


Abstract
The phenotypic adjustments of Mycobacterium tuberculosis are commonly inferred from the analysis of transcript abundance. While mechanisms of transcriptional regulation have been extensively analysed in mycobacteria, little is known about mechanisms that shape the transcriptome by regulating RNA decay rates. The aim of the present study is to identify the core components of the RNA degradosome of M. tuberculosis and to analyse their function in RNA metabolism. Using an approach involving cross-linking to 4-thiouridine-labelled RNA, we mapped the mycobacterial RNA-bound proteome and identified degradosome-related enzymes polynucleotide phosphorylase (PNPase), ATP-dependent RNA helicase (RhlE), ribonuclease E (RNase E) and ribonuclease J (RNase J) as major components. We then carried out affinity purification of eGFP-tagged recombinant constructs to identify protein-protein interactions. This identified further interactions with cold-shock proteins and novel KH-domain proteins. Engineering and transcriptional profiling of strains with a reduced level of expression of core degradosome ribonucleases provided evidence of important pleiotropic roles of the enzymes in mycobacterial RNA metabolism highlighting their potential vulnerability as drug targets.
INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis continues to present a major bacterial threat to global health, with increasing emergence of strains resistant to current drugs and enhanced susceptibility to disease in association with HIV and diabetes (1). Sequencing of the M. tuberculosis genome in 1998 initiated a new era in molecular mycobacteriology with the link to microarray-based transcriptional profiling providing an important route to move from genotype to phenotype analysis (2). While there have been recent advances in proteome and metabolome studies, the transcriptome has provided the basis for the bulk of our current understanding of mycobacterial physiology. This has involved detailed characterization of transcriptional regulators, combining chromatin immunoprecipitation with transcript analysis to generate extensive gene regulatory networks (3,4). Less attention has been given to understanding the dynamics of RNA turnover in mycobacteria. A study by Rustad et al. (5) measured the half-life of mRNA transcripts in M. tuberculosis, documenting decay over a timescale of 5–18 min during the exponential phase of cell growth, with a marked stabilization in response to physiologically relevant stresses, e.g. under hypoxic stress. Understanding the mechanisms that regulate mRNA degradation, and the way that they are influenced by the growth environment, has the potential to provide a key advance in the accurate interpretation of information from studies of steady-state transcript abundance.
mRNA degradation has been extensively characterized using more tractable model organisms such as Escherichia coli and Bacillus subtilis in which it has been shown to involve degradosome complexes built around a membrane-bound multi-domain ribonuclease scaffold: RNase E in the case of most Gram-negative bacteria (6) and RNase Y in model Gram-positive microorganisms (7). RNase E and Y are both endonucleases with preference for single-stranded, 5′-monophosphorylated RNA species (8,9). Other key components include exonucleases: polynucleotide phosphorylase (PNPase), a phosphorolytic enzyme processing RNA in the 3′-to-5′ direction (10), and hydrolytic RNase J, which is present in Gram-positive bacteria and actinobacteria and operates from 5′-to-3′ ends (11). The core degradosome of E. coli also includes RNA helicase RhlB, which can be replaced by orthologous RNA helicases, RhlE and cold shock protein CsdA (12). In B. subtilis, the RhlB helicase function is replaced by RNase Y acting in concert with RNase J1 and RNaseJ2 (7). Degradosomes from E. coli and B. subtilis both include enolase enzymes from the glycolysis pathway, with the additional presence of phosphofructokinase in B. subtilis, suggesting a possible link between RNA turnover and central carbon metabolism. Additional proteins can associate with the core degradosome, such as the Hfq chaperone that regulates small RNA-mediated mRNA decay (6). However, no homologue for Hfq has been identified in M. tuberculosis.
Mutations in PNPase were recently linked with M. tuberculosis resistance to the first-line antituberculosis drug—Pyrazinamide (PZA) (13). The study identified PNPase as molecular target inhibited by pyrazinoic acid (POA), an active compound produced by metabolic conversion of PZA. Single-stranded DNA and RNA polymerization and phosphorolysis activities of PNPase, as well as (p)ppGpp hydrolase activity, were all affected by the POA in vitro. The expression of PNPase Asp67Asn mutant allele, found in clinical isolates of M. tuberculosis, was able to recapitulate PZA resistance in vivo.
RNase E from M. tuberculosis has been partially characterized. The mycobacterial enzyme, like the RNase E present in other Actinobacteria, shows domain shuffling when compared to well characterized gammaproteobacterial enzymes. In Streptomyces coelicolor, the N-terminal domain is reported to be necessary for interaction with PNPase (14). The E. coli protein possesses a C-terminal scaffolding domain instead. The recombinant polypeptide MycRne (6xHis tagged portion of M. tuberculosis RNase E containing amino acids 332–953) was shown to exhibit an endonucleolytic activity dependent on the 5′-phosphorylation status of the RNA substrate (15). In another study, a 6xHis-Flag-tagged recombinant RNase E from Mycobacterium bovis (M. bovis) BCG was used to pull down RNase E interacting proteins. Rather than the anticipated degradosome components, isolation of a protein complex resulted in co-purification with GroEL chaperone, polyphosphate kinase and an acetyltransferase (16). A study describing the identification of a small molecule inhibitor with activity against M. tuberculosis RNase E highlights the potential for targeting RNA turnover in antibacterial drug discovery (17).
The present study is designed to initiate molecular characterization of the mycobacterial degradosome complexes.
MATERIALS AND METHODS
Bacterial strains and culturing conditions
Strains, plasmids and primers used to generate genetic constructs are listed in Supplementary Table S1 (Supplementary File). Saprophytic, fast-growing M. smegmatis mc2155, and laboratory strains of M. bovis BCG Danish 1331 and pathogenic M. tuberculosis H37Rv strains were used in this study. The M. smegmatis and M. bovis BCG strains were cultured under standard laboratory conditions as described in detail elsewhere (18). The M. tuberculosis strain was cultured under the rigours of Biosafety level 3 laboratory in Middlebrook 7H9 broth supplemented with OADC additive (Beckton Dickinson), in roller flasks or on 7H10 solid agar with OADC (Beckton Dickinson). Recombinant protein expression was carried out from tetracycline inducible pKW08 vector, as GFP fusion proteins (18), by adding tetracycline–HCl to the growth media at the final concentration of 50 ng/ml for 48 h for M. tuberculosis. Growth was monitored by measuring the optical density of the cultures. For experiments involving RNA–protein crosslinking, a photoactivatable analogue of uridine, 4-thio-uridine (4SU, Sigma Aldrich), was added to the culture media following the induction with tetracycline, if applicable, at 50 μM final concentration. 4SU was added 16 h before harvesting the cells for slow-growing mycobacterial strains. Upon harvest, the cells treated with 4SU were cross-linked with UV365 nm to induce protein–RNA bond formation by three rounds of irradiation at the dose of 1200 × 100 μJ cm−2, in CL-1000 UV crosslinker (UVP, Inc.).
Sucrose gradient fractionation of RNA-bound proteins
For determination of the protein elements bound to the mycobacterial RNA, exponentially growing, 4SU fed cultures of M. bovis BCG str. Copenhagen were collected, crosslinked and frozen in liquid nitrogen. When needed, the cells were thawed and lysed in lysis buffer containing 20 mM HEPES pH 7.5, 200 mM KCl, 1% Triton X100r, 2 mM DTT, 5 U/ml of DNase turbo using Diagenode sonicator set to high settings for 30 cycles of 30 s ON and 30 s OFF each cycle. The cell lysate was precleared by centrifugation at 21 000 × g for 20 min, passed through a 0.22 μm Ultrafree-MC microcentrifuge filter (Millipore) and overlaid onto a freshly prepared continuous 7–47% sucrose gradient (19). The sample was spun at 39 000 rpm (∼250 000 × g at maximal radius) for 2.5 h with ‘no brake’ deceleration, in a swinging bucket SW41Ti rotor in a Beckman Coulter L90 ultracentrifuge and gradients were collected using AKTA Purifier (GE Healthcare) equipped with UV monitor. For non-denaturing conditions, fractions containing RNA–protein complexes with a sedimentation index lower than that of a 30S subunit of the ribosome were pooled together and supplemented with 50 mM Tris buffer pH 7.5 to dilute the salt content of the sample below 100 mM. In parallel, samples were denatured by adding urea up to 8 M (w/v) and DTT up to 2 mM. Next, they were loaded onto ResourceQ column and RNA–protein complexes were eluted with 50 mM Tris pH 7.5 buffer with a gradient of 100 mM to 1 M NaCl, monitoring the UV254 nm:UV280 nm ratio. Samples were precipitated with pyrogallol red-molybdate (PRM) reagent and analysed by mass spectrometry as previously reported (18).
Protein complexes purification
Potential degradosome components identified as RNA binding proteins in the initial experiment were cloned into pKW08-eGFP tetracycline inducible vector and resulting constructs were used to transform M. bovis BCG str. Copenhagen or M. tuberculosis H37Rv. Protein expression was induced by adding 50 ng/ml of tetracycline–HCl to exponentially grown cultures at OD600 ∼0.6 for ∼48 h. Protein complex purification was done essentially as previously described in details elsewhere (18), except in the case of M. tuberculosis, where the cells were lysed by bead-beating in the MP FastPrep-24 instrument set to 6.0 m/s for 2 cycles of 45 s each, with 5 min intervals on ice in between each cycle. The lysate was precleared by centrifugation at 21 000 × g for 30 min and passed through a 0.2 μm spin filter before removing from biosafety category 3 lab to be processed further exactly as BCG lysates obtained by sonication.
Construction of strains with altered level of RNA degradosome nucleases
Mycobacterium tuberculosis rnj knockout and M. smegmatis gpsI (PNPase) conditional mutant were created following the two-step recombination protocol (20). A 6.1 kb PacI cassette from pGOAL17, carrying the lacZ, aph and sacB genes was inserted into the p2NIL plasmid, next to truncated and out-of-frame ligated copies of either rnj or gpsI genes to create appropriate suicidal delivery vectors used for the gene replacement. M. smegmatis gpsI double cross-over knockouts were screened in the presence of a complementing copy of gpsImtb-egfp fusion gene, expressed from the attB-integrating, tetracycline-regulated pKW08 plasmid. Gene deletion was confirmed by PCR and Southern blotting analysis. Tetracycline dependency of the conditional PNPase mutant strain of M. smegmatis was tested on the tetracycline gradient plates (7H10 with OADC supplement used as a base), with the expected tetracycline concentrations ranging from 0 to 100 ng/ml (Supplementary Figure S1).
To generate strains with depletion of the essential ribonucleases PNPase and RNase E, we have used the CRISPR/Cas9 strategy optimized by Rock et al. (21). The appropriate sgRNA probes were planned according to the published protocol and the list of sgDNA sequences is provided in the supplementary information. The appropriate fragments were cloned into the pLJR965 plasmid, that was used to transform M. tuberculosis H37Rv laboratory strain via standard electroporation procedure. The resulting Cas9 and sgRNA expressing strains were confirmed by PCR amplification for the presence of the Cas9 cassette and tested for the efficiency of silencing by monitoring the growth kinetics of tested strains in the presence of the anhydrotetracycline inducer. For total RNA sequencing, the strains were inoculated at an OD600 of 0.05 and the production of sgRNA was initiated by adding 100 ng/ml of anhydrotetracycline for a period of 7 days, when the cells were harvested by centrifugation and RNA was isolated.
Illumina compatible libraries preparation
For total RNA sequencing, RNA was isolated as described previously with Trizol LS reagent (Invitrogen) (22). Cells were lysed by bead-beating with the MP FastPrep system (MP Biomedicals) using lysing matrix B (2 × 45 s, 6.0 m/s with 5 min intervals on ice). DNase I turbo (Invitrogen by Thermo Fisher Scientific) digestion was performed to remove DNA contaminations according to the manufacturer's protocol. RNA quantity and integrity was assessed using an Agilent 2100 BioAnalyzer per manufacturer's instructions (Agilent RNA 6000 Nano Kit). RNA samples were purified (AMPure XP magnetic beads, Becton Dickinson), rRNA was removed (Ribo-Zero rRNA Removal Kit, Illumina) and sequencing libraries were generated (KAPA Stranded RNA-Seq kit, KAPA Biosystems) according to the detailed instructions provided by the manufacturers. Illumina True Seq v2 indexing system was used to allow multiplex sequencing. The quality and quantity of resulting libraries were examined on an Agilent 2100 BioAnalyzer fitted with DNA 1000 chip. For loading onto sequencing flow cells, the samples were quantified by qPCR with the NEBNext® Library Quant Kit for Illumina (New England Biolabs). NextSeq500 System (Illumina) with the NextSeq 500/550 Mid Output v2 sequencing kit (150 cycles, Illumina) was used to sequence RNA Seq libraries, ensuring ∼5–10 million pair-end reads per sample. Three independent biological replicates were used for each strain and the condition sequenced.
Northern blotting analysis
Levels of some non-coding RNA transcripts were determined by northern blotting analysis. For each sample tested, 2 μg of total RNA was resolved in denaturing 1.2% agarose gel containing formaldehyde in 1× NBC buffer. The gel was transferred onto Hybond+ nylon membrane using the standard laboratory procedures. Next, RNA was cross-linked to the membrane with UV260 nm, stained with methylene blue to confirm transfer efficiency and used for hybridization with γ-32P-ATP labelled, 20–25 bp long oligonucleotide probes complementary to the transcript tested. After extensive washing, the membrane was incubated in a phosphor imaging screen and cassette and the screen was scanned in Typhoon 8600 (GE Healthcare) device to visualize the results.
Bioinformatics data analysis
For RNA sequencing data analysis, raw high-throughput sequencing data were processed with a series of software and bioinformatics scripts. Firstly, sequencing adapters were removed with Cutadapt v. 1.3 (23). This was followed by a window adaptive quality trimming with Sickle script, allowing minimum 30% quality and read length not shorter than 20 bp. Filtered reads were next aligned to appropriate genomes using Bowtie2 short read aligner (24). Assembled genomes references were fetched from the Mycobrowser database and included: M. tuberculosis H37Rv (NC_000962.3) and M. smegmatis mc2 155 (NC_008596). SAMtools (25) and BEDTools (26) software suits were used for data handling, normalization and generation of appropriate bedgraphs. Reads were counted into appropriate features with the help of HTSeq 0.6.1 package (27). Integrative Genomics Viewer (IGV) was used to visualize sequencing results in the genomic context (28). The differential expression was estimated with the online Degust RNA-Seq analysis platform with default parameters (http://degust.erc.monash.edu/, originally designed by D.R. Powell). Statistical analysis of differential gene expression (DGE) was performed by degust and provided as the false discovery rate (FDR) values. Genes with an FDR of <0.05 and a log2 fold change greater than an absolute value of 1.585 (changing three times or more) were considered significantly differential in the current study.
RNA-Seq coverage data was 1× normalized and log-transformed (as log2 ratio) with bamCoverage and bamCompare functions of deepTools (29), respectively. The log2 ratio values were calculated for PNPase depletion (PNP Cas9) versus control (no target) as well as RNase E depletion (RNE Cas9) versus control (no target).
In the case of proteomic data, raw data files fetched from Orbitrap Velos were processed with MaxQuant software (30) against M. tuberculosis protein database Tuberculist.R25 or M. bovis database, when appropriate. Semiquantitative results of label-free analysis were used for ppm estimation and data visualization.
Whenever required, the numerical data obtained from proteomic or transcriptomic analyses was imported into Python Pandas data frame format and Phyton Seaborn software suite with necessary dependencies was used to visualize the results as figure elements.
RESULTS
Characterzation of the mycobacterial RNA-bound proteome
Bacterial mRNAs lack the long polyadenylation tails exploited for isolation of mRNA–protein complexes in eukaryotic mRNA-bound proteome characterization studies (31). To define the RNA-bound proteome of Mycobacterium bovis BCG—an attenuated vaccine strain that is highly homologous to virulent M. tuberculosis and offers the advantage of manipulation under less stringent safety conditions—we exploited a novel RNA enrichment procedure outlined in the Materials and Methods section. Following UV-crosslinking, RNA–protein complexes were purified as depicted in Figure 1A, then analysed by mass spectrometry. A full list of RNA-interacting proteins identified by this approach is provided in Supplementary Table S2 together with semi-quantitative abundance values determined by MaxQuant software (30). To enrich for proteins that interact directly with RNA rather than through protein–protein complexes, the ion-exchange fractionation was repeated under denaturing conditions. MaxQuant-derived protein ‘Intensity’ values were converted to ppm in relation to the total ‘Intensity’ of the entire sample and compared to the intracellular levels of each protein in the M. tuberculosis cell (ppm values from Peptide Atlas (32)). Based on their enrichment factor, 50 proteins found in the RNA–protein pull-downs from non-denaturing (Figure 1B) or denaturing conditions (Figure 1C) were used for data visualization. In addition to ribosomal proteins, RNA polymerase and nucleoid-associated proteins, the list includes four proteins commonly found in degradosomes from other bacteria: ribonuclease E (RNase E; enriched ∼48 times above the average intracellular level under denaturing conditions), ribonuclease J (RNase J; enriched ∼20 times), ATP-dependent RNA helicase (RhlE; enriched ∼17 times) and polynucleotide phosphorylase (PNPase; enriched over 7 times) (Figure 1C). Other proteins prominent in the RNA–protein pull-downs included an uncharacterized Jag-like protein with a RNA-binding KH domain, enzymes associated with carbon metabolism, fatty acid metabolism and uncharacterized proteins (Figure 1B and C, Supplementary Table S2).
Figure 1.

Analysis of the RNA-bound proteome of M. bovis BCG. (A) Experiment schematic. Labelled RNA is UV-crosslinked with protein, which is then purified using a sucrose gradient followed by anion exchange chromatography. Purified proteins were analysed with mass spectrometry. Fifty candidate RNA binding proteins present in preparations under non-denaturing (B) and denaturing (C) conditions were chosen based on their enrichment factor (x enriched) in comparison to the intracellular levels in M. tuberculosis (Supplementary Table S1, Peptide Atlas, (32)). The MaxQuant intensity values were transformed into ppm values by calculating the relative abundance of each protein comparing to the total abundance of the entire sample. Next, the individual ppm values for each protein in the RNA–protein pull-down experiments were divided by corresponding ppm values for intracellular abundance. The putative RNA degradosome constituents enriched under both conditions are annotated with purple font.
Identification of the mycobacterial degradosome
Having identified RNase E, PNPase and RhlE as major components of the mycobacterial RNA-bound proteome we set out to characterize their potential interactions by affinity chromatography followed by mass spectrometry, using C-terminally GFP tagged bait proteins as described before (18). The list of proteins co-purifying with GFP-tagged PNPase or RhlE from M. tuberculosis H37Rv included each of the other candidate degradosome components (Supplementary Table S3). While PNPase was detected amongst the proteins co-purifying with GFP-tagged RNase E, the list, in this case, was dominated by highly abundant proteins including GroEL and DnaK chaperones. Similar findings were reported in a previous RNase E pull-down experiment published by another group (16). The RNase E from E. coli is also known to co-purify with GroEL and DnaK chaperones (33), likely due to an intrinsically disordered scaffolding domain (34), which may attract protein chaperones. While actinobacterial RNase E had undergone domain-shuffling (14), the explanation for interaction with protein chaperones is likely similar to the E. coli enzyme. Other proteins co-purifying with degradosome components include the two KH-domain proteins identified in the RNA-bound proteome (Rv2908c, Rv3920c), and RNase J. Cold shock proteins CspA and CspB were also prominent amongst proteins purified along with RNA and with GFP-tagged degradosome components (Supplementary Table S3, Figure 2A).
Figure 2.

Characterisation of RNA degradosome complexes in M. tuberculosis. (A) Illustration of possible RNA degradosome organisation in mycobacterial species. Chosen core RNA degradosome elements known from model organisms and enriched in RNA-bound proteome were tagged with eGFP and used as baits to pull down their respective intracellular partners with the previously optimised protocol (18). Based on the experimental results, putative core RNA degradosome elements were predicted as PNPase, RNase E, RNase J and RhlE. CspA, CspB RNA chaperones together with KH domain containing proteins Rv3920c (Jag-like) and Rv2908c were predicted as accessory RNA degradosome components. The RNA decay machinery's association with RNA polymerase is also suggested. Thick arrows indicate two-way protein-protein interplay found in the pull-down experiment, while thin arrows represent one-way interactions. (B) Alternative models of the organisation of the mycobacterial RNA degradosome complexes, where the RNase J may form a separate complex, excluding direct interaction with RNase E, or all four core degradosome components form a single complex. Intermediate states and various combinations with accessory proteins are also possible.
The 5′ exonuclease RNase J is a component of the well-characterized B. subtilis degradosome, and to explore its role in M. tuberculosis, we performed a further pull-down with GFP-tagged RNase J as bait. In addition to several abundant cellular proteins, PNPase co-purified with RNase J, along with the weaker signals for RhlE and RNase E (Supplementary Table S3). RNase J and PNPase form a complex as illustrated by size exclusion column UV trace (Supplementary Figure S2).
Enolase is a component of E. coli and B. subtilis degradosomes and was identified in the mycobacterial RNA-bound proteome isolated under the non-denaturing conditions (Supplementary Table S2, Figure 1B). Some PNPase co-purified with the GFP-tagged enolase used as a bait, however, no native enolase co-purified with PNPase-eGFP used as a bait (Supplementary Table S3). Other carbon metabolism enzymes identified in the RNA-bound proteome were essentially absent from mycobacterial degradosome pull-downs.
Protein–protein interaction data (Figure 2A) are consistent with two possible models for the mycobacterial degradosome. In one model, the four core degradosome components form a single complex with a structural organisation that may exclude direct interactions between RNase E and RNase J. In an alternative model, PNPase and RhlE helicase form separate complexes with either RNase E or RNase J (Figure 2B). The absence of enolase from the proposed mycobacterial degradosome complexes is consistent with the lack of an enolase-binding site in the M. tuberculosis RNase E sequence (35). The functional significance of interactions with cold-shock proteins and KH-domain proteins remains to be clarified. Future studies will be required to investigate the association of mycobacterial RNA polymerase with degradosome components observed here.
Based on abundance levels measured by SWATH mass spectrometry, the copy number of PNPase in M. tuberculosis and M. bovis BCG is 5- to 10-fold higher than that of other degradosome components (32), and PNPase may participate in other cellular functions in addition to its role in the degradosome (36).
PNPase and RNase E shape the mycobacterial transcriptome directly and via non-coding RNAs
Polynucleotide phosphorylase is a central component of the mycobacterial degradosome and, considering its cellular abundance and recently published evidence, plays pleiotropic functions in bacteria (36). PNPase may act as an RNA maturation factor and a RNA degradosome component. It is implicated in small RNA stability (37) and transport (36).
Another degradosome ribonuclease, RNase E, is an essential gene and a promising antimicrobial drug target. Similar to PNPase, RNase E plays important roles in bulk RNA processing, including rRNA maturation and regulation of the cellular processes via small RNAs. Activities of both core degradosome enzymes are indispensable for the survival of mycobacterial cells (38,39).
We were unable to isolate either PNPase or RNase E mutants in M. tuberculosis, unless providing merodiploid copies of the M. tuberculosis genes expressed from strong, constitutive promoters. We thus employed the CRISPR/Cas9 interference methodology, recently optimized for mycobacteria by Rock et al. (21), in order to diminish the intracellular pool of PNPase or RNase E in M. tuberculosis. This system relies on inducible and scalable transcriptional repression via RNA-guided gene silencing with the nuclease activity of Cas9 protein inactivated by point mutations (21). Upon seven-day-long induction with anhydrotetracycline, the expression of sgRNAs, targeted against gpsI or rne genes, led to significant repression of their respective targets as judged by total RNA sequencing. We achieved ∼20 and ∼8 times reduction in transcript abundance for gpsI and rne, respectively, compared to the wild type strain carrying a control Cas9 plasmid (Supplementary Figure S3A, B and Supplementary Table S4).
The total RNA profiling of strains defective in expression of degradosome constituents revealed major transcriptional reshaping (Figure 3A, B, Supplementary Figure S3C) that was associated with the growth inhibition of the tested strains. PNP Cas9 strain grown in the presence of the inducer to promote gene silencing, reached 20% the optical density of the un-induced strain, whereas RNE Cas9 reached 38% of the density of its un-induced control (Supplementary Figure S4). Expression of the Cas9 system without the target sgRNA (‘no target’) had negligible negative effect on the cell growth as judged by the optical density of cultures (Supplementary Figure S4). In our experimental setup, the depletion of either PNPase or RNase E affected the expression of about one-quarter of the entire transcriptome. Amongst 35 transcripts encoding essential genes (39) that were significantly depleted in PNP Cas9 and 26 in RNE Cas9, we could find 9 that were common to both strains. Six of the nine encoded proteins that have been implicated in cell division: cell division initiation protein Rv2927c, SepF, FtsQ, RipA, MtrA response regulator and WhiB2 transcription factor. Additionally, transcripts for FtsZ and the chromosomal replication initiator DnaA were reduced in PNP Cas9, whereas transcripts for FtsW, DivIVA and ClpX were depleted from RNE Cas9 strain.
Figure 3.

Transcriptomic profiling of M. tuberculosis strains depleted from essential RNA degradosome nucleases polynucleotide phosphorylase and ribonuclease E. Log2 Fold Change was plotted against log2 Average Expression for Cas9 PNP (A) and Cas9 RNE (B) strains compared to the control strain. Genes were assigned into functional categories (colour coded accordingly to the figure's labels) based on Mycobrowser database records (https://mycobrowser.epfl.ch/). Green horizontal lines mark log2 fold change greater than an absolute value of 1.585 (∼3 times) and red lines indicate log2 fold change greater than an absolute value of 4.907 (∼30 times). Transcripts changing over thirty times have been annotated with gene numbers. Insert figures show the percentage of transcriptional changes observed out of the total number of genes belonging to a given functional category. The results were visualized using Python Seaborn package.
Diminishing the pool of PNPase in the cell had significant impact on the levels of three most abundant non-coding RNA species present in the mycobacterial cell: transfer-messenger RNA (tmRNA, ssrA) (Figures 3A, 4 and 5), rnpB (an RNA component of RNase P) and MTS2823 (a 6S-like molecule) (Figures 3A, 4). Due to the significant depletion of the RNA component of RNase P, the PNP Cas9 strain likely presents a mixture of phenotypes related to the lack of ribonucleolytic activity of both PNPase and RNase P.
Figure 4.

Expression of important non-coding RNAs is affected by the removal of PNPase and RNase E. Expression profiles of important non-coding RNAs that were changed significantly in the total RNA Seq results for PNP Cas9 strain: (A) transfer messenger RNA - tmRNA, (B) RNA component of RNase P – rnpB and (C) MTS2823, mycobacterial 6S-like molecule are shown. The transcripts were depleted to a lesser level from the RNE Cas9 transcriptomes, with only MTS2823 passing the cut-off values set on false discovery rate (0.05) and log2 fold change (|1.585|). 1× normalized expression profiles (1xn.; as bedGraph tracks) are presented together with log2 expression ratios for the chosen genomic regions. Gene annotations and experimentally verified transcription start sites (TSS) are included underneath each genomic segment. TSSs specific to mature ncRNAs are represented by blue arrows.
Figure 5.
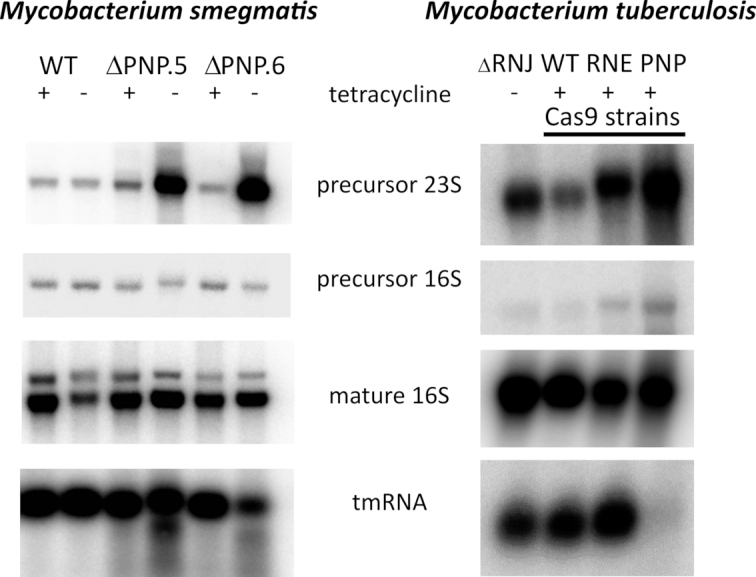
rRNA processing is affected by the removal of PNPase. Total RNA was isolated from a conditional PNPase mutant in M. smegmatis 16 hours after removal of tetracycline inducer, that was used to drive the expression of PNPasemtb-eGFP from the pKW08 vector. In the case of M. tuberculosis, the RNA samples correspond to the total RNA samples used for the RNA-Seq experiment. Northern blotting analysis results are shown for [γ-32P]-ATP-labelled oligonucleotide probes specific to internal transcribed spacer (ITS) regions spanning 16S and 23S rRNA (precursor 16S) or 23S and 5S rRNA (precursor 23S) as well as probes specific to the mature 16S (relative loading control) and tmRNA molecules (non-rRNA control). ΔPNP.5 and ΔPNP.6 are two independently selected PNPase conditional mutant strains from M. smegmatis.
Ms1 non-coding RNA was recently functionally characterized using the M. smegmatis model (40,41). The non-coding RNA was found to interact with PNPase and levels of Ms1 were negatively correlated with levels of PNPase in M. smegmatis. Ms1 and MTS2823 share a high degree of sequence and structure similarity (Supplementary Figure S5) and the M. tuberculosis counterpart likely plays an analogous role as a 6S RNA orthologue in mycobacteria. Using the overexpression strategy, Arnvig et al. previously reported the potential regulon/targetome of the MTS2823 molecule (42), suggesting the PrpR–PrpC–PrpD operon is one of the major targets of the MTS2823 dependent regulation. In our study, the levels of MTS2823 were affected by the conditional removal of either PNPase or RNase E, and were accompanied by enormous overexpression of the PrpBCD system, likely resulting from the deregulation of RNA polymerase complex, that is no longer sequestered by the MTS2823 molecule (Supplementary Table S4).
The RNA-Seq results were used to map potential processing events for the reported non-coding RNAs. Using this approach, we could visualize sharp shift events from positive to a negative log2 ratio within the regions of non-coding RNA expression, especially for the PNPase mutant strain. However, the shift events did not correlate well with the earlier annotated transcription start sites for the three non-coding RNA transcripts tested here, complicating the analysis (Figure 4). Only in the case of tmRNA, the TSS sites mapped ahead of the observed shift in the expression ratio. It is possible that in the case of the other two non-coding RNAs the TSS sites may have been wrongly annotated (41) or alternative TSSs are present.
rRNA processing is affected by the levels of PNPase
A role for degradosome components in the processing and maturation of ribosomal RNA has been demonstrated in bacteria including mycobacteria (11,43). To test for a potential role of mycobacterial PNPase in rRNA maturation, we used northern blot analysis to compare precursor species of 16S and 23S rRNA profiles in the M. smegmatis conditional PNPase mutant deletion strain (Supplementary Figure S1) upon withdrawal of tetracycline inducer (Figure 5). Using radiolabelled oligonucleotide probes complementary to internal transcribed spacer regions of rRNA transcripts, we observed a marked accumulation of precursor 23S rRNA, in relation to the precursor of 16S, supporting a role for PNPase in the maturation of the mycobacterial rRNA. The necessity of PNPase activity for the removal of the internal transcribed spacer region spanning 23S and 5S rRNA fragments was also supported by northern blot analysis of the total RNA isolated from M. tuberculosis strain depleted from PNPase (Figure 5). Exploiting northern blot analysis we also probed for tmRNA, which was significantly downregulated in the absence of PNPase in M. tuberculosis.
Transcriptional shaping driven by the nonessential degradosome-associated 5′-3′ exoribonuclease RNase J
To assess the cellular functions played by RNase J, we constructed a deletion mutant in M. tuberculosis. The strain lacking RNase J grew similar to the wild type strain in liquid 7H9 media, showing slight growth delay in the stationary phase (Supplementary Figure S6), and transcriptional changes associated with its removal were rather moderate, compared to the other two essential degradosome ribonucleases (Supplementary Table S4).
Noticeably, the removal of RNase J led to reduced abundance of a fraction of the DevR regulated genes (Supplementary Table S4). This was consistent with the effects observed upon silencing of PNPase and RNase E (Figure 6A). The RNA degradosome driven silencing of the dormancy regulon will require future investigation.
Figure 6.

Putative DevR and MTS2823 regulons are affected by the depletion of ribonucleases involved in RNA decay in M. tuberculosis. (A) A clustermap of genes belonging to the putative DevR regulon (52) or (B) best-predicted candidates for MTS2823-dependent regulation (42) are shown based on transcriptomic profiles of PNP Cas9, RNE Cas9 and ΔRNJ strains in relation to the strain carrying the pLJR965 vector and the wild type H37Rv parental strain, respectively. Log2 fold change values were visualised and clustered with the help of Phyton Seaborn package and necessary dependencies.
In contrast to PNPase and RNase E, the removal of RNase J promoted stability of the MTS2823 non-coding RNA and we did not observe accumulation of the PrpRCD encoding transcripts in this strain (Figure 6B).
DISCUSSION
Transcriptional adaptation to changing environments encountered during the course of infection has provided a central theme in the exploration of the molecular pathogenesis of M. tuberculosis. While major advances have been made in elucidating mechanisms involved in activating and suppressing transcription, the process of RNA turnover has remained relatively neglected.
We initiated our analysis of components involved in RNA turnover by isolating the set of mycobacterial proteins that interact strongly with RNA. Since bacterial mRNAs lack the polyA tails exploited in isolation of eukaryotic mRNA–protein complexes (31), we adopted an alternative approach to distinguish proteins binding directly to RNA from proteins that participate in RNA-binding complexes. In addition to the degradosome components that provide the focus of the present study, our description of the mycobacterial RNA-bound proteome includes other novel protein targets with potential for future analysis.
Based on their prominence in the RNA-bound proteome, and their identification as degradosome components in other bacteria, we focused on ribonuclease E (RNase E), polynucleotide phosphorylase (PNPase), ATP-dependent RNA helicase RhlE and ribonuclease J (RNase J) as core degradosome components. RNase J—a core component of the B. subtilis degradosome—showed strong two-way interactions with PNPase and RhlE, though not with RNase E. Protein–protein interaction networks based on co-purification with GFP-tagged baits are consistent with the presence of either a single mycobacterial degradosome including all four components but possibly excluding direct interaction between RNase E and RNase J, or two separate complexes involving one or other of the two nucleases (Figure 2). Studies in other microorganisms suggest that degradosomes are dynamic assemblies (44) and may take on multiple forms. PNPase is in excess over both RNase E and RNase J, and RNases E and J appear not to interact directly. Thus, it seems likely that the two upper models in Figure 2 reflect the in vivo situation better than the lower model. The lower model could reflect physiological situations where PNPase becomes limiting.
The mycobacterial degradosome differs from those of E. coli and B. subtilis in lacking an associated enolase. Enolase was detected in the mycobacterial RNA-bound proteome isolated under non-denaturing conditions, but no interactions were observed in pull-down experiments either with GFP-tagged enolase or with GFP-tagged degradosome components.
Four additional RNA-binding proteins co-purified with degradosome subunits. Cold-shock proteins CspA and CspB have been shown to play a role as RNA chaperones in other bacteria and may assist in the delivery of RNA to the degradosome or in unwinding and digestion (45,46). In the mycobacterial genome, CspA is located adjacent to a divergently-oriented DEAD-box RNA helicase (Rv3649 in M. tuberculosis). Neither this helicase nor a third DEAD-box helicase Rv1253 were detected in any of our pull-down fractions. Similarly, annotated ribonucleases other than RNase E and RNase J were not detected in degradosome pull-downs, although RNase PH (Rv1340, BCG_1402) was identified in the RNA-bound proteome isolated under non-denaturing conditions. Rv3920c is described as a Jag-like protein with RNA-binding KH and R3H domains; its function is unknown. In common with similar COG1847 proteins found in some other bacteria, Rv3920c is located in a conserved genome locus that includes the protein component of RNase P and ParAB chromosomal partitioning proteins and is duplicated along with the origin of replication in the genome of M. bovis BCG. Rv2908c is another uncharacterized KH-domain protein which has been found to be highly overexpressed upon infection in macrophages (47). Both proteins are represented in the RNA-bound proteome and co-purify with GFP-tagged degradosome components.
Identification of PNPase as a dominant component of the RNA processing machinery led us to further explore its potential function. As expected from genome-based essentiality screens, we were unable to generate direct deletion constructs in rapid- or slow-growing mycobacteria. A deletion mutant in M. smegmatis demonstrated a marked growth defect even in the presence of a complementing copy of the M. tuberculosis gene, and we were only able to generate complemented deletion strains in M. tuberculosis when a merodiploid copy of PNPase was expressed from a strong promoter on a complementing plasmid. These results suggest that PNPase performs multiple essential functions in mycobacteria and that a quantitative reduction in the level of a functional enzyme has significant detrimental effect. Vulnerability to partial inhibition makes PNPase an attractive target for potential drug discovery.
We have made similar observations regarding the other essential degradosome nuclease—RNase E. With the CRISPR/Cas9 technology recently optimized for use with mycobacteria (21) we were able to look at the effect of PNPase and RNase E depletion on the overall transcriptome. The changes associated with the silencing of either of these enzymes were massive, significantly changing about one-fourth of the bacterial transcriptome (summarized in Supplementary Figure S3C). Importantly, supporting the current opinion regarding the degradosome components, we found the changes associated with the degradosome silencing as a sum of effects driven by the activity of the enzymes and the non-coding RNAs that they regulate. PNPase depletion led to instability of the RNase P RNA component, which possibly causes problems with tRNA maturation and other issues (48). The rRNA molecules that are critical to any domain of life were also affected by the removal of PNPase. Depletion of tmRNA could lead to translation stalling and the accumulation of erroneous, non-functional polypeptides (49). Finally, instability within MTS2823, a 6S RNA-like molecule, may lead to deregulation of RNA polymerase complex, that would impair responses to the growth environment that the bacteria are facing (40,41). Numerous other small non-coding RNAs may contribute to the massive transcriptional reshaping we observe when the essential RNA degradosome components are disrupted. Future studies may reveal the effects of individual non-coding RNAs on the physiology of bacteria as yet another level of complexity to the regulatory pathways that govern the transcriptional adaptation of the super pathogen M. tuberculosis.
The depletion of degradosome nucleases from the mycobacterial cell had a significant effect on the dormancy regulon (Figure 6A). The DevR regulated genes are considered as critical adaptation factors, that contribute to the success of mycobacteria as a pathogen (50). It is likely that the DevR regulon is affected by some yet uncharacterized small RNAs, that are critical for the efficient activation of DevR regulon expression. RNA degradosome nucleases may thus be also critical for the coordinated entry into dormancy.
DATA AVAILABILITY
RNA-bound proteome related mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (51) partner repository with the dataset identifier PXD012661.
Protein complex related mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (51) partner repository with the dataset identifier PXD012662.
The RNA-seq related data have been deposited to the GEO database and are accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126286.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dominik Cysewki, Agata Malinowska, Jacek Olędzki and Agnieszka Fabijańska for LC–MS/MS technical help and discussion; Dominik Strapagiel, Paweł Krawczyk and Dorota Adamska for technical help with sequencing data acquisition and analysis. We thank Jeremy Rock and Sarah Fortune for providing us with the pLJR965 vector and detailed instructions for generation of Cas9 regulated strains in M. tuberculosis.
Author contributions: A.D., D.Y. and P.P. conceived and designed the experiments. D.Y. and P.P. wrote the manuscript. A.D., J.H. and D.Y. corrected the manuscript. P.P. performed experiments, and together with D.Y. and A.D. analysed the results. M.M. and E.N. helped P.P. with the construction of PNPase conditional mutant in M. smegmatis and J.H. helped with RNA degradosome complex isolations from M. tuberculosis. R.P. and A.B. helped P.P. with the generation of RNJ knockout and Cas9 strains in M. tuberculosis. At Biobank Lab, University of Lodz, M.S. performed total RNA sequencing of RNA Seq libraries generated by P.P. for Cas9 strains. J.D. contributed reagents and materials and helped with study design. P.P. performed bioinformatics data analysis of mass spectrometry and RNA sequencing data and created figures included with the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Community's Seventh Framework Programme (Health) [241587, SysteMTb to A.D., D.Y.]; National Science Centre, Poland [2015/19/D/NZ1/02842, SONATA 10 to P.P.]; at IBB PAS, experiments were carried out with the use of CePT infrastructure financed by the European Union: the European Regional Development Fund [Innovative economy 2007–13, Agreement POIG.02.02.00-14-024/08-00]. Funding for open access charge: National Science Centre, Poland [2015/19/D/NZ1/02842, SONATA 10 to P.P.].
Conflict of interest statement. None declared.
REFERENCES
- 1. WHO WHO Global Tuberculosis Report. 2018; World Health Organization. [Google Scholar]
- 2. Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E. 3rd et al.. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998; 393:537–544. [DOI] [PubMed] [Google Scholar]
- 3. Galagan J.E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., Gomes A., Rustad T., Dolganov G., Glotova I. et al.. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013; 499:178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minch K.J., Rustad T.R., Peterson E.J., Winkler J., Reiss D.J., Ma S., Hickey M., Brabant W., Morrison B., Turkarslan S. et al.. The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun. 2015; 6:5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rustad T.R., Minch K.J., Brabant W., Winkler J.K., Reiss D.J., Baliga N.S., Sherman D.R.. Global analysis of mRNA stability in Mycobacterium tuberculosis. Nucleic Acids Res. 2013; 41:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bandyra K.J., Bouvier M., Carpousis A.J., Luisi B.F.. The social fabric of the RNA degradosome. Biochim. Biophys. Acta. 2013; 1829:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lehnik-Habrink M., Newman J., Rothe F.M., Solovyova A.S., Rodrigues C., Herzberg C., Commichau F.M., Lewis R.J., Stulke J.. RNase Y in Bacillus subtilis: a Natively disordered protein that is the functional equivalent of RNase E from Escherichia coli. J. Bacteriol. 2011; 193:5431–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shahbabian K., Jamalli A., Zig L., Putzer H.. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009; 28:3523–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bouvier M., Carpousis A.J.. A tale of two mRNA degradation pathways mediated by RNase E. Mol. Microbiol. 2011; 82:1305–1310. [DOI] [PubMed] [Google Scholar]
- 10. Mohanty B.K., Kushner S.R.. Polynucleotide phosphorylase functions both as a 3′ right-arrow 5′ exonuclease and a poly(A) polymerase in Escherichia coli. PNAS. 2000; 97:11966–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taverniti V., Forti F., Ghisotti D., Putzer H.. Mycobacterium smegmatis RNase J is a 5′-3′ exo-/endoribonuclease and both RNase J and RNase E are involved in ribosomal RNA maturation. Mol. Microbiol. 2011; 82:1260–1276. [DOI] [PubMed] [Google Scholar]
- 12. Khemici V., Toesca I., Poljak L., Vanzo N.F., Carpousis A.J.. The RNase E of Escherichia coli has at least two binding sites for DEAD-box RNA helicases: functional replacement of RhlB by RhlE. Mol. Microbiol. 2004; 54:1422–1430. [DOI] [PubMed] [Google Scholar]
- 13. Njire M., Wang N., Wang B., Tan Y., Cai X., Liu Y., Mugweru J., Guo J., Hameed H.M.A., Tan S. et al.. Pyrazinoic acid inhibits a bifunctional enzyme in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017; 61:e00070-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee K., Cohen S.N.. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol. Microbiol. 2003; 48:349–360. [DOI] [PubMed] [Google Scholar]
- 15. Zeller M.E., Csanadi A., Miczak A., Rose T., Bizebard T., Kaberdin V.R.. Quaternary structure and biochemical properties of mycobacterial RNase E/G. Biochem. J. 2007; 403:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovacs L., Csanadi A., Megyeri K., Kaberdin V.R., Miczak A.. Mycobacterial RNase E-associated proteins. Microbiol. Immunol. 2005; 49:1003–1007. [DOI] [PubMed] [Google Scholar]
- 17. Kime L., Vincent H.A., Gendoo D.M., Jourdan S.S., Fishwick C.W., Callaghan A.J., McDowall K.J.. The first small-molecule inhibitors of members of the ribonuclease E family. Sci. Rep. 2015; 5:8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plocinski P., Laubitz D., Cysewski D., Stodus K., Kowalska K., Dziembowski A.. Identification of protein partners in mycobacteria using a single-step affinity purification method. PLoS One. 2014; 9:e91380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Szczesny R.J., Borowski L.S., Brzezniak L.K., Dmochowska A., Gewartowski K., Bartnik E., Stepien P.P.. Human mitochondrial RNA turnover caught in flagranti: involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010; 38:279–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parish T., Stoker N.G.. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology. 2000; 146:1969–1975. [DOI] [PubMed] [Google Scholar]
- 21. Rock J.M., Hopkins F.F., Chavez A., Diallo M., Chase M.R., Gerrick E.R., Pritchard J.R., Church G.M., Rubin E.J., Sassetti C.M. et al.. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2017; 2:16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pawelczyk J., Brzostek A., Kremer L., Dziadek B., Rumijowska-Galewicz A., Fiolka M., Dziadek J.. AccD6, a key carboxyltransferase essential for mycolic acid synthesis in Mycobacterium tuberculosis, is dispensable in a nonpathogenic strain. J. Bacteriol. 2011; 193:6960–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal. 2011; 17:10–12. [Google Scholar]
- 24. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 31. Baltz A.G., Munschauer M., Schwanhausser B., Vasile A., Murakawa Y., Schueler M., Youngs N., Penfold-Brown D., Drew K., Milek M. et al.. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 2012; 46:674–690. [DOI] [PubMed] [Google Scholar]
- 32. Schubert O.T., Mouritsen J., Ludwig C., Rost H.L., Rosenberger G., Arthur P.K., Claassen M., Campbell D.S., Sun Z., Farrah T. et al.. The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe. 2013; 13:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miczak A., Kaberdin V.R., Wei C.L., Lin-Chao S.. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. PNAS. 1996; 93:3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ait-Bara S., Carpousis A.J.. RNA degradosomes in bacteria and chloroplasts: classification, distribution and evolution of RNase E homologs. Mol. Microbiol. 2015; 97:1021–1135. [DOI] [PubMed] [Google Scholar]
- 35. Marcaida M.J., DePristo M.A., Chandran V., Carpousis A.J., Luisi B.F.. The RNA degradosome: life in the fast lane of adaptive molecular evolution. Trends Biochem. Sci. 2006; 31:359–365. [DOI] [PubMed] [Google Scholar]
- 36. Cameron T.A., Matz L.M., De Lay N.R.. Polynucleotide phosphorylase: Not merely an RNase but a pivotal post-transcriptional regulator. PLos Genet. 2018; 14:e1007654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bandyra K.J., Sinha D., Syrjanen J., Luisi B.F., De Lay N.R.. The ribonuclease polynucleotide phosphorylase can interact with small regulatory RNAs in both protective and degradative modes. RNA. 2016; 22:360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Griffin J.E., Gawronski J.D., Dejesus M.A., Ioerger T.R., Akerley B.J., Sassetti C.M.. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011; 7:e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeJesus M.A., Gerrick E.R., Xu W., Park S.W., Long J.E., Boutte C.C., Rubin E.J., Schnappinger D., Ehrt S., Fortune S.M. et al.. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio. 2017; 8:e02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hnilicova J., Jirat Matejckova J., Sikova M., Pospisil J., Halada P., Panek J., Krasny L.. Ms1, a novel sRNA interacting with the RNA polymerase core in mycobacteria. Nucleic Acids Res. 2014; 42:11763–11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sikova M., Janouskova M., Ramaniuk O., Palenikova P., Pospisil J., Bartl P., Suder A., Pajer P., Kubickova P., Pavlis O. et al.. Ms1 RNA increases the amount of RNA polymerase in Mycobacterium smegmatis. Mol. Microbiol. 2019; 111:354–372. [DOI] [PubMed] [Google Scholar]
- 42. Arnvig K.B., Comas I., Thomson N.R., Houghton J., Boshoff H.I., Croucher N.J., Rose G., Perkins T.T., Parkhill J., Dougan G. et al.. Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 2011; 7:e1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 44. Gorna M.W., Carpousis A.J., Luisi B.F.. From conformational chaos to robust regulation: the structure and function of the multi-enzyme RNA degradosome. Q. Rev. Biophys. 2012; 45:105–145. [DOI] [PubMed] [Google Scholar]
- 45. Jiang W., Hou Y., Inouye M.. CspA, the major cold-shock protein of Escherichia coli, is an RNA chaperone. J. Biol. Chem. 1997; 272:196–202. [DOI] [PubMed] [Google Scholar]
- 46. Bae W., Xia B., Inouye M., Severinov K.. Escherichia coli CspA-family RNA chaperones are transcription antiterminators. PNAS. 2000; 97:7784–7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rachman H., Strong M., Schaible U., Schuchhardt J., Hagens K., Mollenkopf H., Eisenberg D., Kaufmann S.H.. Mycobacterium tuberculosis gene expression profiling within the context of protein networks. Microbes Infect./Institut Pasteur. 2006; 8:747–757. [DOI] [PubMed] [Google Scholar]
- 48. Singh A., Ubaid-ullah S., Ramteke A.K., Batra J.K.. Influence of conformation of M. tuberculosis RNase P protein subunit on its function. PLoS One. 2016; 11:e0153798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Personne Y., Parish T.. Mycobacterium tuberculosis possesses an unusual tmRNA rescue system. Tuberculosis (Edinb.). 2014; 94:34–42. [DOI] [PubMed] [Google Scholar]
- 50. Gengenbacher M., Kaufmann S.H.. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 2012; 36:514–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Magombedze G., Dowdy D., Mulder N.. Latent tuberculosis: models, computational efforts and the pathogen's regulatory mechanisms during dormancy. Front. Bioeng. Biotechnol. 2013; 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-bound proteome related mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (51) partner repository with the dataset identifier PXD012661.
Protein complex related mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (51) partner repository with the dataset identifier PXD012662.
The RNA-seq related data have been deposited to the GEO database and are accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126286.