

Abstract
Background
2,4-dichlorophenoxyacetic acid (2,4-DCPA acid) is a toxic herbicide. Earlier studies to remove 2,4-DCPA acid from water used expensive and/or toxic reagents, resulting in the formation of toxic organic by-products (Org-BPs). This study evaluates the removal of 2,4-DCPA acid from aqueous media using uncatalysed and catalytic ozonation with Fe doped with Ni and Co respectively.
Methods
Mixed metal oxides of Ni and Co loaded on Fe respectively, prepared by co-precipitation and physical mixing were used as catalyst for ozone facilitated oxidation degradation of 2,4-DCPA acid. Their surface properties were determined by employing SEM, BET and NH3-TPD. HPLC, IC and TOC data were used for quantifying substrate and oxidation products.
Results
Conversion of 2,4-DCPA acid increased from 38% in acidic water to 73% in basic water, however, only 26% of the total carbon was removed and 9.5% in the form of Org-BPs. With 7:3 Fe:Ni (Co-ppt) catalyst (surface area 253 m2 g−1; particle size 236 nm), 97% of pollutant was converted. Most importantly, 92% of carbon was removed and Org-BP formation was minimized to 1.5%. With 7:3 Fe:Ni (Mixed) catalyst (surface area 12 m2 g−1; particle size 1274 nm), 68% of 2,4-DCPA acid was converted, while 23% of TOC was removed, however, 66% of Org-BP’s still remained.
Conclusion
In uncatalysed ozonation degradation of 2,4-DCPA acid improved with the increase in hydroxide ion concentration. Ozonation in presence of 7:3 Fe:Ni (Co-ppt) catalyst resulted in highest activity for dechlorination, TOC removal and Org-BP minimization, thus improving the quality of contaminated water.
Keywords: Catalytic ozonation, 7:3 Fe:Ni (co-ppt), Total organic carbon, Organic by-products
Background
In recent years, illegal dumping of hazardous organic waste materials from industrial and commercial facilities has become a major cause of water pollution around the world [1]. Various types of contaminants are found in water and the chlorinated hydrocarbons are considered the most toxic species for environmental and human health. Most of them cannot be easily biodegradable, and are therefore, difficult to remove from water [2]. One such compound, 2,4-DCPA acid a low-cost herbicide has been used extensively in agriculture as a weed killer. It is reasonably toxic and its maximum permissible limit in drinking water is 20 ppm [3]. It has been frequently detected in large amounts in wastewater and soil [4], and a major problem is it’s difficultly to decompose in water. A number of studies have investigated ways to remove 2,4-DCPA acid from water [5–7]. However, in most of these studies expensive and/or toxic reagents had to be used to achieve good degradation of this organic pollutant [8]. Furthermore, most of these studies did not monitor or consider the fate of Org-BPs formed in the water, which sometimes can be more toxic than the organic pollutant itself. Compared to uncatalyzed ozonation, catalysed ozonation is known to enhance degradation efficiency of many organic pollutants in water [9]. Generally, the catalysts used in water treatment by ozonation mainly include expensive and sometimes toxic transition metals [10]. Simple iron oxide based catalysts is seldom used in the areas of water treatment and catalytic ozonation systems in spite of its advantages of low cost, low toxicity and high catalytic activity [11]. This study therefore, demonstrates how iron doped with nickel and cobalt respectively, can be used as catalyst to enhance removal of hazardous pollutant, 2,4-DCPA acid from water by ozonation. The following factors, namely (i) influence of solution pH on 2,4-DCPA acid degradation during ozonation, (ii) effect of Ni loading on the surface properties and catalytic activity of the Fe-Ni catalyst, prepared by co-precipitation and physical mixing methods, (iii) organic by-product formation and TOC removal during ozonation alone and catalytic ozonation.
Methods
Sample preparation
A 100 ppm stock solution of 2,4-Dichlorophenoxyacetic acid (97% AR grade – Sigma Aldrich) was prepared by dissolving 0.1 g of the solid in milli-Q water and making up to 1 l in a volumetric flask.
Catalyst preparation
All chemical used for the preparation of the metal oxides were of analytical grade purchased from Merck SA. Fe alone was used as a reference, and Fe doped with Ni and Co respectively were used for the preparation of mixed metal oxides. The following two methods were used to prepare the catalysts:
Co-precipitation method (referred to as co-ppt catalyst)
To prepare each catalyst material, Fe (NO3)3 ∙ 9H2O and Ni (NO3)2 ∙ 6H2O was accurately weighed out separately, according to amounts shown in Table 1 and dissolved in 200 cm3 of deionized water. The resulting solutions were then transferred into separate 500 cm3 separating funnels. Each salt solution was then added dropwise from each separating funnel to 250 cm3 of a 1 mol dm−3 Na2CO3 solution contained in a 2 dm3 beaker with vigorous stirring over a period of 4–5 h. Required amounts of 0.1 mol dm−3 NaOH solution was introduced intermittently to maintain the pH of the metal salt mixture between 9 and 11. After addition of all the salt solution to the beaker, it was left to stand for 1 h. The resulting precipitate was filtered and washed several times with deionized water until a clear filtrate was obtained. It was then dried at 100 °C for 1 h and calcined in air in a muffle furnace set at 500 °C for 5 h.
Table 1.
Surface characteristics of Fe-Ni catalyst material
| Catalyst | Surface area (m2 g−1) | Particle size (nm) | Pore size (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| 100% Fe | 20 | 304 | 36 | 0.110 |
| 9:1 Fe:Ni (Co-ppt) | 125 | 13 | 12 | 0.059 |
| 8:2 Fe:Ni (Co-ppt) | 135 | 171 | 10 | 0.084 |
| 7:3 Fe:Ni (Co-ppt) | 253 | 236 | 9 | 0.162 |
| 9:1 Fe:Ni (Mixed) | 5 | 1595 | 10 | 0.0085 |
| 8:2 Fe:Ni (Mixed) | 9 | 1404 | 8 | 0.0098 |
| 7:3 Fe:Ni (Mixed) | 12 | 1274 | 2 | 0.0099 |
Physical mixing method (referred to as mixed catalyst)
Weighed out each salt according to Table 1 in a porcelain crucible and mixed thoroughly to obtain a homogeneous mix. The crucible was then placed in a muffle furnace set at 500 °C for 5 h.
Catalyst characterization
Each catalyst was ground to fine powder and characterized using the following techniques: (i) Scanning electron microscope (SEM) measurements, giving information about morphology and location of metallic species on catalyst surface, was carried out using a JOEL JSM-6100 microscope equipped with an energy-dispersive X-Ray spectrometer fitted with a Tungsten filament lamp. The images were taken with an emission current of 100 μA and an accelerator voltage of 12 kV, (ii) N2 adsorption-desorption isotherms of each catalyst was carried out at 77 K on a Micromeritics Gemini 2360 automated single/multiple point BET surface area analyser. The surface area, pore size distribution, pore volume and average pore diameter were determined by the BJH method, and (iii) Temperature programmed desorption (TPD) studies with NH3 was conducted using the AutoChem 2910 (Micromeritics, USA) instrument fitted with a thermal conductivity detector. A 50 mg catalyst sample was initially treated by passing helium over it at a flow rate of 50 mL/min and 200 °C for 2 h. It was then saturated with 10% ammonia, further flushed with helium and thereafter placed in a U-shaped quartz sample tube. TPD analysis was conducted from ambient temperature to 600 °C at a heating rate of 10 °C/min.
Ozonation procedure
Ozone gas was prepared by passing medical grade oxygen (99.9% Purity) through the electric discharge unit of an Ozonox LAB 7000 ozonator instrument. An ozone concentration of 100 ppm was used for all experiments, which was achieved with a generator current of 0.42 A and a constant oxygen flow rate of 200 mL/min. The concentration of ozone was measured by using the iodometric method, which involved bubbling the ozone gas into a KI solution and then titrating the liberated iodine with standard thiosulphate solution using starch as indicator [12]. The ozone reaction was carried out in a cylindrical glass reactor equipped with a sintered porous ceramic gas diffuser (porosity 2) located at the bottom of the reactor to produce fine bubbles and a small magnetic stirrer to ensure homogeneous mixing of ozone, the catalyst and the aqueous substrate mixture. Preliminary experiments showed that ozone reacts very fast with 2,4-DCPA acid in water, therefore, ozonation times of 5, 10, 15 and 20 min were chosen for this work. Ozone gas was continuously bubbled at room temperature for the required time interval into 25 cm3 aqueous solution of 100 ppm 2,4-DCPA acid containing 0.1 g catalyst. The experimental set-up for this work is shown in Fig. 1.
Fig. 1.

Experimental set-up: 1 – medical grade oxygen gas cylinder, 2 – pressure regulator, 3 – ozonator with flowmeter, 4 – reaction vessel with bubbler, stirrer bar and catalyst material, 5 – stirrer, 6 – excess ozone trap containing 0.2 mol dm−3 potassium iodide solution
Instrumental analysis
A HPLC method was developed and validated to monitor the percentage conversion of 2,4-DCPA acid as a function of ozonation time using a Shimadzu 20A high performance liquid chromatograph fitted with a variable wavelength UV detector and a Waters Novapak silica 4 μm column (3.9 mm i.d. and 150 mm length) maintained at 30 °C. The system was run on isocratic mode using a filtered and degassed mobile phase solution of 60/40 (v/v) methanol–water set at a flow rate of 1 mL/min. Linearity of the HPLC instrument was checked by injecting various standard solutions 2,4-DCPA acid prepared by serial dilution from the 100 ppm 2,4-DCPA acid stock solution.
The TOC was determined by sparging 10 mL of the sample under slightly acidic conditions (pH 2) to remove inorganic carbon. The organic carbon in the sample was then digested in a DRB 20 COD reactor with persulphate powder and acid to form carbon dioxide. The carbon dioxide was then allowed to diffuse into a pH indicator reagent contained in an ampule to form carbonic acid. The coloured solution was then measured using a DR 1900 portable Spectrophotometer at a wavelength setting of 430 nm. The amount of colour change is related to the parts per million of organic carbon present in the sample. Calibration of the spectrophotometer was carried out by using a 1000 mg L−1 potassium acid phthalate standard solution.
The concentration of chlorides and low molecular weight organic acids (acetic and formic acids) in each ozonated sample was measured using the Metrohm 761 Compact ion Chromatograph (IC) fitted with a conductivity detector and a Metrohm ASupp 5250/4.0 column. The carbonate eluent used were a mixture of 3.2 Mm Na2CO3 and 1.0 Mm NaHCO3. A 50 mM H2SO4 solution served as the suppressor reagent. Chloride and low molecular weight organic AR grade standards, purchased from Merck SA, was used to check linearity and calibrate the IC instrument. Conductivity of each solution before and after ozonation was measured by a conductivity meter. The pH of the substrate solution before and after ozonation was measured at room temperature using a Metrohm combined pH glass electrode Pt 1000. Calibration of the conductivity and pH electrode was conducted prior to sample measurements.
Results and discussion
Influence of solution pH
To evaluate the effect of pH on the conversion of 2,4-DCPA acid by ozonation, experiments were carried in acidic, neutral and basic water. The conversion profiles and TOC removal efficiency for these experiments are shown in Fig. 2. A comparison of conversion profiles shows that the conversion of 2,4-DCPA acid increases as the pH of the substrate solution increases, suggesting that the pH of the solution during ozonation has a significant effect on the conversion of 2,4-DCPA acid. Similar results were obtained when 2-chlorophenol, 4-chlorophenol and 2,4-dichlorophenol were ozonated at pH 6 and 9 [13].
Fig. 2.

Percent conversion of 2,4-DCPA acid (a) and TOC removal (b) from acidic, neutral and basic water as a function of time
When comparing the three different conditions it is evident that the conversion of 2,4-DCPA acid in acidic water is relatively low for all time intervals and marginally higher in neutral water. In acidic water the decay of ozone, caused by the action of HO− ions [14] is very low, hence, poor production of hydroxyl radicals. The highest conversion of 2,4-DCPA acid is observed in basic water, suggesting that at high pH, the rate of ozone decomposition increases. The improved conversion at higher pH may result from (i) the deprotonation and dissociation of 2,4-DCPA acid followed by reaction with molecular ozone or (ii) an influence from the improved generation of HO• radicals during ozonation. The first step in ozone decay is the reaction between ozone and the hydroxide ion, leading to the formation of a number of intermediate radical species, and finally HO• radicals, and when the solution pH increases, the formation of HO• radicals is favoured. A high pH solution contains more hydroxide ions, which act as initiators for the successful breakdown of ozone. Some of the radicals that are produced during the intermediate stage can lead to further reactions with ozone, thus causing more HO• radicals to be produced [15]:
Our previous study has also shown that decomposition of ozone is significantly enhanced as solution pH increases [16]. In acidic solutions, the ozone molecule itself reacts very slowly with organic compounds, thus leading to poor conversion of the target material, however, at pH levels above 8 it rapidly forms free hydroxyl radicals, which reacts very fast with organic material in water. Organic substances that oxidize marginally in acidic or neutral water will oxidize rapidly at high pH levels. It was found that solution pH from 8 to 10 gave the best results for oxidation of organic molecules in water [17]. Studies conducted on the photo-catalytic oxidation of phenol in water also showed that acidic conditions do not favour substrate degradation, and degradation rate only increases with increasing pH [18].
For all water types, it was found that the amount of TOC removed was always lower than the amount of substrate converted, suggesting that complete mineralization was difficult to achieve with ozonation alone. Fig. 2b clearly shows that TOC removal in basic water is more favoured than in acidic or neutral water, suggesting that mineralization of 2,4-DCPA acid and oxygenated by-products in basic water is more effective. Ozone, when in the presence of large quantities of hydroxide ions, readily decomposes into highly reactive radical species, the main one being the HO• radical, which rapidly reacts with organic intermediates to form CO2 and H2O [19]. Ozonation of halogenated organic compounds has the potential to produce hazardous Org-BP’s, and many of these compound are refractory towards ozone. Therefore, it is necessary to monitor the formation of these Org-BP’s during ozonation of waters containing hazardous organic pollutants. The total Org-BP content of the ozonated samples were measures indirectly by carbon mass balance calculations. Two carboxylic acids, namely acetic and formic acids were detected and monitored by IC analysis. Fig. 3 shows the percentage yields of Org-BP’s and combined yields of the two organic acids formed.
Fig. 3.

Percent yield of Org BP’s (a) and organic acids (b) in acidic, neutral and basic water as a function of time
The results indicate that Org-BP formation is the highest in acidic water and then decreases gradually as the pH of the water increases. During the same time interval it is observed that the organic acids show an opposite trend, that is, in basic water organic acid formation is high, then decreasing gradually, with acidic water generating the lowest amount of organic acids. A comparison of the chloride ion content in Fig. 4 shows that the pattern of chloride ion formation is the same for all water types, increasing in yield from 5 min to 20 min of ozone treatment.
Fig. 4.
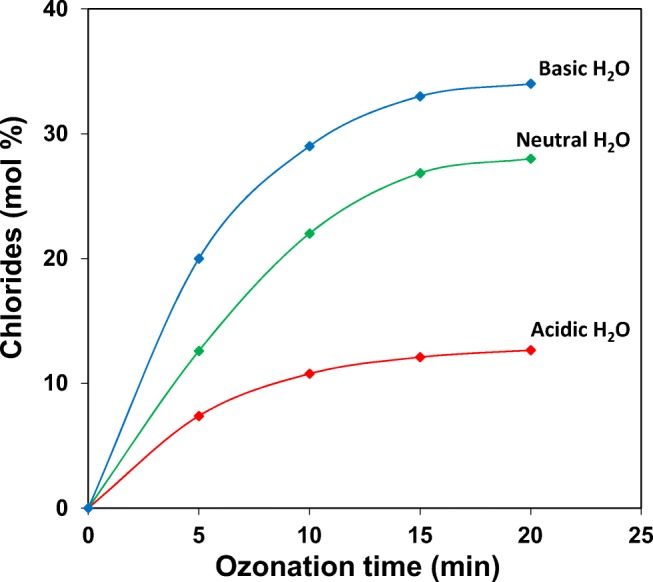
Percent chloride released during ozonation of 2,4-DCPA acid in acidic, neutral and basic water as a function of time
Percentage yield of chloride ion increases with an increase in solution pH, with basic water producing the highest amount of chloride ion, suggesting that dechlorination of 2,4-DCPA acid in water at high pH during ozonation is most effective. The trend observed in the solution conductivity was also consistent with the pattern of chloride ion formation, increasing gradually from 28 μS cm−1 for acidic water, but displaying the highest conductivity of 179 μS cm−1 for basic water, hence confirming that dechlorination is a major reaction step in the ozonation of 2,4-DCPA acid in water.
Catalytic ozonation of 2,4-DCPA acid in water
To further improve 2,4-DCPA degradation, TOC removal and Org-BP minimization, ozonation experiments in the presence of Fe alone and Fe doped with Ni by co-precipitation (Co-ppt) and physical mixing (Mixed) methods were investigated. The results of these experiments are reported and discussed below.
Influence of Ni loading on catalytic activity
The catalytic ozonation of 100 ppm aqueous solution of 2,4-DCPA acid in the presence of Fe alone and Fe combined with different quantities of Ni prepared by co-precipitation method (Co-ppt) was investigated as a function of ozone treatment time. Data for percent conversion of 2,4-DCPA acid and TOC removal in the presence of each catalyst material measured as a function of ozonation time is illustrated in Fig. 5.
Fig. 5.

Percent conversion of 2,4-DCPA acid (a) and TOC removal (b) as a function of ozone treatment time during Fe:Ni (Co-ppt) catalytic ozonation
It is evident from Fig. 5a that the percent conversion of 2,4-DCPA acid increases for all Ni loadings on Fe as ozone treatment time increases, indicating that catalytic ozonation with Fe alone or Fe combined with Ni (Co-ppt) favours the degradation of the toxic pollutant in water. The conversion of the toxic substrate in the presence of Fe alone (Co-ppt) was rapid during the first 5 min of ozone treatment, reaching 32%, and thereafter, increasing steadily to 53% after 20 min of ozonation. In the presence of 9:1 Fe:Ni (Co-ppt) the conversion of 2,4-DCPA acid in water increases more rapidly with ozone treatment time, reaching 42% after 5 min of ozonation and then increasing to 65% after 20 min of ozone treatment. In presence of 8:2 Fe:Ni (Co-ppt), a further improvement in the conversion of 2,4-DCPA acid is observed as a function of ozone aeration time. It quickly reached 50% in 5 min and increased to 77% only after 20 min of ozonation. Catalytic mixture 7:3 Fe:Ni (Co-ppt) converted 2,4-DCPA acid the most, increasing rapidly with ozone treatment time from 60% after 5 min of ozonation to 97% after 20 min of ozonation. It therefore, can be concluded that when the Ni loading on the Fe based catalyst is increased, the conversion of 2,4-DCPA acid increases significantly [20].
TOC results show that there is a gradual increase in organic carbon removal from the substrate solution as ozone treatment time increases. In the presence of Fe the TOC removal only increased by 6% compared to ozonation alone, while in the presence of 9:1 Fe:Ni (Co-ppt), an improvement in TOC removal by 24% was detected. Increasing the Ni loading on Fe by co-precipitation resulted in a further increase by 39% compared to ozonation alone. However, a limiting factor in all these cases is that the amount of TOC removed from the water is much lower than the amount of carbon converted during ozonation. When 7:3 Fe:Ni (Co-ppt) was used in the ozonation process, a significant improvement in TOC removal of 73% was observed, and more importantly the percentage carbon removed compared favourably with the percentage of substrate converted. The results reveal that degree of mineralization increases as Ni loading on Fe is increased. The superior textural surface properties listed in Table 1 may have contributed to the improved conversion of 2,4-DCPA acid and TOC removal from water. The data indicates that the surface characteristics of the Fe:Ni (Co-ppt) catalyst material improves as Ni loadings on Fe is increased.
When 10% Ni is loaded on Fe by co-precipitation, a significant increase in surface area occurs, and when the Ni content on Fe was further increased, its surface area and pore volume further increases, while its average pore size decreases. This could be attributed to the even distribution of Ni on Fe during co-precipitation, which is consistent with results obtained by SEM shown in Fig. 6. These improvements to the catalyst surface properties significantly contributes to the higher catalytic activity of Fe:Ni (Co-ppt) for conversion of 2,4-DCPA acid and TOC removal during ozonation.
Fig. 6.

SEM micrograms for 9:1 Fe:Ni (Co-ppt) and 9:1 Fe:Ni (Mixed) catalyst material
Organic by-product formation during catalytic ozonation
The effect of Ni loading on Fe on Org-BP formation during catalytic ozonation of 2,4-DCPA acid in water was investigated and the results are illustrated in Fig. 7.
Fig. 7.

Percent yield Org-BP’s (a) and organic acids (b) in the presence of Fe-Ni (Co-ppt) as a function of ozone treatment time
In uncatalysed ozone aeration, Org-BP formation is significantly high, and addition of Fe to the ozonation process caused a further increase in Org-BP’s formation. When 10% is loaded on Fe, Org-BP’s decreased by 10% compared to uncatalysed ozonation, while a maximum decrease of 17% was noted in the presence of 7:3 Fe:Ni (Co-ppt). Fig. 8a compares the N2 adsorption-desorption isotherms of 10% and 30% Ni respectively loaded on Fe prepared by the co-precipitation (Co-ppt) method.
Fig. 8.

N2 adsorption-desorption isotherms of (a) Fe:Ni (Co-ppt) and (b) Fe:Ni (Mixed) catalyst material
It is evident that Fe:Ni (Co-ppt) catalyst material show a type IV isotherm with a H3 hysteresis loop, indicating a mesoporous structure and as the Ni content on Fe increases the size of the hysteresis loop increases [21]. 7:3 Fe:Ni (Co-ppt) catalyst shows a broader hysteresis loop ranging from 0.75 to 1.00 of the relative pressure, while the hysteresis loop of 9:1 Fe:Ni (Co-ppt) is much narrower, starting at 0.90 and ending at 1.00, suggesting a less porous structure and thus a smaller surface area and pore volume [22]. The enhanced surface characteristics of the 7:3 Fe:Ni (Co-ppt) catalyst material is a further indication of its improved activity, which could be the reason for its effective minimization of Org-BP’s in water during ozonation.
Ni loaded on Fe by physical mixing
A simpler catalyst preparation method was investigated, were Ni was loaded on Fe by physically mixing the two salts, followed by calcination at 500 °C for 5 h. Data shown in Fig. 9 compares the percentage conversion and TOC removal from a solution containing 100 ppm 2,4-DCPA acid for ozonation alone; and ozonation in the presence of 7:3 Fe:Ni (Co-ppt) and 7:3 Fe:Ni (Mixed) respectively.
Fig. 9.

Comparison of percent conversion of 2,4-DCPA acid (a) and TOC removal (b) for uncatalyzed ozonation and ozonation as a function of time, in the presence of 30% Ni loaded on Fe by co-precipitation and physical mixing
It is evident from Fig. 9 that conversion of 2,4-DCPA acid in ozonation alone is the lowest, reaching only 45% after 20 min of ozone treatment, while TOC removal for the same time interval was 11%. In the presence of 7:3 Fe:Ni (Mixed) catalyst, 2,4-DCPA acid conversion was found to be higher, increasing to 68% after 20 min, while TOC removal was only 23%. The results indicate that in both cases the amount of TOC removed is much lower than percentage of carbon converted, suggesting that complete mineralization could not be achieved in both uncatalyzed ozonation and ozonation in the presence of 7:3 Fe:Ni (Mixed) catalyst. In the case of 7:3 Fe:Ni (Co-ppt) catalyst, a conversion of 97% was achieved after 20 min, while 92% of TOC was removed after 20 min of ozone treatment, indicating nearly complete mineralization of 2,4-DCPA acid and organic by-products. When 30% Co was loaded on Fe by co-precipitation, a decrease in catalytic activity was observed, however, an improvement in mineralization was noted. After 20 min of ozone treatment, 87% of the pollutant was degraded, while 83% of TOC was successfully removed. The solution pH during catalytic ozonation of organic pollutants in water is an important parameter to consider. In our investigation, substrate solution pH changed (i) when Fe:Ni catalyst material was added and (ii) during ozone treatment as illustrated in Table 2.
Table 2.
Initial and final solution pH in the presence of 2,4-DCPA acid measured at different reaction conditions
| Reaction Condition | Initial pH | Final pH | k/min* | R2 |
|---|---|---|---|---|
| Acidic water | 2.70 | 2.40 | 0.0287 | 0.9919 |
| Neutral water | 5.50 | 4.68 | 0.0394 | 0.9546 |
| Basic water | 11.18 | 10.01 | 0.0882 | 0.9749 |
| 100% Fe | 3.69 | 2.94 | 0.0532 | 0.9125 |
| 9:1 Fe:Ni (Co-ppt) | 9.70 | 4.48 | 0.0752 | 0.8988 |
| 7:3 Fe:Ni (Co-ppt) | 8.03 | 3.60 | 0.1637 | 0.9961 |
| 9:1 Fe:Ni (Mixed) | 3.40 | 2.31 | 0.0611 | 0.9200 |
| 7:3 Fe:Ni (Mixed) | 3.23 | 2.12 | 0.0802 | 0.9285 |
100 ppm 2,4-DCPA acid in water; volume = 25 cm3; mass of catalyst = 0.1 g; Temp. = 25 °C. *(n = 3) Relative standard deriation <5%
A recent study on ozone decomposition by alumina in water in the absence of pollutants attributed the increase in solution pH and improved ozone decomposition to contaminants present on the alumina surface [23]. In another study, involving catalytic ozonation of ibuprofen and VOC’s, a small increase in solution pH was observed, followed by a slight decrease. It attributed the slight increase in pH to desorption of contaminants and the decrease in pH to formation of acidic products, however, they suggested further research to verify this hypothesis [24]. In our study, the initial solution pH increased sharply from 3.43 to 9.70 after addition of 9:1 Fe:Ni (Co-ppt) catalyst material. Similar results were obtained when 9:1 Fe:Ni (Co-ppt) was mixed with water in the absence of organic pollutant, suggesting that the catalyst is the only material responsible for increase in initial solution pH. When the Fe-Ni surface is in contact with water it generally becomes hydrated, forming a monolayer of surface hydroxyl groups. The hydroxylated Fe:Ni (Co-ppt) surface then acts as a strong Bronsted base, spontaneously attracting the hydrogen ion from water to form positively charged sites and excess hydroxide ions, causing an increase in solution pH. The mechanism is illustrated in Scheme 1.
Scheme 1.
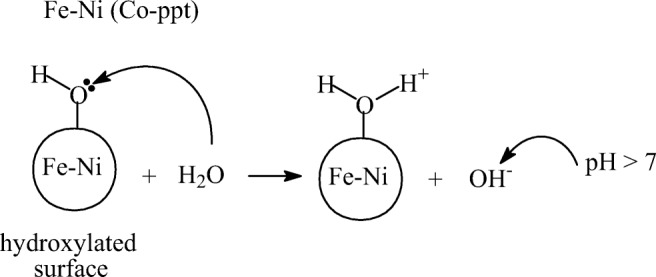
Adsorption of water on Fe-Ni (Co-ppt) catalyst surface showing formation of acidic site
These hydroxide ions and positively charged sites on the catalyst surface is advantageous during ozonation, since ozone, due to its strong polar nature will show a higher affinity for adsorption by its negative end onto the positive catalyst surface and the presence of excess hydroxide ions can lead to enhanced breakdown of ozone and generation of HO• radicals [25]. The released HO• radicals will then rapidly dechlorinate the 2,4-DCPA acid molecule to form the hydroxylated intermediate by-product and large quantities of chloride ions, hence causing a sharp increase in solution conductivity. Therefore, conversion of 2,4-DCPA acid in Fe:Ni (Co-ppt) catalytic ozonation primarily occurs via HO• radicals formed during ozone-catalyst surface interactions. On the other hand, the hydroxylated Fe:Ni (Mixed) surface acts as a strong Bronsted base, spontaneously donating its attached hydrogen to water to form a negatively charged Fe – Co – O− site and excess hydronium ions, causing a decrease in solution pH. This mechanism is illustrated in Scheme 2.
Scheme 2.
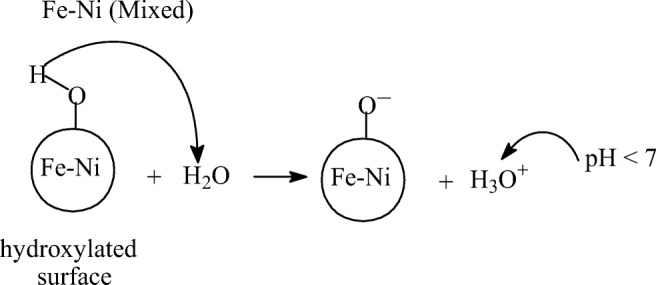
Adsorption of water on Fe-Ni (Mixed) catalyst surface showing formation of basic site
The lower conversion and TOC removal in Fe:Ni (Mixed) catalyst may suggest that the ozone molecule has a poor affinity towards the negative catalytic sites and furthermore, ozone decomposition in acid medium is diminished, hence poor hydroxyl radical formation. To confirm the presence of Bronsted acidic sites on the Fe-Ni catalyst surface, NH3-TPD experiments were conducted for each catalyst material illustrated in Fig. 10.
Fig. 10.

NH3-TPD profile of Fe-Ni (Co-ppt) and Fe-Ni (Mixed) catalyst material
The NH3-TPD profiles show that an increase in Ni loading on Fe from 10% to 30% resulted in an increase in ammonia desorption for Fe:Ni (Co-ppt) catalyst material only. The total acidity for 7:3 Fe:Ni (Co-ppt) was 1541 cm3 g−1 STP, while the acidity for the 7:3 Fe:Ni (Mixed) catalyst material was only 1.83 cm3 g−1 STP. The TCD signal and peak area for 30% Ni loaded on Fe is larger than 10% Ni loaded on Fe indicating a higher concentration of acidic sites on the 7:3 Fe:Ni (Co-ppt) catalyst surface. These acidic sites improve adsorption/desorption of ozone, leading to enhanced decomposition into hydroxyl radicals. The Fe:Ni (Co-ppt) catalyst show a broad peak at 140 °C, suggesting a single type of acidic site. The low temperature peak is an indication of slightly weaker bond strength and hence slightly weaker acidic sites [8]. These acidic sites result in improved catalytic activity, leading to more effective deprotonation reactions, producing hydroxyl radicals. An excess of these hydroxyl radicals then results in increased 2,4-DCPA acid conversion and maximum TOC removal from water. Fe:Ni (Mixed) catalyst material shows no distinct peak, suggesting a negligible amount of acidic sites on the catalyst surface, which explains its poor catalytic action during ozonation of 2,4-DCPA acid in water.
Ozonation processes applied in water treatment for disinfection frequently leads to the generation of hazardous by-products. One study has reported that Org-BP production increases with increasing ozone dosage and/or contact time [26], whereas another has shown that a further increase in contact time can reduce the Org-BP formation [27]. Therefore, it is essential to monitor and find ways to minimize the generation of organic by-products during ozonation of 2,4-DCPA acid in water. Fig. 11 compares the yield (based on mass balance of carbon in mol %) of the Org-BP’s and percent chloride ion released after 20 min for (i) ozonation alone, (ii) ozonation in the presence of 7:3 Fe:Ni (Co-ppt) and (iii) ozonation in the presence of 7:3 Fe:Ni (Mixed).
Fig. 11.

Comparison of Org-BP’s (a) and chloride yield (a) for uncatalyzed ozonation and ozonation in the presence of 30% Ni loaded on Fe by co-precipitation and a simple mixing method as a function time
In ozonation alone, 19% of Org-BP was formed after 20 min of ozone treatment, however, when ozonation was conducted in the presence of 7:3 Fe:Ni (Co-ppt) catalyst material, a significant drop in Org-BP formation to 2% was detected. With 7:3 Fe:Ni (Mixed) catalytic ozonation, Org-BP formation increased to 66% after 20 min of ozone treatment. The results reveal that 7:3 Fe:Ni (Co-ppt) has superior catalytic activity than 7:3 Fe:Ni (Mixed) catalyst material to minimize Org-BP formation during ozonation of 2,4-DCPA acid in water. Fig. 11 also compares the dechlorination efficiency by catalytic ozonation with ozonation alone. It is evident that dechlorination by catalytic ozonation is more effective than ozonation alone. In ozonation alone only 28% chloride was released, however, in the presence of 7:3 Fe:Ni (Mixed) catalyst a slight improvement in chloride yield was detected. Catalytic ozonation with 7:3 Fe:Ni (Co-ppt) resulted in the highest de-chlorination efficiency, reaching 47% after 20 min of ozone treatment. The N2 adsorption-desorption isotherms illustrated in Fig. 8 for both Fe:Co (Co-ppt) and Fe:Co (Mixed) catalyst material showed similar types of isotherms, however, the quantity of nitrogen adsorbed was significantly different. Nitrogen adsorption on both catalyst material showed an increasing trend with Ni loadings on Fe, but the Fe:Ni (Co-ppt) catalyst showed higher nitrogen adsorption than the Fe:Ni (Mixed) catalyst material for all Ni loadings. This pattern of surface behaviour suggests that Fe:Ni (Co-ppt) has better catalytic activity than Fe:Ni (Mixed). The superior surface characteristics of the Fe:Ni (Co-ppt) catalyst material and improved activity could be the main reason for minimizing Org-BP’s formation during catalytic ozonation of 2,4-DCPA acid in water. When 30% of Co was loaded on Fe by co-precipitation, an increase in Org-BP yield was observed. A drop in chloride ion yield occurred after 20 min of ozone treatment, indicating that Fe:Ni (Co-ppt) catalyst has superior characteristics than Fe:Co (mixed) catalyst material for 2,4-DCPA acid degradation, TOC removal and Org-BP minimization in water during ozone treatment.
Kinetics of 2,4-DCPA acid degradation
Figure 12 illustrates the kinetic profiles of 2,4-DCPA conversion under uncatalysed and catalytic ozonation with Ni loaded on Fe, which followed pseudo first-order kinetics with respect organic substrate.
Fig. 12.

Kinetic curves for 2,4-DCPA removal through ozone aeration (a) effect of pH; and (b) effect of Ni loading on Fe
The rate constants and R2 values obtained from the log (C/C0) versus time plots using the data in Fig. 12a, b are appended to Table 2. In uncatalysed ozonation, the rate of conversion increased with increase in hydroxide ion concentration in the solution (k = 0.0882 min−1). This behaviour could be due to the enhanced decomposition of ozone to hydroxyl radicals at high pH. In catalytic ozonation, the rate of 2,4-DCPA conversion increased with increase in Ni loading on Fe. With 7:3 Fe:Ni (Co-ppt) highest rate constant value recorded was 0.1687 min−1. This could be due to the increase in the number of surface active sites on catalyst for 2,4-DCPA, ozone and radical reactions.
Conclusion
The conversion of 2,4-DCPA acid in water increases as the hydroxide ion concentration of the substrate solution increases. Percent conversion of organic pollutant in acidic water is relatively low for all time intervals and marginally higher in neutral water. The highest conversion of 2,4-DCPA acid was achieved in basic water. When solution pH increases, the production of HO• radicals is favoured. In a high pH solution, there are more hydroxide ions present, which initiate ozone decay, to produce HO• radicals, hence enhancing 2,4-DCPA acid dechlorination. Limited mineralization of 2,4-DCPA acid and oxygenated products was achieved in acid and neutral water, however, the amount of TOC removed from basic water compared favourably with the amount of carbon converted, indicating an improvement in mineralization. Org-BP minimization was better in basic water than in acidic water and more organic acids were formed, which was found to be refractory towards ozone. In catalytic ozonation, it was observed that as the Ni content of the Fe based catalyst was increased, its catalytic activity was significantly enhanced, hence improving dechlorination efficiency, TOC removal and Org-BP minimization. The addition of Ni to Fe improved the catalyst surface properties and increased the number of Bronsted acid sites by increasing solution pH, hence contributing to the higher catalytic activity of Fe:Ni (Co-ppt) catalyst material. Ni loaded on Fe by physical mixing resulted in a decrease in catalytic activity, hence a decrease in pollutant conversion, TOC removal and increase in Org-BP formation, compared to Fe:Ni (Co-ppt) catalyst, however, results obtained were better than those obtained for ozonation alone. The poor surface properties of the Fe:Ni (Mixed) catalyst and the fewer number of acidic sites is the main reasons for decrease in catalytic activity. When Ni was replaced with Co as dopant, a slight decrease in 2,4-DCPA acid and TOC removal occured, while Org-BP formation increased sharply. A Ni loading of 30% on Fe prepared by co-precipitation method gave the best results.
Acknowledgements
The authors are thankful for the financial support from Mangosuthu University of Technology, University of Kwa-Zulu Natal and the National Research Foundation for successful completion of this invaluable work.
Author’s contributions
ANG conducted all the lab analysis and drafted this manuscript. All authors contributed to the review and finalization of this manuscript. All authors read and approved the final manuscript.
Compliance with ethical standards
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mazza A, Piscitelli P, Neglia C, Rosa GD, Iannuzzi L. Illegal dumping of toxic waste and its effect on human health in Campania, Italy. Int J Environ Res Public Health. 2015;2(6):6818–6831. doi: 10.3390/ijerph120606818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahnama-Moghadam S, Hillis LD, Lange RA. Chapter 3 - environmental toxins and the heart A2 - Ramachandran, Meenakshisundaram, in Heart and toxins. Boston: Academic Press; 2015. pp. 75–132. [Google Scholar]
- 3.Khoshnood M, Azizian S. Adsorption of 2,4-dichlorophenoxyacetic acid pesticide by graphitic carbon nanostructures prepared from biomasses. J Ind Eng Chem. 2012;18(5):1796–1800. [Google Scholar]
- 4.Bekbölet M, Yenigün O, Yücel I. Sorption studies of 2,4-D on selected soils. Water Air Soil Pollut. 1999;111(1):75–88. [Google Scholar]
- 5.Tsyganok AI, Otsuka K. Selective dechlorination of chlorinated phenoxy herbicides in aqueous medium by electrocatalytic reduction over palladium-loaded carbon felt. Appl Catal B Environ. 1999;22(1):15–26. [Google Scholar]
- 6.Kwan CY. Chu W. a study of the reaction mechanisms of the degradation of 2,4-dichlorophenoxyacetic acid by oxalate-mediated photooxidation. Water Res. 2004;38(19):4213–4221. doi: 10.1016/j.watres.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 7.Rodríguez JL, Valenzuela MA, Poznyak T, Lartundo L, Chairez I. Reactivity of NiO for 2,4-D degradation with ozone: XPS studies. J Hazard Mater. 2013;262:472–481. doi: 10.1016/j.jhazmat.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 8.Guzman-Perez CA, Soltan J, Robertson J. Catalytic ozonation of 2,4-dichlorophenoxyacetic acid using alumina in the presence of a radical scavenger. J Enviro Sci Health B. 2012;47(6):544–552. doi: 10.1080/03601234.2012.665675. [DOI] [PubMed] [Google Scholar]
- 9.Soni KC, Shekar SC. Catalytic oxidation of bis (2-chloroethyl) ether on vanadia titania nanocatalyst. Arab J Chem. 2017:1–11.
- 10.Shahamat YD, Farzadkia M, Nasseri S, Mahvi AH, Gholami M, Esrafili A. Magnetic heterogeneous catalytic ozonation: a new removal method for phenol in industrial wastewater. J Environ Health Sci Eng. 2014;12(1):1–12. doi: 10.1186/2052-336X-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beller M, Bolm C. Transition metals for organic synthesis. Weinheim: Wiley-VCH; 2004. [Google Scholar]
- 12.Rakness K, Gordon G, Langlais B, Masschelein W, Matsumoto N, Richard Y, Robson CM, Somiya I. Guideline for measurement of ozone concentration in the process gas from an ozone generator. Ozone: science. Engineering. 1996;18(3):209–229. [Google Scholar]
- 13.Kuo WS. Synergistic effects of combination of photolysis and ozonation on destruction of chlorophenols in water. Chemosphere. 1999;39(11):1853–1860. doi: 10.1016/s0045-6535(99)00080-6. [DOI] [PubMed] [Google Scholar]
- 14.Straehelin S, Hoigné J. Mechanism and kinetics of decomposition of ozone in water: rate of initiation by hydroxide ions and hydrogen peroxide. Environ Sci Technol. 1982;16:676–681. [Google Scholar]
- 15.Gottschalk C, Libra JA, Saupe A. Wiley-VCH. 2000. Ozonation of water and wastewater. [Google Scholar]
- 16.Gounden AN, Singh S, Jonnalagadda SB. Simultaneous removal of 2,4,6-tribromophenol from water and bromate ion minimization by ozonation. J Hazard Mater. 2018;357:415–423. doi: 10.1016/j.jhazmat.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Staehelin J, Hoigné J. Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions. Environ Sci Technol. 1985;19:1206–1213. doi: 10.1021/es00142a012. [DOI] [PubMed] [Google Scholar]
- 18.Way TY, Wan CC. Heterogeneous photocatalytic oxidation of phenol with titanium dioxide powders. Ind Eng Chem Res. 1991;30:1293–1300. [Google Scholar]
- 19.Staehelin J, Hoigné J. Decomposition of ozone in water. Environ Sci Technol. 1982;16:676–681. doi: 10.1021/es00142a012. [DOI] [PubMed] [Google Scholar]
- 20.Martins RC, Quinta-Ferreira RM. Catalytic ozonation of phenolic acids over a Mn–Ce–O catalyst. Appl Catal B Environ. 2009;90(1):268–277. [Google Scholar]
- 21.Wu Z, Zhang L, Guan Q, Fu M, Ye D. Wu T. catalytic oxidation of toluene over au–co supported on SBA-15. Mater Res Bull. 2015;70:567–572. [Google Scholar]
- 22.Wang Y, Xie Y, Sun H, Xiao J, Cao H, Wang S. 2D/2D nano-hybrids of γ-MnO2 on reduced graphene oxide for catalytic ozonation and coupling peroxymonosulfate activation. J Hazard Mater. 2016;301:6–64. doi: 10.1016/j.jhazmat.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 23.Nawrocki J, Fijołek L. Effect of aluminium oxide contaminants on the process of ozone decomposition in water. Appl Catal B Environ. 2013;142-3:533–537. [Google Scholar]
- 24.Ikhlaq A, Brown DR, Kasprzyk-Hordern B. Catalytic ozonation for the removal of organic contaminants in water on alumina. Appl Catal B Environ. 2015;165:408–418. [Google Scholar]
- 25.Jung H, Ahn Y, Choi H, Kim IS. Catalytic decomposition of ozone and para-chlorobenzoic acid (pCBA) in the presence of nanosized ZnO. Appl Catal B Environ. 2006;66(3–4):288–294. [Google Scholar]
- 26.Silva GHR, Daniel LA, Bruning H, Rulkens WH. Anaerobic effluent disinfection using ozone: by-products formation. Bioresour Technol. 2010;10:6981–6986. doi: 10.1016/j.biortech.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 27.García O, Isarain-Chávez E, Garcia-Segura S, Brillas E, Peralta-Hernández JM. Study of the catalytic ozonation of humic substances in water and their ozonation by-products. Ozone Sci Eng. 1996;18:195–208. [Google Scholar]