


Abstract
The nematode Caenorhabditis elegans contains several types of endogenous small interfering RNAs (endo-siRNAs) produced by RNA-dependent RNA polymerase (RdRP) complexes. Both ‘silencing’ siRNAs bound by Worm-specific Argonautes (WAGO) and ‘activating’ siRNAs bound by the CSR-1 Argonaute require the DRH-3 helicase, an RdRP component. Here, we show that, in the drh-3(ne4253) mutant deficient in RdRP-produced secondary endo-siRNAs, the silencing histone mark H3K9me3 is largely depleted, whereas in the csr-1 partially rescued null mutant strain (WM193), this mark is ectopically deposited on CSR-1 target genes. Moreover, we observe ectopic H3K9me3 at enhancer elements and an increased number of small RNAs that match enhancers in both drh-3 and csr-1 mutants. Finally, we detect accumulation of H3K27me3 at highly expressed genes in the drh-3(ne4253) mutant, which correlates with their reduced transcription. Our study shows that when abundant RdRP-produced siRNAs are depleted, there is ectopic elevation of noncoding RNAs linked to sites with increased silencing chromatin marks. Moreover, our results suggest that enhancer small RNAs may guide local H3K9 methylation.
INTRODUCTION
The RNA interference machinery, and specifically Argonaute proteins, which bind small RNAs, are present both in the cytoplasm and the nucleus in diverse species (1,2). In fission yeast and plants, there is an elaborate connection between siRNAs and heterochromatin (3–5). In animals, Piwi-interacting RNAs (piRNAs), which protect the germline from parasitic elements, are strongly implicated in triggering chromatin-based silencing (1). In the nematode Caenorhabditis elegans, as well as in mammalian cultured cells, exogenous or transgene-based introduction of double-stranded RNA (dsRNA) can initiate transcriptional silencing and/or heterochromatin formation (6–9). Finally, endogenous siRNAs (endo-siRNAs) produced from dsRNA segments arising from large hairpins, overlapping 5′ or 3′ ends of transcripts, or repetitive elements have been detected in oocytes of higher animals (10,11). Whether they also regulate chromatin compaction is an open question.
An open chromatin conformation at enhancer elements is essential for their function and is conserved across species, including C. elegans (12). Enhancers constitute stretches of DNA with characteristic features of actively transcribed chromatin, such as DNase I hypersensitivity, high occupancy by transcriptional machinery, and specific histone modifications (H3K4me1, H3K27ac) (13–15). Intragenic enhancers promote local gene transcription, whereas intergenic enhancers can undergo chromatin looping to promote distal gene transcription up to several thousand base pairs away.
Since their initial discovery (16–18), new features of enhancers have emerged that require further investigation. For example, the field now appreciates that bidirectional transcription at enhancer regions (19–21) results in production of noncoding RNAs (ncRNAs) named enhancer RNAs (eRNAs). This transcription appears to play an important role in enhancer function, most notably by keeping chromatin open (13,15). However, specific examples of eRNA sequences having a functional significance themselves have also been found (15). The most prominent examples of enhancer-derived ncRNAs are RNAs produced from the Ultrabithorax domain in the homeobox gene region in Drosophila (22) and the Evf-2 ncRNA associated with the Dlx-5/6 ultraconserved enhancer in mouse (23). Additionally, super-enhancers are spatially and functionally associated with miRNA genes (24).
It is possible that nuclear siRNAs could be produced from the dsRNA arising from bidirectional transcription at enhancers (15). If such siRNAs were to bind Argonaute (AGO) proteins that guide heterochromatin marks, they might serve as a regulatory mechanism to shut down enhancers in differentiated cells. Putative enhancer elements have only recently been annotated in the C. elegans genome (25,26). Using these annotations, we provide here the first evidence of small RNAs matching enhancers.
The most abundant C. elegans endo-siRNAs are 22G-RNAs, which are produced through amplification of primary siRNAs by RNA-dependent RNA Polymerases (RdRP) (27). A group of redundant Worm-specific AGO proteins (WAGO) bind 22G-RNAs and target repetitive elements, pseudogenes, transposons and some duplicated protein-coding genes for silencing (27). Two nuclear WAGOs are implicated in facilitating transcriptional silencing: NRDE-3 in the soma (28) and HRDE-1 in the germline (29). Both are capable of inducing H3K9me3 at genes complementary to exogenous dsRNA or at transgenes downstream of piRNAs (6,8,28–33). However, the interconnection between 22G-RNA production, H3K9 methylation and transcriptional silencing at endogenous WAGO targets remains unclear (34,35).
Whereas the concept that nuclear Argonaute proteins guide silencing histone modifications is well established, the CSR-1 (Chromosome Segregation and RNAi deficient) Argonaute (36,37) does not fit this stereotype. It is present both in the cytoplasm and the nucleus, it binds 22G-RNAs that are largely antisense to highly and widely expressed genes, and it is associated with nascent transcripts (38) and mature mRNAs complementary to these siRNAs. In contrast to direct gene-silencing roles for WAGOs, CSR-1 has an ‘activating’ or ‘anti-silencing’ role, since nascent transcripts complimentary to CSR-1-bound siRNAs decrease in csr-1-deficient worms (38). Previously, we showed by Global Run-on Sequencing (GRO-seq) that in the partially rescued csr-1 null (strain WM193 (36)) and reduction-of-function drh-3 mutants (see Materials and Methods for strain details) there is both a decrease in nascent transcripts targeted by CSR-1 22G-RNAs and a genome-wide increase in antisense transcription (38). Notably, DRH-3 (Dicer-related helicase) is a component of RdRP complexes that produce 22G-RNAs and is required for generation of both WAGO-bound and CSR-1-bound small RNAs (27).
Here, we report that the decrease in transcription of CSR-1 target genes that occurs upon reduction-of-function of csr-1 and drh-3 (38) correlates with ectopic silencing chromatin marks. Moreover, we describe an increase in noncoding transcription and elevation of H3K9me3 at putative gene enhancers in both csr-1 and drh-3 mutants. Finally, we identify small RNAs originating from enhancers in previously published datasets. Our results support a model whereby ‘activating’ and ‘silencing’ secondary siRNAs in C. elegans have complementary functions required for proper genome-wide chromatin organization. Our data also suggest that, in the absence of abundant 22G-RNAs, small RNAs produced from enhancers may be incorporated into Argonaute complexes capable of guiding H3K9me3 deposition.
MATERIALS AND METHODS
Strains
Strains were maintained at 20°C unless otherwise noted, using standard methods (39). Bristol N2 was the WT strain used. Mutants used in this study were drh-3(ne4253), and the partially rescued csr-1(tm892) (strain WM193). Whereas a null drh-3(tm1217) allele causes sterility, drh-3(ne4253) is fertile at 20°C; it exhibits partial somatic RNAi deficiency and is depleted of most secondary siRNAs (27,36). A partially rescued csr-1(tm892) mutant (strain WM193) was generated by introducing the pie-1p::3xFLAG::csr-1 rescuing transgene into the csr-1(tm892) null background (36). Both drh-3(ne4253) and partially rescued csr-1(tm892) generate male progeny (Him phenotype) (27,36) and lay variable numbers (∼16–50%) of inviable embryos.
For the ChIP-seq experiments, worm populations were synchronized and grown for approximately 40 h after hatching at 20°C on OP-50 Escherichia coli at a density of ∼50 000 animals per 15-cm Petri dish until they reached L3 stage.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed following the modENCODE Protocol from the Lieb lab (40) with some modifications. The worm pellet was fixed with 2% paraformaldehyde for 30 min at 20°C, washed in M9 3 times, and resuspended in RIPA buffer supplemented with protease inhibitors (Thermo Scientific, 78443). DNA fragmentation was performed using Covaris S220 (Peak power 240, Duty factor 20, Cycles/burst 200, 8 min). Next, 1.5–2 mg of crosslinked chromatin extract was incubated at 4°C overnight with a specific antibody and immune complexes were then incubated with 50 μl IgG Dynabeads (Invitrogen) for 3 h at 4°C. DNA was cleaned up with the Qiagen PCR purification kit. We used the following antibodies: 5 μg of anti-H3K27me3 (Diagenode, pAb-195-050), 5 μg of anti-H3K36me3 (Abcam, ab9050), and 5 μg of anti-H3K9me3 (Abcam, ab8898). Validation information for the commercial antibodies is included at the manufacturers’ websites.
Western blotting
Proteins were resolved on precast NuPAGE Novex 12% Bis-Tris gels (Invitrogen) at 4°C for 1 h and transferred to a nitrocellulose membrane (0.45 μm) by semi-dry transfer (BioRad Trans-Blot SD transfer cell) at a constant current of 0.12 A for 1 h. The blotted membranes were blocked with the blocking buffer (5% non-fat dry milk in TBS-T buffer) at room temperature for 1 h. Subsequently, they were incubated with appropriate primary antibodies overnight at 4°C and with secondary antibodies for 2 h at room temperature. Three washes with the TBS-T buffer were done between and after the incubation with the antibodies. The membranes were developed with SuperSignal West Femto kit (ThermoFisher) and imaged using KwikQuant Imager (KindleBiosciences). The H3K9me3, H3K27me3, and H3K36me3 histone modifications were detected with ab8898 (Abcam), 07-449 (Millipore), and ab9050 (Abcam) antibodies, respectively. For control, the membrane was stripped and re-blotted with the anti-H3 antibody (Abcam, ab1791).
Library preparation, sequencing and data processing
ChIP-seq libraries were prepared using the TruSeq Illumina kit (set A – 15034288, set B – 15034289) following the manufacturer's instructions. Sequencing was performed on an Illumina NextSeq500 instrument. The 75-bp single-end Illumina sequencing reads were preprocessed by trimming the adapter sequences with Cutadapt (41). After that, reads were aligned to the WS220/ce10 assembly of the C. elegans genome using Bowtie for Illumina (Galaxy Version 1.1.2) (42) with default settings. The SAMtools (Galaxy Version 1.1.2) (43,44) utility was used to convert the alignments to BAM format. A table containing the number of reads aligned to the genome (Supplementary Table S1) demonstrates sufficient sequencing depth (∼14–28M of reads). Aligned reads were visualized in IGV browser (45,46) to check the overall read distribution shape. Although not a quantitative metric, visible enrichment at known binding regions confirmed a successful ChIP-seq experiment.
Two independent ChIP experiments were performed with each type of antibody. To estimate the concordance between two replicates, we applied Pearson's correlation coefficient that indicated perfect linear correlation (close to 1) between most replicates and good correlation (≥0.68) between H3K9me3 wild type replicates and H3K27me3 drh-3(ne4253) replicates (Supplementary Table S2). Duplicate reads were removed, and the data were processed as described below. Sequencing data have been submitted to the GEO database (submission Series GSE115629).
Analysis of ChIP-seq data
Aligned ChIP-seq reads were TMM-normalized using the edgeR package (47). Read counting at specific loci (genes, enhancers, or non-coding genomic regions) was performed with the GenomicAlignments package (48); only reads with mapping quality 20 or higher were included in subsequent analyses. The resulting ChIP-seq coverage in the immunoprecipitated DNA sample was expressed as log2-transformed RPKM (reads per kilobase per million mapped) value after subtraction of the RPKM value of the corresponding input DNA. Only regions in which the normalized count value in the immunoprecipitated DNA sample was higher than that in the corresponding input DNA in at least one sample in the set were considered. Gene identifiers were converted to Refseq mRNA IDs using the WormBase Converter utility (http://wormbasemanager.sourceforge.net/) and the DAVID Gene ID Conversion Tool (49). Differential enrichment analysis was performed using the edgeR package (47). The peaks were generated with MACS2 peak calling algorithm (Galaxy version 2.0.10.2) (50).
Analysis of GRO-seq data
GRO-seq reads (38) were aligned to the WS220/ce10 assembly of the C. elegans genome using Bowtie for Illumina (Galaxy Version 1.1.2) (42) with default settings. Reads matching ribosomal RNA loci were removed, as described before (51). Read counting at genomic regions was performed with the package GenomicAlignments (48); only reads with mapping quality 20 or higher were included in subsequent analyses. Regions without reads across the sample set were removed. Counts were then normalized using the TMM method, which takes RNA composition bias into account (52), using the edgeR package (53). Coverage was expressed as RPKM and log2-transformed.
Heatmaps
The purpose of heatmaps is the visualization of changes in a histone modification along the gene-coding region in mutants compared to wild type. To generate the heatmaps, the Galaxy platform was used. The aligned reads were normalized to RPKM using the bamCoverage option (bin size = 100 bp, scaling/normalization method = rpkm, no scaling factor, ignoring missing data, reads extension = 250, ignore duplicates = true, center regions with respect to the fragment length = false, minimum mapping quality = 1) and then normalized to input using the bigWigCompare option (method of comparison = ratio, pseudocount = 1.0). Further, mutant coverage representing one of the histone modifications was compared with wild type coverage for the corresponding modification and belonging to the same replicate (method of comparison = log2, pseudocount = 1.0). The mean of two replicates for either H3K27me3 or H3K36me3 was then visualized along the gene bodies using computeMatrix (Distance in bases to which all regions are going to be fit = 1000 bp, Distance upstream/downstream of the region start position = +/-200, Length, in bases, of non-overlapping bins used for averaging the score over the regions length = 50 bp, Convert missing values to 0 = true) option and plotHeatmap (Sort regions = no ordering, Color map to use for the heatmap = bwr, Heatmap width in cm = 10.0, Heatmap height in cm = 20.0, What to show = heatmap and color bar). The options were standard if not specified otherwise. Gene subsets used for the analysis are listed in the Supplementary Table S3.
Analysis of putative enhancer regions and ATAC-seq peaks
Cumulative coordinates of the putative enhancer regions were obtained by combining the coordinates of predicted enhancers from chromatin domains 8, 9 and 10 (26). Among 177845 annotated chromatin regions, 13602 were considered putative enhancers. Enhancer regions at least 1500 bp distal to any annotated transcription start site or transcription termination site were considered distal enhancer regions. Enhancer regions intersecting coordinates of genes <15 kb by at least 50 bp were considered intragenic enhancer regions. The percentage of enhancers with positive value of H3K9me3 coverage after input subtraction in at least one sample was 56%.
The ATAC-seq peak coordinates (25) were downloaded from the NCBI GEO database (GSE89608). Among 56624 genomic regions with open chromatin identified by (25), 9448 regions were defined as ATAC-seq enhancers. Distal and intragenic ATAC-seq peaks were defined as described for the predicted enhancer regions. Positive values of H3K9me3 coverage in at least one sample after input subtraction were detected for 46% of ATAC-seq peaks. The portion of the genome not containing ATAC-seq peaks was divided into genome regions of 2.5 kb and used for comparisons.
Small RNA bioinformatics analyses
Small RNA library sequences were downloaded from the NCBI Gene Expression Omnibus and Sequence Read Archive under the following accessions: GSM454002, GSM455389, GSM455390, GSM455387, GSM455388, SRR030720, SRR030721, SRR030722, SRR030719, SRR030717, SRR5024011, SRR5024012, SRR5024013, SRR5024014, SRR5023999, SRR5024000, SRR5024001, SRR5024002, SRR5024003 and SRR5024004. Fastq files were validated and adapters were trimmed with the programs Fastqc (54) and Cutadapt (41), respectively. The genome and Refseq transcript assemblies for ‘ce10’ were loaded into our custom ‘gene-centric process’ small RNA analysis pipeline (55,56). This program maps small RNAs with Bowtie1 (42) with a maximum of two base mismatches, counts the genomic strand reads against the Ensembl gene transcript models using Bedtools (57) using the longest transcript isoform as the main gene model, and generates WIG plots for coverage visualization on the UCSC Genome Browser (58). Each library's read counts were normalized against library depths (counts per million, cpm), as well as frequency of genomic locations (cpm / number of genomic loci). For further analysis, small RNA read coordinates were intersected with enhancer domain coordinates (26).
RESULTS
Competing regulation of H3K9me3 on highly active genes by the CSR-1 and WAGO pathways
Previously, we observed similar alterations in global transcription in the viable partially rescued csr-1(tm892) and drh-3(ne4253) mutant larvae (L3) compared with wild type animals (38). To establish a connection between transcription changes and chromatin status in these mutants, we performed ChIP-seq with antibodies specific to several histone modifications: H3K27me3, H3K36me3, and H3K9me3.
Consistent with published data (59), H3K9me3 was found predominantly at chromosome arms (Figure 1A), and this distribution profile did not change in the studied mutants. However, we detected an increase in the level of H3K9me3 at CSR-1 target genes in csr-1(WM193) compared to wild type (Figure 1B, left, Figure 1C, D and Supplementary Figure S1), whereas in drh-3(ne4253) we observed a depletion of this silencing mark at the same genes (Figure 1B, left and C). In comparison, H3K9me3 levels were reduced at CSR-1 non-target genes lacking intronic enhancers in both mutants, while enhancer-containing non-target genes became enriched in H3K9me3 (Figure 1B, right plot). The latter group of genes will be discussed in the next section.
Figure 1.

H3K9me3 alterations at CSR-1 target genes in partially rescued csr-1(tm892) and drh-3(ne4253) mutants. (A) On the C. elegans autosomes, H3K9me3 is enriched at the chromosome arms and, on the X chromosome, only at the left arm. Read coverage in wild type worms (L3) normalized to, sequentially, RPKM and input is shown. (B) A box plot (left) demonstrates an increase of H3K9 methylation at CSR-1 target genes in csr-1(WM193), but not in drh-3(ne4253). ChIP-seq data were normalized as described in methods. Significant changes (P < 10−4) are marked by an asterisk: csr-1(WM193): Med = 2.16, P-value = 2.7 × 10−13 (Wilcoxon test); drh-3(ne4253): Med = 1.40, P-value = 4 × 10−4 (Wilcoxon test); wild type: Med = 1.75. A box plot (right) demonstrates a decrease in H3K9me3 at CSR-1 non-target genes not containing enhancers (– enhancer) in both csr-1(WM193) and drh-3(ne4253), and elevated H3K9me3 at non-target genes containing enhancers (+ enhancer). Significant changes (P < 10−4) are marked by an asterisk: No enhancer: csr-1(WM193): Med = 3.50, P-value = 6.1 × 10−15 (Wilcoxon test); drh-3(ne4253). Med = 3.08, P-value < 2 × 10−16 (Wilcoxon test); wild type: Med = 3.70. Enhancer: csr-1(WM193): Med = 1.70, P-value = 2.6 × 10−8 (Wilcoxon test); drh-3(ne4253): Med = 1.45, P-value = 2.5 × 10−7 (Wilcoxon test); wild type: Med = 1.10. (C) A scatter plot shows an enrichment of H3K9me3 in csr-1(WM193) compared to wild type and a depletion of H3K9me3 in drh-3(ne4253) compared to wild type in the large group of CSR-1 target genes (red circles). CSR-1 target genes with detectable H3K9me3 coverage in at least one strain were selected. The fold changes compared to wild type were then calculated and log2 -transformed. Changes in csr-1(WM193) are shown on the Y-axis, while changes in drh-3(ne4253) are shown on the X-axis; this allows simultaneous comparison of the same genes between the two mutants. The dashed lines identify a group of genes (red circles) that are enriched in H3K9me3 in csr-1(WM193) (above the horizontal line) and are depleted in drh-3(ne4253) (to the left of vertical line) compared to wild type. (D) Coverage tracks at two CSR-1 target genes of different sizes and exon/intron composition. In wild type (gray), H3K9me3 is very low (upper plot) or not detected (lower plot), whereas in csr-1(WM193) (green), an increase in H3K9me3 is observed. TMM and CPM normalization was performed followed by input reads subtraction from each sample. The directionality of gene transcription is marked with blue arrows. (E) The global level of H3K9me3 at the L3 stage larvae increases in the csr-1(WM193) mutant compared to wild type. On the contrary, the H3K9me3 level in drh-3(ne4253) is reduced. The levels of H3K27me3 and H3K36me3 do not change globally.
Because both the CSR-1 and the WAGO pathways are deficient in drh-3(ne4253), these results suggest a competition between the ‘activating’ and ‘silencing’ secondary siRNAs, such that the WAGO pathway promotes H3K9me3 and CSR-1 inhibits its deposition at the majority of CSR-1 target genes. This type of competitive relationship has been suggested by other studies utilizing transgenic strains (30,33,60). Consistently, we observed a reduction in H3K9me3 modification abundance in drh-3(ne4253) by western immunoblotting, which indicates that 22G-RNA-dependent methylation of H3K9 is globally affected in this mutant (Figure 1E). On the contrary, in csr-1(WM193), there was a slight increase in H3K9me3 detected by western, suggesting that the WAGO silencing pathway is overactive under these conditions (Figure 1E). We were not able to test whether this increase in H3K9me3 is dependent on DRH-3, because combining drh-3(ne4253) with a partially rescued csr-1(tm892) mutant (strain WM193, (36)) led to early developmental arrest. We did not observe global changes in the levels of H3K27me3 and H3K36me3 in the mutants compared to wild type as assessed by western blotting (Figure 1E).
Our results are consistent with the idea that, in the csr-1 mutant, WAGO-induced H3K9me3 is likely to play a role in inhibiting active genes (61,62). Therefore, CSR-1-bound siRNAs may protect their complementary pre-mRNAs from the WAGO-dependent 22G-RNAs that guide H3K9 methylation.
Increased ncRNA production in partially rescued csr-1(tm892) and drh-3(ne4253) mutants correlates with accumulation of silencing marks at enhancers
Since there is a global increase in antisense transcription detected by GRO-seq in csr-1(WM193) and drh-3(ne4253) mutants (38), we chose to examine a specific CSR-1 target gene, cyclin E (cye-1), as antisense transcription has been previously detected at this locus (63). In agreement with genome-wide analyses (38), we observed a decrease in the number of reads corresponding to sense transcription (+ strand) of the cye-1 gene in csr-1 and drh-3 mutants (Figure 2, compare red and black tracks). We also observed an increase in antisense (– strand) reads corresponding to the large intron 2 of cye-1 (Figure 2, compare orange and gray tracks). Notably, intronic enhancer elements have recently been annotated in C. elegans (26), including the second intron of cye-1. Therefore, the cye-1 antisense transcript may represent an enhancer RNA.
Figure 2.
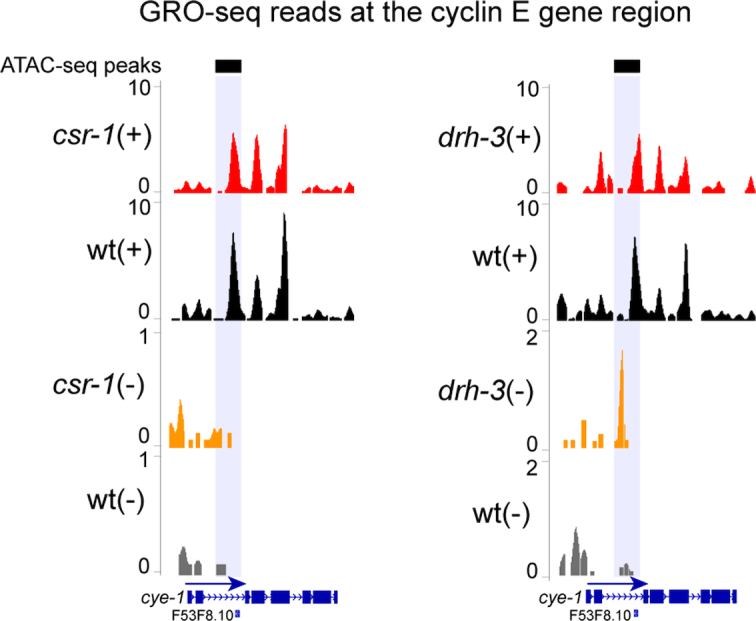
Distribution of GRO-seq reads at the cye-1 gene region. (top) A reduction in GRO-seq read numbers on the plus (+) strand in csr-1(WM193) (left plot) and drh-3(ne4253) (right plot) compared to wild type (red versus black) illustrates a decrease in cye-1 gene transcription. (bottom) On the minus (–) strand, there is an increase in GRO-seq reads in csr-1 (left plot) and drh-3 (right plot) mutants compared to wild type (orange versus gray) at the intronic enhancer region (gray shading) detected by ATAC-seq (black bars) (25). The directionality of transcription is marked with a blue arrow. Normalization was performed as described in methods.
To further investigate transcription associated with other potential enhancer regions, we re-analyzed GRO-seq data (38) with respect to enhancer-mapping information from two recent publications (25,26). In (25), open chromatin regions associated with enhancers were detected by ATAC-seq and the functionality of some was confirmed. In another study (26), enhancer locations were predicted using established histone modification signatures. Our analysis of GRO-seq data identified a global increase in transcription at ATAC-seq-mapped distal (intergenic) enhancers in both csr-1(WM193) and drh-3(ne4253) (Figure 3A). We obtained similar results when analyzing GRO-seq reads at the predicted enhancers (26) (Figure 3B). Indeed, we found increased antisense transcription at a functional upstream regulatory region of the daf-12 gene (64) that has recently been defined as an active L3 stage enhancer by ATAC-seq (25) (Figure 3C).
Figure 3.

Elevated H3K9me3 and transcription at enhancer regions in csr-1(WM193) and drh-3(ne4253). (A) GRO-seq read coverage is increased at the distal enhancer regions (intergenic ATAC-seq peaks) in csr-1 (left panel) and drh-3 (right panel) mutants: top – box plots, bottom – cumulative read distribution plots. P-value = 8.694 × 10−13 and P-value < 2.2 × 10−16 correspond to the left and the right box plots, respectively (Wilcoxon test). The GRO-seq data from (38) were normalized as described in Materials and Methods and the log2 transformed RPKM was plotted. Cumulative plots confirm the box plot data analyses for csr-1 (left) and drh-3 (right) mutants and demonstrate an increase in GRO-seq reads coverage in both mutants at distal enhancer regions (ATAC-seq data from (25)) compared to cumulative GRO-seq reads counted in 2.5 Kb regions from the whole genome with the exception of ATAC-seq peaks (i.e. genomic regions). (B) Box plots demonstrate an increase in total GRO-seq read coverage at the putative enhancers (26) in csr-1 (left) or drh-3 (right) mutant L3 larva. The GRO-seq data (38) were normalized as described in methods, and the log2 transformed RPKM was plotted. P-values < 2.2 × 10−16 (Wilcoxon test). Cumulative plots confirm the box plot data analyses for csr-1 (left) and drh-3 (right) mutants and show an increase in GRO-seq read coverage in both mutants at the predicted enhancers (26). Analysis of cumulative distribution of log2 -transformed fold changes of GRO-seq RPKM values was performed for genome-wide regions not containing putative enhancers (all chromatin domains) and for combined domains 8, 9 and 10 (26) containing putative enhancer regions. (C) GRO-seq reads corresponding to antisense transcription are shown at a region containing a regulatory element detected by ATAC-seq upstream of the daf-12 gene. The directionality of sense transcription is marked with a blue arrow. An increase in the number of antisense reads is shown in drh-3 (blue) and csr-1 (green) mutants compared to wild type (gray). (D) Box plots and cumulative plots show increased H3K9me3 at distal (intergenic) enhancers detected by ATAC-seq (25) in csr-1 and drh-3 mutants compared to wild type. ChIP-seq data normalization was performed as described in methods. The cumulative genome-wide regions not containing ATAC-seq peaks were defined by dividing the genome into windows (genomic regions) of 2.5 kb. P-values < 2.2 × 10−16 (Wilcoxon test).
Importantly, in both csr-1 and drh-3 mutants, we found an increase in H3K9me3 at enhancer regions defined by ATAC-seq (25) (Figure 3D). Therefore, it is possible that elevated ncRNA transcription, including antisense transcription, directly or indirectly leads to chromatin compaction at these regions. Perhaps this mechanism involves dsRNA production and RNAi-induced silencing that depends on non-canonical small RNAs. Notably, the role of nuclear RNAi in suppressing overlapping transcription of convergent genes and read-through antisense transcription has been described in fission yeast (65,66).
Enhancer small RNAs may guide histone modifications in the absence of secondary siRNAs
To examine the mechanism underlying enhancer silencing, we searched for small RNAs originating from enhancers in published datasets (27,36). Intriguingly, there was a larger number of small RNAs mapping to the intronic enhancer of the cyclin E gene in the csr-1(tm892) and drh-3(ne4253) mutant datasets (Figure 4A). Notably, the most prominent peak of sense-oriented small RNAs mapped to the annotated tRNA gene present in this intron (Figure 4A). Some additional enhancer small RNAs mapped to known intronic ncRNAs (Supplementary Figure S2A).
Figure 4.

Small RNA levels increase at enhancer regions in csr-1 and RdRP mutants compared to wild type. (A) Distribution of small RNAs at the cye-1 gene region. Normalization was performed as described in methods. The gene is transcribed from left to right (the directionality of transcription is marked with a blue arrow). The reads on the minus strand (in blue) corresponding to 22G-small RNAs are abundant in wild type (two upper tracks), but are depleted in mutants (two lower tracks). On the contrary, there is a sharp increase in small RNA reads (in red, marked by arrows) from the plus strand at the intronic enhancer region (gray shading (25)) in the mutants compared to wild type. The ATAC-seq peaks (25) are represented by the black squares with extended gray background. (B) Small RNA read coordinates were intersected with enhancer domain coordinates (26). Percentages of small RNA reads mapped to enhancer regions are represented by blue bars. Mutants are grouped with the corresponding wild type. (C) Distribution of small RNAs at the enhancer region on chromosome I in csr-1(tm892) (left) and drh-3(ne4253) (right). Normalization was performed as described in Materials and Methods. The reads corresponding to small RNAs on the minus strand (–) (blue) at the enhancer region (gray shading (25)) are more abundant in the mutants compared to wild type. Consistently, the H3K9me3 peak increases in height and H3K9me3 spreads around the enhancer region in csr-1(WM193) (green) and drh-3(ne4253) (blue) compared to wild type (gray). ATAC-seq peaks (25) are represented by the black squares with extended gray background. (D) The H3K9me3 ChIP-seq data were obtained from a published study (73). Reads were normalized and a log2 fold change was calculated for the early generation, F1, of the hrde-1 mutant and its late generation, F4, compared to the corresponding wild type samples. An increase in H3K9me3 at enhancer regions (26) was detected at an early generation of hrde-1(tm1200) (top plot), and progressive depletion of H3K9me3 (statistically significant only for intronic enhancers) was detected at a late generation of the mutant (bottom plot). (E) Normalized small RNA read coverage was overlapped with all enhancer regions defined in the previous study (26). Fold change was calculated between mutants and wild type for enhancer domains normalized to genome-wide coverage. P-value statistics was generated using Wilcoxon test. Significant changes (P < 10−4) are marked by an asterisk. (F) Small RNAs were sorted by the first nucleotide, and percentages were plotted on the Y axis. Mutants are grouped with the corresponding wild type, and different studies are defined by the square brackets. The CSR-1 IP sample (36) is presented separately.
In addition to the csr-1(tm892) and drh-3(ne4253) datasets, which are only partially depleted of secondary siRNAs, we have also analyzed small RNAs cloned from strong RdRP-deficient mutants that lack either one RdPR, EGO-1, required for germline RNAi (67), or both EGO-1 and RRF-1 RdRPs (27).
In wild type animals, ∼8–11% of small RNA reads map to enhancer domains defined by (26). Moreover, they are present in the CSR-1 IP samples, i.e. are bound by the CSR-1 Argonaute. Interestingly, 22G-RNA depletion in the RdRP mutants is associated with a notable increase in abundance (to 13–19%) of the ‘enhancer’ fraction of small RNAs (Figure 4B and Supplementary Figure S2B). It is possible that less abundant small RNAs are cloned more efficiently under these conditions. However, another possibility is that ‘enhancer’ small RNAs may have a better chance to interact with Argonautes, which lack their more prominent partners. This could explain an increase in H3K9me3 at enhancers in csr-1 and drh-3 mutants (Figures 3D and 4C).
A major player in transcriptional gene silencing downstream of RdRP-produced secondary 22G-RNAs is the nuclear-localized WAGO HRDE-1. HRDE-1 bound to secondary siRNAs facilitates heterochromatinization and silencing at loci targeted by exogenous dsRNA, as well as at a group of native (endogenous) targets (8,29,31,35,68–73). HRDE-1 recruits nuclear RNAi proteins NRDE-1/2/4, which promote deposition of repressive chromatin marks at RNAi-targeted loci and induce co-transcriptional inhibition of RNA Polymerase II (31,68).
To evaluate the relationship between HRDE-1 and H3K9me3 distribution at enhancer regions, we analyzed the H3K9me3 ChIP-seq data from a recent study focused on transgenerational effects of nuclear RNAi on chromatin (73). This study used the hrde-1(tm1200) loss-of-function mutant and analyzed early (F1) or late (F4) generations of homozygous mutant worms. First, we confirmed the global reduction in H3K9me3 coverage at a later (F4) hrde-1(-) generation observed by the authors (Figure 4D, lower plot, gray bars) (73). We also observed a decrease in intronic enhancer-associated H3K9me3 in this generation (Figure 4D, lower plot, pink bars), which correlates with a slight decrease in enhancer-matching small RNAs in this mutant (Figure 4B).
Surprisingly, in the first generation of hrde-1(tm1200) mutant worms (F1), we detected a significant increase in H3K9me3 at enhancers (Figure 4D, upper plot). This effect is similar to the effects of csr-1(WM193) and drh-3(ne4253) (Figure 3D) and suggests that HRDE-1 is not responsible for the ectopic H3K9me3. In the case of csr-1(WM193) and drh-3(ne4253), elevated H3K9me3 correlated with increased numbers of enhancer-matching small RNA. However, this is not the case for hrde-1(tm1200) (Figure 4B). One possible explanation for this discrepancy is a very tight size selection of the cloned 22G-RNAs in the hrde-1(tm1200) study (73) (Supplementary Figure S3A, bottom, and Supplementary Figure S3B), such that all the samples (wild type and mutant) contain fewer small RNAs and completely lack RNAs in the 24+ nucleotide range that feature in the csr-1(tm892) and drh-3(ne4253) samples from two independent studies, (36) and (27), respectively (Supplementary Figure S3). Therefore, our further analyses using small RNA datasets from this study (73) may not fully reflect the distribution of small RNAs at enhancer regions. To investigate the alterations of small RNA read coverage at individual enhancers, we overlapped the genomic coordinates of enhancer regions (26) with the small RNA read coverage and calculated average fold change in RNAi-related mutants compared to wild type. We observed an increase in the number of small RNAs in csr-1 and RdRP-deficient mutants (Figure 4E). Next, we specifically examined intronic enhancers located within previously defined CSR-1 target genes (36). We observed the same directionality of changes in small RNAs within these target genes in the mutants, although the increase in small RNAs was not significant in csr-1(tm892) and ego-1(om97) (Supplementary Figure S2C, left bar-plot). Notably, only 2.7% of the currently recognized CSR-1 target genes contain intronic enhancers, and the vast majority of intronic enhancers are harbored in developmentally-regulated genes (not CSR-1 targets). Small RNAs matching enhancers in these non-target genes demonstrate statistically significant changes in the csr-1 and RdRP null mutants compared to wild type (Supplementary Figure S2C, right bar-plot).
We observed that enhancer-matching small RNAs in the CSR-1 IP, as well as in wild type and csr-1 mutant samples, predominantly start with 5′G (Figure 4F) and are 20–22 nucleotides in length (Supplementary Figure S3A), consistent with the 22G-RNA definition. Intriguingly, 5′U RNAs matching enhancers are also present and they become prevalent in the rrf-1(neC1)ego-1(om97) and drh-3 mutants (Figure 4F), where the function of both RRF-1 and EGO-1 RdRP complexes is compromised (27). This enrichment of 5′U RNAs suggests that RdRP-independent small RNAs may originate from short Pol II transcripts, similarly to the C. elegans 21U-RNAs (74,75), or from degradation products of abundant transcripts, such as rRNA and tRNA, similarly to the S. pombe priRNAs (76). The latter possibility is further supported by the variable length of enhancer small RNAs (Supplementary Figure S3A).
Histone H3K27 methylation is ectopically increased on highly active genes upon drh-3 loss-of-function
Our H3K27me3 and H3K36me3 ChIP-seq results obtained using wild type worms are consistent with previous studies (77–79) and demonstrate that H3K36me3 is linked to actively transcribed genes, while H3K27me3 is enriched in regions with low transcription activity (Figure 5A).
Figure 5.

Ectopic H3K27me3 accumulation at highly expressed genes in drh-3(ne4253). (A) Open chromatin is associated with H3K36me3 histone modification (pink), which opposes silencing histone mark H3K27me3 (blue). Highly expressed genes (among them the CSR-1 targets ncl-1, lin-13, snu-23) are enriched in H3K36me3 and depleted of H3K27me3. H3K27me3 is abundant at low expressed/silent regions. Highly expressed genes are in red, low expressed genes (expression quantile 1 and 2, see Supplementary Figure S4A) are in blue. The peaks were generated with MACS2 peak calling algorithm (Galaxy version 2.0.10.2) (50). (B) ChIP-seq normalized coverage for H3K36me3 (pink) and H3K27me3 (blue) demonstrating a decrease in H3K36me3 in drh-3(ne4253) compared to wild type and an increase in H3K27me3 modification at the highly expressed CSR-1 target gene bet-2. H3K9me3 levels do not change at the presented locus (brown). The directionality of transcription is marked with a blue arrow. (C) Global increase in H3K27me3 and reduction in H3K36me3 at highly expressed CSR-1 target genes in drh-3(ne4253), but not in csr-1(WM193) (scatterplots obtained by differential peak analysis), is concordant with the degree of nascent transcript reduction (bottom box plots, GRO-seq reads from (38)) in the mutants compared to wild type. For histone modification analysis, normalization of both replicas was performed as described in Materials and Methods. Only the subclass of highly active genes (CSR-1 targets) is represented. The colors reflect differential enrichment (red) and depletion (blue) of a histone mark observed in both biological replicates. The GRO-seq data (38) were normalized as described in Materials and Methods, and log2 transformed RPKM was plotted. P-values for GRO-seq data: drh3(ne4253) versus wt, P-value < 2.2 × 10−16; csr-1(WM193) versus wt, P-value = 1.092 × 10−11 (Wilcoxon test). (D) A heatmap demonstrating an increase in the heterochromatic modification H3K27me3 and a decrease in the activating modification H3K36me3 at actively transcribed gene coding regions in drh-3(ne4253) compared to wild type. Each row corresponds to a CSR-1 target gene body between transcription start site (TSS) and transcription termination site (TTS) ±100 bp. Gene length was approximated to be 1000 bp for representation purposes. The mean value from two biological replicates was plotted. (E) A heatmap demonstrating an absence of uniform changes in random sets of genes in drh-3(ne4253) compared to wild type. Each row corresponds to gene coding regions between transcription start site (TSS) and transcription termination site (TTS) ± 100 bp. Gene length was approximated to be 1000 bp for representation purposes. The mean value from two biological replicates is shown. A list of the random set of genes is presented in Supplementary Table S3.
In drh-3(ne4253), transcription reduction of normally highly expressed genes strongly correlates with a reduction in the levels of the active histone modification H3K36me3 and a gain of the opposing repressive histone mark H3K27me3 (Figure 5B). Many highly expressed mRNAs are known to be complementary to CSR-1-bound small RNAs, i.e. they represent CSR-1 target genes (Supplementary Figure S4A) (36). Therefore, the reduced expression of CSR-1 target genes in the drh-3(ne4253) mutant (Figure 5C, bottom left panel) correlates with a global reduction in H3K36me3 accompanied by a gain in H3K27me3 (Figure 5C, top and middle left panels, and Figure 5D).
In order to confirm that changes in the methylation patterns are not random and are linked to transcriptional alterations, we examined two random sets of genes (4000 and 5000) comparable in number to the CSR-1 target gene population. The random gene picking was performed using the R command sample, and the list of random genes can be found in Supplementary Table S3. We did not observe any substantial changes between drh-3(ne4253) and wild type when we compared the mean ChIP-seq coverage for each histone modification at these random sets of genes (Figure 5E and Supplementary Figure S4B). At the same time, we observed that transcriptionally silent or less active regions (pseudogenes and tissue-specific genes, represented by a group of spermatogenesis genes), which become more active in drh-3(ne4253), gain H3K36me3 and lose H3K27me3 in the drh-3(ne4253) mutant (Supplementary Figure S4C and D).
Surprisingly, in the partially rescued csr-1(tm892) null mutant (strain WM193, (36)), the differential enrichment analysis did not reveal dramatic changes in H3K36me3 and H3K27me3 (Figure 5C, top and middle right panels), even though transcription changes similar to those occurring in drh-3(ne4253) take place in this mutant as well (Figure 5C, bottom right panel) (38). We further analyzed these two modifications in csr-1(WM193) by averaging global changes in gene coverage in the mutant compared to wild type and found a statistically significant increase of H3K27me3 at CSR-1 target genes (confirmed with cumulative frequency plot) (Supplementary Figure S4E, H3K27me3). However, it is important to note that the number of CSR-1 target genes in which H3K27me3 read coverage is above 0 in either strain (wild type or csr-1(WM193)) in at least one replicate is very low (∼3%, or 120 genes). Thus, although we have observed an increase in H3K27me3 coverage at a small portion of CSR-1 targets in the csr-1 mutant compared to wild type, the contribution of H3K27me3 to the downregulation of CSR-1 targets in csr-1(WM193) is likely minimal compared to that in drh-3(ne4253). As discussed earlier, H3K9me3 is much more common for CSR-1 target genes (∼25%, or 1000 genes), and its increase correlates with reduced expression of these genes in csr-1(WM193). Although there is also statistically significant change in the H3K36me histone mark shown with the box plot, it is not confirmed with the cumulative plot (Supplementary Figure S4E, H3K36me3).
Overall, our results indicate that a loss of ‘activating’ siRNA function leads to ectopic H3K9me3 at CSR-1 target genes, likely dependent on WAGO (Figures 1 and 6A). However, a loss of both ‘activating’ and ‘silencing’ siRNAs results in dramatic redistribution of H3K36me3 and H3K27me3 histone modifications along gene bodies (Figures 5 and 6A). Since WAGO siRNAs are depleted in the drh-3(ne4253) mutant, we favor the possibility that additional mechanisms are responsible for ectopic deposition of H3K27me3.
Figure 6.

Models describing connections between small RNAs and chromatin at actively transcribed genes (A) and at enhancer regions (B). RdRP complexes that include the DRH-3 helicase (not shown) produce 22G-RNAs (red), which are loaded onto CSR-1 or WAGO Argonautes. Enhancer small RNAs (purple) are generated in RdRP-independent manner. (A) 22G-RNAs in complex with CSR-1 Argonaute bind nascent transcripts of highly active genes, thus protecting the latter from WAGO-bound 22G-RNAs that guide H3K9me3, which is associated with silencing. Either active transcription facilitated by CSR-1 Argonaute or silencing by WAGO (when CSR-1 is deficient) prevent H3K27me3-modulated silencing through additional mechanisms. (B) CSR-1-associated 22G-RNAs bind some enhancer-derived transcripts, protecting them from silencing by other mechanisms. When CSR-1 or RdRP-produced 22G-RNAs are limited, other Argonautes may load enhancer small RNAs that accumulate in these regions. This may lead to silencing through H3K9 methylation driven by Argonaute proteins and associated methyltransferases.
DISCUSSION
Nuclear RNAi in C. elegans has been associated with secondary 22G-RNAs generated in the cytoplasm that then move to the nucleus to execute their function. It has been largely studied using exogenous dsRNA for silencing initiation and reporter sequences for nuclear silencing readout (6,8,28–33,68).
Our work links deregulation of the nuclear 22G-RNA pathways to ectopic H3K9 and H3K27 methylation at endogenous genes. Both histone marks can be artificially induced on active genes targeted by exogenous dsRNA (8,31,80), and several H3K9 methyltransferases have been connected to WAGO-induced silencing (30,72,80,81). However, it was not possible to reliably correlate nuclear Argonaute function in inhibiting endogenous genes with H3K9 methylation (34,35). Our results connect an increase in H3K9me3 to reduced transcription of CSR-1 target genes when CSR-1 is limited (38) (Figure 6A). Our data also support earlier models suggesting that under these conditions WAGO may interact with pre-mRNAs normally engaged by CSR-1 (61,62,82). Consistent with this possibility, a deep analysis of small RNAs present in WAGO IP samples identified a considerable amount of 22G-RNAs matching typical CSR-1 targets in wild type worms (83), and dramatic misrouting of 22G-RNAs normally bound by CSR-1 to HRDE-1 takes place in the absence of piRNAs (84).
Given the role of HRDE-1-bound 22G-RNAs (29,35,68–73) in maintaining normal levels of H3K9me3, we were surprised to find an increase in H3K9me3 at enhancer elements in drh-3 and hrde-1 mutants. Although we found small RNAs matching enhancers in CSR-1 IP samples (27,36), the number of small RNAs produced from enhancers in csr-1(WM193) and RdRP mutants exceeds the number of small RNAs precipitated with CSR-1 IP. Also, the majority of enhancer small RNAs in RdRP null and drh-3(ne4253) mutants start with uridine. These results indicate that most small RNAs matching enhancers are not 22G-RNAs. This is consistent with our finding that HRDE-1 is not specifically involved in depositing H3K9me3 at enhancers, at least in an early generation of mutants (F1). On the contrary, it appears to antagonize H3K9me3 at these genomic loci.
A logical explanation of these results is that, in the absence of 22G-RNAs, enhancer small RNAs may get loaded onto some silencing Argonautes that are working together with H3K9 methyltransferases (Figure 6B). There is a great variety of Argonautes in C. elegans, and it remains to be seen which ones are involved. In terms of RdRP-independent enhancer small RNA biogenesis, our results support the possibility of: (i) dsRNA formation and Dicer-dependent primary siRNA production, or (ii) a 21U-RNA-like biogenesis mechanism that involves trimming of transcripts starting with uridine. The first possibility is consistent with the described role of Dicer in promoting H3K9me2 at loci with overlapping ncRNA transcription in mammalian cells (85). The second possibility is reminiscent of priRNA production in S. pombe (76). Also, it is known that tRNA fragments can be bound by Argonautes (86), and we identify tRNA-derived and ncRNA-derived small RNAs at the intronic enhancers in C. elegans.
We would like to emphasize that in this study, as well as in our earlier GRO-seq publication (38), we used the partially rescued csr-1(tm892) mutant strain WM193 (36), which is homozygous viable. Moreover, we used L3 stage worms with intact germlines. These conditions are distinct from those utilized by Desai and colleagues for studying cytoplasmic CSR-1 function in the early embryo (87) and also from those in our report on the role of CSR-1 in histone mRNA maturation, which employed csr-1(RNAi) in a sensitized background during post-embryonic development (88).
A surprising finding that should stimulate further inquiry is that the most dramatic histone modification re-distribution involving H3K36me3 and H3K27me3 was observed in the drh-3(ne4253) mutant background, where both CSR-1 and WAGO pathways are largely inactive. This suggests that either ectopic 22G-RNA-independent silencing of CSR-1 target genes takes place in this case (Figure 6A), or that residual 22G-RNAs present in drh-3(ne4253) (27) play some role. The latter possibility may involve WAGO-1, which was connected to small RNA-guided H3K27me3 deposition (80). It is still possible that histone modification changes in drh-3(ne4253) reflect a reduction in CSR-1-bound 22G-RNAs. Although we do not observe dramatic alterations in the H3K27me3/H3K36me3 landscapes in csr-1(WM193), this strain may be less compromised in the CSR-1 pathway function than drh-3(ne4253).
In any case, it is likely that the observed elevation of antisense RNA expression at CSR-1 target genes is functionally important. First, it may lead to dsRNA formation and nuclear RNAi initiation through Dicer-dependent siRNA production. Alternatively, nuclear antisense or other ncRNA may participate in promoting H3K27me3 by acting as a scaffold for histone modifying complexes in scenarios similar to those described in other systems (89).
Overall, we uncovered an unexpected complexity in the ways that small RNAs affect chromatin in C. elegans. In addition to providing evidence for competition between the CSR-1 and WAGO pathways in regulation of the most active genes in the nucleus, we describe their involvement in shaping enhancer chromatin and connect nuclear RNAi to regulation of H3K27 methylation on a genome-wide scale.
DATA AVAILABILITY
GEO accession number: GSE115629; UCSC genome browser session: https://genome.ucsc.edu/cgi-bin/hgTracks?db=ce10&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chrI%3A1%2D201000&hgsid=721327325_uphqYy4cv0zAXGasJOWm7Uq1q4LA.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Craig Mello's lab (UMass Worcester) for providing RNAi mutant strains, and we thank Daniel Cifuentes, Dmitry Kretov and members of the BU RNA Club for fruitful discussions.
Author contributions: E.S.G. conducted experiments and analyzed data, R.E. analyzed data, Q.M. and N.C.L. analyzed small RNA data, A.G. supervised the project. E.S.G. and A.G. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences of the National Institutes of Health [R01GM107056 to A.G.]; National Institute of Aging of the National Institutes of Health [R01AG052465 to N.C.L.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Some strains used in this study were obtained from the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs [P40 OD010440]. The BU Microarray and Sequencing Resource Core Facility is supported by the UL1TR001430 grant awarded to the BU Clinical and Translational Science Institute. Funding for open access charge: Boston University funds (provided to A.G.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Cecere G., Grishok A.. A nuclear perspective on RNAi pathways in metazoans. Biochim. Biophys. Acta. 2014; 1839:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gagnon K.T., Li L., Chu Y., Janowski B.A., Corey D.R.. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014; 6:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holoch D., Moazed D.. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015; 16:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pikaard C.S., Mittelsten Scheid O.. Epigenetic regulation in plants. Cold Spring Harbor Perspect. Biol. 2014; 6:a019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grewal S.I. RNAi-dependent formation of heterochromatin and its diverse functions. Curr. Opin. Genet. Dev. 2010; 20:134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burton N.O., Burkhart K.B., Kennedy S.. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. PNAS. 2011; 108:19683–19688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grishok A., Sinskey J.L., Sharp P.A.. Transcriptional silencing of a transgene by RNAi in the soma of C. elegans. Genes Dev. 2005; 19:683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gu S.G., Pak J., Guang S., Maniar J.M., Kennedy S., Fire A.. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat. Genet. 2012; 44:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gullerova M., Proudfoot N.J.. Convergent transcription induces transcriptional gene silencing in fission yeast and mammalian cells. Nat. Struct. Mol. Biol. 2012; 19:1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tam O.H., Aravin A.A., Stein P., Girard A., Murchison E.P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R.M. et al.. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008; 453:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watanabe T., Totoki Y., Toyoda A., Kaneda M., Kuramochi-Miyagawa S., Obata Y., Chiba H., Kohara Y., Kono T., Nakano T. et al.. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008; 453:539–543. [DOI] [PubMed] [Google Scholar]
- 12. Gaudet J., McGhee J.D.. Recent advances in understanding the molecular mechanisms regulating C. elegans transcription. Dev. Dyn. 2010; 239:1388–1404. [DOI] [PubMed] [Google Scholar]
- 13. Henriques T., Scruggs B.S., Inouye M.O., Muse G.W., Williams L.H., Burkholder A.B., Lavender C.A., Fargo D.C., Adelman K.. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018; 32:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Long H.K., Prescott S.L., Wysocka J.. Ever-Changing landscapes: transcriptional enhancers in development and evolution. Cell. 2016; 167:1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li W., Notani D., Rosenfeld M.G.. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 2016; 17:207–223. [DOI] [PubMed] [Google Scholar]
- 16. Banerji J., Olson L., Schaffner W.. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell. 1983; 33:729–740. [DOI] [PubMed] [Google Scholar]
- 17. Gillies S.D., Morrison S.L., Oi V.T., Tonegawa S.. A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell. 1983; 33:717–728. [DOI] [PubMed] [Google Scholar]
- 18. Mercola M., Wang X.F., Olsen J., Calame K.. Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science. 1983; 221:663–665. [DOI] [PubMed] [Google Scholar]
- 19. De Santa F., Barozzi I., Mietton F., Ghisletti S., Polletti S., Tusi B.K., Muller H., Ragoussis J., Wei C.L., Natoli G.. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010; 8:e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim T.K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J., Harmin D.A., Laptewicz M., Barbara-Haley K., Kuersten S. et al.. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010; 465:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Melgar M.F., Collins F.S., Sethupathy P.. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol. 2011; 12:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lipshitz H.D., Peattie D.A., Hogness D.S.. Novel transcripts from the Ultrabithorax domain of the bithorax complex. Genes Dev. 1987; 1:307–322. [DOI] [PubMed] [Google Scholar]
- 23. Feng J., Bi C., Clark B.S., Mady R., Shah P., Kohtz J.D.. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006; 20:1470–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki H.I., Young R.A., Sharp P.A.. Super-Enhancer-Mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017; 168:1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daugherty A.C., Yeo R.W., Buenrostro J.D., Greenleaf W.J., Kundaje A., Brunet A.. Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res. 2017; 27:2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evans K.J., Huang N., Stempor P., Chesney M.A., Down T.A., Ahringer J.. Stable Caenorhabditis elegans chromatin domains separate broadly expressed and developmentally regulated genes. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E7020–E7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu W., Shirayama M., Conte D. Jr, Vasale J., Batista P.J., Claycomb J.M., Moresco J.J., Youngman E.M., Keys J., Stoltz M.J. et al.. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol. Cell. 2009; 36:231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guang S., Bochner A.F., Pavelec D.M., Burkhart K.B., Harding S., Lachowiec J., Kennedy S.. An Argonaute transports siRNAs from the cytoplasm to the nucleus. Science. 2008; 321:537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buckley B.A., Burkhart K.B., Gu S.G., Spracklin G., Kershner A., Fritz H., Kimble J., Fire A., Kennedy S.. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature. 2012; 489:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ashe A., Sapetschnig A., Weick E.M., Mitchell J., Bagijn M.P., Cording A.C., Doebley A.L., Goldstein L.D., Lehrbach N.J., Le Pen J. et al.. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell. 2012; 150:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guang S., Bochner A.F., Burkhart K.B., Burton N., Pavelec D.M., Kennedy S.. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010; 465:1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luteijn M.J., van Bergeijk P., Kaaij L.J., Almeida M.V., Roovers E.F., Berezikov E., Ketting R.F.. Extremely stable Piwi-induced gene silencing in Caenorhabditis elegans. EMBO J. 2012; 31:3422–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shirayama M., Seth M., Lee H.C., Gu W., Ishidate T., Conte D. Jr, Mello C.C.. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012; 150:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kalinava N., Ni J.Z., Peterman K., Chen E., Gu S.G.. Decoupling the downstream effects of germline nuclear RNAi reveals that H3K9me3 is dispensable for heritable RNAi and the maintenance of endogenous siRNA-mediated transcriptional silencing in Caenorhabditis elegans. Epigenet. Chromatin. 2017; 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ni J.Z., Chen E., Gu S.G.. Complex coding of endogenous siRNA, transcriptional silencing and H3K9 methylation on native targets of germline nuclear RNAi in C. elegans. BMC Genomics. 2014; 15:1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Claycomb J.M., Batista P.J., Pang K.M., Gu W., Vasale J.J., van Wolfswinkel J.C., Chaves D.A., Shirayama M., Mitani S., Ketting R.F. et al.. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell. 2009; 139:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yigit E., Batista P.J., Bei Y., Pang K.M., Chen C.C., Tolia N.H., Joshua-Tor L., Mitani S., Simard M.J., Mello C.C.. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell. 2006; 127:747–757. [DOI] [PubMed] [Google Scholar]
- 38. Cecere G., Hoersch S., O’Keeffe S., Sachidanandam R., Grishok A.. Global effects of the CSR-1 RNA interference pathway on the transcriptional landscape. Nat. Struct. Mol. Biol. 2014; 21:358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974; 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Celniker S.E., Dillon L.A., Gerstein M.B., Gunsalus K.C., Henikoff S., Karpen G.H., Kellis M., Lai E.C., Lieb J.D., MacAlpine D.M. et al.. Unlocking the secrets of the genome. Nature. 2009; 459:927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Didion J.P., Martin M., Collins F.S.. Atropos: specific, sensitive, and speedy trimming of sequencing reads. Peer J. 2017; 5:e3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang D.W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. 2007. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cecere G., Hoersch S., Jensen M.B., Dixit S., Grishok A.. The ZFP-1(AF10)/DOT-1 complex opposes H2B ubiquitination to reduce Pol II transcription. Mol. Cell. 2013; 50:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robinson M.D., Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Robinson M.D., Stirzaker C., Statham A.L., Coolen M.W., Song J.Z., Nair S.S., Strbenac D., Speed T.P., Clark S.J.. Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010; 20:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brown J., Pirrung M., McCue L.A.. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017; 33:3137–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chirn G.W., Rahman R., Sytnikova Y.A., Matts J.A., Zeng M., Gerlach D., Yu M., Berger B., Naramura M., Kile B.T. et al.. Conserved piRNA Expression from a Distinct Set of piRNA Cluster Loci in Eutherian Mammals. PLos Genet. 2015; 11:e1005652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sytnikova Y.A., Rahman R., Chirn G.W., Clark J.P., Lau N.C.. Transposable element dynamics and PIWI regulation impacts lncRNA and gene expression diversity in Drosophila ovarian cell cultures. Genome Res. 2014; 24:1977–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quinlan A.R. BEDTools: The Swiss-Army tool for genome feature analysis. Curr. Protoc. Bioinformatics. 2014; 47:doi:10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Speir M.L., Zweig A.S., Rosenbloom K.R., Raney B.J., Paten B., Nejad P., Lee B.T., Learned K., Karolchik D., Hinrichs A.S. et al.. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016; 44:D717–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu T., Rechtsteiner A., Egelhofer T.A., Vielle A., Latorre I., Cheung M.S., Ercan S., Ikegami K., Jensen M., Kolasinska-Zwierz P. et al.. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011; 21:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee H.C., Gu W., Shirayama M., Youngman E., Conte D. Jr, Mello C.C.. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell. 2012; 150:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seth M., Shirayama M., Gu W., Ishidate T., Conte D. Jr, Mello C.C.. The C. elegans CSR-1 argonaute pathway counteracts epigenetic silencing to promote germline gene expression. Dev. Cell. 2013; 27:656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wedeles C.J., Wu M.Z., Claycomb J.M.. Protection of germline gene expression by the C. elegans Argonaute CSR-1. Dev. Cell. 2013; 27:664–671. [DOI] [PubMed] [Google Scholar]
- 63. Grishok A., Sharp P.A.. Negative regulation of nuclear divisions in Caenorhabditis elegans by retinoblastoma and RNA interference-related genes. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:17360–17365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Antebi A., Culotti J.G., Hedgecock E.M.. daf-12 regulates developmental age and the dauer alternative in Caenorhabditis elegans. Development. 1998; 125:1191–1205. [DOI] [PubMed] [Google Scholar]
- 65. Gullerova M., Proudfoot N.J.. Cohesin complex promotes transcriptional termination between convergent genes in S. pombe. Cell. 2008; 132:983–995. [DOI] [PubMed] [Google Scholar]
- 66. Zofall M., Fischer T., Zhang K., Zhou M., Cui B., Veenstra T.D., Grewal S.I.. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature. 2009; 461:419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smardon A., Spoerke J.M., Stacey S.C., Klein M.E., Mackin N., Maine E.M.. EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr. Biol.: CB. 2000; 10:169–178. [DOI] [PubMed] [Google Scholar]
- 68. Burkhart K.B., Guang S., Buckley B.A., Wong L., Bochner A.F., Kennedy S.. A pre-mRNA-associating factor links endogenous siRNAs to chromatin regulation. PLos Genet. 2011; 7:e1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kalinava N., Ni J.Z., Gajic Z., Kim M., Ushakov H., Gu S.G.. C. elegans heterochromatin factor SET-32 plays an essential role in transgenerational establishment of nuclear RNAi-Mediated epigenetic silencing. Cell Rep. 2018; 25:2273–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lev I., Seroussi U., Gingold H., Bril R., Anava S., Rechavi O.. MET-2-dependent H3K9 methylation suppresses transgenerational small RNA inheritance. Curr. Biol. 2017; 27:1138–1147. [DOI] [PubMed] [Google Scholar]
- 71. Ni J.Z., Kalinava N., Chen E., Huang A., Trinh T., Gu S.G.. A transgenerational role of the germline nuclear RNAi pathway in repressing heat stress-induced transcriptional activation in C. elegans. Epigenet. Chromatin. 2016; 9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ni J.Z., Kalinava N., Mendoza S.G., Gu S.G.. The spatial and temporal dynamics of nuclear RNAi-targeted retrotransposon transcripts in Caenorhabditis elegans. Development. 2018; 145:dev167346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Weiser N.E., Yang D.X., Feng S., Kalinava N., Brown K.C., Khanikar J., Freeberg M.A., Snyder M.J., Csankovszki G., Chan R.C. et al.. MORC-1 Integrates Nuclear RNAi and transgenerational chromatin architecture to promote germline immortality. Dev. Cell. 2017; 41:408–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cecere G., Zheng G.X., Mansisidor A.R., Klymko K.E., Grishok A.. Promoters recognized by forkhead proteins exist for individual 21U-RNAs. Mol. Cell. 2012; 47:734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gu W., Lee H.C., Chaves D., Youngman E.M., Pazour G.J., Conte D. Jr, Mello C.C.. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell. 2012; 151:1488–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Halic M., Moazed D.. Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell. 2010; 140:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gaydos L.J., Wang W., Strome S.. Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science. 2014; 345:1515–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schmitges F.W., Prusty A.B., Faty M., Stutzer A., Lingaraju G.M., Aiwazian J., Sack R., Hess D., Li L., Zhou S. et al.. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell. 2011; 42:330–341. [DOI] [PubMed] [Google Scholar]
- 79. Yuan W., Xu M., Huang C., Liu N., Chen S., Zhu B.. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011; 286:7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mao H., Zhu C., Zong D., Weng C., Yang X., Huang H., Liu D., Feng X., Guang S.. The nrde pathway mediates Small-RNA-Directed histone H3 Lysine 27 trimethylation in caenorhabditis elegans. Curr. Biol. 2015; 25:2398–2403. [DOI] [PubMed] [Google Scholar]
- 81. Spracklin G., Fields B., Wan G., Becker D., Wallig A., Shukla A., Kennedy S.. The RNAi inheritance machinery of caenorhabditis elegans. Genetics. 2017; 206:1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shen E.Z., Chen H., Ozturk A.R., Tu S., Shirayama M., Tang W., Ding Y.H., Dai S.Y., Weng Z., Mello C.C.. Identification of piRNA binding sites reveals the argonaute regulatory landscape of the C. elegans germline. Cell. 2018; 172:937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. de Albuquerque B.F., Placentino M., Ketting R.F.. Maternal piRNAs are essential for germline development following de novo establishment of Endo-siRNAs in Caenorhabditis elegans. Dev. Cell. 2015; 34:448–456. [DOI] [PubMed] [Google Scholar]
- 84. Phillips C.M., Brown K.C., Montgomery B.E., Ruvkun G., Montgomery T.A.. piRNAs and piRNA-Dependent siRNAs protect conserved and essential C. elegans genes from misrouting into the RNAi pathway. Dev. Cell. 2015; 34:457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. White E., Schlackow M., Kamieniarz-Gdula K., Proudfoot N.J., Gullerova M.. Human nuclear Dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat. Struct. Mol. Biol. 2014; 21:552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sobala A., Hutvagner G.. Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip. Rev. RNA. 2011; 2:853–862. [DOI] [PubMed] [Google Scholar]
- 87. Gerson-Gurwitz A., Wang S., Sathe S., Green R., Yeo G.W., Oegema K., Desai A.. A small RNA-Catalytic argonaute pathway tunes germline transcript levels to ensure embryonic divisions. Cell. 2016; 165:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Avgousti D.C., Palani S., Sherman Y., Grishok A.. CSR-1 RNAi pathway positively regulates histone expression in C. elegans. EMBO J. 2012; 31:3821–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Meller V.H., Joshi S.S., Deshpande N.. Modulation of chromatin by noncoding RNA. Annu. Rev. Genet. 2015; 49:673–695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GEO accession number: GSE115629; UCSC genome browser session: https://genome.ucsc.edu/cgi-bin/hgTracks?db=ce10&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chrI%3A1%2D201000&hgsid=721327325_uphqYy4cv0zAXGasJOWm7Uq1q4LA.