


Abstract
Inspired by DNA mimic proteins, we have introduced aromatic foldamers bearing phosphonate groups as synthetic mimics of the charge surface of B-DNA and competitive inhibitors of some therapeutically relevant DNA-binding enzymes: the human DNA Topoisomerase 1 (Top1) and the human HIV-1 integrase (HIV-1 IN). We now report on variants of these anionic foldamers bearing carboxylates instead of phosphonates. Several new monomers have been synthesized with protecting groups suitable for solid phase synthesis (SPS). Six hexadecaamides have been prepared using SPS. Proof of their resemblance to B-DNA was brought by the first crystal structure of one of these DNA-mimic foldamers in its polyanionic form. While some of the foldamers were found to be as active as, or even more active than, the original phosphonate oligomers, others had no activity at all or could even stimulate enzyme activity in vitro. Some foldamers were found to have differential inhibitory effects on the two enzymes. These results demonstrate a strong dependence of inhibitory activity on foldamer structure and charge distribution. They open broad avenues for the development of new classes of derivatives that could inhibit the interaction of specific proteins with their DNA target thereby influencing the cellular pathways in which they are involved.
INTRODUCTION
During the last 25 years, chemists have investigated synthetic DNA analogues with Watson–Crick base-pairing abilities. These DNA mimics are generally composed of the four natural nucleobases and of a backbone different from natural deoxyribose-phosphate (1–3). They thus resemble DNA but also feature essential differences that can result in improved behaviors, which represent a fundamental principle in molecular mimicry. For instance, peptide nucleic acids (4–7) (PNAs) and locked nucleic acids (8–10) (LNAs) can bind to complementary DNA sequences better than DNA itself and have therefore been developed to interfere with nucleic acid base-pairing in biological contexts (5,11–13). This vast field focuses on one part only of the nucleic acid interactome: their interactions with other nucleic acids.
Another part of the nucleic acid interactome lies with the recognition of their surface by proteins. Protein-nucleic acid interactions are at the core of multiple cellular processes and provide numerous opportunities for therapeutic intervention. Inhibitors of protein-nucleic acid interactions include selective DNA ligands (14–16), ‘interfacial’ inhibitors that bind the protein-nucleic acid interface (17–20), and nucleic acids themselves (21–23). However, until recently, synthetic DNA analogues that would reproduce the surface features of double stranded DNA (instead of its base-pairing ability) in order to bind to DNA-binding proteins and eventually achieve competitive inhibition of protein-DNA interactions have rarely been considered (24). Yet some naturally occurring proteins called DNA mimic proteins (25–32) do exactly that by displaying helical arrays of anionic amino acid residues (Figure 1G, H), thus hinting at the development of artificial systems with similar properties.
Figure 1.

DNA mimics design. (A) Formulae of amino acid monomers QPho, mQPho and Q5Pho. (B) Formulae of mQPhoQPho and mQPhoQ5Pho repeat units. (C–E) Top views and side views of molecular models of the structures of: (C) an eight-base-pair B-DNA; (D) (mQPhoQPho)8 and (E) (mQPhoQ5Pho)8. Phosphorus atoms are shown as large spheres. The same color code is used in (D) and (E) as in the formulae shown in (A). (F–I) Surface rendering showing negative charges (in red) of the structures of B-DNA (F), DNA mimic proteins TAFII230 (G) and HI1450 (H) and (mQPhoQ5Pho)8 (I). All structures are shown at the same scale. The structures of B-DNA, HI1450 (PDB# 1NNV) (28) and TAFII230 (PDB# 1TBA) (29) are derived from crystal data. The structure of (mQPhoQ5Pho)8 is an energy minimized model.
Along this line, we reported foldamer-based DNA analogues that mimic the negative charge surface of double-stranded B-DNA (33). Oligoamides composed of an alternation of 8-amino-2-quinolinecarboxylic acid (QPho, Q5Pho, Figure 1A) and 8-aminomethyl-2-quinolinecarboxylic acid (mQPho) adopt stable single helical conformations at the surface of which phosphonate side chains form a double helical array that matches the positions of phosphates in B-DNA. This resemblance opened the prospect to bind some non-sequence selective DNA-binding proteins, i.e. proteins that mainly recognize the shape and charge distribution of nucleic acids. The anionic foldamers eventually proved to perform much better than anticipated in that they actually inhibited several DNA-binding enzymes: the foldamers bind to these enzymes better than the DNA substrate itself. In particular, we observed unprecedented inhibitory activity for two therapeutically relevant enzymes: the human Topoisomerase 1 (Top1) (34) which mediates the relaxation of supercoiled DNA, and the human immunodeficiency virus 1 integrase (HIV-1 IN) (35) which catalyzes the insertion of the HIV-1 DNA formed after reverse transcription into the human genome. Inhibition by 32 unit-long foldamers − the equivalent of sixteen DNA base pairs − occurred at sub-micromolar concentrations, matching or even exceeding the performance of the best inhibitors of these enzymes, camptothecin (36) for Top1 and raltegravir (37) for HIV-1 IN, and offering access to a novel mechanism of inhibition. Indeed, camptothecin and raltegravir typically block the DNA-protein complex (19,20), whereas the foldamers compete with the DNA substrate. In contrast, other DNA-binding enzymes were inhibited to a lower extent (Topoisomerase 2, Flap endonuclease 1) or not inhibited at all (deoxyribonuclase 1, S1 nuclease, benzonase®) (33).
These discoveries called for further developments in particular regarding the structural basis of foldamer-mediated enzyme inhibition. It should be noted that our initial efforts aimed at making the foldamers resemble B-DNA as closely as possible, but that their inhibitory activity emerged precisely because of differences from the DNA structure: a ‘perfect’ mimic would not be more active as DNA itself, which is a rather poor inhibitor of Top1 and HIV-1 IN. Thus, phosphonates were selected as anion bearing functionalities and Q5Pho was introduced in (mQQ5Pho)n sequences because it confers a smaller minor groove and a larger major groove to the mimics that match the grooves of DNA better than QPho in (mQQPho)n sequences (Figure 1B–E) (33). However, the need for all these features to achieve inhibition was not evidenced. For instance, DNA mimic proteins have a less obvious resemblance to B-DNA (Figure 1G, H) (24,30,31), and possess no phosphate or phosphonate anions since negative charges are borne by aspartate and glutamate carboxylate functions. Furthermore, an intriguing and essential aspect concerns selective enzyme inhibition: can two enzymes be differentially inhibited by a given DNA mimic even though they act on the same kind of double-stranded DNA substrate?
The initial objectives of the current study were thus multiple: (i) to develop a solid phase synthesis of foldamer-based DNA mimics as a more suitable approach to the preparation of multiple variants than the initial segment doubling solution phase strategy (33); (ii) to assess the ability of carboxylic acid functionalized foldamers inspired from DNA mimic proteins to inhibit Top1 and HIV-1 IN; and (iii) to assess the dependence of enzyme inhibition on the length and position of the side chains. In the following, we report the synthesis and characterization of several new monomers and hexadecameric oligomers, including, in one case, crystallographic structural evidence of the anionic form. We discovered that carboxylate side chains can impart enhanced inhibitory activity in some cases, or no inhibition in others, or even cause enzyme activation. Importantly, we found a sequence that has differential inhibitory effects on the two enzymes. Altogether, these results further validate the novel concept of foldamer-mediated DNA surface mimicry. The variety of DNA-binding enzymes and the feasibility of foldamer modifications augur well for the development of this approach and open new avenues in molecular mimicry for protein surface recognition.
MATERIALS AND METHODS
Chemicals and reagents were used as commercially supplied without any further purification unless otherwise stated. Low loading Wang resin (0.38 mmol/g) was purchased from Novabiochem. Ghosez reagent was purchased from Sigma Aldrich. N,N-diisopropylethylamine (DIPEA) was distilled over calcium hydride. Anhydrous THF and CH2Cl2 for solid phase synthesis were dispensed from an MBRAUN SPS-800 solvent purification system. Reactions requiring anhydrous conditions were performed under nitrogen atmosphere. Melting points were determined using a Buchi B-540 melting point apparatus.
Synthesis
The detailed synthesis and characterization of monomers and oligomers are described in the Supplementary Information.
Nuclear magnetic resonance
NMR spectra were recorded on an Avance II NMR spectrometer (Bruker Biospin) with a vertical 7.05T narrow-bore/ultrashield magnet operating at 300 MHz for 1H observation, 75 MHz for 13C observation and 121.4 MHz for 31P observation by means of a 5-mm direct BBO H/X probe with Z gradient capabilities. Chemical shifts are reported in ppm and are referenced against residual solvent signals of CDCl3 (δH: 7.26, δC: 77.0), DMSO-d6 (δH: 2.50, δC: 39.4), HDO (δH: 4.8). 31P NMR signals are referenced to PPh3O at 27 ppm. Peak multiplicities are noted as follows: singlet (s), broad singlet (bs), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), doublet of quartets (dq) and multiplet (m). Data processing was performed with Bruker TOPSPIN 2.1 software.
RP-HPLC analysis and purification
RP-HPLC quality acetonitrile and MilliQ water were used for RP-HPLC analyses and purification.
HPLC analyses of the oligomers were performed on a JASCO AS-2055 chromatography system equipped with a reverse-phase Varian Pursuit C18 column (250 × 4.6 mm, 5 μm) and a multichannel UV/VIS detector JASCO UV-2077. Oligomers were analysed using a linear gradient of 15 min starting from 0% of B and 100% of A to 20% of B and 80% of A with a flow of 1 ml/min. Solvents A and B were prepared as follows: a stock 50 mM aqueous triethyl-ammonium acetate buffer solution at pH 8.7 was prepared by dissolving 3 ml of glacial acetic acid in 950 ml water and, while mixing, adding freshly distilled triethylamine (6.5 ml). After adjusting the pH to 8.7 with triethylamine, the volume was finally adjusted to 1 l with water. Solvent A was prepared by dilution of the stock buffer solution with water 1:3 (vol/vol) to a final concentration of 12.5 mM triethyl-ammonium acetate, pH 8.7, in water. Solvent B was prepared by dilution of the stock buffer solution with acetonitrile 1:3 (vol/vol) to a final composition of 12.5 mM triethyl-ammonium acetate, pH 8.7. Semi-preparative purification of oligomers was performed on a semi-preparative HPLC using a Varian Pursuit C18 column (250 × 15 mm, 5 μm) with a flow of 4 ml/min. Oligomers were purified using linear gradients starting from 95% of A and 5% of B to different ratios of A/B which are stated for each oligomer in the Supplementary Information. Monitoring by UV detection was carried out at 254 or 300 nm using a diode array detector. Monomers were analysed with a Nucleodur C8 column (120 × 4.6 mm, 5 μm) by using a linear gradient of 20 min from 30% of D and 70% of C to 100% of D (where solvent C was H2O + 0.1% TFA and solvent D was ACN + 0.1% TFA) with a flow of 1 ml/min.
High resolution mass spectrometry analysis and exact mass measurements
Detection of compounds was performed using a thermo Orbitrap Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, USA). Electrospray ionization source (HESI-II) was used for ionization of the target compounds in positive and negative ion modes. Instrument calibration in positive mode and negative mode were done prior to sample injection (ion calibration solution 88323 and 88324, for positive and negative ions, respectively, Thermo Scientific Pierce). The tuning parameters were set to avoid fragmentation and keep ions intact as follows: ESI voltage, +3.3 kV/−3.0 kV; sheath gas pressure, 30.0 arbitrary units and capillary temperature, 373 K. The RF of the ion guides were optimized to obtained the maximum transmission efficiencies. For the compounds of interest, a scan range of m/z 500–4000 was chosen. The automatic gain control (AGC) target for a maximum capacity in C-trap was set at 2 × 106 ions for a maximum injection time of 250 ms.
Crystallization
For crystallization experiments, a lyophilized powder of 4 was dissolved in ultra-pure water and ammonium bicarbonate. Since oligomer 4 does not possess stereogenic centres, it was expected to fold as a racemic mixture of right-handed (P) and left-handed (M) helices. Final concentration of the racemic solution to start crystallization screening was 8 mg/ml. Crystallization trials were performed using standard aqueous hanging drop vapor diffusion methods (38) in 24-well Linbro-style plates, at 293 K. Screening of crystallization conditions was carried out using commercial sparse matrix screen JBScreen Basic 1 to 4, from Jena Bioscience (39,40). For each screening condition, drops were prepared using 0.75 μl of solution of racemic 4 and an equal volume of the crystallization reagent on a silanized glass slide, which was then suspended over a reservoir solution. Clusters of small, light yellow crystals were observed after 5 days in one of the 96 screening conditions. X-ray quality crystallogenesis was optimized by increasing the drop size by equilibration of a mixture containing 1.0 μl of 8 mg/ml solution of 4 and 1.5 μl of a reservoir solution of 18% w/vol polyethylene glycol 8000, 100 mM MES buffer, pH 6.5, and 200 mM calcium acetate; against 500 μl of the reservoir solution. Crystals (Supplementary Figure S1) used for the X-ray diffraction measurements grew to a size of 0.3 × 0.05 × 0.05 mm in 10 days.
Data collection, structure solution and refinement
For low temperature X-ray diffraction measurements, a single crystal was mounted using a cryo-loop after quick soaking on Paratone-N oil and flash-frozen. Diffraction data were collected at the IECB X-ray facility (UMS 3033) on a micro-focus, rotating anode Rigaku FRX diffractometer, with Cu Kα radiation (λ = 1.5417 Å) and a hybrid pixel detector (PILATUS 200K), at 100 K. The crystal diffracted to a maximum resolution of 0.95 Å. Diffraction data were processed and scaled using the CrystalClear-SM 1.36 package (41). The symmetry of the crystal was triclinic with space group 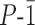 , Z’ = 1 and unit cell parameters: a, 20.85 Å; b, 21.73 Å; c, 31.33 Å; α, 83.61°; β, 75.07°; and γ, 77.15°. The structure was solved by direct method using the program SHELXT (42). The phase set calculated allowed identifying most of 4, including all side chains. The structure was refined by full-matrix least-squares method on F2 with SHELXL-2014 (43) within the Olex2 suite (44). For all non-hydrogen atoms attempts to introduce anisotropic displacement parameters were made. AFIX 116 constraints were used for the mQAc and Q5Ac atoms. In case of side chains and solvent molecules, the non-hydrogen atoms were refined with anisotropic or isotropic displacement parameters. Hydrogen atoms were included for 4 in idealized positions using HFIX and refined with a riding model. For solvent molecules, positions of hydrogen atoms were not determined. After several attempts to model the disordered water molecules, the PLATON/SQUEEZE procedure (45) was implemented to treat the regions with highly disordered solvent molecules. The total potential solvent accessible void volume was 4772.1 Å3 and the number of electron count per cell was 1361. DFIX, DELU, SIMU and ISOR instructions were used to model displacement parameters and the geometry of 4. The FVAR function was used during refinement of occupancy factors of disordered parts. The final cif file was checked using IUCR’s checkcif algorithm. The coordinates and structure factors have been deposited in the the Cambridge Crystallographic Data Centre (CCDC). The accession numbers and refinement statistics are provided in Supplementary Tables S1 and S2.
, Z’ = 1 and unit cell parameters: a, 20.85 Å; b, 21.73 Å; c, 31.33 Å; α, 83.61°; β, 75.07°; and γ, 77.15°. The structure was solved by direct method using the program SHELXT (42). The phase set calculated allowed identifying most of 4, including all side chains. The structure was refined by full-matrix least-squares method on F2 with SHELXL-2014 (43) within the Olex2 suite (44). For all non-hydrogen atoms attempts to introduce anisotropic displacement parameters were made. AFIX 116 constraints were used for the mQAc and Q5Ac atoms. In case of side chains and solvent molecules, the non-hydrogen atoms were refined with anisotropic or isotropic displacement parameters. Hydrogen atoms were included for 4 in idealized positions using HFIX and refined with a riding model. For solvent molecules, positions of hydrogen atoms were not determined. After several attempts to model the disordered water molecules, the PLATON/SQUEEZE procedure (45) was implemented to treat the regions with highly disordered solvent molecules. The total potential solvent accessible void volume was 4772.1 Å3 and the number of electron count per cell was 1361. DFIX, DELU, SIMU and ISOR instructions were used to model displacement parameters and the geometry of 4. The FVAR function was used during refinement of occupancy factors of disordered parts. The final cif file was checked using IUCR’s checkcif algorithm. The coordinates and structure factors have been deposited in the the Cambridge Crystallographic Data Centre (CCDC). The accession numbers and refinement statistics are provided in Supplementary Tables S1 and S2.
Top1-catalyzed relaxation assays
Top1 catalytic activity was assessed in plasmid DNA relaxation assays according to the specifications of the supplier (Topogen, Colombus, OH, USA) with minor modifications. For each reaction, 100 ng of (–)-pcDNA3.1 supercoiled plasmid was incubated with ∼3 ng of purified human recombinant Top1 in 1× reaction buffer (10 mM Tris–HCl, pH 7.9, 1 mM EDTA, 150 mM NaCl, 0.1% bovine serum albumin, 0.1 mM spermidine, 5% glycerol). Reactions were performed in 10 μl final volume for 15 min at room temperature and stopped by the addition of 1 μl of 5% SDS and 2 μl of BlueJuice™ Gel Loading Buffer (Life Technologies, Saint-Aubin, France). Reaction mixtures were then electrophoresed in 1% agarose gels for 2 h at 25 V in 0.5× TAE buffer. Gels were stained by incubation in 0.5× TAE solution containing 2 μg/ml ethidium bromide for 5 min and destained by incubation in fresh 0.5× TAE buffer containing 1 mM MgSO4 for 30 min. Reaction products were then visualized by UV-transillumination and quantified using the ImageJ software. Percentages of relaxed DNA (R) were normalized to reactions conducted with Top1 alone (set at 100%) and plotted as a function of foldamer concentrations. Results are the mean ± standard deviation of at least three independent experiments.
HIV-1 IN-catalyzed integration assays
HIV-1 IN was purified from bacteria using the same procedure as previously described (46). Typical concerted integration assays were carried as previously described (47) using both pBSK-Zeo target and HIV-1 U5 containing donor DNA (48). For this purpose, we used 5′-end-labeled donor DNA (10 ng), circular target DNA plasmids (50 ng) and purified IN previously diluted to 2 μM in 1 M NaCl, 20 mM HEPES pH 7, 10 mM DTT. Then 200 nM IN was incubated for 30 min on ice with 10 ng of donor DNA and 50 ng of acceptor plasmid in 5 μl final volume in the presence or lack of foldamers. Reaction was then started by adding 5 μl of the reaction buffer (final concentrations 100 nM IN, 15% DMSO, 8% PEG, 10 mM MgCl2, 20 μM ZnCl2, 100 mM NaCl, 10 mM DTT). After the reaction, integration products were loaded onto 1.4% agarose gel. The gel was then dried and autoradiography was performed. Quantification of the integration activity was performed using the ImageJ software with the following procedure: the bands corresponding to the free substrate (S), the donor/donor (d/d), linear FSI (FSI) and circular HSI + FSI (HSI + FSI) were quantified. Results are the mean ± standard deviation of at least three independent experiments.
RESULTS AND DISCUSSION
Design and synthesis
Previously described sequence 1 (Figure 2) bearing phosphonate side chains in position 4 of both Q and mQ units serves as the reference compound for this study. It was shown to have excellent inhibitory properties of both HIV-1 IN and Top1. Several notable differences exist between this compound and B-DNA. Phosphonates can be either mono- or di-anionic depending on the local environment (33). In addition, single point attachments of the side chains to the foldamer backbone allow for more flexible positioning of the anions than for doubly connected DNA phosphates. Also, contrary to B-DNA and to (mQQ5Pho)n sequences (Figure 1B, E, I), the two grooves of (mQQ4)n differ marginally in width (Figure 1B, D): the Q4-mQ connectivity differs from the mQ-Q4 connectivity by a single methylene unit. Finally, the single helical foldamer main chain may in principle be kinked upon rotation about a single bond and may thus possess larger flexibility than double stranded helical B-DNA.
Figure 2.
Formulae of DNA mimic hexadecaamides 1–6.
Sequences 2–6 (Figure 2) were designed by analogy with carboxylate functionalized DNA-mimic proteins. They represent variations of side chain nature and charge state (2, 6), side chain position (3, 4, 5), and side chain length (4, 5) with the hypothesis that longer side chains may adjust their conformation to form salt bridges or hydrogen bonds with the targeted enzyme. Although no accurate prediction could be made, we expected that at least some of these variations would modulate foldamer inhibitory activity, possibly in a way that can be correlated to structure.
For the preparation of these sequences, we anticipated that SPS would be less labor intensive than the previously reported solution phase synthesis, whose main advantage is to be easily amenable to scale up. SPS of Qn sequences bearing different proteinogenic side chains has been reported before (49–51) and we thought its adaptation to (mQQ)n sequences would not require much development. The SPS of 1–6, entailed the preparation of the nine monomers shown in Figure 3. These bear Fmoc amine main chain protection and acid labile tert-butyl ester side chain protection instead of the tert-butyl-carbamate amine protection and diethyl-phosphonate ester protection used in solution (33). All of them are new except QOAc and Q5Ac (49,50). Of note are the different ether or carbon-carbon bond main chain-side chain linkages. These do not reflect a specific design but are simply a consequence of the synthetic route followed (49,50).
Figure 3.

Formulae of Fmoc-main chain amine and tert-butyl-side chain acid protected monomers for SPS.
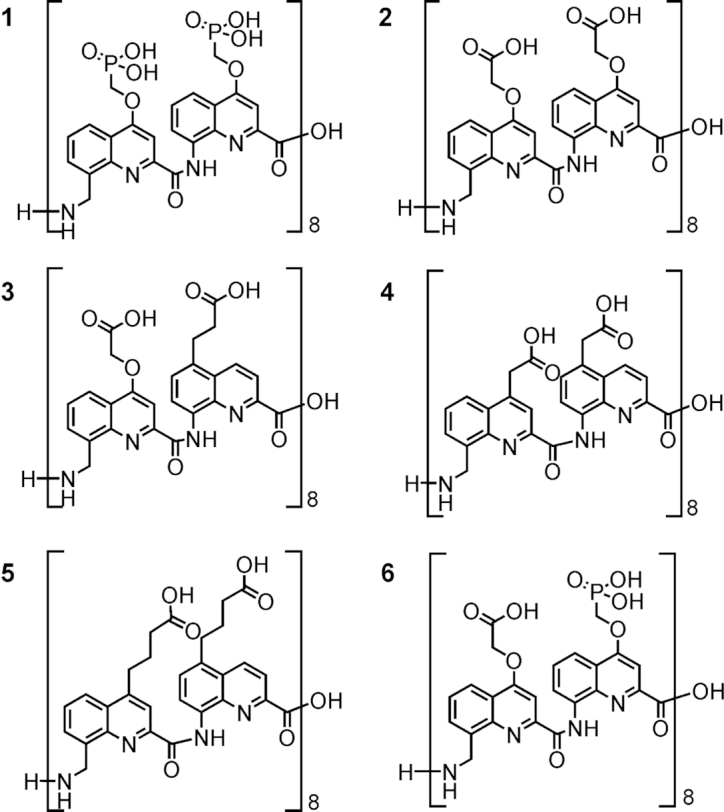
The syntheses of protected Fmoc(QPho) and Fmoc(mQPho) are shown in Figure 4 as representative examples (see also Supplementary Information). Precursors 7 and 10 were prepared on a multi gram scale following previously reported procedures (52). Side chain introduction under Mitsunobu conditions, hydrogenolysis of the benzyl ester and hydrogenation of the 8-nitro or 8-cyano group followed by installation of Fmoc and purification through silica-gel chromatography afforded to the final compounds ready to use in SPS in ca. 50% overall yield and >98% purity (RP-HPLC, Supplementary Figures S48 and S49). A crystal structure of protected monomer Fmoc(mQPho) is shown in Figure 4C.
Figure 4.
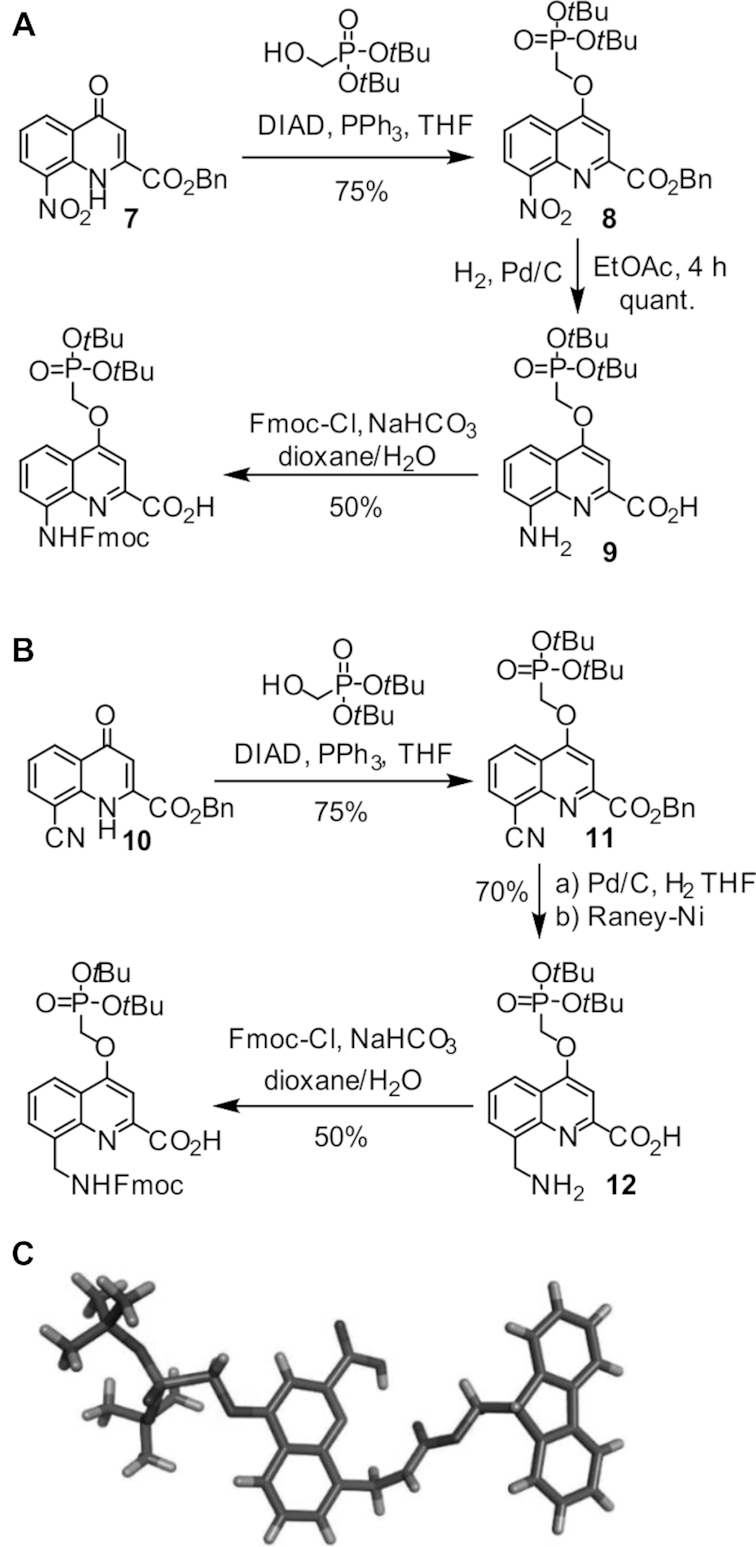
Synthetic pathway to (A) Fmoc(QPho) and (B) Fmoc(mQPho) monomers. Crystal structure of Fmoc(mQPho) monomer (C) crystallized from dichloromethane/ether. Included solvent molecules have been removed for clarity. DIAD = diisopropyl azodicarboxylate; Fmoc = fluorenylmethyloxycarbonyl.
Drawing on our experience in microwave assisted oligoamide foldamer SPS (49–51), we prepared oligomers 1–6 on a low loading Wang resin via acid chloride activation of the monomers under neutral conditions with the Ghosez reagent (1-chloro-N,N,2-trimethylpropenylamine) (53). Removal from the resin with concomitant side chain tert-butyl deprotection was performed by treatment with 95:2.5:2.5 TFA/H2O/iPr3SiH (vol/vol/vol) (TFA = trifluoroacetic acid; iPr3SiH = triisopropylsilane). The crude materials were analyzed and purified by RP-HPLC providing foldamers 1–6 in good purity (RP-HPLC purity > 95%, Supplementary Figures S43–S47). All foldamers were characterized by 1H NMR in H2O/D2O, HPLC, and mass spectrometry (see Supplementary Information). Semi-preparative RP-HPLC proved to be an efficient analytical method to purify anionic foldamers providing the sequences in high purity. As a representative example, Figure 5 shows analytical data for 4 before and after purification. As a proof of the helical folding in water, NMR spectra displayed sharp amide and aromatic protons which are distributed over a wide range of chemical shift (from 12 to 9 ppm for amide protons and from 9 to 8 ppm for aromatic protons, Figure 5C and D), in agreement to our previous reported studies (49,54).
Figure 5.

(A, B) RP-HPLC profiles of (A) crude and (B) pure oligomer 4. (C, D) Carboxamide and aromatic regions of the 1H NMR spectra of (C) crude and (D) pure oligomer 4 in H2O/D2O 9:1 (vol/vol), 50 mM NH4HCO3 at 298 K. RP-HPLC = reversed phase high-performance liquid chromatography.
Structural characterization
Structural evidence of anionic oligomer helical folding was previously established in aqueous solution by NMR investigations of an octameric sequence (33). Folding typically results in the spreading of NMR resonances over a broad range of chemical shift values despite the repetitive nature of the sequence, as can be seen in Figure 5D and in Supplementary Figures S9–S13. The sharp lines are a good indication of the absence of foldamer aggregation even at mM concentrations, as could be expected for a polyelectrolyte. Solid state evidence of the helical conformations had also been obtained by X-ray crystallographic analyses. These structures were obtained from crystals grown in organic solvents using side chain protected oligomers. However, earlier attempts to grow crystals from unprotected anionic oligomers such as 1 have been unsuccessful. We assigned this difficulty to the relative length of the phosphonate side chain, which appears to be detrimental to crystal growth from water (50). This hypothesis was verified when attempting to grow crystals of the new carboxylate-functionalized oligomers: crystal growth from water was successful, but only in the case of hexadecaamide (mQAcQ5Ac)8 which bears the shortest side chains. Crystals that diffracted at atomic resolution were obtained and the structure could be resolved in centrosymmetrical space group 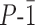 (Figure 6C and Supplementary Figure S1). The asymmetric unit contained a molecule 4 folded as a single helix with no noticeable difference from the structures obtained from organic solvents. As with other helical aromatic amides (50,54), and unlike with α-helical peptides (55,56), electrostatic repulsion between the multiple charges is not conducive of a denaturation, or even of a structural change. This is despite the fact that charges are close enough to influence their apparent pKa values, as was measured for phosphonate side chains (33).
(Figure 6C and Supplementary Figure S1). The asymmetric unit contained a molecule 4 folded as a single helix with no noticeable difference from the structures obtained from organic solvents. As with other helical aromatic amides (50,54), and unlike with α-helical peptides (55,56), electrostatic repulsion between the multiple charges is not conducive of a denaturation, or even of a structural change. This is despite the fact that charges are close enough to influence their apparent pKa values, as was measured for phosphonate side chains (33).
Figure 6.

Side views and top views shown at the same scale of: (A) crystal structure of 8-bp B-DNA duplex d(CGCTAGCG)2 (PDB ID: 250D) (70); (B) molecular overlay of B-DNA (ochre) and DNA mimic 4 (gray); (C) crystal structure of DNA mimic 4; (D) molecular overlay of DNA mimic 4 (gray) and a molecular model of (mQPhoQ5Pho)8 (ochre) and (E) a molecular model of (mQPhoQ5Pho)8. In (B) and (D), foldamers or DNA are shown in stick representations, except the side chain carboxylate carbon atoms, and the phosphonate and phosphate phosphorus atoms that are shown as spheres. Two molecules of (mQPhoQ5Pho)4 as observed in the crystal (33) have been stacked to produce the model of (mQPhoQ5Pho)8. Hydrogen atoms have been removed for clarity. In (E), ethyl ester functions at the side chains, tert-butyl-carbamate and benzyl ester functions at the N terminus and C terminus, respectively, have been removed for clarity.
The structure of 4 also contained six Ca2+ ions used as an additive in crystallization drop to screen anion repulsions between adjacent molecules (Supplementary Figures S2, S4 and S5). Divalent metal ions often serve that purpose in the crystal structures of nucleic acids (57–59) and other poly-anionic species (60).
The helically folded structure of (mQAcQ5Ac)8 matched the predicted model of (mQPhoQ5Pho)8 (Figure 6D, E) especially in the central part of the sequence, and the crystal structure of protected (mQPhoQPho)8 obtained from organic solvents (Supplementary Figure S6). It confirmed the resemblance between these oligomers and B-DNA (Figure 6A–C). In particular, there is a good match of the minor and major groove widths when side chains are in position 5 of Q units (Supplementary Figure S3). Even solid state packing was found to be analogous to the packing of related DNA structures (Supplementary Figure S5). Nevertheless, the shorter side chains of mQAc and Q5Ac are conducive of an overall diameter significantly smaller than that of B-DNA (Figure 6A–C).
Inhibition of DNA-processive enzymes
We then assessed the effect of the foldamers on the catalytic transesterification of scissile phosphodiester bonds of recombinant Top1 and HIV-1 IN using dedicated in vitro assays, as previously described (33) (Figure 7, Table 1). These enzymes share a common strand-transferase activity. Despite scant overall sequence similarity, Top1 also shares a conserved catalytic pentad in its tertiary structure with a number of other integrases (61). We compared the effect of the new variants to that of reference oligomer 1 (mQPhoQPho)8 which completely inhibits Top1-mediated DNA relaxation at 1 μM concentration (Figure 7C) and HIV-1 IN integration at 10 μM concentration (Figure 7D). The results showed that the position and nature of the anions strongly affected the inhibitory activity of the DNA mimics and that, in many cases, they did so differently for the two enzymes.
Figure 7.

Enzyme inhibition. (A, B) Representative gel electrophoresis of (A) the inhibition of Top1-mediated relaxation of supercoiled circular DNA and (B) in vitro HIV-1 IN integration (200 nM of IN) of a radio-labelled 246-base-pair (bp) viral DNA donor (10 nM) into a 2,700 bp pZeo plasmid acceptor (2.8 nM). In (A), lane 1 is DNA alone, control lane 2 is DNA + Top1 and lane 3 is the same as lane 2 in the presence of 50 μM camptothecin (CPT). The control lane 2 shows full Top1 activity. Camptothecin (CPT) is a classical Top1 inhibitor. In (B), for each foldamer, the left lane is the control lane (i.e. all things equal but without any foldamer). The position and structures of the donor substrate and different products obtained after half-site (HSI), full-site (FSI) and donor/donor integration are shown. (C, D) Quantitation of (C) Top1 inhibition expressed as a percentage of relaxation, compared to the control, and (D) in vitro HIV-1 IN integration. In (D), the circular FSI + HSI and linear FSI products were quantified on gels using the ImageJ software and the circular and linear HSI + FSI is reported as the percentage of heterointegration quantified in the absence of foldamers. Results are the means of at least triplicate experiments ± standard deviation.
Table 1.
Inhibition of Top1 and HIV-1 IN by anionic foldamers 1–6. IC50 values (concentrations of each DNA mimic that induce 50% inhibition of the catalytic activity) and standard deviations were extracted from Figure 7A and B. nd: not determined due to the lack of inhibitory activity in the range of concentration investigated
| IC50 (μM) | ||
|---|---|---|
| Top1 | HIV-1 IN | |
| 1 (mQPhoQPho)8 | 0.225 ± 0.04 | 2.0 ± 0.4 |
| 2 (mQOAcQOAc)8 | nd. | nd. (>10) |
| 3 (mQOAcQ5Prop)8 | nd. | 2.2 ± 0.2 |
| 4 (mQAcQ5Ac)8 | nd. (activation) | nd. (activation) |
| 5 (mQButQ5But)8 | 0.83 ± 0.06 | 0.80 ± 0.02 |
| 6 (mQOAcQPho)8 | 0.94 ± 0.12 | 0.08 ± 0.03 |
A first striking result was the complete inactivity of oligomer 2 for both enzymes. Considering that 2 differs from 1 only by the carboxylate vs. phosphonate nature of the anions, this result points to an essential role of this parameter. When only half of the phosphonates are replaced by carboxylates, i.e. in 6 (mQOAcQPho)8, activity was only moderately restored for Top1 (IC50 of 0.2 μM for 1 and 0.94 μM for 6). In contrast, activity was fully restored and even enhanced for HIV-1 IN (IC50 of 0.08 μM). This latter result suggests that optimal affinity arises at a given charge balance – neither not enough nor too many negative charges are desirable – and that this parameter is enzyme-dependent. Indeed, the IC50 values of 6 for the two enzymes differ by more than one order of magnitude, inhibition of HIV-1 IN being the strongest, whereas 1 inhibits Top 1 at a lower concentration than HIV-1 IN. Nevertheless, one should keep in mind that these comparisons are valid for what concerns the active concentration of 1 and 6, but are not strictly quantitative in that the enzyme concentration and the substrate nature and concentration differ in the two assays.
It is worth stressing here how remarkable is the very occurrence of strong competitive inhibition of a DNA-binding enzyme by a DNA-mimic. The assays used to assess enzyme activity are carried out in the presence of B-DNA substrates in considerably larger amounts, if one considers both their length and concentration, than the enzyme and the foldamer used for inhibition. Even though processive enzymes may not possess very high binding to DNA to allow for their scrolling along DNA sequences (to be the best of our knowledge, the exact dissociation constant for Top1 is not known and the non-Michaelis-Menten nature of HIV-1 IN, due to its multiple oligomeric active states has not allowed for the precise determination of its affinity for DNA to date) competitive inhibition can only be achieved if the foldamer binds much stronger.
A second striking and differential effect came from changing side chain position. Shifting some carboxylate side chains of inactive 2 from position 4 to position 5 of the quinoline rings, as in 3 (mQOAcQ5Prop)8, did not restore Top1 inhibition but it did restore HIV-1 IN inhibition. Thus carboxylate oligomer 3 is as active against HIV-1 IN as phosphonate oligomer 1. The effect is also observed when the carboxylate side chains are made longer. Oligomer 5 (mQButQ5But)8 inhibits HIV-1 IN slightly better than 3. It is also the only all-carboxylate oligomer that shows some inhibition of Top1 (IC50 of 0.83 μM). In addition to 6, oligomers 3 and 5 can thus also be considered to be more selective for HIV-1 IN.
Finally, a peculiar effect was observed when side chains are made shorter, as in 4. This compound did not show any inhibition. On the contrary, it activated Top1 and HIV-1 IN above 0.1 μM. The fact that it occurred with both proteins in completely different assays suggests it is not an artifact. The reduced diameter of this oligomer (Figure 6C) hints at a non-optimal fit with the proteins’ DNA binding site. Weaker binding would then not be conducive of competitive inhibition but it might nevertheless promote active conformations of the proteins for binding to DNA, resulting in an activation. Such an activation was indeed reported using short DNA duplexes (47), and might be associated with the presence of other DNA binding sites. Indeed, several DNA binding sites have been characterized in HIV-1 IN. The viral DNA binds the catalytic core domain while the target DNA binds both the catalytic core and the carboxyterminal domain which possesses a non-sequence specific DNA binding property and which is thus probably involved in foldamer binding.
The wide range of activities of the new oligomers demonstrate the feasibility of selective enzyme inhibition for two enzymes that have DNA as a common substrate. HIV-1 IN inhibition shows remarkable dependence on all the parameters varied (side chain position, length, and nature of the anion) whereas Top1 inhibition appears to depend mostly on the presence of the phosphonate ions.
Unfortunately, the lack of quantitative binding assays does not allow one to directly relate the observed inhibitions to protein-foldamer affinity. The results presented here also call for further screening of foldamer variations. For example, aromatic tyrosine-like or tryptophan-like foldamer side chains might also interact favorably with positively charged residues at the protein surface and further enhance selectivity. Higher throughput foldamer syntheses (e.g. automation of SPS), would facilitate such plans and is currently being developed.
CONCLUSION
Nucleic acid surface mimicry appears to have high potential for modulating protein binding to DNA. Yet it has been vastly overlooked until now, perhaps out of lack of suitable molecular scaffolds, and also because the focus of DNA mimicry has been centered on Watson-Crick base pairing ability (1–13). This situation contrasts with the significant advances in protein surface mimicry for the purpose of inhibiting protein-protein interactions (62–64). For example, α-helix mimicry and stabilization constitutes a field of its own in which foldamer-based approaches prove to be successful (65–69). The successes met in protein epitope mimicry may thus be a source of inspiration for nucleic acid surface mimicry. Along this line, the results presented above consolidate our claim that helically folded aromatic oligoamides are valid and original scaffolds to reproduce some of the charge surface features of B-DNA. The identification of foldamer side chain patterns that elicit selective inhibition properties, that is, HIV-1 IN versus Top1, is an important milestone. This is especially true considering two proteins that act on a similar B-DNA substrate, i.e. at the exclusion of a well-defined DNA sequence binding site. More generally, the strong dependence of inhibition on the spatial distribution and nature of anionic side chains point to the existence of protein-specific foldamer-protein interactions, and thus to the possibility of further tailoring activity and selectivity. Next steps will be to determine the structural requirements for these foldamers to achieve stronger and more selective inhibition, a process that would require the use of crystal structure data (34,35) as well as molecular modelling. Targeting Top1 or HIV-1 IN by a new mechanism of action, namely competitive inhibition, using such foldamers would represent an alternative to existing drugs that are used in the clinic to poison these two recombinases in the treatment of cancer or AIDS, respectively. Such an alternative might help counteracting drug resistance that develops in both pathologies. In the long term, our results also point out towards the use of foldamers to inhibit the activity of other key cellular DNA-interacting enzymes that are specifically deregulated in human diseases or that are difficult to target because of their ubiquitous nature (i.e. transcription factors) and/or the absence of pharmacological inhibitors. Identification of those potential targets is currently under way.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under accession numbers 1527431 and 1474392 for 4 and Fmoc(mQPho), respectively.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. S.J. Dawson and Dr. C. Chollet for preliminary experiments and Dr. B. Kauffmann for assistance with crystallographic measurements and resolution. This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform at IECB, CNRS UMS3033, INSERM US001, Université de Bordeaux.
Notes
Present address: Daniel Carbajo, Department of Biological Chemistry Institute of Advanced Chemistry of Catalonia, IQAC-CSIC, Barcelona 08034, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-11-BS07-013-01]; European Research Council under the European Union's Seventh Framework Programme [ERC-2012-AdG-320892 and PEOPLE-2011-IEF-300948]; Ligue Contre Le Cancer (Comité Languedoc-Roussillon); SIRIC Montpellier Cancer [INCa_Inserm_DGOS_12553]; SIDACTION (AO2016, VIH2016721002). Funding for open access charge: University of Munich fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Leumann C.J. DNA Analogues: From supramolecular principles to biological properties. Bioorg. Med. Chem. 2002; 10:841–854. [DOI] [PubMed] [Google Scholar]
- 2. Gryaznov S.M. Oligonucleotide N3′→P5′ phosphoramidates as potential therapeutic agents. Biochim. Biophys. Acta. 1999; 1489:131–140. [DOI] [PubMed] [Google Scholar]
- 3. Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim. Biophys. Acta. 1999; 1489:141–158. [DOI] [PubMed] [Google Scholar]
- 4. Nielsen P.E., Egholm M., Berg R.H., Buchardt O.. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991; 254:1497–1500. [DOI] [PubMed] [Google Scholar]
- 5. Nielsen P.E. Nucleic Acid Backbone Structure Variations: Peptide Nucleic Acids. 2014; Chichester: Wiley. [Google Scholar]
- 6. Li W.J., Shi H.H., Dong B., Nie K.X., Liu Z.C., He N.Y.. Recognition mechanisms and applications of peptide nucleic acids targeting double-stranded DNA. Curr. Med. Chem. 2016; 23:4681–4705. [DOI] [PubMed] [Google Scholar]
- 7. Sharma C., Awasthi S.K.. Versatility of peptide nucleic acids (PNAs): role in chemical biology, drug discovery, and origins of life. Chem. Biol. Drug Des. 2017; 89:16–37. [DOI] [PubMed] [Google Scholar]
- 8. Koshkin A.A., Singh S.K., Nielsen P., Rajwanshi V.K., Kumar R., Meldgaard M., Olsen C.E., Wengel J.. LNA (locked nucleic acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998; 54:3607–3630. [Google Scholar]
- 9. Obika S., Nanbu D., Hari Y., Morio K.-i., In Y., Ishida T., Imanishi T.. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3, -endo sugar puckering. Tetrahedron Lett. 1997; 38:8735–8738. [Google Scholar]
- 10. Hagedorn P.H., Persson R., Funder E.D., Albaek N., Diemer S.L., Hansen D.J., Moller M.R., Papargyri N., Christiansen H., Hansen B.R. et al.. Locked nucleic acid: modality, diversity, and drug discovery. Drug Discov. Today. 2018; 23:101–114. [DOI] [PubMed] [Google Scholar]
- 11. Veedu R.N., Wengel J.. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010; 7:536–542. [DOI] [PubMed] [Google Scholar]
- 12. Gupta A., Mishra A., Puri N.. Peptide nucleic acids: advanced tools for biomedical applications. J. Biotechnol. 2017; 259:148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bala A., Gorski L.. Application of nucleic acid analogues as receptor layers for biosensors. Anal. Methods. 2016; 8:236–244. [Google Scholar]
- 14. Zhang Y., Sicot G., Cui X., Vogel M., Wuertzer C.A., Lezon-Geyda K., Wheeler J., Harki D.A., Muzikar K.A., Stolper D.A. et al.. Targeting a DNA binding motif of the EVI1 protein by a pyrrole–imidazole polyamide. Biochemistry. 2011; 50:10431–10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang J.S., Meier J.L., Dervan P.B.. Design of sequence-specific DNA binding molecules for DNA methyltransferase inhibition. J. Am. Chem. Soc. 2014; 136:3687–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asamitsu S., Kawamoto Y., Hashiya F., Hashiya K., Yamamoto M., Kizaki S., Bando T., Sugiyama H.. Sequence-specific DNA alkylation and transcriptional inhibition by long-chain hairpin pyrrole-imidazole polyamide-chlorambucil conjugates targeting CAG/CTG trinucleotide repeats. Bioorg. Med. Chem. 2014; 22:4646–4657. [DOI] [PubMed] [Google Scholar]
- 17. Pommier Y., Marchand C.. Interfacial inhibitors of protein-nucleic acid interactions. Curr. Med. Chem. Anticancer Agents. 2005; 5:421–429. [DOI] [PubMed] [Google Scholar]
- 18. Pommier Y., Kiselev E., Marchand C.. Interfacial inhibitors. Bioorg. Med. Chem. Lett. 2015; 25:3961–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Canc. 2006; 6:789–802. [DOI] [PubMed] [Google Scholar]
- 20. Mouscadet J.-F., Tchertanov L.. Raltegravir: molecular basis of its mechanism of action. Eur. J. Med. Res. 2009; 14(Suppl. 3):5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morishita R., Gibbons G.H., Horiuchi M., Ellison K.E., Nakama M., Zhang L., Kaneda Y., Ogihara T., Dzau V.J.. A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferation in vivo. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:5855–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klaus M., Prokoph N., Girbig M., Wang X., Huang Y.-H., Srivastava Y., Hou L., Narasimhan K., Kolatkar P.R., Francois M. et al.. Structure and decoy-mediated inhibition of the SOX18/Prox1-DNA interaction. Nucleic Acids Res. 2016; 44:3922–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hecker M., Wagner A.H.. Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 2017; 144:29–34. [DOI] [PubMed] [Google Scholar]
- 24. Yüksel D., Bianco P.R., Kumar K.. De novo design of protein mimics of B-DNA. Mol. Biosyst. 2016; 12:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee C.-H., Shih Y.-P., Ho M.-R., Wang A.H.J.. The C-terminal D/E-rich domain of MBD3 is a putative Z-DNA mimic that competes for Zα DNA-binding activity. Nucleic Acids Res. 2018; 46:11806–11821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang M.F., Lin S.J., Ko T.P., Liao Y.T., Hsu K.C., Wang H.C.. The monomeric form of Neisseria DNA mimic protein DMP19 prevents DNA from binding to the histone-like HU protein. PLoS One. 2017; 12:e0189461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Melkina O.E., Goryanin II, Zavilgelsky G.B.. The DNA-mimic antirestriction proteins ArdA ColIB-P9, Arn T4, and Ocr T7 as activators of H-NS-dependent gene transcription. Microbiol. Res. 2016; 192:283–291. [DOI] [PubMed] [Google Scholar]
- 28. Parsons L.M., Yeh D.C., Orban J.. Solution structure of the highly acidic protein HI1450 from Haemophilus influenzae, a putative double-stranded DNA mimic. Proteins. 2004; 54:375–383. [DOI] [PubMed] [Google Scholar]
- 29. Liu D., Ishima R., Tong K.I., Bagby S., Kokubo T., Muhandiram D.R., Kay L.E., Nakatani Y., Ikura M.. Solution structure of a TBP–TAFII230 Complex: Protein mimicry of the minor groove surface of the TATA box unwound by TBP. Cell. 1998; 94:573–583. [DOI] [PubMed] [Google Scholar]
- 30. Wang H.-C., Ho C.-H., Hsu K.-C., Yang J.-M., Wang A.H.J.. DNA mimic proteins: functions, structures, and bioinformatic analysis. Biochemistry. 2014; 53:2865–2874. [DOI] [PubMed] [Google Scholar]
- 31. Dryden D.T.F. DNA mimicry by proteins and the control of enzymatic activity on DNA. Trends Biotechnol. 2006; 24:378–382. [DOI] [PubMed] [Google Scholar]
- 32. Wang H.-C., Chou C.-C., Hsu K.-C., Lee C.-H., Wang A.H.J.. New paradigm of functional regulation by DNA mimic proteins: recent updates. IUBMB Life. 2019; 71:539–548. [DOI] [PubMed] [Google Scholar]
- 33. Ziach K., Chollet C., Parissi V., Prabhakaran P., Marchivie M., Corvaglia V., Bose P.P., Laxmi-Reddy K., Godde F., Schmitter J.-M. et al.. Single helically folded aromatic oligoamides that mimic the charge surface of double-stranded B-DNA. Nat. Chem. 2018; 10:511–518. [DOI] [PubMed] [Google Scholar]
- 34. Redinbo M.R., Stewart L., Kuhn P., Champoux J.J., Hol W.G.J.. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998; 279:1504–1513. [DOI] [PubMed] [Google Scholar]
- 35. Hare S., Gupta S.S., Valkov E., Engelman A., Cherepanov P.. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010; 464:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hsiang Y.-H., Lihou M.G., Liu L.F.. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989; 49:5077–5082. [PubMed] [Google Scholar]
- 37. Hazuda D.J., Felock P., Witmer M., Wolfe A., Stillmock K., Grobler J.A., Espeseth A., Gabryelski L., Schleif W., Blau C. et al.. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000; 287:646–650. [DOI] [PubMed] [Google Scholar]
- 38. McPherson A. Introduction to Macromolecular Crystallography. 2009; 2nd edn. Hoboken: Wiley-Blackwell. [Google Scholar]
- 39. Jancarik J., Kim S.H.. Sparse matrix sampling: a screening method for crystallization of proteins. J. Appl. Cryst. 1991; 24:409–411. [Google Scholar]
- 40. Cudney R., Patel S., Weisgraber K., Newhouse Y., McPherson A.. Screening and optimization strategies for macromolecular crystal growth. Acta Cryst. D. 1994; 50:414–423. [DOI] [PubMed] [Google Scholar]
- 41. Rigaku CrystalClear-SM Expert and CrystalStructure. 2011; Tokyo. [Google Scholar]
- 42. Sheldrick G. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. A. 2015; 71:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheldrick G. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015; 71:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dolomanov O.V., Bourhis L.J., Gildea R.J., Howard J.A.K., Puschmann H.. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009; 42:339–341. [Google Scholar]
- 45. Spek A. Structure validation in chemical crystallography. Acta Cryst. D. 2009; 65:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Valkov E., Gupta S.S., Hare S., Helander A., Roversi P., McClure M., Cherepanov P.. Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic. Acids. Res. 2009; 37:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lesbats P., Métifiot M., Calmels C., Baranova S., Nevinsky G., Andreola M.L., Parissi V.. In vitro initial attachment of HIV-1 integrase to viral ends: control of the DNA specific interaction by the oligomerization state. Nucleic Acids Res. 2008; 36:7043–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Faure A., Calmels C., Desjobert C., Castroviejo M., Caumont-Sarcos A., Tarrago-Litvak L., Litvak S., Parissi V.. HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 2005; 33:977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baptiste B., Douat-Casassus C., Laxmi-Reddy K., Godde F., Huc I.. Solid phase synthesis of aromatic oligoamides: application to helical water-soluble foldamers. J. Org. Chem. 2010; 75:7175–7185. [DOI] [PubMed] [Google Scholar]
- 50. Hu X., Dawson S.J., Mandal P.K., de Hatten X., Baptiste B., Huc I.. Optimizing side chains for crystal growth from water: a case study of aromatic amide foldamers. Chem. Sci. 2017; 8:3741–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dawson S.J., Hu X., Claerhout S., Huc I.. Chapter thirteen - solid phase synthesis of helically folded aromatic oligoamides. Methods Enzymol. 2016; 580:279–301. [DOI] [PubMed] [Google Scholar]
- 52. Qi T., Deschrijver T., Huc I.. Large-scale and chromatography-free synthesis of an octameric quinoline-based aromatic amide helical foldamer. Nat. Protoc. 2013; 8:693–708. [DOI] [PubMed] [Google Scholar]
- 53. Ghosez L., Haveaux B., Viehe H.G.. Alkyl and aryl α-chloro enamines. Angew. Chem. Int. Ed. 1969; 8:454–455. [Google Scholar]
- 54. Hu X., Dawson S.J., Nagaoka Y., Tanatani A., Huc I.. Solid-Phase synthesis of water-soluble helically folded hybrid α-Amino Acid/Quinoline Oligoamides. J. Org. Chem. 2016; 81:1137–1150. [DOI] [PubMed] [Google Scholar]
- 55. Mirtič A., Grdadolnik J.. The structure of poly-l-lysine in different solvents. Biophys. Chem. 2013; 175–176:47–53. [DOI] [PubMed] [Google Scholar]
- 56. Cieślik-Boczula K. Alpha-helix to beta-sheet transition in long-chain poly-l-lysine: Formation of alpha-helical fibrils by poly-l-lysine. Biochimie. 2017; 137:106–114. [DOI] [PubMed] [Google Scholar]
- 57. Hays F.A., Teegarden A., Jones Z.J.R., Harms M., Raup D., Watson J., Cavaliere E., Ho P.S.. How sequence defines structure: a crystallographic map of DNA structure and conformation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:7157–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Narayana N., Weiss M.A.. Crystallographic analysis of a sex-specific enhancer element: sequence-dependent DNA structure, hydration, and dynamics. J. Mol. Biol. 2009; 385:469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mandal P.K., Venkadesh S., Gautham N.. Structure of d(CGGGTACCCG)4 as a four-way Holliday junction. Acta Cryst. F. 2011; 67:1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cnudde S.E., Prorok M., Dai Q., Castellino F.J., Geiger J.H.. The crystal structures of the calcium-bound con-G and con-T[K7γ] dimeric peptides demonstrate a metal-dependent helix-forming motif. J. Am. Chem. Soc. 2007; 129:1586–1593. [DOI] [PubMed] [Google Scholar]
- 61. Cheng C., Kussie P., Pavletich N., Shuman S.. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell. 1998; 92:841–850. [DOI] [PubMed] [Google Scholar]
- 62. Zerbe K., Moehle K., Robinson J.A.. Protein epitope mimetics: from new antibiotics to supramolecular synthetic vaccines. Acc. Chem. Res. 2017; 50:1323–1331. [DOI] [PubMed] [Google Scholar]
- 63. Plante J.P., Burnley T., Malkova B., Webb M.E., Warriner S.L., Edwards T.A., Wilson A.J.. Oligobenzamide proteomimetic inhibitors of the p53-hDM2 protein-protein interaction. Chem. Commun. 2009; 5091–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ernst J.T., Becerril J., Park H.S., Yin H., Hamilton A.D.. Design and application of an α-Helix-Mimetic scaffold based on an oligoamide-foldamer strategy: antagonism of the Bak BH3/Bcl-xL Complex. Angew. Chem. Int. Ed. 2003; 42:535–539. [DOI] [PubMed] [Google Scholar]
- 65. Wilson A.J. Helix mimetics: Recent developments. Prog. Biophys. Mol. Biol. 2015; 119:33–40. [DOI] [PubMed] [Google Scholar]
- 66. Shaginian A., Whitby L.R., Hong S., Hwang I., Farooqi B., Searcey M., Chen J., Vogt P.K., Boger D.L.. Design, synthesis, and evaluation of an alpha-helix mimetic library targeting protein-protein interactions. J. Am. Chem. Soc. 2009; 131:5564–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rodriguez J.M., Hamilton A.D.. Benzoylurea oligomers: synthetic foldamers that mimic extended α Helices. Angew. Chem. Int. Ed. 2007; 46:8614–8617. [DOI] [PubMed] [Google Scholar]
- 68. Sadowsky J.D., Fairlie W.D., Hadley E.B., Lee H.-S., Umezawa N., Nikolovska-Coleska Z., Wang S., Huang D.C.S., Tomita Y., Gellman S.H.. (α/β+α)-Peptide antagonists of BH3 Domain/Bcl-xL recognition: toward general strategies for foldamer-based inhibition of protein-protein interactions. J. Am. Chem. Soc. 2007; 129:139–154. [DOI] [PubMed] [Google Scholar]
- 69. Grison C.M., Miles J.A., Robin S., Wilson A.J., Aitken D.J.. An α-helix-mimicking 12,13-helix: designed α/β/γ-foldamers as selective inhibitors of protein-protein interactions. Angew. Chem. Int. Ed. 2016; 55:11096–11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Urpi L., Tereshko V., Malinina L., Huynh-Dinh T., Subirana J.A.. Structural comparison between the d(CTAG) sequence in oligonucleotides and trp and met repressor-operator complexes. Nat. Struct. Biol. 1996; 3:325–328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under accession numbers 1527431 and 1474392 for 4 and Fmoc(mQPho), respectively.