


Abstract
Carboxysomes, protein-coated organelles in cyanobacteria, are important in global carbon fixation. However, these organelles are present at low copy in each cell and hence must be segregated to ensure transmission from one generation to the next. Recent studies revealed that a DNA partition-like ParA–ParB system mediates carboxysome maintenance, called McdA-McdB. Here, we describe the first McdA and McdB homolog structures. McdA is similar to partition ParA Walker-box proteins, but lacks the P-loop signature lysine involved in ATP binding. Strikingly, a McdA-ATP structure shows that a lysine distant from the P-loop and conserved in McdA homologs, enables ATP-dependent nucleotide sandwich dimer formation. Similar to partition ParA proteins this ATP-bound form binds nonspecific-DNA. McdB, which we show directly binds McdA, harbors a unique fold and appears to form higher-order oligomers like partition ParB proteins. Thus, our data reveal a new signature motif that enables McdA dimer formation and indicates that, similar to DNA segregation, carboxysome maintenance systems employ Walker-box proteins as DNA-binding motors while McdB proteins form higher order oligomers, which could function as adaptors to link carboxysomes and provide for stable transport by the McdA proteins.
INTRODUCTION
Many bacteria contain intracellular protein-coated microcompartments (BMCs) that encapsulate functionally related enzymes (1). These microcompartments serve as bacterial organelles by sequestering and localizing specific metabolic pathways. To date, 23 distinct classes of BMCs have been identified. The best studied BMC is the carboxysome, which is found in cyanobacteria and some chemoautotrophs (2–8). This organelle houses ribulose-l,5-bisphosphate carboxylase/oxygenase (RuBisCO) and carbonic anhydrase, thereby greatly enhancing carbon fixation by increasing CO2 levels in the vicinity of RuBisCO. As cyanobacteria that contain carboxysomes are responsible for >25% of all global carbon fixation there has been a keen interest in understanding how these organelles are formed and maintained. Interestingly, each cell contains only a few carboxysomes suggesting they must somehow be actively segregated to daughter cells to be distributed from one generation to the next. Indeed, studies show that carboxysomes align longitudinally along the cell and this presumed positioning is thought to be key to carboxysome inheritance (9). However, while recent work has uncovered the composition and mechanism of assembly of these organelles (4–8), the means by which they are transmitted to daughter cells has been somewhat unclear.
Recent studies by MacCready et al. in the cyanobacteria Synechococcus elongatus showed that a putative Walker-box ParA-like protein collaborates with a previously unknown protein to actively position carboxysomes (10). They called these proteins maintenance of carboxysome distribution (Mcd) proteins, McdA and McdB (10). The McdB protein was shown via two hybrid studies to interact with carboxysome shell proteins. The McdA contains a putative Walker-box ATPase fold that appears similar to Walker-box ParA ATPases, which are used by the most common of the three types of prokaryotic plasmid DNA partition systems (11). The two less abundant plasmid partition systems employ actin and tubulin-like NTPases. In these systems the NTPases form polymers to mediate DNA segregation (12–20). Initial data suggested that Walker-box ParA proteins also form polymers for segregation (17). However, recent studies including in vitro reconstitution experiments, super resolution analyses and crystallographic analyses support a non-polymer, diffusion-ratchet like mechanism (21–30). For partition, these Walker-box systems require, in addition to the ParA ATPase, a DNA centromere site and centromere binding ParB protein (11,21).
A distinguishing feature of the Walker-box systems is that, when complexed with ATP, their ParA NTPases bind and use the bacterial nucleoid DNA as a substratum to transport and thus equi-partition replicated plasmid DNA cargo (23–32). Thus, ATP binding to ParA acts as the key trigger for Walker box partition. The basis for this trigger was revealed by biochemical and structural studies, which showed that ParA proteins are monomeric in their apo forms and dimerize in the presence of ATP (32–34). Dimer formation is required for ParA binding to nonspecific DNA and hence the nucleoid. ParA dimerization is mediated by so-called signature lysine residues present in the Walker A motifs of these proteins, which forms inter-monomer contacts to the ATP bound at the other subunit of the dimer (33). Partition ParB proteins serve as the connection between the ParA motor and the cargo DNA to be transported. They do so by binding with high specificity and cooperativity to DNA centromeres, which consist of repeat elements. These are 16 base pair (bp) elements in many well-studied chromosomal centromeres and consist of repeat elements of sometimes varying length and sequence in plasmid centromeres (34–42). For example, the P1 plasmid centromere consists of heptamer and hexamer boxes (36,37). Binding of ParB proteins to centromeres leads to the formation of a higher order structure called the partition complex (35). Studies on P1 ParB and F plasmid SopB implicated not only the DNA binding domains but also their C-terminal dimerization domains as DNA binding elements (41,42). Indeed, studies by the Bouet and Dillingham labs revealed that chromosomal ParB proteins form complex, bridged 3D structures, with the C-terminal domains mediating bridging and DNA condensation (43,44). This large ParB-DNA cargo binds the ParA–ATP dimer and stimulates its ATP hydrolysis as the ParA proteins are themselves, weak ATPases. Structures and biochemical analyses indicate that ParB proteins bind at the dimer interface, between ParA subunits, and employ arginine residue(s) that appear to aid in ParA ATP hydrolysis (32,33,45). ATP hydrolysis drives ParA into a monomer state, which dissociates from the DNA. Repeated cycles of ParA binding and release from the nucleoid with concomitant capture of the ParB-DNA cargo somehow leads to equidistribution of the cargo. One hypothesis is that the ParA proteins equilibrate to DNA dense regions that form at the two poles of the dividing nucleoids, allowing for each daughter cell to retain a cargo DNA via a piggy back mechanism (31).
The carboxysome retention machinery appears similar to Walker-box DNA segregation systems (10). In particular, the McdA protein, which like partition ParA proteins has a putative Walker-box fold, appears to bind the nucleoid DNA in the presence of ATP and data suggests it also uses the nucleoid as a track to move its molecular cargo, in this case, carboxysomes (10). However, as noted by McCready et al., the S. elongatus McdA protein and its homologs, although harboring Walker A and B motifs notably lack the signature lysine residue that mediates dimerization of partition ParA proteins (10). Indeed, a hallmark feature of a large group of ATPases, which includes not only partition ParAs but proteins involved in nitrogen fixation and spatial regulation of cell division, is the presence of two lysines within the phosphate binding P-loop of the Walker A motif. One lysine is near the carboxy end of the motif, which is common to all Walker A motifs, and the second lysine, the signature lysine, is located at the beginning of the motif (KGGXXK(S/T), where the signature lysine is underlined) and is unique for these proteins (46–52). Walker A motifs with this signature lysine have been termed ‘deviant’ Walker A motifs to contrast them with classical Walker A sequences, which lack this lysine. Consistent with the central role of the signature lysines in these proteins, studies have shown that their mutation leads to significantly impaired partition (49,50). As noted (10), the absence of the deviant Walker lysine residue in McdA proteins suggests the possibility that they may function in a manner distinct from partition ParAs. Moreover, the McdB proteins show no homology to partition ParB proteins or any other structurally characterized protein. However, the data indicating that they bind carboxysome components, suggests that they could serve as the link between the carboxysome cargo and the McdA motor (10). To gain insight into the molecular architecture of the Mcd components, we determined structures of homologs of the S. elongatus McdA and McdB proteins and also performed biochemical analyses. These studies reveal a new type of signature motif for forming a nucleotide sandwich dimer and show that McdB harbors a unique fold that can form higher order structures that may be functionally akin to the ParB-DNA partition complexes. Overall the data suggest that the Mcd proteins form a segregation system that likely functions similarly to prokaryotic Walker-box DNA segregation systems.
MATERIALS AND METHODS
Expression and purification of Cyanothece McdB, McdA and McdA(38A) proteins
Artificial genes (codon optimized for E. coli expression) encoding the McdB and McdA homologs from the Cyanothece PCC7424 plasmid (herein referred to as McdB and McdA) were purchased from Genscript and cloned into pET15b between the BamHI and NdeI restriction sites. This resulted in the placement of thrombin cleavable N-terminal His-tags on each expressed protein. The McdA(D38A) mutant was made with QuikChange and the mutant protein was expressed and purified as for the wild type (WT) McdA protein. The mcdA(D38A ) encoding plasmid was used to construct a McdA(D38A-K151A) double mutant. The double mutant protein was expressed and purified using the same method as the other McdA protein. The McdB truncation was generated by Genscript and cloned into pET15b between the BamHI and NdeI restriction sites. The expressed McdB truncation was purified as per the WT McdB protein.
For protein production, the expression plasmids for the constructs were transformed into E. coli C41(DE3) cells. Protein expression of McdB and McdA proteins was induced when the cells reached an OD600 of ∼0.5 by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM for 4 h at 37°C. The resultant cell pellets were resuspended in Buffer A (25 mM Tris pH 7.5, 300 mM NaCl, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol (βME)) and a microfluidizer was used to disrupt the cells. The expressed McdB and McdA proteins were found in the soluble fractions and purified using cobalt and nickel-nitrilotriacetic acid (Ni-NTA) column chromatography respectively. Proteins were further purified by Superdex 75 size exclusion column (SEC) chromatography. His-tags were removed using thrombin cleavage capture kits (Qiagen). Prior to crystallization, the proteins were concentrated using centricon concentrators.
Crystallization and structure determination of the Cyanothece McdA
For crystallization experiments of McdA, the proteins were concentrated just prior to setups as all McdA proteins precipitated over time. To obtain apo McdA crystals the protein was concentrated to 40 mg/ml and subjected to crystallization trials using multiple screens via the hanging drop vapor diffusion at room temperature. Data quality crystals of apo McdA were obtained by mixing the protein 1:1 with a reservoir of 0.1 M (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) HEPES pH 7.5, 0.5 M lithium sulfate and 2 M ammonium sulfate. The same crystals were produced of the protein with or without the His-tag. Crystals were cryo-preserved by dipping them in the crystallization solution supplemented with 20% (v/v) glycerol for 2 s prior to placement in the cryo-stream. Crystals of the McdA bound to β,γ-imidoadenosine 5′-triphosphate (AMPPNP) were grown using protein (either with or without the His-tag) at 30 mg/ml mixed with 5 mM AMPPNP and 2 mM MgCl2 and combining this mixture 1:1 with a crystallization reagent consisting of 0.1 M HEPES pH 7.5, 1.5 M sodium thiocyanate. For data collection the crystals were cryo-preserved by dipping them for 2 s in the crystallization reagent mixed with 25% (v/v) glycerol. Crystals of McdA(D38A)-ATP were obtained from a solution of the protein at 35 mg/ml, 5 mM ATP, 2 mM MgCl2 and mixing this solution 1:1 with the crystallization reagent, 100 mM imidazole pH 8.0, 20% (v/v) PEG 300, 1 M ammonium sulfate, 10% (v/v) glycerol. The crystals were cryo-preserved straight from the drop.
To obtain phases, selenomethione substituted McdA protein was produced using the methionine inhibitory pathway (53). The protein was purified and crystallized as for the WT apo McdA. Multiple wavelength anomalous diffraction (MAD) data were collected at the advanced light source (ALS) beamline 8.3.1 on a selenomet-McdA apo crystal to 2.68 Å resolution. X-ray intensity data were processed with MOSFLM and scaled with SCALA in CCP4 (54,55). Autosol in Phenix (56) was used to identify selenomethionine sites, perform density modification and calculate an experimental electron density map. The model building program O (57) was used to construct the structure, which consisted of 2 McdA subunits in the crystallographic asymmetric unit (ASU). Interestingly, one subunit was in the apo form and one bound to ADP. The model was subjected to several rounds of iterative model building in O and refinement in Phenix (56,57). The final model has Rwork/Rfree values of 18.1%/22.2% to 2.68 Å resolution. The structure includes residues 1–251 of each subunit, 1 ADP molecule and 83 water molecules. One McdA subunit (minus solvent and ADP) was used in molecular replacement (MR) as a search model to solve the McdA-AMPPNP structure in MolRep. The McdA-AMPPNP crystals are P6422 and similar to the ‘apo’ McdA structure, this structure contains one apo subunit and one bound to AMPPNP. Phenix was used to refine the structure to final Rwork/Rfree of 19.5%/22.2% to 2.50 Å resolution. The final model includes residues 1–251 of each subunit, 1 AMPPNP molecule, 1 Mg2+ ion and 27 solvent molecules. The McdA(D38A)-ATP crystals take the trigonal space group, P3121 and contain two subunits in the ASU. The structure was solved by MolRep using a starting model consisting of one McdA subunit. Two solutions were obtained (there is a dimer in the ASU) and the structure was fit in O and refined in Phenix to final Rwork/Rfree of 18.2%/20.4% to 1.70 Å resolution. The final model includes residues 1–256 of each subunit of the dimer, two ATP molecules, 2 Mg2+ ions and 253 water molecules. See Table 1 for all McdA data collection and refinement statistics.
Table 1.
Data collection and refinement statistics for McdA structures
| McdA-ADP-Apo | McdA-AMPPNP | McdA(D38A)-ATP | |
|---|---|---|---|
| Data collection | |||
| Pdb code | 6NON | 6NOO | 6NOP |
| Space group | P6422 | P6422 | P3121 |
| Cell dimensions | |||
| a, b, c (Å) | 155.4,155.4, 180.7 | 154.9, 154.9, 181.3 | 115.7,115.7, 76.6 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0,120.0 | 90.0, 90.0,120.0 |
| # Unique reflections | 61015 | 81993 | 60006 |
| # Total reflections | 310596 | 497408 | 384150 |
| Resolution (Å) | 134.5 (2.68)* | 134.1 (2.50) | 76.75 (1.70) |
| R sym | 0.072 (0.660) | 0.061 (1.282) | 0.026 (0.379) |
| Rpim | 0.050 (0.486) | 0.021 (0.750) | 0.010 0.263) |
| I / σI | 16.5 (1.9) | 20.2 (1.0) | 31.0 (2.4) |
| Redundancy | 4.7 (3.2) | 6.1 (6.0) | 6.4 (2.6) |
| CC(1/2) | 0.997 (0.404) | 0.999 (0.356) | 1.00 (0.903) |
| Refinement | |||
| Resolution (Å) | 134.5 (2.68) | 134.1 (2.50) | 76.75 (1.70) |
| R work/Rfree | 18.1/22.2 | 19.5/22.2 | 18.2/20.4 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.009 | 0.008 | 0.007 |
| Bond angles (°) | 1.294 | 1.194 | 1.141 |
| Ramachandran analyses | |||
| Favored (%) | 96.3 | 95.0 | 98.0 |
| Disallowed (%) | 0.0 | 0.0 | 0.0 |
*Values in parentheses are for highest-resolution shell.
Crystallization and structure determination of the Cyanothece McdB
The purified McdB protein was utilized in multiple crystal screens using the hanging drop vapor diffusion method. Data qualify crystals were produced with protein at 20 mg/ml and mixing it 1:1 with a reservoir consisting of 0.1 M sodium acetate pH 5.0, 1.8 M ammonium sulfate. The crystals were produced at room temperature and took from 3 days to a week to grow to full size. To obtain phases, selenomethionine substituted protein was produced using the methionine inhibitory pathway (53). For expression of soluble selenomethionine substituted McdB, the protein was induced overnight at 15°C by adding 0.5 mM IPTG when the cells had reached an OD600 of 0.5. The expressed protein was purified as for the WT. Isomorphous crystals were obtained of the selenomethionine substituted crystals and SAD data were collected at ALS beamline 8.3.1. Crystals were cryo-preserved by adding the crystal to a 2 ul drop of the crystallization solution supplemented with 25% (v/v) glycerol for 2 s before placement in the cryo-stream. The data were processed with MOSFLM and scaled with SCALA (54,55). The crystals take the trigonal space group, P3221. Autosol in Phenix was used to identify selenomethionine sites, carry out density modification and calculate an experimental electron density map. Although only one selenomethionine site was present for each subunit, the high solvent content of the crystals resulted in an interpretable electron density map after density modification. The model building program O (57) was used to construct the structure, which consisted of two McdB dimers in the ASU. The model was subjected to several rounds of iterative model building in O and refinement in Phenix (56,57). The final structure consists of residues 77–156 for each of the four subunits in the ASU and has final Rwork/Rfree values of 22.1%/26.7% to 3.45 A resolution. See Table 2 for McdB data collection and refinement statistics.
Table 2.
Data collection and refinement statistics for the McdB structure
| Structure | McdB |
|---|---|
| Data collection | |
| Pdb code | 6NOY |
| Space group | P3221 |
| Cell dimensions | |
| a, b, c (Å) | 95.5, 95.5,177.1 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| # Unique Reflections | 23328 |
| # Total Reflections | 106842 |
| Resolution (Å) | 75.0 (3.45)* |
| R sym | 0.091 (1.25) |
| Rpim | 0.047 (0.653) |
| I / σI | 16.5 (1.9) |
| Redundancy | 8.5 (8.8) |
| CC(1/2) | 0.999 (0.776) |
| Refinement | |
| Resolution (Å) | 75.0 (3.45) |
| R work/Rfree | 22.1/26.7 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 0.891 |
| Ramachandran analyses | |
| Favored (%) | 97.1 |
| Disallowed (%) | 0.0 |
*Values in parentheses are for highest-resolution shell.
Fluorescence polarization (FP) experiments: DNA and protein–protein interactions.
FP experiments (58) were performed using a PanVera Beacon 2000 FP system at 25°C. For the experiments, McdA(D38A) was titrated into 0.990 ml buffer composed of Tris–HCl pH 7.5, 5% (v/v) glycerol and 150 mM NaCl, 5 mM MgCl2 and 10 mM ATP and containing 1 nM (final concentration) fluorescently labelled DNA, termed F-DNA (with the sequence 5′-GTGAGTACTCAC-3′). To investigate the effect of salt concentration on McdA(D38A) binding, the same experiment was performed but the salt concentration was varied. The data were fit using Kaleidagraph. To assess the ability of McdB to bind to McdA, McdA(D38A) was first titrated into the F-DNA to the point of saturation, then McdB protein was added to the reaction cell. The apparent Kd for this binding was obtained using Kaleidagraph. Control binding experiments were conducted examining McdB binding to F-DNA and McdA(D38A) binding to F-DNA in the absence of ATP/Mg2+. Each binding curve is a representative analysis from at least three technical replicates. The standard error was determined from the binding affinities of the separate experiments.
Fluorescence polarization ATP Binding Experiments
FP experiments to examine McdA(D38A) and McdA(D38A-K151A) binding to ATP were performed using a PanVera Beacon 2000 FP system at 25°C with a fluorescently labeled ATP, Fluorescein-12-ATP (Perkin Elmer). This ATP has a fluorescein attached at the N7 atom of the adenine ring by a long linker that the McdA-ATP structure indicates should still allow binding. For the experiments, McdA(D38A) was titrated into 0.990 ml buffer composed of Tris–HCl pH 7.5, 5% (v/v) glycerol and 150 mM NaCl, 5 mM MgCl2 and containing 1 nM (final concentration) fluorescently labeled ATP (Fluorescein-12-ATP). The data were fit using Kaleidagraph.
Glutaraldehyde crosslinking experiments
To assess the molecular weight (MW) of McdB we performed glutaraldehyde crosslinking. Purified McdB at 2 mg/ml in 50 mM HEPES pH 7.0, 200 mM NaCl was crosslinked with 0.1% glutaraldehyde (final concentration) and time points collected. The reaction was quenched by addition of SDS running buffer and the time point samples were run on SDS page gels to assess changes in MW.
Size exclusion chromatography (SEC) analysis of McdB
For SEC analyses of McdB, the purified protein was concentrated to 2 mg/ml and loaded onto a Superdex 200 size exclusion column (GE Healthcare) and eluted with a buffer containing 20 mM Tris–HCl, pH 7.5, 300 mM NaCl, 5% (v/v) glycerol and 0.5 mM βME. The elution volume was compared to a series of protein standards to calculate the apparent molecular weight. The standards used were cytochrome c oxidase (12.4 kDa), carbonic anhydrase (29 kDa), albumin (66 kDa) and alcohol dehydrogenase (150 kDa).
RESULTS AND DISCUSSION
Biochemical analysis of purified Cyanothece McdA-McdB proteins
Studies illuminating the carboxysome maintenance machinery, composed of McdA and McdB, were conducted in S. elongatus (10). However, we could not express the McdA and McdB from S. elongatus in high yields in a soluble form, hence we used homologous proteins from Cyanothece that were solubly expressed and could be purified to homogeneity. The Cyanothece McdA protein showed 54% sequence identity to the S. elongatus McdA protein. Its putative partner McdB was located on the operon encoding the McdA protein and similarly organized as the mcdA and mcdB genes in the S. elongatus operon (10). Consistent with the finding that McdB proteins show very low sequence similarity, the Cyanothece McdB protein was less than 20% identical to the S. elongatus protein. However, the Cyanothece McdB protein is of similar length to S. elongatus McdB (Cyanothece McdB is 156 residues and the S. elongatus protein encompasses 152 residues) also, secondary structural predictions suggest that both McdB homologs are primarily helical.
We utilized our purified Cyanothece McdA and McdB proteins in biochemical studies to examine protein-protein and protein-DNA interactions. To analyze DNA binding by the Cyanothece McdA, we sought a McdA mutant that cannot hydrolyse ATP. Previous studies on partition ParA proteins showed that mutation of the key catalytic residue corresponding to Asp38 in McdA, allows ATP binding without significant ATP hydrolysis (45,47). Thus, we generated the McdA(38A) mutant protein and employed it in fluorescence polarization (FP) DNA binding assays. These experiments revealed that the McdA(D38A) protein bound DNA in the presence of ATP/Mg2+ with a Kd of 1.8 ± 0.4 μM, while it showed weak, nonsaturable binding without ATP (Figure 1A). Partition ParA proteins bind nucleoid DNA in a sequence nonspecific manner and structures of ParA-DNA complexes show that the proteins interact with the DNA using basic residues to contact the phosphate backbone and make no base specific contacts (28,32). If the McdA protein interacts with DNA using basic surface residues similar to ParA proteins the expectation would be that the binding affinity should increase under reducing salt conditions, as has been observed for partition ParA proteins (22,32). The FP analyses revealed a significant enhancement in McdA(D38A) binding under lower salt concentrations; the Kds for binding at 150 mM NaCl, 75 mM NaCl and 30 mM NaCl were 1.8 ± 0.4 μM, 0.7 ± 0.1 μM and 50 ± 0.4 nM, respectively (Figure 1B).
Figure 1.

Fluorescence polarization (FP) binding analyses of purified Mcd homologs. (A) FP binding curves showing that McdA(D38A), an McdA mutant that cannot hydrolyze ATP, binding to nonspecific DNA (5′-CGTGTAAGATTTCCTGACACG-3′) in the presence (red) and absence (blue) of ATP. McdA(D38A) in the presence of ATP binds DNA saturably with a Kd of 1.8 ± 0.4 μM, while in the absence of ATP no saturable binding is observed. The y-axis is millipolarization units (mP) and the x-axis is the concentration of McdA(D38A). (B) Analysis of McdA(D38A) binding to F-DNA in buffers with different salt concentrations. The binding curves were done in 150 mM NaCl (red), 75 mM NaCl (blue) and 30 mM NaCl (green). The y-axis is normalized mP units, (A – A0)/(Amax – A0), and the x-axis is the concentration of McdA(D38A) in μM. (C) McdB shows no binding to DNA. The y-axis is mP and the x-axis is the concentration of McdB. (D) FP experiment showing that addition of McdB into prebound McdA(D38A)-DNA reveals a second binding event with an apparent Kd of of 1.1 ± 0.3 μM. For this analysis, McdA(D38A) was first titrated into the F-DNA containing mixture until saturation (a concentration of 10 μM McdA(D38A)). Then McdB was titrated, which produced the plot shown. The y-axis is normalized mP units, (A – A0)/(Amax – A0), and the X-axis is concentration of McdB.
To test for a direct interaction between the Cyanothece McdA and McdB proteins, we utilized our FP binding assay. First, we assessed whether the McdB protein binds DNA. The FP experiment revealed no DNA binding by McdB (Figure 1C). We next titrated the McdB into a cell containing a complex of bound McdA(D38A)–ATP–DNA. If the McdB interacts with the McdA(D38A)–ATP–DNA complex the expectation would be a saturable increase in millipolarization upon McdB addition. Indeed, titration of low μM amounts of the McdB into the McdA(D38A)–ATP–F-DNA complex revealed a binding event with an apparent Kd of 1.1 ± 0.3 μM (Figure 1D; Supplementary Figure S1).
Crystal structure of the McdA protein reveals homology to prokaryotic partition Walker box ParA proteins.
To gain insight into how McdA functions at the molecular level, we next solved the structure of apo McdA by MAD. The structure was refined to Rwork/Rfree values of 18.1%/22.2% to 2.68 Å resolution (Figure 2A; Table 1). The structure consists of a central core of β-strands surrounded by α-helices and is similar overall to partition ParA protein structures. Indeed, the most structurally homologous protein as revealed by Dali searches was the Thermus thermophilus Soj partition ParA protein, which can be superimposed into the McdA structure with a root mean squared deviation (rmsd) of 1.9 Å for 224 Cα atoms (Supplementary Figure S2). There are two McdA subunits in the crystallographic asymmetric unit (ASU). Interestingly, one of the subunits is in the apo form while the second contains a clearly bound ADP, which apparently co-purified with the protein. The ADP in the McdA-ADP complex is contacted by residues in the McdA Walker motifs. As observed in other Walker box ATPase, the McdA Walker A motif includes a P-loop, which interacts with the nucleotide phosphate moieties, the Walker A′ sequence contains the putative catalytic residue and the Walker B motif harbors acidic residues that complex the Mg2+, contributing to catalysis. In the McdA-ADP structure, the phosphate moieties of the ADP are bound at the N-terminus of the Walker A helix (α1) (Figure 2B). The amide nitrogens of the glycines residues in the P-loop fold around and contact the phosphates. Additional contacts to the phosphates are mediated by the C-terminal lysine in the P-loop, Lys15, and threonine residues, Thr16 and Thr17. A magnesium ion is bound by Walker B residues. The adenine base is sandwiched, on one side by Phe182 and Arg183 and the other side by the Tyr221 side chain. Specificity for the adenine base is provided by a hydrogen bond between the ATP adenine N6 to the backbone carbonyl of Leu216 (Figure 2B). There are no contacts to the ribose hydroxyls, which project into the solvent from the binding pocket.
Figure 2.

Crystal structures of McdA. (A) Ribbon diagram of apo McdA structure with secondary structure elements labeled and the Walker A (P-loop), Walker A′ and Walker B motifs colored light purple, cyan and purple, respectively. Also labeled and shown as sticks are the side chains of residues Ser10 and Lys18. In deviant Walker A motifs both residues are lysines. This and all cartoon and ribbon diagram figures were made using PyMOL (67). (B) Ribbon diagram of the McdA-ADP structure. The Fo-Fc map calculated before addition of the ADP molecule is shown as a blue mesh and contoured at 4.3σ. Also labeled and shown as sticks are residues that contact the ADP. (C) Overlay of the McdA apo and ADP bound structures. (D) Close up of the overlaid P-loop residues in Figure 2C honing in on the ADP induced structural changes. Notable is the large rotation of residues Gly12-Gln13 and the resultant shift of the Phe182 side chain. In the apo state, the Gln13 side chain assumes the position of the ADP ribose. Upon ADP binding it is ejected from the P-loop region, with the result that the Phe182 side chain rotates and acquires a position that allows it to stack with the adenine ring of the ADP.
Superimposition of the apo and ADP bound McdA structures resulted in an rmsd of 0.8 Å indicating structural differences. Examination of the overlaid structures revealed subtle differences in the loop between β6 and α9 in the two structures, but these appear to be due to different crystal packing environments of the apo and ADP bound McdA monomers. By contrast, conformational differences not dependent upon crystal packing are observed within the nucleotide binding pocket, in particular in the P-loop, and appear triggered by ADP binding (Figure 2C). Specifically, ADP binding leads to a restructuring of the P-loop whereby the flexible glycine residues Gly11 and Gly12, undergo large rotations upon nucleotide binding to drive Gln13 from its position in the apo structure where it would clash with the ribose moiety of the bound ADP (Figure 2D). This structural change creates the phosphate binding pocket within the C-terminal region of the P-loop and also forces the side chain of Phe182 to shift to avoid clash with the repositioned Gln13 side chain. The new location of the Phe182 is then positioned for stacking with the adenine ring of ADP (Figure 2D). Thus, nucleotide docking onto McdA leads to a coordinated reconstruction of the pocket to allow ligand binding.
PISA analyses (59) of the crystal packing revealed no protein-protein interfaces consistent with a dimer or higher order oligomer for either the apo or ADP bound state; the largest interface in the crystal between proteins buried <490 Å3 of protein surface from solvent, which is far below the buried surface area observed in physiologically relevant dimers (Figure 3). The only observed cross contact between subunits was from the side chain of Asp86 (Figure 3), which interacts with the ribose hydroxyl of the adjacent dimers ADP molecule. However, sequence alignments (Figure 4) of multiple McdA homologs revealed the Asp86 is not conserved among these proteins arguing against a key role for this residue in McdA function.
Figure 3.

Comparison of McdA-ADP and McdA-AMPPNP structures. Shown side by side are the only possible dimer structures in these crystals (which bury less than 490 Å3 of protein surface from solvent). One subunit is colored cyan and the other green. The ADP and AMPPNP molecules are shown as sticks and labeled. Right, electron density for the AMPNPs, calculated before addition of the AMPPNP molecules to the structure, are shown as blue mesh and contoured at 4.3σ. Density for the ADP molecules are shown in Figure 2B. The only cross contact between subunits in the dimer is made by the Asp86 side chain.
Figure 4.

Sequence alignment of McdA homologs. Following are the codes and the source of the protein. WP_012599601, the Cyanothece sp. PCC 7424, the homolog solved in this study; WP_106458588, Aphanothece hegewaldii; REJ50457, Microcystis wesenbergii TW10; WP_073623093, Calothrix sp HK-06; WP_106866653, MULTISPECIES unclassified Cyanobacteria: WP_069073201, Nostoc sp. KVJ20; WP_035159995, Calothrix sp. 336/3; RCJ39576, Nostoc minutum NIES-26; WP_106301017, Chamaesiphon polymorphus; WP_103141075, Nostoc sp. CENA543; WP_038019401, Synechococcus sp. PCC7335; WP_100904358, Nostoc flagelliforme; WP_106866341, unclassified Cyanobacteria (miscellaneous); WP_106331468, filamentous cyanobacterium Phorm 6; WP_103325575, sp. PC7822; WP_106370190, Chlorogloea sp. CCALA 695; WP_012599601, Synechococcus elongatus PCC7942. Conserved residues among all McdAs are highlighted in yellow and indicated by an asterisk under the sequences. Residues that contact adenine nucleotides are colored magenta. Secondary structural elements, helices and strands, are indicated on top of the sequence alignments. The signature lysine position in deviant Walker A motifs is boxed (serine or threonine in McdAs) and the McdA signature lysine is colored cyan. Indicated in blue under the secondary structural elements are the Walker A, Walker A′ and Walker B motifs which are also colored as in Figure 2A.
McdA(D38A)-ATP structure reveals a nucleotide sandwich dimer with a new signature motif
Our FP data demonstrated that DNA binding by McdA requires that it be in the ATP bound state, similar to partition ParA proteins (22–35). However, whether McdA undergoes a similar dimer switch as partition ParA proteins is unclear as it does not harbor the deviant Walker A motif used by partition proteins for this purpose. To gain insight into this question we next obtained a McdA-AMPPNP structure (Figure 3). The structure was solved by molecular replacement to 2.50 Å resolution and final Rwork/Rfree values of 19.5%/22.2%. The crystal form was the same as the apo McdA structure and similarly contained one apo McdA subunit and one AMPPNP bound McdA subunit. While electron density was clear for the bound AMPPNP (Figure 3), it acted analogously to ADP, making the same set of contacts with the protein and inducing the same conformation changes in the P-loop region. But AMPPNP binding, like ADP, did not induce the formation of a McdA dimer.
Because previous studies on partition ParA proteins have shown that ATP nucleotide analogs are often not functional mimics for ATP (27,60–61), we utilized the McdA(D38A) protein, which cannot hydrolyze ATP, in crystallization trials with ATP/Mg2+. A McdA(D38A)-ATP structure was obtained to 1.7 Å resolution with final Rwork/Rfree values of 18.2%/20.4% (Materials and Methods). Again, the same interactions are made to the ATP adenine, ribose and phosphates as observed in the ADP and AMPPNP structures. However, in this structure, a tight McdA dimer is observed with a buried surface area of 1150 Å3 (Figure 5A). Strikingly, superimpositions reveal that the McdA(D38A)-ATP dimer overlays well with the nucleotide sandwich dimer forms of deviant Walker A proteins bound to ATP, such as MinD-ATP, TP228 ParA-ATP and ArsA-ATP (Figure 5B); the rmsds for these dimer superimpositions are between 2.3 and 2.5 Å, which is similar to the rmsds resulting from overlays of the single subunits of these proteins alone.
Figure 5.
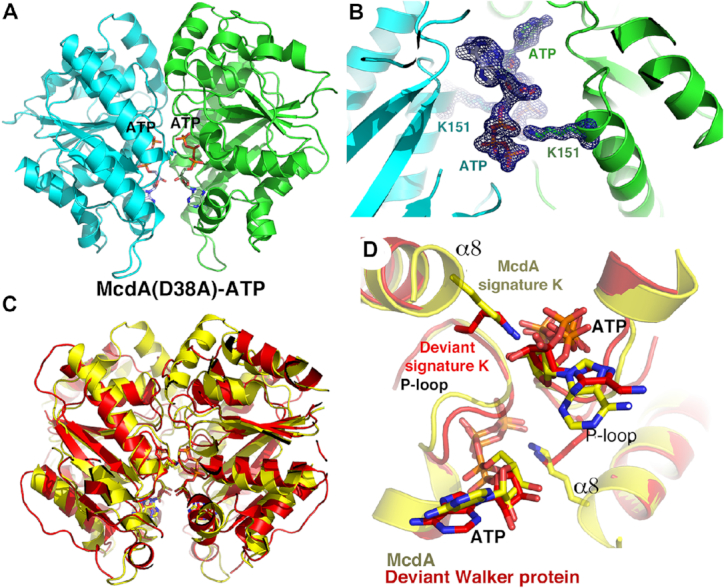
The McdA(D38A)-ATP structure reveals a nucleotide sandwich dimer that employs a novel McdA signature lysine. (A) Ribbon diagram of the McdA(D38A)-ATP nucleotide sandwich dimer. One subunit in cyan and the other green. The ATP molecules are shown as sticks and labeled. (B) Close up of the ATP binding site with the side chains of Lys151 shown. Included is an omit Fo – Fc electron density map (blue mesh) calculated after removal of both the ATP and Lys151 side chains. The map is contoured at 4.5σ. (C) Superimposition of the McdA dimer (yellow) onto the MinD-ATP dimer (pdb code: 3Q9L) structure (red). The structures superimpose with an rmsd of 2.5 for 448 corresponding Cα atoms. (D) Close up of the ATP binding pocket of the structures overlaid in Figure 4C showing how the signature lysines of the MinD Deviant Walker A motif overlays remarkably well with the McdA signature lysines, Lys151, which emanate from the N-terminus of McdA α8.
The McdA(D38A)-ATP structure, thus, reveals that the McdA protein can adopt a similar nucleotide sandwich dimer as deviant Walker A ATPases bound to ATP, despite lacking the deviant Walker A signature lysines. Examination of the structure revealed that the McdA protein employs a lysine, Lys151, outside the Walker A motif to contact the ATP bound in the adjacent subunit of the dimer. This lysine is located far from the Walker A motif, at the N-terminus of McdA α8. Remarkably, however comparison of the McdA(D38A)-ATP dimer with that of the deviant Walker A MinD-ATP structure shows that McdA residue Lys151 overlays onto the deviant signature lysine from the MinD structure, making the same cross contacts to the oxygen atom connecting the β and γ phosphates (Figure 5C and D). This oxygen is substituted by a nitrogen in AMPPNP, likely explaining why AMPPNP does not induce the dimer switch. Notably, multiple sequence alignments of McdA homologs show that the lysine corresponding the Lys151 is conserved among McdA homologs, (Figure 4). The structure thus predicts that Lys151 is important in ATP binding and formation of the nucleotide sandwich dimer. To test this structure-based prediction biochemically, we employed a fluorescently labeled ATP molecule, fluorescein-12-ATP, which has a fluorescein attached to the N7 atom of the adenine by a long linker. Modeling indicates that this molecule should be capable of binding McdA. The structure also predicts that a K151A mutation should impair but not prevent binding. FP studies revealed that the McdA(D38A) (used to prevent ATP hydrolysis during the experiment) resulted in a Kd of 0.5 ± 0.05 μM while the McdA(D38A-K151A) protein showed a significantly reduced binding affinity of 3.6 ± 0.5 μM (Supplementary Figure S3). Thus, given its role replacing the signature lysine in the deviant Walker A motifs of partition ParA proteins, we herein refer to Lys151 as the ‘McdA signature lysine’.
McdA sequence alignments also highlight other residues, in addition to Lys151, that the structures indicate as important. In particular, Phe182 and Arg183, which contact the adenine ring of the ADP/ATP substrates are also conserved in McdA homologs while Tyr221 is either a tyrosine or phenylalanine. As expected, Walker B box acidic residues are also invariant as are Walker A′ residues (Figure 4). A number of amino acids appear to be conserved because of their importance to the protein fold such as leucines, prolines and glycines. But there are several residues that point into the solvent in the apo and ADP bound states, implying a role other than structural. Strikingly, when these residues are mapped onto the McdA nucleotide sandwich dimer structure, they are observed to make contacts to the other subunit of the dimer. For example, the side chains of Glu147 contacts the ribose hydroxyl of the ATP bound in the other subunit and Arg158 packs with residues in the dimer interface (Supplementary Figure S4). Interestingly, conserved residue, Arg183, not only stacks with the adenine bound within its ATP binding pocket but also reaches over and contacts the ribose ring oxygen of the ATP molecule bound in the other subunit of the dimer. Thus, this residue simultaneously contacts both ATP molecules in the dimer. Why Trp186 and Gly188, which lie on the loop between β6 and α9, are conserved in unclear. Perhaps these residues could be involved in DNA or McdB binding. There are several basic patches on the surface of the McdA nucleotide sandwich dimer that may be involved in nucleoid interaction. But, the sequence alignment reveals that none of these residues are absolutely conserved making it difficult to predict the regions of McdA that are responsible for DNA binding. However, the strict conservation of residues that are involved in nucleotide sandwich dimerization in the McdA homologs suggests that these proteins would form the same dimer.
Crystal structure of the Cyanothece McdB protein reveals a unique fold
Our McdA structures showed that it is structurally similar to partition ParA proteins and while it is missing the signature lysine in the deviant Walker A motif, it utilizes a lysine residue outside the Walker A motif in an analogous manner. In addition, our biochemical data show that the ATP bound form binds nonspecifically to DNA. McdB proteins, by contrast, show no homology to any partition ParB protein or, in fact, any previously structurally characterized protein. Thus, to gain insight into this protein we crystallized the Cyanothece McdB and solved its structure by SAD. The structure was refined to final Rwork/Rfree values of 22.1%/26.7% (Table 2). Each McdB protomer is comprised of an N-terminal strand followed by three α-helices. Notably, helix 3 forms a long coiled coil, which mediates tight dimerization. There are four McdB subunits in the crystal ASU and they form the same coiled coil dimers. Each McdB subunit has the following topology: Strand (residues 77–83)-α1(90–102)-α2(102–120)-α3 coiled coil (122–156) (Figure 6A). Supporting that the McdB dimer is physiologically relevant, its formation by the coiled coil interaction buries a large 2781 Å2 of protein surface from solvent. The McdB structure appears to be unique; Dali searches failed to uncover structures in the database that showed strong homology to the McdB structure. Interestingly, only one protein family, the high mobility group (HMG) of DNA binding proteins from eukaryotes, showed homology, albeit weak (rmsd for 120 Cα atoms of 4.4 Å for 4QR9) (62–64) (Supplementary Figure S5). HMG domains have a similar three helical arrangement as each subunit of McdB, but they are monomeric and unlike the HMG proteins McdB does not bind DNA.
Figure 6.
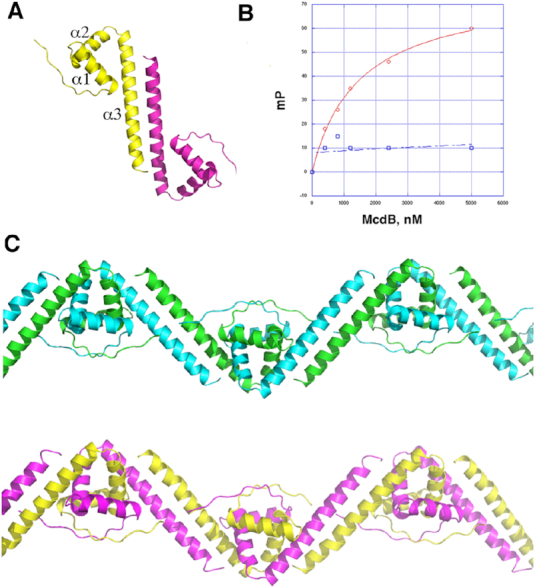
Crystal structure of Cyanothece McdB protein. (A) Structure of the McdB dimer. One subunit is colored yellow and the other magenta and secondary structural elements are labeled. (B) FP binding curves examining the ability of WT McdB (red) to bind to a preformed McdA(D38A)–DNA complex (as in Figure 1D) and the McdB truncation mutant (with residues 1–55 truncated) (blue). The y-axis is in mP and the x-axis is the concentration of the McdB protein. (C) McdB dimers form order oligomers in the crystal structure. Shown are the two McdB oligomers in the crystallographic asymmetric unit, which are formed via interdigitation of helices 1 and 2 of each subunit with helices 1 and 2 of an adjacent subunit.
As noted, although McdB proteins are similar in length, they show limited sequence homology. However, analysis of McdB sequences with the program Coils (65) revealed that they are all predicted to contain coiled coils in their C-terminal region, which is the most conserved region among the proteins (Figure 7). Thus, McdB homologs likely all harbor coiled coil C-terminal domains but, like partition ParB proteins, may have differences in the remainder of their structures. The N-terminal region of the Cyanothece McdB, from resides 1–76, is disordered in the four subunits and only contain a few conserved residues (Figure 7). Interestingly, partition ParBs also have N-terminal disordered regions attached to their centromere DNA binding domains. These N-terminal ParB domains bind ParA proteins and a TP228 ParA-ATP-ParB N-terminal domain structure showed that the ParB N-terminal region folds upon ParA binding (32,33). Removal of the ParB N-terminal domain caused disruption of the ParB-ParA interaction (33). Thus, to test the possibility that the N-terminal region of McdB may similarly interact with its McdA partner protein, we truncated the first 55 residues of the Cyanothece McdB and performed FP studies. These experiments revealed that this McdB truncation, indeed, significantly impaired McdA binding (Figure 6B).
Figure 7.

Sequence alignment of McdB homologs. Following are the codes and the source of the protein. WP_012599602, the Cyanothece sp. PCC 7424 McdB protein, the homolog solved in this study; WP_106458589, Aphanothece hegewaldii; WP_123230018, Microcystis aeruginosa; WP_045356818, Microcystis aeruginosa; WP_103672767, Microcystis aeruginosa: WP_035985573, Leptolyngbya sp. KIOST-1; WP_073607357, Phormidium tenue; WP_039728201, Lyngbya confervoides; WP_023075037, Leptolyngbya sp. Heron Island J; WP_017292327, Leptolyngbya boryana. Residues that are completely conserved are highlighted in yellow and indicated by an asterisk under the sequence. Secondary structural elements obtained from the Cyanothece McdB structure are indicated over the sequences and labeled. The N-terminal disordered region is indicated in red.
Partition ParB proteins form oligomers and the crystal packing of McdB structure revealed that the McdB dimers combine to form higher order oligomers (Figure 6C). In these interactions, McdB helices 1 and 2 intercalate with helices 1 and 2 from a neighboring molecule to form an extended oligomer. Notably, residues that are conserved in McdB homologs cluster at these interfaces (Figure 7; Supplementary Figure S6A and B). The formation of higher order oligomers by partition ParB proteins allows them to function as stable scaffolds to bind their cargo DNA as well as to engage with the ParA motor proteins (14,19,42,43). The McdB oligomers might act in a similar fashion. However, to assess the oligomeric state of McdB in solution we employed size exclusion chromatography (SEC). The SEC analysis was performed on the his-tagged McdB (MW ∼ 19 kDa). The SEC experiment revealed a peak for McdB that ran at a calculated MW of 80 kDa, which would correspond to the MW of a McdB tetramer. The SEC profile, however, revealed a bump near the void volume, where McdB protein was found, consistent with the formation of higher order (>110 kDa) oligomers (Supplementary Figure S7A and B). Because coiled coil proteins are not globular, they tend to run anomalously in SEC. But these result suggest McdB exists as higher order oligomers in solution. To provide another assessment of possible McdB oligomer formation, we performed glutaraldehyde crosslinking. We previously employed this method to assess the molecular weight of the RacA protein, which forms dimers and a few tetramers, consistent with our structural and biochemical data (Supplementary Figure S7C) (66). Using this same method we found that the McdB was crosslinked to higher order oligomers in solution (Supplementary Figure S7D). Further studies, however, will be needed to address the specific oligomer formation by McdB proteins.
In conclusion, we describe the first structures of Mcd proteins. These structures revealed a newly described lysine signature motif in the McdA protein that is used in a similar way to the signature lysine present in deviant Walker A P-loops to form a nucleotide sandwich dimer. We also show that McdA binds DNA in an ATP dependent manner, supporting the notion that, also like partition ParAs, McdA proteins bind and use the nucleoid DNA for cargo transport. By contrast, the McdB has a novel fold, completely distinct from partition ParB proteins. It appears to share some features with ParBs, such as the ability to form higher order oligomers and the presence of an N-terminal region that binds its partner protein. These structures of Mcd proteins can serve as a foundation for future studies aimed at achieving a molecular understanding of BMC cargo segregation.
DATA AVAILABILITY
Coordinates and structure factor amplitudes for the McdA and McdB structures have been deposited with the Protein Data Bank under the Accession codes 6NON, 6NOO, 6NOP and 6NOY.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Advanced Light Source (ALS) and their support staff. The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, and Material Science Division of the US Department of Energy at the Lawrence Berkeley National Laboratory. We would like to thank Dr. Anthony Vecchiarelli for initial discussions and the Schumacher lab for support and discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Nanaline H. Duke endowed professorship (to M.A.S.); US National Institutes of Health (NIH) [R35GM130290 to M.A.S.]. The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Axen S.D., Erbilgin O., Kerfeld C.A.. A taxonomy of bacterial microcompartment loci constructed by a novel scoring method. PLoS Comput. Biol. 2014; 10:e1003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kerfeld C.A., Sawaya M.R., Tanaka S., Nguyen C.V., Phillips M., Beeby M., Yeates T.O.. Protein structures forming the shell of primitive bacterial organelles. Science. 2005; 309:936–938. [DOI] [PubMed] [Google Scholar]
- 3. Rae B.D., Long B.M., Badger M.R., Price G.D.. Structural determinants of the outer shell of β-carboxysomes in Synechococcus elongatus PCC 7942: Roles for CcmK2, K3-K4,CcmO and CcmL. PLoS One. 2012; 7:e43871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rae B.D., Long B.M., Badger M.R., Price G.D.. Functions, compositions and evolution of the two types of carboxysomes: polyhedral microcompartments that facilitate CO2 fixation in cyanobacteria and some proteobacteria. Microbiol. Mol. Biol. Rev. 2013; 77:357–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turmo A., Gonzalez-Esquer C.R., Kerfeld C.A.. Carboxysomes: metabolic models for CO2 fixation. FEMS Microbiol. Lett. 2017; 364:doi:10.1093/femsle/fnx176. [DOI] [PubMed] [Google Scholar]
- 6. Kerfeld C.A., Melnicki M.R.. Assembly, function and evolution of cyanobacterial carboxysomes. Curr. Opin. Plant Biol. 2016; 31:66–75. [DOI] [PubMed] [Google Scholar]
- 7. Aussignargues C., Paasch B.C., Gonzalez-Esquer R., Erbilgin O., Kerfeld C.A.. Bacterial microcompartment assembly: the key role of encapsulation peptides. Commun. Integr. Biol. 2015; 23:e1039755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cornejo E., Abreu N., Komeili A.. Compartmentalization and organelle formation in bacteria. Curr. Opin. Cell Biol. 2014; 26:132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Savage D.F., Afonso B., Chen A.H., Silver P.A.. Spatially ordered dynamics of the bacterial carbon fixation machinery. Science. 2010; 327:1258–1261. [DOI] [PubMed] [Google Scholar]
- 10. MacCready J.S., Hakim P., Young E.J., Hu L., Liu J., Osteryoung K.W., Vecchiarelli A.G., Ducat D.C.. Protein gradients on the nucleoid position the catbon-fixing organelles of cyanobacteria. Elife. 2018; 7:e39723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerdes K., Møller-Jensen J., Bugge Jensen R.. Plasmid and chromosome partitioning: surprises from phylogeny. Mol. Microbiol. 2000; 37:455–466. [DOI] [PubMed] [Google Scholar]
- 12. Møller-Jensen J., Borch J., Dam M., Jensen R.B., Roepstorff P., Gerdes K.. Bacterial mitosis: ParM of plasmid R1 moves plasmid DNA by an actin-like insertional polymerization mechanism. Mol. Cell. 2003; 12:1477–1487. [DOI] [PubMed] [Google Scholar]
- 13. Garner E.C., Campbell C.S., Weibel D.B., Mullins R.D.. Reconstitution of DNA segregation driven by assembly of a prokaryotic actin homolog. Science. 2007; 315:1270–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schumacher M.A., Glover T., Brzoska A.J., Jensen S.O., Dunham T.D., Skurray R.A., Firth N.. Segrosome structure revealed by a complex of ParR with centromere DNA. Nature. 2007; 450:1268–1271. [DOI] [PubMed] [Google Scholar]
- 15. Ni L., Xu W., Kumaraswami M., Schumacher M.A.. Plasmid protein TubR uses a distinct mode of HTH-DNA binding and recruits the prokaryotic tubulin homolog TubZ to effect DNA partition. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11763–11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gayathri P., Fujii T., Møller-Jensen J., van den Ent F., Namba K., Löwe J.. A bipolar spindle of antiparallel ParM filaments drives bacterial plasmid segregation. Science. 2012; 338:1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gerdes K., Howard M., Szardenings F.. Pushing and pulling in prokaryotic DNA segregation. Cell. 2010; 141:927–942. [DOI] [PubMed] [Google Scholar]
- 18. Bharat T.A., Murshudov G.N., Sachse C., Löwe J.. Structures of actin-like ParM filaments show architecture of plasmid-segregating spindles. Nature. 2015; 523:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schumacher M.A. Bacterial plasmid partition machinery: a minimalist approach to survival. Curr. Opin. Struct. Biol. 2012; 22:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fink G., Löwe J.. Reconstitution of a prokaryotic minus end-tracking system using TubRC centromeric complexes and tubulin-like protein TubZ filaments. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:1845–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barillà D. Driving apart and segregating genomes in Archaea. Trends Microbiol. 2016; 24:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schumacher M.A., Tonthat N.K., Lee J., Rodriguez-Castaneda F.A., Chinnam N.B., Kalliomaa-Sanford A.K., Ng I.W., Barge M.T., Shaw P.L., Barillà D.. Structures of archaeal DNA segregation machinery reveal bacterial and eukaryotic linkages. Science. 2015; 349:1120–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bouet J.Y., Ah-Seng Y., Benmeradi N., Lane D.. Polymerization of SopA partition ATPase: regulation by DNA binding and SopB. Mol. Microbiol. 2007; 63:468–481. [DOI] [PubMed] [Google Scholar]
- 24. Castaing J.P., Bouet J.Y., Lane D.F. Plasmid partition depends on interaction of SopA with non-specific DNA. Mol. Microbiol. 2008; 70:1000–1011. [DOI] [PubMed] [Google Scholar]
- 25. Vecchiarelli A.G., Han Y.W., Tan X., Mizuuchi M., Ghirlando R., Biertümpfel C., Funnell B.E., Mizuuchi K.. ATP control of dynamic P1 ParA-DNA interactions: a key role for the nucleoid in plasmid partition. Mol. Microbiol. 2010; 78:78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vecchiarelli A.G., Mizuuchi K., Funnell B.E.. Surfing biological surfaces: exploiting the nucleoid for partition and transport in bacteria. Mol. Microbiol. 2012; 86:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vecchiarelli A.G., Havey J.C., Ing L.L., Wong E.O., Waples W.G., Funnell B.E.. Dissection of the ATPase active site of P1 ParA reveals multiple active forms essential for plasmid partition. J. Biol. Chem. 2013; 288:17823–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chu C.H., Yen C.Y., Chen B.W., Lin M.G., Wang L.H., Tang K.Z., Hsiao C.D., Sun Y.J.. Crystal structures of HpSoj-DNA complexes and the nucleoid-adaptor complex formation in chromosome segregation. Nucleic Acids Res. 2019; 47:2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vecchiarelli A.G., Neuman K.C., Mizuuchi K.. A propagating ATPase gradient drives transport of surface-confined cellular cargo. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:4880–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vecchiarelli A.G., Taylor J.A., Mizuuchi K.. Reconstituting ParA/ParB-mediated transport of cargo DNA. Methods Cell Biol. 2015; 128:243–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le Gall A., Cattoni D.I., Guilhas B., Mathieu-Demazière C., Oudjedi L., Fiche J-B, Rech J., Abrahamsson S., Murray H., Bouet J.Y., Nollmann M.. Bacterial partition complexes segregate within the volume of the nucleoid. Nat. Commun. 2016; 7:12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H., Schumacher M.A.. Structures of partition protein ParA with nonspecific DNA and ParB effector reveal molecular insights into principles governing Walker-box segregation. Genes Dev. 2017; 31:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barillà D., Carmelo E., Hayes F.. The tail of the ParG DNA segregation protein remodels ParF polymers and enhances ATP hydrolysis via an arginine finger-like motif. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livny J., Yamaichi Y., Waldor M.K.. Distribution of centromere-like parS sites in bacteria: insights from comparative genomics. J. Bacteriol. 2007; 189:8693–8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hayes F., Barilla D.. The bacterial segrosome: a dynamic nucleoprotein machine for DNA trafficking and segregation. Nat. Rev. Microbiol. 2006; 4:133–143. [DOI] [PubMed] [Google Scholar]
- 36. Funnell B.E., Gagnier L.. The P1 plasmid partition complex at parS. J. Biol. Chem. 1993; 268:3616–3624. [PubMed] [Google Scholar]
- 37. Hayes F., Austin S.. Topological scanning of the P1 plasmid partition site. J. Mol. Biol. 1994; 243:190–198. [DOI] [PubMed] [Google Scholar]
- 38. Lin D.C., Grossman A.D.. Identification and characterization of a bacterial chromosome partitioning site. Cell. 1998; 92:675–685. [DOI] [PubMed] [Google Scholar]
- 39. Pillet F., Sanchez A., Lane D., Anton Leberre V., Bouet J.Y.. Centromere binding specificity in assembly of the F plasmid partition complex. Nucleic Acids Res. 2011; 39:7477–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bartosik A.A., Jagura-Burdzy G.. Bacterial chromosome segregation. Acta. Biochim. Pol. 2005; 52:1–34. [PubMed] [Google Scholar]
- 41. Schumacher M.A., Funnell B.E.. Structures of ParB bound to DNA reveal mechanism of partition complex formation. Nature. 2005; 438:516–519. [DOI] [PubMed] [Google Scholar]
- 42. Schumacher M.A., Piro K.M., Xu W.. Insights into plasmid DNA segregation revealed by structures of SopB and SopB-DNA complexes. Nucleic Acid Res. 2010; 38:4514–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fisher G.L.M., Pastrana C.L., Higman V.A., Koh A., Taylor J.A., Butterer A., Craggs T., Sobott F., Murray H., Crump M.P. et al.. The structural basis for dynamic DNA binding and bridging interactions which condense the bacterial centromere. eLife. 2017; 6:e28086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanchez A., Cattoni D.L., Walter J-G., Rech J., Parmeggiani A., Nollmann M., Bouet J.Y.. Stochastic self-assembly of ParB proteins builds the bacterial DNA segregation apparatus. Cell Syst. 2015; 1:163–173. [DOI] [PubMed] [Google Scholar]
- 45. Ah-Seng Y., Rech J., Lane D., Bouet J.Y.. Defining the role of ATP hydrolysis in mitotic segregation of bacterial plasmids. PLoS Genet. 2013; 9:e1003956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schumacher M.A., Ye Q., Barge M.T., Zampini M., Barillà D., Hayes F.. Structural mechanism of ATP induced polymerization of the partition factor ParF: implications for DNA segregation. J. Biol. Chem. 2012; 287:26146–22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leonard T.A., Butler P.J., Lowe J.. Bacterial chromosome segregation: structure and DNA binding of the Soj dimer-a conserved biological switch. EMBO J. 2005; 24:270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koonin E.V. A superfamily of ATPases with diverse functions containing either classical or deviant ATP-binding motif. J. Mol. Biol. 1993; 229:1165–1174. [DOI] [PubMed] [Google Scholar]
- 49. Lutkenhaus J., Sunaramoorthty M.. MinD and role of the deviant Walker A motif, dimerization and membrane binding in oscillation. Mol. Microbiol. 2003; 48:295–303. [DOI] [PubMed] [Google Scholar]
- 50. Fu H-L., Ajees A., Rosen B.P., Bhattacharjee H.. Role of signature lysines in the deviant Walker A motifs of the ArsA ATPase. Biochemistry. 2010; 49:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Geordiadis M.M., Komiya H., Chakrabarti P., Woo D., Kornuc J.J., Rees D.C.. Crysallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science. 1992; 257:1653–1659. [DOI] [PubMed] [Google Scholar]
- 52. Schindelin H., Kisker C., Schlessman J.L., Howard J.B., Rees D.C.. Structure of ADPxAlF4(-)-stabilized nitrogenase and its implications for signal transduction. Nature. 1997; 387:370–376. [DOI] [PubMed] [Google Scholar]
- 53. Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997; 276:523–530. [PubMed] [Google Scholar]
- 54. Leslie A.G. The integration of macromolecule diffraction data. Acta Crystallogr. D. Biol. Crystallogr. 2006; 26:48–57. [DOI] [PubMed] [Google Scholar]
- 55. Potterton E., Briggs P., Turkenburg M., Dodson E.. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D. Biol. Crystallogr. 2003; 59:1131–1137. [DOI] [PubMed] [Google Scholar]
- 56. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.M., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Phyton-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jones T.A., Zou J-Y., Cowan W., Kjeldgaard M.. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991; A47:110–119. [DOI] [PubMed] [Google Scholar]
- 58. Lundblad J.R., Laurance M., Goodman R.H.. Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol. Endocrinol. 1996; 10:607–612. [DOI] [PubMed] [Google Scholar]
- 59. Krissinel E., Hendrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 60. Dunham T.D., Xu W., Funnell B.E., Schumacher M.A.. Structural basis for ADP-mediated transcriptional regulation by P1 and P7 ParA. EMBO J. 2009; 28:1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Davey M.J., Funnell B.E.. The P1 plasmid partition protein ParA. A role for ATP in site-specific DNA binding. J. Biol. Chem. 1994; 269:29908–29913. [PubMed] [Google Scholar]
- 62. Malarkey C.S., Churchill M.E.. The high mobility group box: the ultimate utility player of a cell. Trends Biochem. Sci. 2012; 37:553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baxevanis A.D., Landsman D.. The HMG-1 box protein family: classification and functional relationships. Nucleic Acids Res. 1995; 23:1604–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stros M., Launholt D., Grasser K.D.. The HMG-box: a versatile protein domain occuring in a wide varitey of DNA-binding proteins. Cell. Mol. Life Sci. 2007; 64:2590–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lupas A., Van Dyke M., Stock J.. Predicting coiled coils from protein sequences. Science. 1991; 252:1162–1164. [DOI] [PubMed] [Google Scholar]
- 66. Schumacher M.A., Lee J., Zeng W.. Molecular insights into DNA binding and anchoring by the Bacillus subtilis sporulation kinetochore-like RacA protein. Nucleic Acids Res. 2016; 44:5438–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Delano W.L. The PyMOL Molecular Graphics System. 2002; San Carlos: DeLano Scientific. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factor amplitudes for the McdA and McdB structures have been deposited with the Protein Data Bank under the Accession codes 6NON, 6NOO, 6NOP and 6NOY.