


Abstract
Utilization of non-AUG alternative translation start sites is most common in bacteria and viruses, but it has been also reported in other organisms. This phenomenon increases proteome complexity by allowing expression of multiple protein isoforms from a single gene. In Saccharomyces cerevisiae, a few described cases concern proteins that are translated from upstream near-cognate start codons as N-terminally extended variants that localize to mitochondria. Using bioinformatics tools, we provide compelling evidence that in yeast the potential for producing alternative protein isoforms by non-AUG translation initiation is much more prevalent than previously anticipated and may apply to as many as a few thousand proteins. Several hundreds of candidates are predicted to gain a mitochondrial targeting signal (MTS), generating an unrecognized pool of mitochondrial proteins. We confirmed mitochondrial localization of a subset of proteins previously not identified as mitochondrial, whose standard forms do not carry an MTS. Our data highlight the potential of non-canonical translation initiation in expanding the capacity of the mitochondrial proteome and possibly also other cellular features.
INTRODUCTION
As semi-autonomous organelles mitochondria have their own DNA and translational machinery; however, mitochondrial DNA encodes only for a few polypeptides. The majority of mitochondrial proteins are nuclear-encoded, translated in the cytosol and imported into mitochondria (1). The best-characterized mechanism of mitochondrial targeting involves specialized translocases which deliver proteins containing the N-terminal mitochondrial targeting signal (MTS) to mitochondrial matrix, inner (IMM) or outer (OMM) membrane or intermembrane space (IMS) as unfolded precursors (2); reviewed in (3)). In most cases the presequence with the MTS is cleaved after transport by mitochondrial processing peptidases (4). Similar mechanisms are used to transport proteins to peroxisomes, endoplasmic reticulum (ER) or chloroplasts in plants (5). Many mitochondrial proteins do not contain an N-terminal presequence constituting the MTS, but apparently harbor unidentified or ill-defined internal targeting sequences and the mechanisms of their translocation to mitochondria remain elusive (6).
According to proteomics analyses, combined with genomics and bioinformatics, yeast and human mitochondria contain 1187 and 1837 proteins, respectively, which are annotated as mitochondrial in the Saccharomyces Genome Database (SGD) and the MitoMiner Database (1,7,8). In addition, several hundred unrecognized proteins are likely to reside in the mitochondrial compartment (9). This prediction is supported by a recent mitoproteome survey of yeast Saccharomyces cerevisiae that has identified over 3400 proteins of mitochondria and mitochondria-associated fractions and classified 901 high-confidence mitochondrial proteins (10). One solution to the question of cryptic mitochondrial isoforms may be provided by alternative protein targeting to multiple cellular compartments (reviewed in (11–14)). Although several cases of proteins with multiple cellular destinations are well documented, it has become evident that this process is more common than it was assumed previously. Such protein variants with different features that expand protein diversity may be generated by alternative mechanisms, including transcription initiation, mRNA splicing and translation initiation.
Several cases of alternative translation initiation codons used to generate multiple protein isoforms from a single gene have been reported in different eukaryotic organisms (reviewed in (15–17)). A more specific case concerns utilization of alternative translation initiation sites (aTIS), particularly at non-AUG codons. Non-AUG initiation has been reported for a number of proteins in higher eukaryotes and may represent a widespread phenomenon (16,18–25). Also in yeast non-AUG triplets can be used at low efficiency to initiate translation (26,27). A few identified examples include tRNA synthetase Ala1, tRNA synthase Grs1 and thiol peroxidase Hyr1/Gpx3, which localize to mitochondria as extended isoforms produced by translation initiation from upstream non-AUG codons, while their major forms synthesized from the canonical AUG codon are devoid of an MTS and localize to the nucleus or cytoplasm (28–30). A distinct case of non-canonical initiation is observed for mitochondrial acetyl-CoA carboxylase Hfa1 that is generated exclusively through a non-AUG start codon (31). Finally, extended mitochondrial isoforms of Bpl1, Faa2 and Sam2 have been predicted based on comparative analyses (31). There are also rare cases of alternative isoforms translated from two in-frame AUG codons. In yeast, extended forms of two aminoacyl-tRNA synthetases Hts1 and Vas1 have been shown to localize to mitochondria, while their canonical versions are cytoplasmic (32,33). Conversely, the Osm1 protein produced from the annotated start localizes to the ER, whereas a shorter variant translated from the downstream codon is targeted to mitochondria (34).
As the frequency of translation initiation at non-canonical codons is usually rather low (27), detection of these alternative forms might be difficult especially in large-scale analyses due to uneven distribution of the two variants between compartments. Such so-called eclipsed distribution, where the location of the minor isoform is masked by the more dominant one, may often lead to overlooking variants with very low expression levels in proteome analyses (12). Despite its regulatory potential, the prospect of a more general utilization of aTISs in dual protein targeting has not been systematically explored. This may be partially due to the removal of import presequences, so products of upstream translation cannot be easily detected in mitochondrial proteome studies. On the other hand, peptides from N-terminal extensions that may remain after cleavage will not match any sequences available in the databases and will remain unidentified. Only recently genomewide Ribo-seq data has become available, providing global translation mapping that enabled identification of potential upstream translational events.
In this study we present an extensive in silico analysis of genomewide occurrence of alternative translation start codons in S. cerevisiae. We concentrated on non-AUG initiators that are located upstream of annotated canonical codons in-frame with the main products and in principle can generate N-terminally extended (NTE) protein isoforms. Next, focusing on candidates that gain a predictable MTS within the NTE region we generated a comprehensive list of >600 unique proteins that are predicted with high probability to localize to mitochondria only as extended variants. We validated these predictions for several selected cases using genetic, bioimaging and biochemical approaches and confirmed that the overwhelming majority of these candidates are indeed present in mitochondria. Our combined bioinformatics and experimental data show that utilization of upstream near-cognate start codons in yeast, and most likely also in other organisms, is more widespread than previously anticipated and that that such a mechanism may significantly contribute to enhancing the complexity of the cellular proteome. Many of protein non-canonical N-terminal extensions are likely to be conserved in higher eukaryotes, and mutations in these hitherto uncharacterized domains may contribute to human disease, mitochondrial or otherwise.
MATERIALS AND METHODS
α-Complementation and Split-Intein-GFP assays
For the α-complementation assay strains were incubated at 30°C for 72 h on SCGal-Ura-Leu+Xgal plates. For the Split-Intein-GFP assay strains were analysed by confocal fluorescence microscopy. Yeast cultures grown in SCD-Ura-Leu medium to OD600 = 1 were incubated with 500 nM MitoTracker Red CMXRos (ThermoFisher Scientific) for 30 min, pelleted, washed twice and resupended in the same medium. Cell suspensions were mounted on slides and fluorescence images were taken with Zeiss LSM700 confocal microscope with the 100× objective oil lens and processed with Zen 2009 software.
Cell fractionation and western blotting
Yeast cells were fractionated according to the modified yeast mitochondria isolation protocol (35). Sixty OD of cells were harvested by centrifugation, washed twice with distilled water and resuspend in 6 ml of 100 mM Tris-SO4 pH 9.4, 10 mM DTT buffer and incubated with shaking for 15 min at 30°C. The pellet was washed once in 1.2 M sorbitol, 20 mM KPi pH 7.4, resuspend in the same buffer with 10 mg/ml Zymolyase T20. Cells were spheroplasted at 30°C for 30 min and spheroplasts were haversted by centrifugation for 5 min at 3000 g at room temperature. From this point, all operations were carried out in ice-cold buffers. Spheroplasts were washed with ice-cold 0.6 M sorbitol, 10 mM Tris–HCl pH 7.4 with protease inhibitor Complete ULTRA Tablets (Roche) and homogenized by 4–5 strokes using Potter homogenizer. Homogenate was centrifuged at 3000 × g for 5 min at 4°C and the supernatant was divided into four fractions: 1/6 was precipitated with 10% TCA and resuspend in 130 μl Urea sample buffer (6 M urea, 6% SDS, 125 mM Tris–HCl pH 6.8, 0,01%, 50 mM DTT, Bromophenol Blue) (total fraction, T); 1/6 and 2/3 were centrifuged at 18 000 × g for 15 min at 4°C and pellets were resuspend in 130 μl or 60 μl of Urea sample buffer, respectively (mitochondrial fraction, M and dense mitochondria, D); the supernatant from the mitochondrial fraction was TCA-precipitated and resuspend in 130 μl of Urea sample buffer (cytosolic fraction, C). Protein samples (20 μl of each fraction) were resolved by 8%, 10% or 12% SDS-PAGE and analysed by western blotting followed by autoradiography. Western blotting was performed using peroxidase-anti-peroxidase antibody (P1291, 1:3000, Sigma) to detect the TAP-tag, anti-HA (12CA5, 1:1000, Roche) to detect the HA-tag and polyclonal custom-raised anti-Ilv2, anti-Sgt2 and anti-Trr1 specific antibodies. Secondary commercial anti-mouse IgG horseradish peroxidase-conjugated antisera (31 430, 1:25 000, Thermo Fisher Scientific) were used against anti-HA antibodies and anti-rabbit IgG horseradish peroxidase-conjugated antisera (A0545, 1:10 000, Sigma) against anti-Ilv2, anti-Sgt2 and anti-Trr1 antibodies.
Crude mitochondrial fractions of has1Δ strains expressing Has1wt and Has1mf variants were isolated as described (36). Protein concentration was determined by the Bradford assay and 5 μg of mitochondrial protein extracts were analysed by SDS-PAGE and western blotting. MTCO1 MTCO2 MTCO3 (ab110270, ab110271, ab110259; 1:1000, Abcam, UK) were used as primary antibodies to detect Cox1, Cox2 and Cox3 proteins, respectively; anti-VDCA1 antibody (ab14734, 1:1000, Abcam) was used as a loading control and anti-mouse-HRP (W4021, 1:3333, Promega) as a secondary antibody.
Bioinformatics analyses
NTE screen
N-terminally extended protein (NTE) forms were predicted using the MitoCrypt R package developed for this purpose and available under the UTR: https://github.com/Miswi/MitoCrypt. The major utilities of this package include aTIS prediction, MTS prediction software wrapping and ribosomeA-site profile plotting. Saccharomyces cerevisiae genome sequence FASTA and gene annotation GTF (General Transfer Format) files were extracted from Ensembl (ensembl.org) release 94 repository (37). Sequences, genomic coordinates and other features of potential NTE isoforms were extracted using custom R script employing commonly used general purpose R packages (dplyr, tibble) as well as bioinformatic R packages designed for sequence data manipulation (Biostrings, refGenome) and all their dependencies (38–42). The search for all potential TIS was performed by filtering out all upstream in-frame near-cognate triplets: CUG, GUG, UUG, ACG, AUA, AUC, AUU (27) and upstream AUG (uAUG) not separated from the major ORF by an in-frame stop codon. N-terminally truncated (NTT) isoforms were found by searching all ORFs for internal in-frame AUG codons. An array of consecutive potential isoforms was created by translating genomic DNA sequences for the coordinates of the extended regions in reverse, storing all possible candidates based on analyzed aTIS and concatenating them with the original annotated translated ORF sequence. Transcriptome heterogeneity data based on (43) was added as additional information for further analysis. The list of high-confidence genes was generated by filtering out unconfirmed features based on keywords: ‘merged’, ‘deleted’ and ‘dubious ORFs’ without indication of translation in the Ribo-seq data. Isoforms not covered by transcriptome data (43) were excluded from further Ribo-seq analysis. The output of the MitoCrypt package is displayable with genome browsers, e.g. Integrative Genomics Viewer (44). Throughout every step of data processing and analysis, data is stored in tabular format based on UCSC BED (Browser Extensible Data) format (45).
Ribo-seq data analysis
Analysis of Ribo-seq data was performed on antibiotic-free, RNAse I datasets for yeast in exponential growth phase (46–49). Sub-codon resolution ribosome occupancy profiles were generated using Scikit-Ribo package, which enables precise footprint mapping (50). This method also allows discrimination between uORFs and NTEs, as only in-frame reads are used for further analysis. Profiles from all datasets were concatenated as RPKM (ribosomes per kilobase per million mapped reads) profiles to normalize for different sequencing depth. Ribosome occupancy profiles plots were generated with the custom R script and ggplot packages (51). For TIS calling, ribosome profiles ranging from the furthermost potential aTIS downstream of the annotated AUG start codon to the annotated stop codon for all yeast ORFs were generated. Genes with too low ribosome occupancy (arbitrarily chosen at 1 RPKM) were removed from further analysis. Each profile was subjected to the R-implemented algorithm described in Supplemental Figure S1. Mean ribosome occupancies were tested using permutation t-test (10 000 repetitions) with a 0.05 P-value threshold. When major isoforms were tested only the first 50 codons were taken into account to avoid underestimation of translation efficiency.
RESULTS
Translation of numerous yeast ORFs may initiate at codons upstream of annotated start sites
In yeast alternative protein isoforms generated by translation from upstream canonical and non-AUG codons have been described in the case of only a few specific proteins (28–31,33,52,53). To evaluate whether utilization of such codons may have a widespread character we performed a transcriptome-wide in silico screen of all known yeast ORFs to identify all potential upstream aTISs (Figure 1A). This search was conducted by an algorithm that scanned in-frame (by three nucleotides) upstream of each SGD-annotated start codon (6664 yeast ORFs listed in SGD) and until a stop codon, threshold traversal length or end of the transcript was reached (Figure 1B). Each upstream AUG or near-cognate non-AUG codon differing from the canonical start codon by a single nucleotide and able to initiate translation in yeast (CTG, TTG, GTG, ACG, ATC, ATA and ATT) (27,54) was ascribed as a potential aTIS that could produce protein N-terminal extensions (NTE). All translated ORFs were then concatenated with known protein sequences. This genomewide search revealed that 4457 of 6664 genes contained putative upstream aTISs that potentially may generate additional 12 953 protein isoforms (totally 19 618 isoforms) (Figure 1C and Supplemental Figure S1B; Table S1). We narrowed down the number of annotated yeast ORFs by filtering out merged, deleted or dubious ORFs (see Supplementary Materials and Methods), ending up with 6110 genes. Out of these high-confidence expressed genes 4065 may encode NTE proteins generating additional 11 607 isoforms (Figure 1C and Supplemental Figure S1B). Surprisingly, we observed that in 212 cases alternative translation may initiate at upstream AUG codons, which are not annotated in SGD as TISs.
Figure 1.

Alternative initiation start sites (aTIS) are common in yeast cells. (A) Outline of the in silico (left panel) and in vivo (right panel) pipeline used in this work. Alternative protein isoforms, which are translated from the listed non-AUG or upstream AUG (uAUG) codons for NTE and downstream AUG (dAUG) codons for NTT, are identified by the algorithm depicted in (B). This is followed by MTS prediction for each isoform using MitoProt II, TargetP and MitoFates, and ribosome profiling (Ribo-seq) analysis. Mitochondrial localization of selected mitochondria-targeted NTE candidates is validated using two in vivo approaches, followed by detailed case studies (bottom panel). (C) Bioinformatics screen results. The columns contain number of genes included in the screen and genes/proteins resulting from the screen as follows: NTE/NTT-generating genes, NTE/NTT-containing isoforms, NTE/NTT proteins with NTE/NTT-MTS gain and NTE/NTT-MTS loss (see text). The rows refer to all annotated ORFs in SGD, high-confident ORFs with merged, deleted or dubious ORFs filtered out and ORFs utilised in translation according to the Ribo-seq analysis. (D) Number of translation start sites (TIS) observed in ribosome profiling analysis. (E) Number of predicted and utilized start codons, canonical AUG, dAUG, uAUG, upstream non-AUG and all upstream TIS. (F) Density plot of relative translation level from alternative TIS. Fraction of upstream (NTE, red color) and downstream (NTT, blue color) translation is calculated for each gene that utilizes at least one NTE as a ratio of RPKM upstream/downstream of the canonical TIS. Only NTE proximal to the canonical TIS and the first 50 codons of the CDS (coding sequence) are considered to account for the ‘codon ramp’ that causes artificially higher ribosome density at CDS 5′ ends. (G) Distribution of the relative translation level from canonical and non-AUG codons for NTE proteins with one additional aTIS. (H) Scatter plot of transcript and translation level relative to absolute transcript and translation level of each gene. Fraction of translation is calculated as in (F); fraction of transcription represents a ratio of mTIFs (major transcript isoform; (43)) encompassing given isoform to the total number of mTIFs for a given gene. Red and blue dots correspond to downstream and upstream protein isoforms, respectively. Transcript isoform is considered to contain particular TIS when it covers at least 5 nucleotides upstream of the TIS. (I) Efficiency of codon usage for proteins translated from one TIS, which are supported by the Ribo-seq analysis. (J) MTS probability of mitochondria-targeted N-terminally extended (MT-NTE) proteins. The numbers show the maximal MTS value, predicted using MitoProt II, TargetP or MitoFates. (K) Venn diagram depicting the number of mitochondria-associatedand high-confidence mitochondrial proteins according to (10) and ‘MTS gain’ proteins identified in this study.
Internal start codons downstream of major TIS may also serve as aTIS (55,56). Using similar bioinformatics approach as for NTE we established that 2672 genes have the potential to generate N-terminally truncated (NTT) proteins by translation from downstream AUG (dAUG) codons, giving rise to 4049 isoforms (Supplemental Figure S1B and Table S1). We reckon that the majority of dAUG codons serve as internal methionine codons and their utilization as internal aTIS is probably mediated by the leaky scanning mechanism, which strongly depends on translation initiation start context (57). We therefore performed context analysis for each canonical and non-canonical translation initiation start site by comparing sequences of each TIS and aTIS (region from –6 to +4 relative to TIS) to the reference context sequence derived from the 10% most efficiently translated ORFs (Supplemental Figure S1C and Supplementary Materials and Methods). We found that each upstream non-AUG codon context score differed slightly (Supplemental Figure S1D), as was the case for downstream and upstream AUGs (Supplemental Figure S1E), and aTIS codon context scores were usually slightly lower than for canonical AUG codons (Supplemental Figure S1F). Interestingly, in some cases aTIS is located only one amino acid upstream (five cases) or downstream (93 cases) of the canonical codon. Context analysis showed that only 11% of AUG aTISs have a significantly weaker context than canonical TISs, while 54% have higher context scores (20% with the fold > 2) (Supplemental Table S2). This surprising observation calls for a mechanism by which the pre-initiation complex (PIC) discriminates between adjacent initiation codons, thus affecting the dynamics of translation initiation.
To restrict the list of NTE-containing candidates in our bioinformatics screen and substantiate its output, we used available ribosome profiling (Ribo-seq) data (46,47) to assess translation upstream of annotated TISs for ORFs from the high-confidence list. To this end, we extracted P-sites (peptidyl site, second binding site for tRNA in the ribosome) of elongating ribosomes and ribosome profiles using the Scikit-Ribo package (50) (Supplemental Figure S1). This analysis showed that out of 5,265 genes with sufficient ribosome occupancy distribution along transcripts to support translation of 6094 isoforms, 1150 genes may produce NTE proteins, giving rise to additional 1132 isoforms. In addition, the Ribo-seq data revealed 295 genes with predicted N-terminal truncations (301 isoforms) (Figure 1C; Supplemental Figure S1B). The main part of alternative isoforms is synthesized from one TIS; utilization of two TISs is also common, whereas three active TISs are relatively rare (Figure 1D). Translated NTEs vary vastly in length, but the majority are shorter than 25 amino acids with some notable exceptions that are well beyond 50 amino acids (Supplemental Table S1). Usage of closely spaced TIS codons is not surprising as it can be inferred from N-terminal proteomics data (58,59). As expected, we found translation evidence for only ∼10% of predicted NTE forms. Utilization of upstream aTIS codons in general also oscillates ∼10%, except for the rarest AUG and more frequently used CUG codons (Figure 1E). In agreement with the notion that non-AUG-initiated translation is less efficient than from the canonical codon, the ribosome density upstream of AUG is in most cases rather low, but there are several instances for which it is relatively abundant (Figure 1F, G and Supplemental Table S1). In turn, the AUG-derived NTT fraction shows the opposite relationship, where usage of aTIS is predominant, while translation form annotated TIS is mostly not observed (Figure 1F). This is probably due to the use of the elongating ribosome profiling Ribo-seq datasets (see Disscusion). The NTE and the restricted NTT data support the leaky scanning model, where downstream codons are mostly used as initiation start sites, even if two TISs are present. Nevertheless, for a number of genes utilization of the upstream aTIS predominates over the canonical one, which is manifested by the clear bimodal distribution of the fraction of upstream translation (Figure 1H). We envisage that upstream aTIS may serve as a major initiation site by titrating initiating ribosomes. Notably, it appears that as many as 479 proteins are exclusively expressed as NTE forms from upstream TISs, of which 468 are non-canonical codons (Figure 1D, F and I). On the other hand, from 212 cases with previously unannotated upstream AUG codons only 15 are utilized according to Ribo-seq data. However, 11 proteins (Adh4, Asg7, Crg1, Mgm1, Msf1, Rnr3, Rsm18, Saf1, Sen34, Srp73 and Ste11) appear to be translated entirely from upstream AUG rather than from the SGD-annotated start site (Figure 1I). We found no correlation between codon context score and Ribo-seq data for the complete Ribo-seq dataset (Pearson correlation coefficient (PCC), r = 0.159). For proteins with one or more additional aTIS, correlation between fraction of translation and codon context score is slightly higher, but still not significant (PCC, r = 0.289).
We conclude that occurrence of upstream translation initiation may generate at least several hundreds of N-terminally extended or truncated protein variants with potential physiological functions. There is also a possibility of rare instances of translation at additional alternative initiation codons that were not included in our screen and will add to the number of estimated alternative NTE isoforms. For example, a CTT codon has been reported to act as an upstream aTIS in the case of Hyr1 (30).
Identification of mitochondrial targeting signals in N-terminally extended isoforms
To assess the functional capacity of N-terminal extensions and truncations, we focused on those that contain the mitochondrial targeting signal (MTS) and therefore may target NTE proteins to mitochondria. Using MitoProt II, TargetP and MitoFates prediction software for mitochondrial localization (60–62) we indexed all NTE and NTT isoforms with a ‘MTS gain’ parameter (see Supplementary Materials and Methods) that designates which otherwise non-mitochondrial proteins may be transported into mitochondria as extended or truncated alternative variants. This approach allowed classification of 517 proteins (1199 isoforms) that gain MTS through their NTE and 227 proteins (275 isoforms) of NTT ‘MTS gain’ proteins (Figure 1C, Supplemental Figure S1B and Supplemental Table S1). We envisage that a large fraction of NTE proteins may localize to mitochondria, especially those isoforms that acquire a relatively high MTS probability (543 with MTS > 0.8 and 301 with MTS > 0.9) (Figure 1J), 25 of which are in good Kozak context (score > 0.8). Moreover, translation of 127 isoforms for 123 proteins is corroborated by Ribo-seq data, making their synthesis more probable.
Our search accurately identified the mitochondrial isoforms of Ala1, Grs1, Hfa1 and Hyr1, which were reported to localize to mitochondria as NTE proteins produced from upstream non-AUG codons (28–31). From the list of our 517 candidates only 88 are annotated in SGD as mitochondrial, but 364 were identified in the latest mitoproteome analysis, of which 59 belong to the high-confidence class I (10) (Figure 1K). Considering that only 34% of recently classified mitochondrial proteins (10) have the predicted N-terminal MTS with a probability >0.4, we checked how many of the remaining proteins may gain the MTS through their N-terminal extension. Indeed, more than 14% of NTE-containing mitochondrial proteins with a low MTS belong to the ‘MTS gain’ category (319 proteins/675 isoforms). This constitutes a significant fraction, especially as NTE isoforms are translated for 83 of these proteinsaccording to the Ribo-seq data. A small number (6.2%) of proteins with a low but recognizable MTS probability in their annotated variant have NTE isoforms with a much higher MTS probability that are also included in the MTSgain group. It is possible that they are targeted to mitochondria not as canonical forms, but as extended variants. Interestingly, our analysis also revealed that 206 proteins with a predicted MTS > 0.4 (84 of these are annotated as mitochondrial according to the SDG), lose their MTS as N-terminally extended variants (623 predicted ‘MTS loss’ isoforms). However, these isoforms still may localize to mitochondria using internal mitochondrial targeting signal (iMTS), which is recognised by Tom70 (63). As expected, many more ‘MTS loss’ cases were found in the NTT group (421 proteins/565 isoforms).
MT-NTE proteins localize to mitochondria
Assessment of localization of protein variants in a nonstandard location is hampered by the presence of a large amount of the same protein in its primary site of action. To validate mitochondrial localization of predicted mitochondria-targeted N-terminally extended (MT-NTE) proteins we studied dual distribution of selected candidates in yeast cells using a modified version of a β-galactosidase α-complementation approach (64). The presence of a protein in a particular cellular location is assayed by co-expression of E. coli β-galactosidase ω-fragment targeted exclusively to this compartment with the protein of interest fused to the α-fragment of β-galactosidase. Co-localization of the fusion proteins leads to a reconstitution of β-galactosidase activity that is detected by blue colony colour on X-gal plates. For our purpose, yeast strains expressing mitochondria-targeted β-galactosidase ω-fragment and candidate proteins fused C-terminally to β-galactosidase α fragment, both under the control of the galactose-inducible GAL10 promoter, were tested on X-gal galactose-containing plates (Figure 2).
Figure 2.

Mitochondrial localization of selected candidates tested by genetic assays. (A) List of tested candidates. Canonical MTS and NTE MTS columns show the predicted value of the MTS (mean of scores predicted by MitoProt II, TargetP and MitoFates) when the protein is translated from the major AUG or alternative non-AUG codon, respectively. NTE length represents the length of N-terminal extensions of putative non-AUG initiated isoforms, numbers in bold denote the isoform with the highest MTS probability score, shown in NTE MTS column). Class refers to classification of proteins in the mitochondrial proteome (10). (B) Representative α-complementation assays with 12 candidate proteins. Schematic illustration of the α-complementation assay (top left panel). Co-localization in the same compartment of the candidate protein fused to the α fragment of β-galactosidase with its ω fragment results in blue colonies (left panel), whereas lack of co-localization gives white colonies (right panel). Cyt, cytosol; Mit, mitochondria. Kgd2 and Aco1 were used as positive mitochondrial controls, while the cytosolic α fragment w/o functional MTS and a lacZα fusion variant of the nuclear/cytosolic protein Gic1 served as negative controls. (C) Split-Intein-GFP subcellular localization assay of the representative 12 screened proteinslisted in (A). Schematic illustration of the Split-Intein-GFP assay (top left panel). The candidate protein, expressed from a strong promoter, and fused to the HA-tagged N-terminus of GFP, followed by the N-intein domain of a split intein, is co-expressed with a mitochondrial reporter containing a cleavable MTS followed by the HA-tagged C-intein domain of a split intein and the C-terminal domain of GFP. A functional MTS N-terminally appended to the tester-GFP-N-intein will target the tester to mitochondria and allow for reconstitution of GFP fluorescence, visualized by fluorescence microscopy, even when only a small fraction of the candidate protein is localized to mitochondria (right panel). Kgd2 and Aco1 were used as positive mitochondrial controls, while the N-intein w/o MTS and Gic1 served as negative controls. Mitochondria (red) were counterstained with Mitotracker. (D) Ribosome density A-site profiles along mRNAs coding for tested proteins. Shown is the region spanning codons from –75 to +175 upstream or downstream of annotated AUG codon, respectively. Every potential upstream start codon is indicated with a vertical line in a color corresponding to the heatmap (below each graph), which represents MTS values predicted by MitoProt II for each isoform.
To ensure that our test is sufficiently sensitive to distinguish between cytosolic and mitochondrial protein localisation, we checked three control proteins: aconitase Aco1 dually localized to cytosol and mitochondria, exclusively mitochondrial dihydrolipoyl transsuccinylase Kgd2 and exclusively cytoplasmic/nuclear Rho GTPase activator Gic1. These candidates, fused the β-galactosidase α-fragment at their C-termini, were tested with both mitochondrial or cytosolic-targeted ω-fragment of β-galactosidase. In agreement with documented cellular localisation of these proteins, Aco1-ω-cyto, Aco1-ω-mito, Kgd2-ω-mito and Gic1-ω-cyto formed blue colonies, whereas Kgd2-ω-cyto and Gic1-ω-mito were white (Supplementary Figure S2A). We then tested 52 MT-NTE candidates that fall into different functional categories, including DNA replication, transcription, RNA processing, translation, ribosome biogenesis, nucleosome and histone function, cell cycle organization, and signalling (Figure 2A and Supplemental Figure S2B). A subset of particularly interesting factors are proteins involved in RNA processing and ribosome synthesis and function, as these are underrepresented in mitochondria. Out of the 51 tested candidates, 42 strains developed blue colony color with varying intensities on X-gal containing plates (Figure 2B and Supplemental Figure S2C), consistent with mitochondrial localization of the tested proteins. Surprisingly, four proteins (Fun26, Mbp1, Pex11 and Pmt2) with a negative score have been qualified as class 3 in the most recent mitoproteome (10), but their fraction present in mitochondria may be too low for detection in the α-complementation assay or the α-fragment tag may not be available for trans-complementation in these fusion variants. Strains expressing either the α fragment alone without the functional MTS or fused to Gic1, which is located in the cytoplasm and the nucleus and is absent from our list of candidates, were used as negative controls and remained white. In turn, strains expressing the α fragment fused to mitochondrial dihydrolipoyl transsuccinylase Kgd2 or aconitase Aco1 dually localized to cytosol and mitochondria (65), which served as positive controls, became blue. As an additional control, predicted NTE-MTSs from Bpl1 and Hfa1 were fused to the β-galactosidase α-fragment and tested for reconstitution of active β-galactosidase in the presence of mitochondrially localized ω-fragment. The enzymatic activity of β-galactosidase on X-gal plates was restored in both cases to a similar extent as when the α-fragment was fused to the full length Bpl1 or Hfa1 proteins. The NTE-less α-fragment was unable to support blue colony development on X-gal plates (negative control), while the Aco1 MTS fused to the α-fragment (positive control) yielded blue colonies (Supplemental Figure S2D). This demonstrates that MTSs within NTE can act as a mitochondrial localization signal and, like bona fide MTS sequences, are sufficient to direct unrelated proteins to mitochondria.
The α-complementation assay produces a quick and simple readout, but is only an indirect indicator of presence of the α fragment fusion proteins in mitochondria. To verify possible mitochondrial localization of a subset of the candidates that scored positively in the β-galactosidase reconstitution assay we applied a Split-Intein-GFP approach. This method is based on a reconstitution of a split GFP reporter by protein trans-splicing mediated by fusions to the DnaE split intein derived from Nostoc punctiforme and has been used previously to screen mitochondrial proteins in mammals (66,67). We constructed strains expressing both the candidate and mitochondrial reporter proteins. The tester constructs represent the MTS-X-(N)GFP-(N)intein fusions, where selected candidates with their putative MTSs (MTS-X) are C-terminally extended with the N-terminal fragment of GFP, followed by the N-intein domain of the DnaE split-intein. The mitochondrial MTSCoq3-(C)intein-(C)GFP detector construct consists of the Coq3 cleavable mitochondrial import sequence followed by the C-intein and the C-terminal domain of GFP (Figure 2C). Only a functional MTS will target the (N)GFP-(N)intein construct to mitochondria and allow for reconstitution of GFP fluorescence. This assay permits for specific detection of low abundance mitochondrial proteins with a dual distribution in intact organelles in live cells. Using this approach, we tested 21 non-AUG initiated candidates fused to (N)GFP-(N)intein and co-expressed with the (C)intein-(C)GFP detector. All tested proteins showed a green fluorescence pattern matching the Mitotracker dye (Figure 2C and Supplemental Figure S2E) indicating that they partition into the mitochondrial compartment in vivo. These results are in full agreement with the data obtained using the α-complementation approach.
To confirm mitochondrial localization of MT-NTE proteins we also used a complementary biochemical approach that involved fractionation of mitochondria followed by western blotting, for 18 proteins, selected based on their strong ‘MTS gain’ (Figure 3A and Supplemental Figure S3A). Four of the selected MT-NTE proteins (Adh4, Trr1, Trz1 and Hyr1) have been described previously as mitochondrial (30,68–70). We carried out fractionation of yeast extracts from strains expressing C-terminally TAP-tagged proteins using a modified method based on a published protocol (35). TAP-tagged proteins were detected with peroxidase-anti-peroxidase (PAP) antibodies and Trr1 with specific antibodies. Specific antibodies were also used against mitochondrial Ilv2 and cytoplasmic Sgt2 to estimate the purity of each fraction (Figure 3B and Supplemental Figure S3B). Sgt2 also served as an additional non-mitochondrial control, since it has the capacity to produce NTE variants, but lacks an MTS. All of the tested proteins, except the Sgt2 control, were detected in the mitochondrial fraction, with the majority visible exclusively or predominantly in the ‘dense’ fraction (concentrated eight times). This observation is consistent with the notion that protein isoforms with dual distribution are usually present in mitochondria at a very low level and therefore are not easily detectable. This is most likely due to inefficient non-AUG-initiated translation of mitochondria-targeted NTE proteins. However, at this stage we cannot exclude the possibility that the additional population of mitochondrial proteins results from unproductive translocation of mainly non-mitochondrial major protein variants to this compartment. To address this issue, we checked localization of major forms of two candidates, Mtr4 and Lsm1, using strains expressing only their HA-tagged AUG-initiated variants (Figure 3C). As expected, the major forms of these proteins were not present in mitochondria, supporting the concept that in general only the MTS-containing non-AUG NTE isoforms are subject to mitochondrial targeting. Interestingly, in two cases, Trz1 and Mtr4, we observed that the mitochondrial isoforms migrated slightly slower than their cytosolic counterparts (Figure 3B). We predict that mitochondrial variants probably undergo post-translational modifications that retard their migration (reviewed in (71)). Alternatively, the pre-sequence of these proteins is not effectively removed following mitochondrial transport and the larger proteins correspond to NTE forms.
Figure 3.

Mitochondrial localization of selected candidates tested by a biochemical assay (A) List of tested candidates. Description as for Figure 2A. (B) Western blot analysis of total (T) yeast extracts and fractionated into cytosolic (C) and mitochondrial (M) or dense mitochondrial (D, 8 times concentrated) fractions. C-terminally TAP-tagged proteins were detected using peroxidase-anti-peroxidase (PAP) antibodies; Trr1, Ilv2 (mitochondrial control) and Sgt2 (cytoplasmic control) with specific antibodies. (C) AUG-initiated variants of Mtr4 and Lsm1 do not localize to mitochondria. Western blot analysis of cytosolic (C) and mitochondrial (M, D) fractions from yeast strains expressing N-terminally HA-tagged proteins from the major AUG codon using anti-HA (haemagglutinin) antibody. (D) Ribosome density A-site profiles along mRNAs coding for tested proteins. Description as for Figure 2D.
Altogether, we tested 65 different candidates and confirmed mitochondrial localization of 57 proteins, out of which 48 have the strong ‘MTS gain’ as NTE variants. Only 6 of these proteins were previously annotated in SGD as mitochondrial, but as many as 39 were more recently identified in the mitochondrial proteome, with 8 in a high-confidence class (10). Inspection of ribosome density profiles for the set of positive candidates reveals that in several cases translation may occur, albeit to different extent, upstream of the canonical AUG codon (Figures 2D, 3D and Supplemental Figures S2F, S3C). Based on these profiles we predict that upstream translation may often reflect the synthesis of NTE proteins, which is most evident for proteins such as Adh4, Bpl1, Gpp1 or Trz1. As observed previously, ribosome density upstream of AUG is generally low, but there are several instances for which ribosome occupancy is relatively abundant (see Adh4, Aur1, Bpl1, Clb1, Eaf1, Mch4 or Rnh1 in Figures 2D and 3D), suggesting that expression of some MTS-containing NTE variants that are probably targeted to mitochondria may be higher than expected.
Functional validation of MT-NTE proteins and mitochondrial respiration phenotype
Since lack of proteins with mitochondrial function may lead to a respiratory deficient phenotype, i.e. impaired growth on a non-fermentable carbon source, we checked the ability of several strains not expressing MT-NTE proteins to grow on glycerol-containing media. Out of 10 tested strains, each deleted for a specific non-essential gene, only two, lsm1Δ and lsm6Δ, showed respiratory growth defects. In addition, strains expressing only the major AUG-initiated form of essential Trz1 and Mtr4 proteins failed to grow on glycerol (Supplemental Figure S5; (70)). Observations for Trz1 and Lsm1 are in agreement with previous reports showing that lack of these proteins leads to mitochondrial defects (70,72). Also autophagy-related Atg1 protein that belongs to the MT-NTE group has been reported to have a mitochondrial function (73).
To further explore the link between expression of MT-NTE protein forms and mitochondrial activity, we focused on two specific cases. Since the class comprising factors related to ribosome synthesis and function is well-represented among ‘MTS gain’ proteins, we tested the mitochondrial function of Has1, the essential DEAD-box RNA helicase involved in the biogenesis of both ribosomal subunits (74) that scored positive in both genetic assays. We created the has1Δ strain expressing either the wild-type HAS1 gene containing the NTE (has1Δ HAS1wt) or carrying a point mutation (deletion) at position –1 from the annotated AUG start codon resulting in a Has1 MTS misframe variant (has1Δ HAS1mf). Only translation from the canonical AUG start codon will produce Has1 protein, since translation of the misframe variant from aTIS eliminates utilization of AUG due to several stop codons produced immediately downstream of the frameshift site. Essentially, HAS1mf does not give rise to any proper aTIS-initiated protein but only an oligopeptide consisting of an MTS followed by few amino acids (Figure 4A and Supplemental Figure S4A). The two strains were tested for growth on non-fermentable glycerol media, with the expectation that lack of the Has1 form translated from the upstream non-AUG codon caused by the misframe mutation will prevent Has1 mitochondrial localization and cause respiratory deficiency. However, we were unable to detect a respiratory growth defect for the has1Δ HAS1mf strain (Figure 4B). Even so, the steady state level of mitochondria-encoded cytochrome c oxidase subunits was markedly reduced in the misframe mutant, suggesting that the mutation may decrease rather than abolish the expression of NTE Has1 (Figure 4C). To test this possibility, we investigated the mitochondrial localization of the Has1 misframe variant using both the α-complementation and Split-Intein-GFP approaches. We observed a clear decrease in blue colour intensity (Figure 4D) and a marked reduction in GFP signal (Figure 4E) in the case of cells expressing HAS1mf. We performed quantitative analysis to estimate the degree of colocalization between the red and green fluorophores. Colocalization coefficients (Pearson's correlation coefficient (PCC) and Mander's overlap coefficient (MOC)) of cells expressing Has1wt and Has1mf variants reflect reduction of Has1 localization in mitochondria as a result of the frameshift mutation (Figure 4E). These data indicate that mutating the N-terminal extension encoding an MTS translated from a non-AUG initiation codon does not prevent targeting of Has1 to mitochondria, but apparently leads to a decrease in the mitochondrial pool of this protein. To check whether depletion of Has1 in mitochondria may result from reduced Has1 expression that is caused by the misframe mutation, we analysed the level of Has1 protein in yeast cells expressing the HA-tagged Has1wt-N-GFP-N-intein or Has1mf-N-GFP-N-intein reporters from the native promoter. The expression of both variants was comparable (Supplemental Figure S4B), indicating that the observed mitochondria-specific defects are not due to the reduced level of the essential Has1 protein. Also lack of a general growth defect of the has1Δ strain complemented with the misframe variant does not support this possibility.
Figure 4.
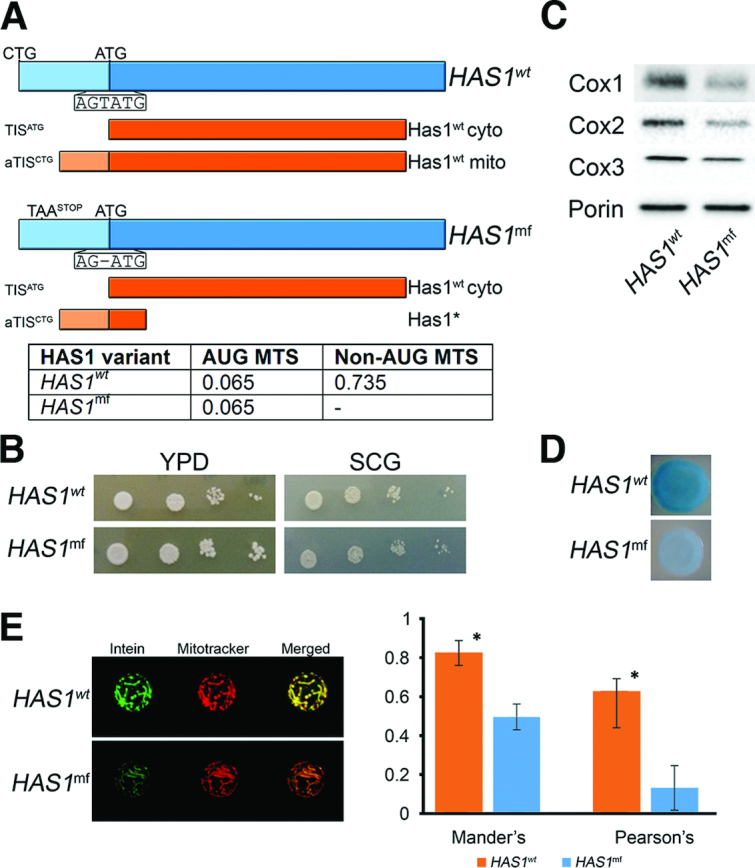
N-terminally extended isoform of Has1 localizes to mitochondria more efficiently than the major form (A) Scheme of Has1wt and Has1mf variants. Expression of HAS1wt may produce both AUG (cytosolic) and non-AUG (mitochondrial) initiated isoforms, whereas HAS1mf gives only the protein starting at the AUG codon due to a frameshift mutation (deletion) at position –1 that results in an upstream stop codon in the AUG frame. In the aTIS frame STOP codons downstream of the AUG result in translation of truncated, dysfunctional peptide (Has1*), which is probably degraded. (upper panel) Table with MTS score (MitoProtII) of AUG and NTE isoforms for Has1wt and Has1mf. (lower panel). (B) The has1Δ strains expressing Has1wt or Has1mf are respiratory competent and grow on glycerol-containing media. SCG, glycerol-containing media; YPD, glucose-containing media. (C) Expression of Has1mf leads to the reduced levels of mitochondria-encoded cytochrome c oxidase subunits. Western blot analysis of Cox1, Cox2 and Cox3 using specific antibodies in has1Δ HAS1wt and has1Δ HAS1mf strains. Porin was used as loading control. (D) α-Complementation assay of Has1wt and Has1mf variants. Description as for Figure 2B. (E) Split-Intein-GFP subcellular localization assay of Has1wt and Has1mf proteins. Description as for Figure 2C. The graph on the right represents Mander's and Pearson's colocalization coefficients obtained from 3D stack imaging. Results are the mean values (n = 5) and error bars represent SD; * denotes statistical significance (Student's t-test, *P < 0.05).
We also analysed the 3′ tRNA endonuclease Trz1, which has been shown to localize and function in both the nucleus and mitochondria (70,75). Mutants in the essential TRZ1 gene are respiratory deficient, including a strain expressing only a major form translated from the AUG codon. To verify that only the NTE isoform is responsible for the mitochondrial function of Trz1 we generated a set of trz1Δ strains expressing different plasmid-encoded Trz1 variants: TRZ1ATG (expression of Trz1AUG from the canonical AUG codon), TRZ1TTC and TRZ1TTC-TAP (expression of Trz1UUC or Trz1UUC-TAP from the most upstream predicted non-AUG codon together with Trz1AUG or Trz1AUG-TAP from the AUG codon) and TRZ1-TAPTTC/GCG with the AUG codon mutated to GCG (expression of Trz1UUC-TAP) (Figure 5A). The viability of the strain expressing Trz1UUC-TAP from the TRZ1-TAPTTC/GCG construct was sustained by the presence of the TRZ1ATG plasmid providing the untagged nuclear Trz1AUG. Testing the mitochondrial phenotype of these strains (Figure 5B) showed that, as expected, the presence of both Trz1 variants in the cell, Trz1AUG and Trz1UUC, allowed for efficient respiration and normal growth on glycerol media, whereas cells expressing only Trz1AUG were petite and did not grow on glycerol. Also, wild-type-like cells carrying both TRZ1ATG and TRZ1TTC constructs turned petite following 5-FOA-mediated loss of the TRZ1TTC plasmid. Next we validated localization of Trz1 variants by western blot of TAP-tagged proteins. In agreement with previous observations, the major Trz1AUG-TAP form was not targeted to mitochondria and only the NTE variant, expressed either from the TRZ1TTC-TAP or TRZ1-TAPTTC/GCG constructs, localized to mitochondria (Figure 5C). Notably, isoforms synthesised from the TRZ1TTC-TAP construct, Trz1UUC and Trz1AUG, were found in all fractions; again the mitochondrial Trz1 variant migrates slightly slower, while the TRZ1-TAPTTC/GCG-derived Trz1UUC was present predominantly in mitochondria. The residual amount of cytosolic Trz1 may represent Trz1UUC that had not been transported to mitochondria.
Figure 5.

Trz1 localizes to mitochondria as the NTE variant translated from the non-AUG codon. (A) Schematic illustration of different constructs encoding Trz1 variants (top panel) and their expected localization by western analysis of fractionated yeast extracts (down panel). TTC and ATG denote the upstream non-canonical and standard translation start codons, respectively; TAP – tandem affinity purification tag; p41XADH – yeast expression vectors used for TRZ1 constructs. (B) Lack of the Trz1UUC isoform translated from the upstream non-canonical codon results in respiratory deficient phenotype. Growth tests on glucose- or glycerol-containing plates of controls (WT BY4741 and TRZ1/trz1Δ BY4743) and trz1Δ strains expressing Trz1 variants, indicated on the right. (C) Cellular localization of Trz1 variants by western analysis of cytosolic (C) and mitochondrial (M or D, dense, eight times concentrated) fractions. Description as for Figure 3B.
Thus, as reported previously for Hyr1 (30), it is the MTS present in the NTE of Trz1 and likely also Has1 that is required for or enhances their mitochondrial localization and function. We propose that this mechanism of mitochondrial targeting is relatively widespread and concerns the majority of otherwise non-mitochondrial proteins that gain the MTSs through their N-terminal extensions that are generated by translation from the near-cognate start codons.
Conservation of aTIS-related N-terminal extensions in higher eukaryotes
Our genomewide search of yeast genes with a probable capacity to encode MT-NTE variants produced a surprisingly high number of likely candidates. Our subsequent analyses using genetic approaches, reporter constructs and high purity subcellular fractionation studies supported genuine mitochondrial localization for the majority of tested cases. We therefore wondered whether there is evidence for conservation of mitochondrial isoforms in higher eukaryotes. As genomewide in silico analysis of mammalian genomes for putative candidates is beyond the scope of this work due to a number of additional parameters to be taken into account (transcript/splicing isoforms, exon skipping, intron retention etc.), we focused on selected yeast ‘MTS gain’ proteins that have documented human homologs in the ENSEMBL database (37). We investigated two possible scenarios (i) mitochondrial localization of the protein isoform documented in human or mouse databases (MTS major form); (ii) mitochondrial localization of a plausible aTIS-generated N-terminally extended isoform. The possible aTIS variants were identified using PreTIS (76) and mitochondrial localization prediction was carried out using MitoProtII, TargetP and MitoFates.
This analysis revealed that out of 71 examined candidates more than a half are likely to be localized to mitochondria, 30 proteins as major forms and 18 as NTE isoforms. To strengthen our prediction for MT-NTE protein isoforms we performed TI-seq analysis using available datasets for harringtonine- or lactomidomycin-treated extracts from human cell lines (76–78). This approach confirmed that the majority (15) of MT-NTE isoforms are indeed translated from alternative TIS (Supplemental Table S3). Several of these candidates play important roles in processes linked to mitochondrial biogenesis, e.g. ribosome maturation or mitochondrial fatty acid synthesis. This result indicates that our yeast data can serve as a powerful tool to identify proteins targeted to mitochondria, which have not been previously detected in this compartment. Identification of these cryptic mitochondrial variants has important implications for our understanding of mitochondrial function and disease.
DISCUSSION
Alternative transcription, splicing and translation provide effective means to boost the capacity of cellular proteome. These often non-canonical processes generate additional protein variants with unique localization, interacting partners, altered stability or function (77). It appears that an intriguing concept of normal mRNAs coding for more than one protein could be more general than previously anticipated (78). In addition, short proteins and peptides can be encoded in transcripts considered previously as noncoding (79,80). Recent advances in high-throughput techniques, especially ribosome profiling, have allowed quantitative transcriptome-wide mapping of translational events, revealing nearly pervasive translation outside of annotated start ORFs that includes widespread utilization of aTISs (20,81–87). Unexpectedly, non-AUG codons constitute approximately half of all aTISs and >70% of upstream aTISs. The major mechanism accountable for alternative translation initiation is probably leaky scanning that allows a portion of ribosomes to initiate at weaker codons, usually in a suboptimal context and located upstream of a strong TIS (57). Alternative protein isoforms may be also generated from different TISs due to the existence of multiple transcription start sites (88,89). It is, however, difficult to unequivocally assign a physiologically relevant upstream aTIS without corroborating evidence, particularly for start codons with very low apparent initiation activity.
Analysis of available ribosome profiling data is hampered by experimental bias caused by technical setbacks during sample preparation (e.g. read length, distance from the start codon, and utilization of specific antibiotics and nucleases, untemplated nucleotide addition). Also data with harringtonine or lactomidomycin pre-treatment, which immobilises initiating ribosomes, are not yet available for yeast cells. In addition, identification of protein isoforms based on Ribo-seq in S. cerevisiae is impeded by high gene density and very limited datasets for initiating ribosomes that allow precise recognition of start codons. To overcome these drawbacks and exclude other translational processes occurring at the transcript 5′ end, mainly upstream ORFs (uORFs), we used only Scikit-Ribo selected in-frame reads, since uORF stop codons preclude generation of such reads. Still, even with only elongating ribosomes, it is possible to distinguish TISes, especially for N-terminal extensions. The non-drug Ribo-seq data are sufficient for NTE analysis, but not for the majority of NTTs, as their unequivocal detection requires much higher ribosome occupancy than on upstream canonical start codons. To evaluate whether NTT variants are produced, the output of our analyses was limited to the most evident cases with the highest probability of NTT utilisation.
Bioinformatics screens, Ribo-seq data analyses and proteomics approaches, especially mapping of protein N-termini, demonstrated that translation from upstream or downstream aTISs, in addition to the most common expression of uORFs, may also produce extended or truncated protein isoforms with potentially different properties (18,19,21,23–25,56,83,86,87,90–99). These analyses were mainly conducted for higher eukaryotes and analogous data was not available for yeast until the most recent study, which reported that utilization of near-cognate translation initiation resulting in the expression of uORFs or N-terminally extended proteins is controlled by DEAD-box RNA helicase Ded1 (87). Notably, regulation of uORF and NTE protein expression from aTISs may be particularly relevant in stress conditions (e.g. oxidative and ER stress, integrated stress response, starvation) or during the cell cycle (e.g. meiosis) (81,87,100–104).
While the role of uORFs in multiple aspects of translation regulation is relatively well described, the impact of N-terminal extensions generated by upstream non-canonical translation is still underappreciated. In principle, such NTEs may confer various auxiliary functions, but to date mainly NTE-mediated localization to different cellular compartments, usually mitochondria, has been predicted, or in a few cases demonstrated, in mammals, plants and to a limited extent also in yeast (18,19,28–33,56). Our in silico screen to identify NTE-containing proteins in yeast supported by Ribo-seq data demonstrates that more than a thousand genes may undergo translation from upstream aTISs producing additional protein isoforms, of which several hundred are potentially targeted to mitochondria via the NTE-provided MTS. We have confirmed mitochondrial localization of the majority of analysed candidates with the NTE-provided MTSs, and therefore it is likely that many other proteins from our screen with predicted MTS are also targeted to this compartment. In addition to altered localization N-terminal extensions may create additional signals, structural motifs and functional domains. Such a role in forming a membrane anchoring domain has been suggested in the case of the long Kgd4 isoform (105,106), which was also identified in our in silico screen as the non-AUG initiated NTE-containing protein.
Although Gene Ontology term analysis of isoforms with ‘MTS gain’ did not reveal significant differences between genome frequency and frequency in analysed sets, many of these proteins have plausible roles in mitochondria. Interestingly, several of potentially mitochondria-targeted NTE proteins are associated with the ribosome function. Indeed, at least 34 proteins among candidates with ‘MTS gain’ (see Supplemental Table S4) are annotated as involved in different steps of ribosome maturation or activity according to SGD. While >200 factors have been described to be required for the formation of cytosolic ribosomes, biogenesis of mitochondrial ribosomes is still poorly described and thus far only a handful of proteins have a well-documented role in this process (74). Other interesting categories related to RNA metabolism include transcription by RNA polymerase II, pre-mRNA splicing, RNA processing and decay. At this moment it is difficult to predict exactly how these factors contribute to the metabolism of mitochondrial RNAs, but at least some proteins have been reported to have a mitochondrial function. In addition to Hts1, Vas1, Ala1 and Grs1 enzymes that aminoacylate tRNAs in mitochondria (28,29,32,33), other cases include endonuclease Trz1 that participates in the processing of mitochondrial transcripts (70), Rnh1 (Ribonuclease H1, RNase H1) that cleaves RNA–DNA hybrids and has been implicated in the resolution of R-loops in mitochondria (107); a DEAH box RNA spliceosomal helicase Prp43 that has been postulated to function in mitochondria in apoptosis (108) and Jsn1, a cytoplasmic member of the Puf RNA binding family proteins that also facilitates localization of Arp2/3 to mitochondria (109). Notably, human homologs of some of these factors, namely tRNA endonuclease RNAse ZL, RNase H1 and helicase DHX15, have a dual nuclear-mitochondrial localization and a described mitochondrial function (110–113). Our initial survey of mitochondrial association of human homologs of selected ‘MTS gain’ yeast proteins suggests that generation of differentially localized variants generated via aTIS is to some extent conserved. Unsurprisingly, mammalian cells appear to use a wider range of mechanisms to produce alternative isoforms. Our data therefore also serves as a powerful prediction tool for hitherto undetected mammalian mitochondrial isoforms and may have direct consequences for the identification of mitochondrial disorders, especially considering that MTS-containing isoforms are usually expressed at low levels, preventing their SNPS to be recognized as pathogenic. In summary, our work results in a sizable expansion of the mitochondrial proteome and provides an essential resource for the investigation of possible cryptic mitochondrial localization and function of a variety of proteins with different cellular activities.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Teresa Żołądek and Roman Szczęsny (IBB, Polish Academy of Sciences) for p415-ADH, p416-ADH, p426-ADH and p413-ADH vectors; Ophry Pines (Hebrew University Medical School, Israel) for pGalYep51-1-(Aco1-α) and pGalSu9(1–69)ω plasmids; Aneta Kaniak-Golik (IBB, Polish Academy of Sciences), Karolina Łabędzka-Dmoch and Michał Koper (University of Warsaw), Leila Polus and Ali Julfiker Masud (University of Oulu) for technical assistance and advice; Gretchen Edwalds-Gilbert (Scripps College, USA) for critically reading the manuscript.
Authors contributions: J.K. and A.J.K. conceived and directed the studies and wrote the manuscript, with the contribution of G.M., A.M. and M.S. G.M. participated in the planning of the in silico screen and carried out the α-complementation and Split-Intein-GFP experiments. G.M. and J.A. carried out the Has1 analyses. A.M. performed the conservation analysis and cellular fractionations and western blot analyses, with the assistance of L.W. and A.C. L.Z. and O.N. performed aTIS in silico screen. M.S. carried out Ribo-seq analyses.
Notes
Present address: Geoffray Monteuuis, Gene and Stem Cell Therapy Program, Centenary Institute, The University of Sydney, Camperdown, Australia.
Present address: Lidia Wrobel, Department of Medical Genetics, Cambridge Institute for Medical Research, University of Cambridge, Wellcome/MRC Building, Addenbrooke's Hospital, Hills Road, Cambridge CB2 0XY, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Polish-Swiss Research Programme [PSPB-183/2010]; Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund [TEAM POIR.04.04.00-00-5C33/17-00 to J.K.]; Academy of Finland, Sigrid Juselius Foundation and AFM-Téléthon grants and by Biocenter Oulu (to A.J.K.). Experiments were carried out with the use of CePT infrastructure financed by the European Union – the European Regional Development Fund (Innovative economy 2007–2013 [POIG.02.02.00-14-024/08-00]. Funding for open access charge. Foundation for Polish Science [TEAM POIR.04.04.00-00-5C33/17-00].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gonczarowska-Jorge H., Zahedi R.P., Sickmann A.. The proteome of baker's yeast mitochondria. Mitochondrion. 2017; 15–21. [DOI] [PubMed] [Google Scholar]
- 2. von Heijne G., Steppuhn J., Herrmann R.G.. Domain structure of mitochondrial and chloroplast targeting peptides. Eur. J. Biochem. 1989; 180:535–545. [DOI] [PubMed] [Google Scholar]
- 3. Sokol A.M., Sztolsztener M.E., Wasilewski M., Heinz E., Chacinska A.. Mitochondrial protein translocases for survival and wellbeing. FEBS Lett. 2014; 588:2484–2495. [DOI] [PubMed] [Google Scholar]
- 4. Gakh O., Cavadini P., Isaya G.. Mitochondrial processing peptidases. Biochim. Biophys. Acta. 2002; 1592:63–77. [DOI] [PubMed] [Google Scholar]
- 5. Kunze M., Berger J.. The similarity between N-terminal targeting signals for protein import into different organelles and its evolutionary relevance. Front. Physiol. 2015; 6:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neupert W., Herrmann J.M.. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007; 76:723–749. [DOI] [PubMed] [Google Scholar]
- 7. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R. et al.. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith A.C., Robinson A.J.. MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 2016; 44:D1258–D1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calvo S.E., Mootha V.K.. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010; 11:25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morgenstern M., Stiller S.B., Lübbert P., Peikert C.D., Dannenmaier S., Drepper F., Weill U., Höß P., Feuerstein R., Gebert M. et al.. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 2017; 19:2836–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Silva-Filho M.C. One ticket for multiple destinations: dual targeting of proteins to distinct subcellular locations. Curr. Opin. Plant Biol. 2003; 6:589–595. [DOI] [PubMed] [Google Scholar]
- 12. Regev-Rudzki N., Pines O.. Eclipsed distribution: a phenomenon of dual targeting of protein and its significance. Bioessays. 2007; 29:772–782. [DOI] [PubMed] [Google Scholar]
- 13. Carrie C., Giraud E., Whelan J.. Protein transport in organelles: dual targeting of proteins to mitochondria and chloroplasts. FEBS J. 2009; 276:1187–1195. [DOI] [PubMed] [Google Scholar]
- 14. Yogev O., Pines O.. Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta. 2011; 1808:1012–1020. [DOI] [PubMed] [Google Scholar]
- 15. Touriol C., Bornes S., Bonnal S., Audigier S., Prats H., Prats A.C., Vagner S.. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol. Cell. 2003; 95:169–178. [DOI] [PubMed] [Google Scholar]
- 16. Kearse M.G., Wilusz J.E.. Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 2017; 31:1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sriram A., Bohlen J., Teleman A.A.. Translation acrobatics: how cancer cells exploit alternate modes of translational initiation. EMBO Rep. 2018; 19:e45947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kochetov A.V., Sarai A., Rogozin I.B., Shumny V.K., Kolchanov N.A.. The role of alternative translation start sites in the generation of human protein diversity. Mol. Genet. Genomics. 2005; 273:491–496. [DOI] [PubMed] [Google Scholar]
- 19. Wamboldt Y., Mohammed S., Elowsky C., Wittgren C., de Paula W.B., Mackenzie S.A.. Participation of leaky ribosome scanning in protein dual targeting by alternative translation initiation in higher plants. Plant Cell. 2009; 21:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ivanov I.P., Firth A.E., Michel A.M., Atkins J.F., Baranov P.V.. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 2011; 39:4220–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tzani I., Ivanov I.P., Andreev D.E., Dmitriev R.I., Dean K.A., Baranov P.V., Atkins J.F., Loughran G.. Systematic analysis of the PTEN 5′ leader identifies a major AUU initiated proteoform. Open Biol. 2016; 6:150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang L., Morris J., Wan J., Moore C., Fujita Y., Gillaspie S., Aube E., Nanda J., Marques M., Jangal M. et al.. Competition between translation initiation factor eIF5 and its mimic protein 5MP determines non-AUG initiation rate genome-wide. Nucleic Acids Res. 2017; 45:11941–11953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diaz de Arce A.J., Noderer W.L., Wang C.L.. Complete motif analysis of sequence requirements for translation initiation at non-AUG start codons. Nucleic Acids Res. 2017; 46:985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yeom J., Ju S., Choi Y., Paek E., Lee C.. Comprehensive analysis of human protein N-termini enables assessment of various protein forms. Sci. Rep. 2017; 7:6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zitomer R.S., Walthall D.A., Rymond B.C., Hollenberg C.P.. Saccharomyces cerevisiae ribosomes recognize non-AUG initiation codons. Mol. Cell. Biol. 1984; 4:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang C.P., Chen S.J., Lin C.H., Wang T.L., Wang C.C.. A single sequence context cannot satisfy all non-AUG initiator codons in yeast. BMC Microbiol. 2010; 10:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang H.L., Yeh L.S., Chen N.K., Ripmaster T., Schimmel P., Wang C.C.. Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. J. Biol. Chem. 2004; 279:49656–49663. [DOI] [PubMed] [Google Scholar]
- 29. Chang K.J., Wang C.C.. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J. Biol. Chem. 2004; 279:13778–13785. [DOI] [PubMed] [Google Scholar]
- 30. Kritsiligkou P., Chatzi A., Charalampous G., Mironov A.J., Grant C.M., Tokatlidis K.. Unconventional targeting of a thiol peroxidase to the mitochondrial intermembrane space facilitates oxidative protein folding. Cell Rep. 2017; 18:2729–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suomi F., Menger K.E., Monteuuis G., Naumann U., Kursu V.A., Shvetsova A., Kastaniotis A.J.. Expression and evolution of the non-canonically translated yeast mitochondrial acetyl-CoA carboxylase Hfa1p. PLoS One. 2014; 9:e114738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Natsoulis G., Hilger F., Fink G.R.. The HTS1 gene encodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae. Cell. 1986; 46:235–243. [DOI] [PubMed] [Google Scholar]
- 33. Chatton B., Walter P., Ebel J.P., Lacroute F., Fasiolo F.. The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl-tRNA synthetases. J. Biol. Chem. 1988; 263:52–57. [PubMed] [Google Scholar]
- 34. Williams C.C., Jan C.H., Weissman J.S.. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science. 2014; 346:748–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meisinger C., Pfanner N., Truscott K.N.. Isolation of yeast mitochondria. Methods Mol. Biol. 2006; 313:33–39. [DOI] [PubMed] [Google Scholar]
- 36. Meisinger C., Sommer T., Pfanner N.. Purification of Saccharomcyes cerevisiae mitochondria devoid of microsomal and cytosolic contaminations. Anal. Biochem. 2000; 287:339–342. [DOI] [PubMed] [Google Scholar]
- 37. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S. et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wickham H., François R., Henry L., Müller K.. dplyr: A Grammar of Data Manipulation. 2018; R package version 0.7.8. [Google Scholar]
- 39. Müller K., Wickham H.. tibble: Simple Data Frames. 2018; R package version 1.4.2. [Google Scholar]
- 40. Pagès H., Aboyoun P., Gentleman R., DebRoy S.. Biostrings: Efficient Manipulation of Biological Strings. 2018; R package version 2.48.0. [Google Scholar]
- 41. Kaisers W. refGenome: Gene and Splice Site Annotation Using Annotation Data from Ensembl and UCSC Genome Browsers. 2017; R package version 1.7.3. [Google Scholar]
- 42. R Core Team R: a language and environment for statistical computing. R Foundation for Statistical Computing. 2018; Vienna: http://www.R-project.org/. [Google Scholar]
- 43. Pelechano V., Wei W., Steinmetz L.M.. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013; 497:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J.. The ucsc table browser data retrieval tool. Nucleic Acids Res. 2004; 32:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerashchenko M.V., Gladyshev V.N.. Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res. 2014; 42:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gerashchenko M.V., Gladyshev V.N.. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 2017; 45:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nedialkova D.D., Leidel S.A.. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell. 2015; 161:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nissley D.A., Sharma A.K., Ahmed N., Friedrich U.A., Kramer G., Bukau B., O’Brien E.P.. Accurate prediction of cellular co-translational folding indicates proteins can switch from post- to co-translational folding. Nat. Commun. 2016; 7:10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fang H., Huang Y.F., Radhakrishnan A., Siepel A., Lyon G.J., Schatz M.C.. Scikit-ribo enables accurate estimation and robust modeling of translation dynamics at codon resolution. Cell Syst. 2018; 6:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2016; NY: Springer. [Google Scholar]
- 52. Chiu M., Mason T.L., Fink G.R.. HTS1 encodes both the cytoplasmic and mitochondrial histidyl-tRNA synthetase of Saccharomyces cerevisiae: mutations alter the specificity of compartmentation. Genetics. 1992; 132:987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Turner R.J., Lovato M., Schimmel P.. One of two genes encoding glycyl-tRNA synthetase in Saccharomyces cerevisiae provides mitochondrial and cytoplasmic functions. J. Biol. Chem. 2000; 275:27681–27688. [DOI] [PubMed] [Google Scholar]
- 54. Hinnebusch A.G., Ivanov I.P., Sonenberg N.. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016; 352:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anaganti S., Hansen J.K., Ha D., Hahn Y., Chertov O., Pastan I., Bera T.K.. Non-AUG translational initiation of a short CAPC transcript generating protein isoform. Biochem. Biophys. Res. Commun. 2009; 380:508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kazak L., Reyes A., Duncan A.L., Rorbach J., Wood S.R., Brea-Calvo G., Gammage P.A., Robinson A.J., Minczuk M., Holt I.J.. Alternative translation initiation augments the human mitochondrial proteome. Nucleic Acids Res. 2013; 41:2354–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002; 299:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Van Damme P., Støve S.I., Glomnes N., Gevaert K., Arnesen T.. A Saccharomyces cerevisiae model reveals in vivo functional impairment of the Ogden syndrome N-terminal acetyltransferase NAA10 Ser37Pro mutant. Mol. Cell. Proteomics. 2014; 13:2031–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Helsens K., Van Damme P., Degroeve S., Martens L., Arnesen T., Vandekerckhove J., Gevaert K.. Bioinformatics analysis of a Saccharomyces cerevisiae N-terminal proteome provides evidence of alternative translation initiation and post-translational N-terminal acetylation. J. Proteome Res. 2011; B10:3578–3589. [DOI] [PubMed] [Google Scholar]
- 60. Claros M.G., Vincens P.. Computational method to predict mitochondrially imported proteins and their targeting sequences. FEBS J. 1996; 241:779–786. [DOI] [PubMed] [Google Scholar]
- 61. Emanuelsson O., Brunak S., von Heijne G., Nielsen H.. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007; 2:953–971. [DOI] [PubMed] [Google Scholar]
- 62. Fukasawa Y., Tsuji J., Fu S.C., Tomii K., Horton P., Imai K.. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol. Cell. Proteomics. 2015; 14:1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Backes S., Hess S., Boos F., Woellhaf M.W., Gödel S., Jung M., Mühlhaus T., Herrmann J.M.. Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J Cell Biol. 2018; 217:1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Karniely S., Rayzner A., Sass E., Pines O.. Alpha-complementation as a probe for dual localization of mitochondrial proteins. Exp. Cell Res. 2006; 312:3835–3846. [DOI] [PubMed] [Google Scholar]
- 65. Regev-Rudzki N., Karniely S., Ben-Haim N.N., Pines O.. Yeast aconitase in two locations and two metabolic pathways: seeing small amounts is believing. Mol. Biol. Cell. 2005; 16:4163–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ozawa T., Takeuchi T.M., Kaihara A., Sato M., Umezawa Y.. Protein splicing-based reconstitution of split green fluorescent protein for monitoring protein-protein interactions in bacteria: improved sensitivity and reduced screening time. Anal. Chem. 2001; 73:5866–5874. [DOI] [PubMed] [Google Scholar]
- 67. Ozawa T., Sako Y., Sato M., Kitamura T., Umezawa Y.. A genetic approach to identifying mitochondrial proteins. Nat. Biotechnol. 2003; 21:287–293. [DOI] [PubMed] [Google Scholar]
- 68. Young E.T., Pilgrim D.. Isolation and DNA sequence of ADH3, a nuclear gene encoding the mitochondrial isozyme of alcohol dehydrogenase in Saccharomyces cerevisiae. Mol. Cell Biol. 1985; 5:3024–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vögtle F.N., Burkhart J.M., Rao S., Gerbeth C., Hinrichs J., Martinou J.C., Chacinska A., Sickmann A., Zahedi R.P., Meisinger C.. Intermembrane space proteome of yeast mitochondria. Mol. Cell Proteomics. 2012; 11:1840–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Skowronek E., Grzechnik P., Späth B., Marchfelder A., Kufel J.. tRNA 3′ processing in yeast involves tRNase Z, Rex1, and Rrp6. RNA. 2014; 20:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stram A.R., Payne R.M.. Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol. Life Sci. 2016; 73:4063–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Merz S., Westermann B.. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009; 10:R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang Y., Qi H., Taylor R., Xu W., Liu L.F., Jin S.. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007; 3:337–346. [DOI] [PubMed] [Google Scholar]
- 74. Fernández-Pevida A., Kressler D., de la Cruz J.. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip. Rev. RNA. 2015; 6:191–209. [DOI] [PubMed] [Google Scholar]
- 75. Chen Y., Beck A., Davenport C., Shattuck D., Tavtigian S.V.. Characterization of TRZ1, a yeast homolog of the human candidate prostate cancer susceptibility gene ELAC2 encoding tRNase Z. BMC Mol. Biol. 2005; 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Reuter K., Biehl A., Koch L., Helms V.. PreTIS: A tool to predict non-canonical 5′ UTR translational initiation sites in human and mouse. PLoS Comput. Biol. 2016; 12:e1005170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. de Klerk E., ’t Hoen P.A.. Alternative mRNA transcription, processing, and translation: insights from RNA sequencing. Trends Genet. 2015; 31:128–139. [DOI] [PubMed] [Google Scholar]
- 78. Mouilleron H., Delcourt V., Roucou X.. Death of a dogma: eukaryotic mRNAs can code for more than one protein. Nucleic Acids Res. 2016; 44:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pueyo J.I., Magny E.G., Couso J.P.. New peptides under the s(ORF)ace of the genome. Trends Biochem. Sci. 2016; 41:665–678. [DOI] [PubMed] [Google Scholar]
- 80. Plaza S., Menschaert G., Payre F.. In search of lost small peptides. Annu. Rev. Cell Dev. Biol. 2017; 33:391–416. [DOI] [PubMed] [Google Scholar]
- 81. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee S., Liu B., Lee S., Huang S.-X., B. S., Qian S.-B.. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fritsch C., Herrmann A., Nothnagel M., Szafranski K., Huse K., Schumann F., Schreiber S., Platzer M., Krawczak M., Hampe J. et al.. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012; 22:2208–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ingolia N.T., Brar G.A., Stern-Ginossar N., Harris M.S., Talhouarne G.J., Jackson S.E., Wills M.R., Weissman J.S.. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014; 8:1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Michel A.M., Andreev D.E., Baranov P.V.. Computational approach for calculating the probability of eukaryotic translation initiation from ribo-seq data that takes into account leaky scanning. BMC Bioinformatics. 2014; 15:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang X., Hou J., Quedenau C., Chen W.. Pervasive isoform-specific translational regulation via alternative transcription start sites in mammals. Mol. Syst. Biol. 2016; 12:875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guenther U.P., Weinberg D.E., Zubradt M.M., Tedeschi F.A., Stawicki B.N., Zagore L., Brar G.A., Licatalosi D.D., Bartel D.P., Weissman J.S. et al.. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature. 2018; 559:130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rojas-Duran M.F., Gilbert W.V.. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA. 2012; 18:2299–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kurihara Y., Makita Y., Kawashima M., Fujita T., Iwasaki S., Matsui M.. Transcripts from downstream alternative transcription start sites evade uORF-mediated inhibition of gene expression in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:7831–7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oyama M., Kozuka-Hata H., Suzuki Y., Semba K., Yamamoto T., Sugano S.. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol. Cell Proteomics. 2007; 6:1000–1006. [DOI] [PubMed] [Google Scholar]
- 91. Fournier C.T., Cherny J.J., Truncali K., Robbins-Pianka A., Lin M.S., Krizanc D., Weir M.P.. Amino termini of many yeast proteins map to downstream start codons. J. Proteome Res. 2012; 11:5712–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Menschaert G., Van Criekinge W., Notelaers T., Koch A., Crappé J., Gevaert K., Van Damme P.. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell Proteomics. 2013; 12:1780–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Koch A., Gawron D., Steyaert S., Ndah E., Crappé J., De Keulenaer S., De Meester E., Ma M., Shen B., Gevaert K. et al.. A proteogenomics approach integrating proteomics and ribosome profiling increases the efficiency of protein identification and enables the discovery of alternative translation start sites. Proteomics. 2014; 14:2688–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Van Damme P., Gawron D., Van Criekinge W., Menschaert G.. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol. Cell Proteomics. 2014; 13:1245–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hsu P.Y., Calviello L., Wu H.L., Li F.W., Rothfels C.J., Ohler U., Benfey P.N.. Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E7126–E7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sendoel A., Dunn J.G., Rodriguez E.H., Naik S., Gomez N.C., Hurwitz B., Levorse J., Dill B.D., Schramek D., Molina H. et al.. Translation from unconventional 5’ start sites drives tumour initiation. Nature. 2017; 541:494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang P., He D., Xu Y., Hou J., Pan B.F., Wang Y., Liu T., Davis C.M., Ehli E.A., Tan L. et al.. Genome-wide identification and differential analysis of translational initiation. Nat. Commun. 2017; 8:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Spealman P., Naik A.W., May G.E., Kuersten S., Freeberg L., Murphy R.F., McManus J.. Conserved non-AUG uORFs revealed by a novel regression analysis of ribosome profiling data. Genome Res. 2018; 28:214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. van der Horst S., Snel B., Hanson J., Smeekens S.. Novel pipeline identifies new upstream ORFs and non-AUG initiating main ORFs with conserved amino acid sequences in the 5′ leader of mRNAs in Arabidopsis thaliana. RNA. 2019; 25:292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gerashchenko M.V., Lobanov A.V., Gladyshev V.N.. Genomewide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:7394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Andreev D.E., O’Connor P.B., Fahey C., Kenny E.M., Terenin I.M., Dmitriev S.E., Cormican P., Morris D.W., Shatsky I.N., Baranov P.V.. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. Elife. 2015; 4:e03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M.. The integrated stress response. EMBO Rep. 2016; 17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Starck S.R., Tsai J.C., Chen K., Shodiya M., Wang L., Yahiro K., Martins-Green M., Shastri N., Walter P.. Translation from the 5′ untranslated region shapes the integrated stress response. Science. 2016; 351:aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Van Dalfsen K.M., Hodapp S., Keskin A., Otto G.M., Berdan C.A., Higdon A., Cheunkarndee T., Nomura D.K., Jovanovic M., Brar G.A.. Global proteome remodeling during ER stress involves Hac1-driven expression of long undecoded transcript isoforms. Dev. Cell. 2018; 46:219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Heublein M., Burguillos M.A., Vögtle F.N., Teixeira P.F., A. I., C. M., Ott M.. The novel component Kgd4 recruits the E3 subunit to the mitochondrial α-ketoglutarate dehydrogenase. Mol. Biol. Cell. 2014; 25:3342–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Heublein M. Mitochondrial Energy Metabolism: Kgd4 is a novel subunit of the α-ketoglutarate dehydrogenase complex. 2014; PhD thesisDepartment of Biochemistry and Biophysics, Stockholm University. [Google Scholar]
- 107. El Hage A., Webb S., Kerr A., Tollervey D.. Genome-wide distribution of RNA-DNA hybrids identifies RNase H targets intRNA genes, retrotransposons and mitochondria. PLoS Genet. 2014; 10:e100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Heininger A. Characterisation of the yeast RNA helicase Prp43. 2016; PhD thesisSweden: University of Gottingen. [Google Scholar]
- 109. Fehrenbacher K.L., Boldogh I.R., Pon L.A.. A role for Jsn1p in recruiting the Arp2/3 complex to mitochondria in budding yeast. Mol. Biol. Cell. 2005; 16:5094–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Cerritelli S.M., Frolova E.G., Feng C., Grinberg A., Love P.E., Crouch R.J.. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell. 2003; 11:807–815. [DOI] [PubMed] [Google Scholar]
- 111. Rossmanith W. Localization of human RNase Z isoforms: dual nuclear/mitochondrial targeting of the ELAC2 gene product by alternative translation initiation. PLoS One. 2011; 6:e19152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mosallanejad K., Sekine Y., Ishikura-Kinoshita S., Kumagai K., Nagano T., Matsuzawa A., Takeda K., Naguro I., Ichijo H.. The DEAH-box RNA helicase DHX15 activates NF-κB and MAPK signaling downstream of MAVS during antiviral responses. Sci. Signal. 2014; 7:ra40. [DOI] [PubMed] [Google Scholar]
- 113. Holmes J.B., Akman G., Wood S.R., Sakhuja K., Cerritelli S.M., Moss C., Bowmaker M.R., Jacobs H.T., Crouch R.J., Holt I.J.. Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9334–9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.