI protocol and the development of allodynia verified 10 days after the surgery (data not shown). These animals were injected with (S)-LCM (20μg in 5μl, i.th.) and spinal cords were harvested one hour later to isolate pre- and post-synaptic fractions as described before (Figure 4). By western blot analysis, we measured the pre-synaptic levels of CRMP2, CRMP2 pS522, CaV2.2 and NaV1.7 in contralateral (non-injured) compared to ipsilateral (injured) sides from the same animal, treated as indicated with (S)-LCM (Figure 8). In accordance with previous findings [36,64], pre-synaptic CRMP2 phosphorylation and CaV2.2 levels were increased in the ipsilateral side in SNI. We found that (S)-LCM decreased pre-synaptic CRMP2 phosphorylation levels in both the contra- and ipsi-lateral sides of SNI rats (Figure 8). This was accompanied by a concomitant decrease of CaV2.2 and NaV1.7 pre-synaptic levels in both the contra- and ipsi-lateral sides of the SNI (Figure 8). To control if this decreased pre-synaptic localization for CaV2.2 and NaV1.7 could be a consequence of decreased synaptic activity induced by (S)-LCM (Figure 5), we tested if presynaptic localization of the R-type voltage-gated calcium (CaV2.3) channel or cannabinoid receptor 1 (CB1R) was also be altered by (S)-LCM. We did not observe any difference in pre-synaptic content for CaV2.3 and CB1R after treatment with (S)-LCM (Figure 8). To confirm that these effects were pre-synaptic, we examined if the DRG expression levels of CRMP2, CaV2.2 or NaV1.7 could be altered by (S)-LCM. We did not detect any changes (Supplementary Figure 1). These results show that CRMP2 phosphorylation level controls the pre-synaptic levels of the nociceptive ion channels CaV2.2 and NaV1.7.
Figure 8. Linking CRMP2 phosphorylation to regulation of nociceptive ion channels in a model of neuropathic pain.

Adult rats (n=6 per group) were used for these experiments 10 days after the SNI. (A) Representative immunoblots showing the presynaptic expression of CRMP2, CRMP2 pS522, CaV2.2, NaV1.7, CaV2.3 and cannabinoid receptor 1 (CB1R) in the lumbar dorsal horn of the spinal cord of SNI rats injected with (S)-Lacosamide (20μg in 5 μl, i.th.) compared to vehicle (0.1% DMSO in saline). Spinal cords (ipsilateral (i.e., injured) and contralateral (i.e., non-injured) side)) were harvested from rats 1 hour following treatment. Flotilin is used as a loading control. (B) Bar graph showing decreased CRMP2 p522 concomitant with decreased CaV2.2 and NaV1.7 levels at the presynaptic sites of lumbar dorsal horn of the spinal cord in (S)-lacosamide treated animals. Mean ± SEM, *p<0.05, Kruskal-Wallis compared to the DMSO-treated contralateral side. #p<0.05, Kruskal-Wallis compared to the DMSOtreated ipsilateral side.
Discussion
The results reported here establish a role CRMP2 in regulating central synaptic transmission to substantia gelatinosa neurons. We found that, of several CRMP2 post-translational modifications including SUMOylation and phosphorylation, depolarization-evoked calcium influx in sensory neurons was contingent on CRMP2 modification by Cdk5 at Serine 522. As a consequence, Cdk5-phosphoryalted CRMP2 had a facilitatory effect on the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in dorsal horn neurons, implicating a presynaptic role for CRMP2 in spinal neurotransmission. Pharmacological antagonism with a CRMP2 phosphorylation inhibitor ((S)-LCM) decreased CRMP2 phosphorylation, and consequently, lowered the presynaptic localization of CaV2.2 and NaV1.7, two ion channels with strong links to nociceptive signaling. (S)-LCM also decreased the frequency of sEPSCs as well as spinal glutamatergic neurotransmission. Finally, we found that decreasing CRMP2 phosphorylation inhibited depolarization-evoked CGRP release in spinal cord. Increased CRMP2 phosphorylation in rats with SNI was decreased by intrathecal administration of (S)-LCM resulting in a loss of pre-synaptic localization of CaV2.2 and NaV1.7, thus linking CRMP2 phosphorylation to convergent regulation of nociceptive ion channels. Taken together, our findings highlight a novel role for phosphorylated CRMP2 in regulating presynaptic excitatory neurotransmission in the spinal cord.
While there is extensive literature on how phosphorylation of CRMP2 controls its canonical functions of neurite outgrowth/branching (see review by Khanna [17] and Ip [65]), how posttranslational modifications of CRMP2 control ion channel trafficking are only now beginning to be understood [66,22,23,20,11,34,10,25]. While our recent work has focused on Cdk5, a ‘priming’ kinase in the context of CRMP2 [34,67,48,36,22], the current work suggests that RhoK phosphorylation site may also be important in shaping calcium influx. The decrease in calcium influx observed in the presence of a CRMP2 mutated at its RhoK site may be linked to kainate receptor activation [68]. As reported by Marques and colleagues, Kainate (KA) receptors delay neuronal maturation by downregulating the density of calcium channels at the neuronal membrane through the phosphorylation of CRMP2 at T555, thereby reducing overall calcium activity [68]. While CRMP2 phosphorylation on T555 was not detected in the dorsal horn of the spinal cord and was not changed in neuropathic pain [36], this phosphorylation site is also known to participate in axonal degeneration observed in multiple sclerosis [69]. This suggests that CaV2.2 could be inhibited by increased CRMP2 phosphorylation on T555 in multiple sclerosis and could impair spinal excitatory neurotransmission.
Our data also provide new insights into the regulation of CaV2.2 and NaV1.7 by CRMP2 in primary sensory neurons. We previously reported that CRMP2 phosphorylation by Cdk5 and SUMOylation are required for NaV1.7 function while CRMP2 phosphorylation by the Src family kinase Fyn (at Y32) inhibits NaV1.7 [22]. The results here show that CRMP2 phosphorylation on S522 is required for CaV2.2 function and CRMP2 phosphorylation on T555 inhibits CaV2.2. CRMP2 phosphorylation on Y32 and SUMOylation (on K374) had no effect on CaV2.2 function thus showing that these modifications have an exclusive function in regulating NaV1.7. Conversely, the phosphorylation of CRMP2 on T555 has an exclusive regulatory function for CaV2.2. Thus, while CRMP2 is important for the physiological and pathological function of both channels, differential manipulation of CRMP2’s post-translational modification state may offer a selective advantage in targeting neuronal NaV1.7 versus CaV2.2 channels and how they affect spinal neurotransmission and nociceptive plasticity. Whilst a decrease in presynaptic Cav2.2 may underlie the (S)-LCM phenotype, our data shows that decreasing presynaptic NaV1.7 is also of relevance here. CaV2.2’s role in neurotransmission is to trigger the release of synaptic vesicles but its activation relies on a prior depolarization event, triggered by opening of voltage gated sodium channels, particularly NaV1.7 in lamina I/II of the dorsal horn of the spinal cord. Along with CaV2.2, NaV1.7 is another major determinant of synaptic transmission that happens to be regulated by CRMP2 phosphorylation on the S522 site. Because (S)-LCM targets both channels via convergent regulation of CRMP2, we cannot not exclude the possibility of a concomitant decrease in the presynaptic localization of NaV1.7 as contributing to the effect of (S)-LCM.
Central terminals of primary afferent fibers terminate in the dorsal horn of spinal cord and most nociceptive Aδ and C-fibers terminate superficially in laminae I – II, with a smaller number reaching the deeper laminae [4]. As a result, the spinal cord is a crucial site for integration of sensory transmission, and it is here that the incoming nociceptive signals undergo convergence and modulation. It is also known that the majority of the primary afferents synapsing onto the dorsal horn of the spinal cord, regardless of their diameters, utilize glutamate as the excitatory neurotransmitter [70]. Moreover, as alluded to earlier spinal presynaptic neurotransmission relies on DRG neuron action potential firing which is dependent on voltage gated Na+ channels function as well as on neurotransmitter release which is dependent on voltage-gated calcium channel activation, although we cannot exclude possible contribution from calcium-induced calcium release from intracellular stores as inwardly rectifying Ca2+dependent I(CRAC) (Ca2+-release activated current), representing store-operated calcium entry (SOCE) exist in sensory neurons and have been linked to injury and neuronal excitability [71]. We further investigated whether CRMP2 phosphorylation in primary afferent sensory neurons could affect the spinal synaptic neurotransmission. The phosphomimetic CRMP2 (S522D) mutant did not exhibit increased depolarization-mediated calcium entry despite an increase in frequency of sEPSCs. A possible explanation for this discrepancy may be the dependence on synaptic activity for pathological function of CRMP2 phosphorylation. Synaptic activity decreases CRMP2 phosphorylation and its interaction with CaV2.2 [11,72], thus a lack of synaptic activity in culture conditions might mask an effect on increased calcium influx when CRMP2 phosphorylation is forced (as in the S522D mutant); in other words, CRMP2 phosphorylation may already be at its maximum. Our data showed that preventing CRMP2 phosphorylation on S522 resulted in a decrease, while increasing CRMP2 phosphorylation at S522 increased the frequency of sEPSCs. These observations point to a regulatory function of phosphorylated CRMP2 in altering glutamatergic transmission. In line with this argument, pharmacological antagonism, with (S)-LCM, of CRMP2 phosphorylation by Cdk5 also decreased the frequency of sEPSCs.
Recently, Zhang and colleagues reported that both dephosphorylation of CRMP2 at T514 and deSUMOylation at K374 enhanced amplitude and frequency of miniature excitatory postsynaptic currents (mEPSCs) in hippocampal neurons, thereby promoting formation and maturation of dendritic spines [73]. Here, our data provide new insight into the regulation of central spinal synaptic neurotransmission by CRMP2 in primary sensory neurons. Dephosphorylation of CRMP2 at S522 decreased the frequency, but not the amplitude, of sEPSCs, highlighting a presynaptic effect of CRMP2, which may rely on the modulation of trafficking/localization of CaV2.2 and NaV1.7 by CRMP2 in primary sensory neurons. No change of the amplitude and frequency of mEPSCs is consistent with an action potential-dependent inhibition, which also relies on trafficking/localization of CaV2.2 and NaV1.7, with dephosphorylation of CRMP2 at S522 on central spinal presynaptic neurotransmission, rather than on basal vesicle quantal release. These findings are also in line with a recent report demonstrating that CaV2.2 enables voltage-dependent neurotransmitter secretion [74]. Several factors can account for reduced presynaptic neurotransmitter release, including but not limited to vesicle depletion, inactivation of release sites, and decreased presynaptic calcium influx [75]. The paired-pulse ratio (PPR) protocol measures the short term plasticity characteristics of neurons, and is a widely used approach for assessing the synaptic sites of drug action [76]. Because the PPR is inversely related to synaptic neurotransmitter release probability, our results indicate that (S)-LCM can, at least in part, decrease presynaptic glutamate release. Moreover, we observed a significant depression of the first EPSC in the presence of (S)-LCM in all neurons tested (data not shown), accompanied by a significant increase of PPR in the responsive neurons, suggesting that the CRMP2 dephosphorylation also causes a decrease in release probability at the first response, which may be related to the effects of CRMP2 on presynaptic CaV2.2 trafficking.
CGRP is found mainly in small DRG cells and unmyelinated axons (C-fibers) but also in some medium-sized and a few large DRG cells and in myelinated axons of Aδ- and even Aβ-fibers. CGRP also coexists with glutamate in primary afferent terminals[77]. Moreover, CGRP has long been served as a molecular marker of peptidergic nociceptive neurons and expected to play an important role in pathophysiological nociceptive pain [78]. Our data showed that dephosphorylation of CRMP2 by (S)-LCM inhibited depolarization evoked CGRP release in spinal cord, suggesting the analgesic modulation of CRMP2 phosphorylation.
Conclusion
We conclude that gain of CRMP2 phosphorylation in neuropathic pain, increases sEPSC frequency dependent on CaV2.2 and NaV1.7. This facilitates excitatory neurotransmitter release and could underlie allodynia. This suggests that CRMP2 phosphorylation is an important event regulating pathological pain through the sensitization of nociceptive afferents. (S)-LCM can be used to further study CRMP2 functions but designing novel targeting strategies to inhibit CRMP2 phosphorylation by Cdk5 will have great potential for the treatment of chronic neuropathic pain.
Supplementary Material
{kind=link}
Acknowledgements –
This work was supported by a grant from the National Natural Science Foundation of China (81603088) to J.Y., National Institutes of Health awards (R01NS098772 from the National Institute of Neurological Disorders and Stroke and R01DA042852 from the National Institute on Drug Abuse to R.K.); and a Neurofibromatosis New Investigator Award from the Department of Defense Congressionally Directed Military Medical Research and Development Program (NF1000099) to R.K. S.S.B. and N.Y.N.P. were supported by funds to the Undergraduate Biology Research Program from the University of Arizona’s Senior Vice President for Research’s office, and the University of Arizona’s Native American Cancer Prevention Program (N.Y.N.P.).
Footnotes
Conflict of interest – There is no conflict of interest for any of the authors.
References
- 1.Costigan M, Scholz J, Woolf CJ (2009) Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 32:1–32. doi: 10.1146/annurev.neuro.051508.135531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moulin D, Boulanger A, Clark AJ, Clarke H, Dao T, Finley GA, Furlan A, Gilron I, Gordon A, Morley-Forster PK, Sessle BJ, Squire P, Stinson J, Taenzer P, Velly A, Ware MA, Weinberg EL, Williamson OD, Canadian Pain S (2014) Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain research & management 19 (6):328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price TJ, Basbaum AI, Bresnahan J, Chambers JF, De Koninck Y, Edwards RR, Ji RR, Katz J, Kavelaars A, Levine JD, Porter L, Schechter N, Sluka KA, Terman GW, Wager TD, Yaksh TL, Dworkin RH (2018) Transition to chronic pain: opportunities for novel therapeutics. Nature reviews Neuroscience. doi: 10.1038/s41583-018-0012-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Todd AJ (2010) Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 11 (12):823–836. doi: 10.1038/nrn2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Mello R, Dickenson AH (2008) Spinal cord mechanisms of pain. Br J Anaesth 101 (1):8–16. doi: 10.1093/bja/aen088 [DOI] [PubMed] [Google Scholar]
- 6.Latremoliere A, Woolf CJ (2009) Central sensitization: a generator of pain hypersensitivity by central neural plasticity. The journal of pain : official journal of the American Pain Society 10 (9):895–926. doi: 10.1016/j.jpain.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Divito CB, Underhill SM (2014) Excitatory amino acid transporters: roles in glutamatergic neurotransmission. Neurochem Int 73:172–180. doi: 10.1016/j.neuint.2013.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolphin AC (2012) Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nature reviews Neuroscience 13 (8):542–555. doi: 10.1038/nrn3311 [DOI] [PubMed] [Google Scholar]
- 9.Khanna R, Zougman A, Stanley EF (2007) A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. Journal of biochemistry and molecular biology 40 (3):302–314 [DOI] [PubMed] [Google Scholar]
- 10.Chi XX, Schmutzler BS, Brittain JM, Hingtgen CM, Nicol GD, Khanna R (2009) Regulation of N-type voltage-gated calcium (CaV2.2) channels and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. JCell Sci 23:4351–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R (2009) An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. The Journal of biological chemistry 284 (45):31375–31390. doi: 10.1074/jbc.M109.009951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, Meroueh SO, Hingtgen CM, Brustovetsky N, Ji RR, Hurley JH, Jin X, Shekhar A, Xu XM, Oxford GS, Vasko MR, White FA, Khanna R (2011) Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(+) channel complex. Nature medicine 17 (7):822–829. doi: 10.1038/nm.2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francois-Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X, Wang Y, Ju W, Largent-Milnes TM, Khanna M, Vanderah TW, Khanna R (2015) A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156 (7):1247–1264. doi: 10.1097/j.pain.0000000000000147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moutal A, Li W, Wang Y, Ju W, Luo S, Cai S, Francois-Moutal L, Perez-Miller S, Hu J, Dustrude ET, Vanderah TW, Gokhale V, Khanna M, Khanna R (2017) Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. British journal of pharmacology. doi: 10.1111/bph.13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R, Meroueh SO, Vasko MR, White FA, Khanna R (2012) CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Molecular pain 8 (1):54. doi: 10.1186/1744-8069-8-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ripsch MS, Ballard CJ, Khanna M, Hurley JH, White FA, Khanna R (2012) A peptide uncoupling CRMP-2 from the presynaptic Ca2+ channel complex demonstrate efficacy in animal models of migraine and AIDS therapy-induced neuropathy. Translational neuroscience 3 (1):1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khanna R, Wilson SM, Brittain JM, Weimer J, Sultana R, Butterfield A, Hensley K (2012) Opening Pandora’s jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol 7 (6):749–771. doi: 10.2217/fnl.12.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM (2015) CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases. Molecular psychiatry 20 (9):1037–1045. doi: 10.1038/mp.2015.77 [DOI] [PubMed] [Google Scholar]
- 19.Moutal A, Francois-Moutal L, Brittain JM, Khanna M, Khanna R (2014) Differential neuroprotective potential of CRMP2 peptide aptamers conjugated to cationic, hydrophobic, and amphipathic cell penetrating peptides. Front Cell Neurosci 8:471. doi: 10.3389/fncel.2014.00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dustrude ET, Wilson SM, Ju W, Xiao Y, Khanna R (2013) CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. The Journal of biological chemistry 288 (34):24316–24331. doi: 10.1074/jbc.M113.474924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brustovetsky T, Pellman JJ, Yang XF, Khanna R, Brustovetsky N (2014) Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. The Journal of biological chemistry 289 (11):7470–7482. doi: 10.1074/jbc.M113.518472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dustrude ET, Moutal A, Yang X, Wang Y, Khanna M, Khanna R (2016) Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proceedings of the National Academy of Sciences of the United States of America 113 (52):E8443–E8452. doi: 10.1073/pnas.1610531113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dustrude ET, Perez-Miller S, Francois-Moutal L, Moutal A, Khanna M, Khanna R (2017) A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels (Austin):1–13. doi: 10.1080/19336950.2017.1299838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francois-Moutal L, Dustrude ET, Wang Y, Brustovetsky T, Dorame A, Ju W, Moutal A, PerezMiller S, Brustovetsky N, Gokhale V, Khanna M, Khanna R (2018) Inhibition of the Ubc9 E2 SUM-Oconjugating enzyme-CRMP2 interaction decreases NaV1.7 currents and reverses experimental neuropathic pain. Pain. doi: 10.1097/j.pain.0000000000001294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ju W, Li Q, Wilson SM, Brittain JM, Meroueh L, Khanna R (2013) SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels 3 (7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moutal A, Dustrude ET, Largent-Milnes TM, Vanderah TW, Khanna M, Khanna R (2017) Blocking CRMP2 SUMOylation reverses neuropathic pain. Molecular psychiatry. doi: 10.1038/mp.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernandez F, Eickholt BJ, Nikolic M, Sutherland C (2006) Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. The Journal of biological chemistry 281 (24):16591–16598. doi: 10.1074/jbc.M513344200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120 (1):137–149. doi: 10.1016/j.cell.2004.11.012 [DOI] [PubMed] [Google Scholar]
- 29.Arimura N, Inagaki N, Chihara K, Menager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K (2000) Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. The Journal of biological chemistry 275 (31):23973–23980. doi: 10.1074/jbc.M001032200 [DOI] [PubMed] [Google Scholar]
- 30.Varrin-Doyer M, Vincent P, Cavagna S, Auvergnon N, Noraz N, Rogemond V, Honnorat J, Moradi-Ameli M, Giraudon P (2009) Phosphorylation of collapsin response mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration. The Journal of biological chemistry 284 (19):13265–13276. doi: 10.1074/jbc.M807664200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uchida Y, Ohshima T, Yamashita N, Ogawara M, Sasaki Y, Nakamura F, Goshima Y (2009) Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. The Journal of biological chemistry 284 (40):27393–27401. doi: 10.1074/jbc.M109.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ (2007) Protein kinases as potential targets for the treatment of pathological pain. Handbook of experimental pharmacology (177):359–389 [DOI] [PubMed] [Google Scholar]
- 33.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y (2005) Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes to cells : devoted to molecular & cellular mechanisms 10 (2):165–179. doi: 10.1111/j.1365-2443.2005.00827.x [DOI] [PubMed] [Google Scholar]
- 34.Brittain JM, Wang Y, Eruvwetere O, Khanna R (2012) Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS letters 586 (21):3813–3818. doi: 10.1016/j.febslet.2012.09.022 [DOI] [PubMed] [Google Scholar]
- 35.Moutal A, Cai S, Luo S, Voisin R, Khanna R (2018) CRMP2 is necessary for Neurofibromatosis type 1 related pain. Channels 12 (1):47–50. doi: 10.1080/19336950.2017.1370524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moutal A, Luo S, Largent-Milnes TM, Vanderah TW, Khanna R (2018) Cdk5-mediated CRMP2 phosphorylation is necessary and sufficient for peripheral neuropathic pain. Neurobiology of Pain. doi: 10.1016/j.ynpai.2018.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hergenroeder GW, Redell JB, Choi HA, Schmitt L, Donovan W, Francisco GE, Schmitt K, Moore AN, Dash PK (2018) Increased Levels of Circulating Glial Fibrillary Acidic Protein and Collapsin Response Mediator Protein-2 Autoantibodies in the Acute Stage of Spinal Cord Injury Predict the Subsequent Development of Neuropathic Pain. J Neurotrauma. doi: 10.1089/neu.2018.5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moutal A, Yang X, Li W, Gilbraith KB, Luo S, Cai S, Francois-Moutal L, Chew LA, Yeon SK, Bellampalli SS, Qu C, Xie JY, Ibrahim MM, Khanna M, Park KD, Porreca F, Khanna R (2017) CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide. Pain 158 (12):2301–2319. doi: 10.1097/j.pain.0000000000001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moutal A, Chew LA, Yang X, Wang Y, Yeon SK, Telemi E, Meroueh S, Park KD, Shrinivasan R, Gilbraith KB, Qu C, Xie JY, Patwardhan A, Vanderah TW, Khanna M, Porreca F, Khanna R (2016) (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain 157 (7):1448–1463. doi: 10.1097/j.pain.0000000000000555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang SH, Yu J, Lou GD, Tang YY, Wang RR, Hou WW, Chen Z (2016) Widespread pain sensitization after partial infraorbital nerve transection in MRL/MPJ mice. Pain 157 (3):740–749. doi: 10.1097/j.pain.0000000000000432 [DOI] [PubMed] [Google Scholar]
- 41.Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, Ross S, Lowell BB, Wang Y, Goulding M, Ma Q (2014) Identification of spinal circuits transmitting and gating mechanical pain. Cell 159 (6):1417–1432. doi: 10.1016/j.cell.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moutal A, Sun L, Yang X, Li W, Cai S, Luo S, Khanna R (2018) CRMP2-Neurofibromin Interface Drives NF1-related Pain. Neuroscience 381:79–90. doi: 10.1016/j.neuroscience.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moutal A, Wang Y, Yang X, Ji Y, Luo S, Dorame A, Bellampalli SS, Chew LA, Cai S, Dustrude ET, Keener JE, Marty MT, Vanderah TW, Khanna R (2017) Dissecting the role of the CRMP2-neurofibromin complex on pain behaviors. Pain 158 (11):2203–2221. doi: 10.1097/j.pain.0000000000001026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dustrude ET, Perez-Miller S, Francois-Moutal L, Moutal A, Khanna M, Khanna R (2017) A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels 11 (4):316–328. doi: 10.1080/19336950.2017.1299838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moutal A, Li W, Wang Y, Ju W, Luo S, Cai S, Francois-Moutal L, Perez-Miller S, Hu J, Dustrude ET, Vanderah TW, Gokhale V, Khanna M, Khanna R (2018) Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. British journal of pharmacology 175 (12):2244–2260. doi: 10.1111/bph.13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie JY, Chew LA, Yang X, Wang Y, Qu C, Wang Y, Federici LM, Fitz SD, Ripsch MS, Due MR, Moutal A, Khanna M, White FA, Vanderah TW, Johnson PL, Porreca F, Khanna R (2016) Sustained relief of ongoing experimental neuropathic pain by a CRMP2 peptide aptamer with low abuse potential. Pain 157 (9):2124–2140. doi: 10.1097/j.pain.0000000000000628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moutal A, Eyde N, Telemi E, Park KD, Xie JY, Dodick DW, Porreca F, Khanna R (2016) Efficacy of (S)-Lacosamide in preclinical models of cephalic pain. Pain Rep 1 (1). doi: 10.1097/PR9.0000000000000565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moutal A, Francois-Moutal L, Perez-Miller S, Cottier K, Chew LA, Yeon SK, Dai J, Park KD, Khanna M, Khanna R (2016) (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Molecular neurobiology 53 (3):1959–1976. doi: 10.1007/s12035-015-9141-2 [DOI] [PubMed] [Google Scholar]
- 49.Ibrahim MM, Patwardhan A, Gilbraith KB, Moutal A, Yang X, Chew LA, Largent-Milnes T, Malan TP, Vanderah TW, Porreca F, Khanna R (2017) Long-lasting antinociceptive effects of green light in acute and chronic pain in rats. Pain 158 (2):347–360. doi: 10.1097/j.pain.0000000000000767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R, Meroueh SO, Vasko MR, White FA, Khanna R (2012) CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol Pain 8:54. doi: 10.1186/1744-8069-8-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD, Khanna R (2009) Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci 122 (Pt 23):4351–4362. doi: 10.1242/jcs.053280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pacchioni AM, Vallone J, Worley PF, Kalivas PW (2009) Neuronal pentraxins modulate cocaine-induced neuroadaptations. The Journal of pharmacology and experimental therapeutics 328 (1):183–192. doi: 10.1124/jpet.108.143115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duan JH, Hodgdon KE, Hingtgen CM, Nicol GD (2014) N-type calcium current, Cav2.2, is enhanced in small-diameter sensory neurons isolated from Nf1+/− mice. Neuroscience 270:192–202. doi: 10.1016/j.neuroscience.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson SM, Toth PT, Oh SB, Gillard SE, Volsen S, Ren D, Philipson LH, Lee EC, Fletcher CF, Tessarollo L, Copeland NG, Jenkins NA, Miller RJ (2000) The status of voltage-dependent calcium channels in alpha 1E knock-out mice. JNeurosci 20 (23):8566–8571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW (2012) Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149 (5):1112–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McIntosh JM, Corpuz GO, Layer RT, Garrett JE, Wagstaff JD, Bulaj G, Vyazovkina A, Yoshikami D, Cruz LJ, Olivera BM (2000) Isolation and characterization of a novel conus peptide with apparent antinociceptive activity. The Journal of biological chemistry 275 (42):32391–32397. doi: 10.1074/jbc.M003619200 [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Brittain JM, Jarecki BW, Park KD, Wilson SM, Wang B, Hale R, Meroueh SO, Cummins TR, Khanna R (2010) In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein-2 identifies a pocket important in modulating sodium channel slow inactivation. The Journal of biological chemistry 285 (33):25296–25307. doi: 10.1074/jbc.M110.128801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Xiao X, Zhang XM, Zhao ZQ, Zhang YQ (2012) Estrogen facilitates spinal cord synaptic transmission via membrane-bound estrogen receptors: implications for pain hypersensitivity. J Biol Chem 287 (40):33268–33281. doi: 10.1074/jbc.M112.368142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schou WS, Ashina S, Amin FM, Goadsby PJ, Ashina M (2017) Calcitonin gene-related peptide and pain: a systematic review. The journal of headache and pain 18 (1):34. doi: 10.1186/s10194-017-0741-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nitzan-Luques A, Devor M, Tal M (2011) Genotype-selective phenotypic switch in primary afferent neurons contributes to neuropathic pain. Pain 152 (10):2413–2426. doi: 10.1016/j.pain.2011.07.012 [DOI] [PubMed] [Google Scholar]
- 62.Lee SE, Kim JH (2007) Involvement of substance P and calcitonin gene-related peptide in development and maintenance of neuropathic pain from spinal nerve injury model of rat. Neurosci Res 58 (3):245–249. doi: 10.1016/j.neures.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 63.Decosterd I, Woolf CJ (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87 (2):149–158 [DOI] [PubMed] [Google Scholar]
- 64.Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, Orendacova J, Yaksh TL (2002) Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Experimental brain research 147 (4):456–463. doi: 10.1007/s00221-002-1217-3 [DOI] [PubMed] [Google Scholar]
- 65.Ip JP, Fu AK, Ip NY (2014) CRMP2: Functional Roles in Neural Development and Therapeutic Potential in Neurological Diseases. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. doi: 10.1177/1073858413514278 [DOI] [PubMed] [Google Scholar]
- 66.Chew LA, Khanna R (2018) CRMP2 and voltage-gated ion channels: potential roles in neuropathic pain. Neuronal Signaling 2:16. doi: 10.1042/NS20170220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moutal A, Eyde N, Telemi E, Park KD, Xie JY, Dodick DW, Porreca F, Khanna R (2016) Efficacy of (S)-Lacosamide in preclinical models of cephalic pain. Pain Rep 1 (1):1–10. doi: 10.1097/PR9.0000000000000565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marques JM, Rodrigues RJ, Valbuena S, Rozas JL, Selak S, Marin P, Aller MI, Lerma J (2013) CRMP2 tethers kainate receptor activity to cytoskeleton dynamics during neuronal maturation. The Journal of neuroscience : the official journal of the Society for Neuroscience 33 (46):18298–18310. doi: 10.1523/JNEUROSCI.3136-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petratos S, Ozturk E, Azari MF, Kenny R, Lee JY, Magee KA, Harvey AR, McDonald C, Taghian K, Moussa L, Mun Aui P, Siatskas C, Litwak S, Fehlings MG, Strittmatter SM, Bernard CC (2012) Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain : a journal of neurology 135 (Pt 6):1794–1818. doi: 10.1093/brain/aws100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Y, Danbolt NC (2014) Glutamate as a neurotransmitter in the healthy brain. Journal of neural transmission 121 (8):799–817. doi: 10.1007/s00702-014-1180-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH (2011) Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 31 (10):3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadoyama K, Matsuura K, Nakamura-Hirota T, Takano M, Otani M, Matsuyama S (2015) Changes in the expression of collapsin response mediator protein-2 during synaptic plasticity in the mouse hippocampus. Journal of neuroscience research 93 (11):1684–1692. doi: 10.1002/jnr.23626 [DOI] [PubMed] [Google Scholar]
- 73.Zhang J, Zhao B, Zhu X, Li J, Wu F, Li S, Gong X, Cha C, Guo G (2018) Phosphorylation and SUMOylation of CRMP2 regulate the formation and maturation of dendritic spines. Brain research bulletin 139:21–30. doi: 10.1016/j.brainresbull.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 74.Chai Z, Wang C, Huang R, Wang Y, Zhang X, Wu Q, Wang Y, Wu X, Zheng L, Zhang C, Guo W, Xiong W, Ding J, Zhu F, Zhou Z (2017) CaV2.2 Gates Calcium-Independent but Voltage-Dependent Secretion in Mammalian Sensory Neurons. Neuron 96 (6):1317–1326 e1314. doi: 10.1016/j.neuron.2017.10.028 [DOI] [PubMed] [Google Scholar]
- 75.Fioravante D, Regehr WG (2011) Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21 (2):269–274. doi: 10.1016/j.conb.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomoyose O, Kodama D, Ono H, Tanabe M (2014) Presynaptic inhibitory effects of fluvoxamine, a selective serotonin reuptake inhibitor, on nociceptive excitatory synaptic transmission in spinal superficial dorsal horn neurons of adult mice. J Pharmacol Sci 126 (2):136–145 [DOI] [PubMed] [Google Scholar]
- 77.Meng J, Wang J, Lawrence G, Dolly JO (2007) Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. Journal of cell science 120 (Pt 16):2864–2874. doi: 10.1242/jcs.012211 [DOI] [PubMed] [Google Scholar]
- 78.McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ (2013) Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 78 (1):138–151. doi: 10.1016/j.neuron.2013.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.