

Abstract
In all domains of life, the nucleobases of tRNA can be methylated. These methylations are introduced either by enzymes or by the reaction of methylating agents with the nucleophilic centers of the nucleobases. Herein, we present a systematic approach to identify the methylation sites within RNA in vitro and in vivo. For discrimination between enzymatic tRNA methylation and tRNA methylation damage in bacteria, we used nucleic acid isotope labeling coupled mass spectrometry (NAIL‐MS). With NAIL‐MS, we clearly observed the formation of 7‐methylguanosine, 3‐methyluridine, and 6‐methyladenosine during exposure of bacteria to the alkylating agent methyl methanesulfonate (MMS) in vivo. These damage products were not reported to form in tRNA in vivo, as they were masked by the enzymatically formed modified nucleosides in previous studies. In addition, we found formation of the known damage products 1‐methyladenosine and 3‐methylcytidine in vivo. With a dynamic NAIL‐MS setup, we observed tRNA repair by demethylation of these two RNA modifications in vivo. Furthermore, we saw the potential repair of 6‐methyladenosine but not 7‐methylguanosine in bacterial tRNA.
Keywords: isotopic labeling, mass spectrometry, nucleosides, RNA demethylation, tRNA
Introduction
RNA, and especially tRNA, contains a vast variety of modified nucleosides. From the natural RNA modifications known to date (≈160), 70 contain a methylated base or ribose.1 These methylations are enzymatically incorporated at defined positions of the RNA. In addition, nucleobases display several nucleophilic centers that are prone to reactions with electrophiles, and thus, non‐enzymatic methylation and alkylation have been reported.2 Currently, it is not possible to distinguish enzymatic methylation from direct chemical methylation.
Assessing the nucleophilicity of nucleosides in vitro
The reactivity of each position within the nucleobase is best assessed by exposing the nucleic acid or, even simpler, the nucleoside to the electrophile of interest in vitro. The reaction of DNA with various monoalkylating reagents was summarized in 1983.2 The conclusion of all analyzed studies was “that all simple, direct‐acting alkylating agents react with nucleic acid in vivo on the same sites as in vitro”.2 For cytidine and thymidine/uridine, alkylation of N3 was observed. In adenosine, the N1 position was most reactive, but N3 and N7 alkylation was also found, especially if adenosine was base paired. Guanosine was mainly alkylated at position 7 but also mildly at positions 2, 3, and 6. Figure 1 A shows the common alkylation sites of the canonical nucleosides as defined in the literature. The alkylation of nucleosides has been exploited in vitro for the introduction of functional groups such as coumarin derivatives with3 and without4 clickable moieties for further functionalization (Figure 1 A). Here, mainly uridine and thymidine (at position N3) were found to be alkylated under the alkaline reaction conditions.
Figure 1.
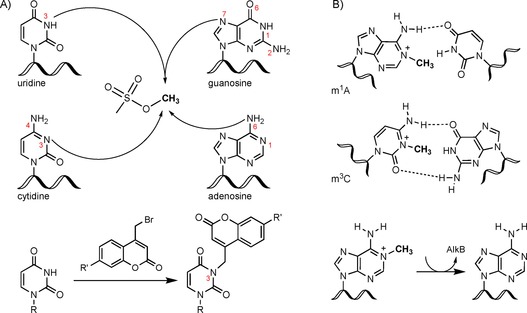
Nucleic acid alkylation sites; the impact on base pairing and repair by AlkB. A) Nucleophilic sites in nucleobases as indicated with the alkylating agent MMS or bromomethylcoumarins (e.g., R′=azide or propargyl). B) Methylation of N1 in adenine (m1A) and N3 in cytosine (m3C) disrupts base pairing and is, thus, repaired by AlkB in bacteria.
Beyond the nucleophilic centers of the canonical nucleosides, some modified nucleosides of RNA contain additional nucleophilic sites. These are exploited for the detection of modified ribonucleosides by reaction with various electrophiles, as recently reviewed.5
RNA damage and repair
In vivo, alkylation of DNA bases is linked directly to genomic instability, whereas the alkylation of RNA bases is believed to have a less dramatic effect. Interestingly, methylation of adenosine in RNA is as efficiently recognized and repaired as DNA alkylation damage. The key players of ribonucleic acid demethylation in both bacteria and mammals are the α‐ketoglutarate‐dependent dioxygenases AlkB and AlkBH, respectively.6 Previously reported in vitro tests showed that methylation of position 1 of adenine and position 3 of cytosine was quickly repaired by AlkB, as these positions were necessary for correct base pairing7 (Figure 1 B). The authors also showed that MS2 phage RNA, inactivated by methylation, became reactivated upon bacterial expression of AlkB, and thus, the importance of RNA methylation repair was presented in vivo. Later studies identified repair of the same methylated bases in mRNA and tRNA. Here, the negative effects on translation and aminoacylation caused by methylation damage were rescued by AlkB.8 Intriguingly, in this study only 3‐methylcytidine (m3C) and 1‐methyladenosine (m1A) were shown to be repaired in tRNA, but not 7‐methylguanosine (m7G). One reason might be that m7G is commonly found in many bacterial tRNAs (e.g., m7G48 in tRNAs Arg, Phe, and Val).1 It is possible that bacteria cannot distinguish natural m7G from damage‐derived m7G, and thus, m7G is not a substrate of demethylation by AlkB. The authors also described a pulse‐chase study based on radioactively labeled adenine, which clearly proved m1A repair by AlkB in vivo after previous induction of AlkB.8 m1A and m3C in bacterial small RNA (mostly tRNA) were identified to account for 99 % of the nucleic acid lesions formed upon exposure with methyl methanesulfonate (MMS).9 As a result of RNA turnover, RNA biosynthesis, and dilution effects, it was not possible to measure the RNA repair in vivo. Nevertheless, the authors demonstrated an accumulation of m1A in the AlkB‐deficient strain, which again hinted towards active repair of the lesion by AlkB.
Nucleic acid isotope labeling as a tool to observe nucleic acid repair in vivo
Radioactive labeling of RNA previously allowed observation of m1A demethylation in vivo.8 Although the radioisotope labeling approach was ideal to follow the fate of a biological mark, no further studies to explore the demethylation of nucleic acids were conducted. This is most likely due to the regulatory hurdles of running radioisotope laboratories. In 2017, we implemented isotope‐labeling techniques that could be done without radioisotopes. The prerequisite for these studies was the complete labeling of the nucleic acid with heavy, non‐radioactive isotopes such as carbon‐13, nitrogen‐15, and sulfur‐34 and access to a mass spectrometer. The combination of the different labeling media allowed the creation of a pulse‐chase experiment, which was used to observe repair of phosphorothioates in bacterial DNA.10 In the same year, we adapted the approach to RNA modification analysis in yeast.11 We followed the modification density of tRNA as a function of the growth phase, and we identified the underlying mechanisms for several modified nucleosides.
Herein, we present a systematic approach to identify the methylation sites within RNA both in vitro and in vivo. For discrimination of enzymatic RNA methylation and RNA methylation damage, we use nucleic acid isotope labeling coupled mass spectrometry (NAIL‐MS). With the presented method, we show RNA demethylation in vivo by overcoming the biases introduced by RNA turnover, RNA transcription, and dilution. With the strength of NAIL‐MS, it is possible to observe the dynamics of RNA modifications and to study RNA repair in vivo.
Results and Discussion
In vitro methylation of nucleosides reveals the most nucleophilic centers
Systematic assessment of nucleoside reactivity requires defined reaction conditions, a reliable way to stop the reaction, and an analytical system for quantification of the reaction products. With the goal to compare the in vitro damage products of the ribonucleosides with the damage products found in tRNA in vivo, we decided to use reaction conditions similar to those of the in vivo environment. The ribonucleosides were incubated in aqueous conditions at pH 7, 100 mm ionic strength, and 37 °C for 60 min. As quenchers, we tested several sulfur‐containing nucleophiles for their ability to suppress reaction of canonical nucleosides with the methylating agent MMS. Here, we found dithiothreitol (DTT) at pH 8 in 100‐fold excess to be a successful quencher that completely suppressed further methylation after its addition.
An equimolar mixture of the canonical nucleosides cytidine, uridine, guanosine, and adenosine was exposed to increasing doses of MMS at pH 7. After the reaction was quenched by the addition of DTT in alkaline buffer, the stable isotope labeled internal standard from yeast was added,11 and the number of methylated nucleosides was determined. The quantities of each methylation were normalized to the respective canonical they were derived from and were plotted as damage in percent (Figure 2 A for 1‐methyladenosine and Figure S1 in the Supporting Information for the other nucleosides). Under equimolar conditions, 0.15±0.09 % of all adenosines were methylated at the N1 position (m1A). A tenfold excess amount of MMS led to the formation of 0.62±0.02 % m1A, and a 100‐fold excess amount led to nearly 1 % m1A (0.97±0.2 %). Overall, the N1 of adenosine was one of the most nucleophilic positions and was easily methylated at neutral pH. Figure 2 B summarizes the found methylation sites within RNA nucleosides. Position 7 of guanosine was slightly more reactive than position 1 in adenosine. In the same order of magnitude, methylation of position 3 in cytidine and uridine was observed. We also detected the formation of methylation products at positions 1, 2, and 6 in purine nucleosides; however, these sites were only methylated once every 10 000 nucleosides. As expected, no reaction of position 5 in pyrimidines, position 3 in purines, or at the 2′‐OH of the ribose was observed in our studies.
Figure 2.
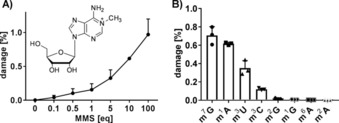
In vitro methylation of nucleosides (error bars represent the standard deviation of three replicate experiments). A) Formation of 1‐methyladenosine (m1A) during exposure of adenosine with increasing amounts of MMS starting from 0.1 equivalents MMS per A (tenfold excess A) up to 100 equivalents MMS per A (100‐fold excess MMS). B) Quantities of methylated nucleosides formed after exposing a mixture of canonical nucleosides to a tenfold excess amount of MMS (10 equiv.). Abbreviations: m7G, 7‐methylguanosine; m1A, 1‐methyladenosine; m3U, 3‐methyluridine; m3C, 3‐methylcytidine; m2G, 2‐methylguanosine; m1G, 1‐methylguanosine; m6A, 6‐methyladenosine; m2A, 2‐methyladenosine.
Distinguishing enzymatic methylation marks from RNA damage in vivo
From our in vitro data, the nucleosides 7‐methylguanosine (m7G), 1‐methyladenosine (m1A), 3‐methyluridine (m3U), and 3‐methylcytidine (m3C) seemed to be the most prominent damage products within RNA. To our knowledge, m1A and m3C have not been reported as natural modifications in Escherichia coli tRNAs,12 whereas m7G is a natural tRNA modification and 3‐methyluridine is a natural ribosomal RNA modification in E. coli.1 Therefore, it was not yet possible to determine reliably the quantities of, for example, N7 methylation damage of guanosine in vivo.13 NAIL‐MS overcame this limitation. The prerequisite for a discriminatory NAIL‐MS experiment was the availability of a heavy isotope‐labeling medium, which could label either the damaged or the natural product (e.g., here, methylation or thiolation10). Feeding organisms with CD3‐methionine led to the formation of CD3‐SAM and transfer of heavy methyl marks onto the RNA (Figure S2) by the respective enzymes.
With this labeling tool, we could study the methylation damage formation of position 7 in guanosine in the presence of enzymatically formed m7G by NAIL‐MS. For this purpose, the bacteria were cultured in CD3‐methionine‐containing M9 medium, which resulted in the formation of heavy, enzymatically methylated nucleosides, for example, CD3‐m7G (m/z 301). Exposure to MMS led to the formation of nucleoside damage products, for example, CH3‐m7G (m/z 298), which is three mass units lighter than the enzymatic CD3‐m7G. With this NAIL‐MS‐based RNA‐methylome discrimination assay (concept shown in Figure 3 A), we could distinguish all enzymatically methylated nucleosides (m/z+3) from the MMS‐derived methylation marks (regular CH3, m/z±0) by mass spectrometry.
Figure 3.
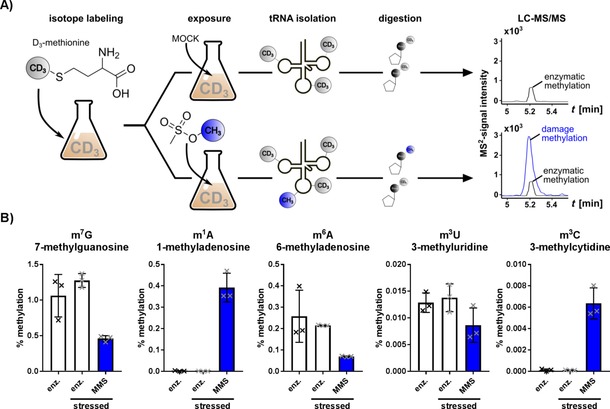
Concept sketch of the RNA methylome discrimination assay. A) CD3‐methionine‐containing medium is used for metabolic labeling of native tRNA methylations. Enzymatic methylation (black) and MMS‐derived methylation (blue) are distinguished by mass spectrometry. B) Methylation sites in canonical ribonucleosides found in vivo and sorted by abundance (left: high; right: low). The abundance of methylated nucleosides from unstressed cells is compared to that of enzymatically derived nucleosides (white) and MMS‐derived nucleosides (blue bars) from MMS‐exposed cells. Error bars represent the standard deviation of three biological replicates.
With the goal to determine the most nucleophilic centers of canonical ribonucleosides in vivo, we first defined the median lethal dose (LD50) of MMS in minimal M9 medium in E. coli (BW25113) by colony counting after exposure for 60 min (Figure S3).
The bacteria were grown in the presence of CD3‐methionine and were exposed to the LD50 of MMS for 60 min. The total RNA was extracted, and the total tRNA was purified by sizeexclusion chromatography14 and subsequently digested to nucleosides for mass spectrometry analysis. The mass spectrometer was programmed to detect all canonical nucleosides and their methylated derivatives. The enzymatically and MMS‐methylated nucleosides were distinguished by their +3 difference in the m/z values of the precursor and product ions. Exemplary mass transitions are given for all possible m7G isotopomers in Table 1, and the complete list is given in Table S2. The quantities of each methylated nucleoside were normalized to the abundance of its respective canonical nucleoside, and thus, the percentage methylation of a specific site was calculated. The summarized results in Figure 3 B show that five methylation sites were found in vivo, namely, m7G, m1A, 6‐methyladenosine (m6A), m3U, and m3C. No other MMS products could be detected (Figure S4). The highest number of damage was found for guanosine with 0.46±0.034 % m7G per guanosine and adenosine with 0.39±0.05 % m1A per adenosine. With our RNA methylome discrimination, we thus revealed position 7 of guanosine to be the main target of methylation damage in tRNA in vivo. This finding is in accordance with the reported reactivity and our observed reactivity of the N7 position in vitro. However, owing to the natural abundance of m7G in tRNA,1 this damage was overlooked in past studies. Another, yet unreported, in vivo damage site is position 6 of adenosine (m6A). The damage is with 0.07±0.002 % per adenosine less prominent than that in the main alkylation sites m7G and m1A. Nevertheless, m6A is more prominent than the in vitro found methylation products m3U and m3C, which account for less than 0.008 % damage per respective canonical. We also found the formation of m3U, which is usually a prominent ribosomal RNA modification in E. coli. In our hands, m3U was also formed as a damage product in tRNA. In summary, we found the same major reaction products in the in vitro and in vivo reactions of MMS with nucleobases (m1A and m7G). The different reactivity observed for cytidine and uridine was most likely caused by lower accessibility of the N3 position as a result of base pairing in vivo. We also observed base pairing as the main reason for the formation of m6A in vivo. Although in vitro the N1 of adenosine was found to be the most reactive center, it was blocked because of base pairing in vivo, and thus, the N6 position became an easily accessible nucleophile for reaction with MMS. In our hands, we did not see statistically significant adaptation of enzymatically introduced tRNA modifications after MMS exposure.
Table 1.
Mass transitions of 7‐methylguanosine isotopomers in methylation discrimination NAIL‐MS.
| Nucleoside | Labeling[a] | Precursor[b] | Product[c] | t R [min] |
|---|---|---|---|---|
| guanosine | unlabeled | 284 | 152 | 3.7 |
| SIL‐IS[d] | 299 | 162 | 3.7 | |
| 7‐methylguanosine | unlabeled[e] | 298 | 166 | 2.0 |
| CD3‐label[f] | 301 | 169 | 2.0 | |
| SIL‐IS | 314 | 177 | 2.0 |
[a] Used labeling technique. [b] Precursor ion m/z. [c] Product ion m/z. [d] SIL‐IS: stable isotope labeled internal standard prepared from a complete 13C/15N‐labeled E. coli culture. [e] From MMS damage. [f] Labeling with CD3‐methionine to detect enzymatic methylation.
Observing the repair of methylation damage in E. coli tRNA by pulse‐chase NAIL‐MS
The positions within the nucleobases that are damaged by MMS methylation are mainly needed for correct base pairing, and thus, their enzymatic repair is crucial for cell homeostasis and survival. The repair of m1A was shown by radioisotope labeling in vivo.8 To our knowledge, there are no reports that present evidence for the in vivo demethylation of the other MMS damaged products. In the past, we used a combination of isotope labeling in bacteria10 and yeast11 and pulse‐chase experiments to determine the fate of nucleic acid modifications. Here, we adapted these NAIL‐MS assays to follow the fate of damaged tRNAs and their damage‐derived methylated nucleosides in E. coli. The assay was started in unlabeled medium, and thus, all canonical nucleosides and all methylated nucleosides were unlabeled. These unlabeled E. coli cultures were exposed to MMS, and thus, canonical nucleosides in the tRNA received an unlabeled methyl group. In this assay, damage methylation and enzymatic methylation could not be distinguished. After 60 minutes′ exposure, MMS was removed by medium exchange. The new medium contained nitrogen‐15, which resulted in labeling of +5 for purines, +3 for cytidine, and +2 for uridine in the newly transcribed tRNAs. Furthermore, we used CD3‐methionine, which is typically used to study additional methylation of the original tRNAs (mass increase of +3) in case of enzymatic adaptation of the tRNA modification profile. The assay was set up as outlined in Figure 4 A. Using mass spectrometry, the MMS‐exposed tRNA and the newly transcribed tRNA could be clearly distinguished, and the abundance of modified nucleosides in the damaged, original tRNA could be quantified. An exemplary list for the mass transitions for all observed m7G isotopomers is given in Table 2 and for the other nucleosides in Table S3.
Figure 4.
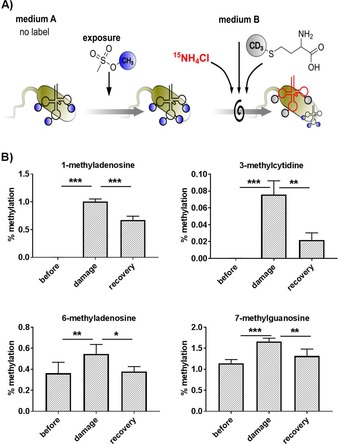
A) Concept of a pulse‐chase NAIL‐MS assay to study the repair of methylated nucleosides by using isotope labeling to distinguish MMS‐damaged tRNAs (black, from medium A) and newly transcribed tRNAs (red, from medium B). B) Abundance of methylated nucleosides before MMS exposure, after 1 h of exposure to MMS, and after 24 h of recovery. Error bars represent the standard deviation of three biological replicates. P values of student t‐test are indicated as * p<0.05, ** p<0.01, *** p<0.005.
Table 2.
Mass transitions of 7‐methylguanosine isotopomers in pulse‐chase NAIL‐MS for RNA repair observation.
| Nucleoside | Labeling[a] | Precursor[b] | Product[c] | t R [min] |
|---|---|---|---|---|
| guanosine | unlabeled[d] | 284 | 152 | 3.7 |
| 15N‐label[e] | 289 | 157 | 3.7 | |
| SIL‐IS[f] | 299 | 162 | 3.7 | |
| 7‐methylguanosine | unlabeled | 298 | 166 | 2.0 |
| CD3‐label[g] | 301 | 169 | 2.0 | |
| 15N/CD3‐label[e] | 306 | 174 | 2.0 | |
| SIL‐IS | 314 | 177 | 2.0 |
[a] Used labeling technique. [b] Precursor ion m/z. [c] Product ion m/z. [d] No labeled metabolites, original nucleosides. [e] tRNA transcribed in the presence of 15N, de novo nucleosides. [f] SIL‐IS: stable isotope labeled internal standard prepared from a complete 13C/15N‐labeled E. coli culture. [g] Labeling with CD3‐methionine to detect enzymatic methylation of original nucleoside, methylated after stress.
After MMS exposure for 1 h, the damage products m1A and m3C were formed with around 1 % damaged adenosines and only 0.08 % damaged cytidines (Figure 4 B, top row) in tRNA. The abundance of m1A decreased in the damaged tRNA over the 24 h recovery period. The reason for the decrease in m1A abundance was most likely active demethylation (potentially by AlkB8). With our experimental setup of the NAIL‐MS experiment, we could clearly distinguish damaged tRNA from freshly transcribed tRNAs. Thus, the decrease in m1A was not caused by dilution effects from tRNA transcription but by a reduction in m1A in the damaged tRNA. We observed even more pronounced removal of m3C from the damaged tRNAs during the recovery timeframe. With our NAIL‐MS assay, we report for the first time, repair of m3C in vivo. The repair was more efficient for m3C than for m1A (Figure 4 B), and this was most likely caused by the low abundance of m3C sites in the tRNAs. Unfortunately, the abundance of m3U in the samples was too low for quantification. Thus, we could not follow the fate and potential repair of m3U in vivo. For m6A and m7G (Figure 4 B, bottom), which occur from enzymatic and MMS methylation in E. coli tRNAs, we observed a significant increase after MMS exposure. This increase was most likely due to active methylation by MMS, as shown in the RNA methylome discrimination assay (Figure 3 B). In the discrimination assay, we observed that roughly 30 % of all m7G and m6A was derived from direct MMS methylation (Figure 3 B). In our pulse‐chase assay, for which damage and enzymatic methylation are not distinguishable, we observed an increase in m7G and m6A of again approximately 30 % (Figure 4 B, bottom).
For both m7G and m6A, we detected a decrease during the recovery period, which hinted towards demethylation repair of these damaged nucleosides. Intriguingly, the abundance of m6A dropped from the elevated damage level to the pre‐exposure level. For m7G, the abundance remained elevated relative to the starting abundance. This indicated that the main methylation damage in bacterial tRNA, m7G, was repaired more slowly than m1A, m3C, and m6A or, potentially, not at all.
Intrigued by this finding, we wanted to study the behavior of m6A and m7G in the original, but damaged tRNAs over time. For comparison, we took unstressed E. coli and analyzed the behavior of modifications in the original tRNAs by pulse‐chase NAIL‐MS as shown in Figure 4 A. Figure 5 A shows the principle of the analysis. To assess the impact of tRNA degradation and tRNA transcription on the modification content, we plotted the ratio of original canonicals and new canonicals over time. With the presented setup of the NAIL‐MS experiment, we observed dilution of the original nucleosides, which was caused by a combination of tRNA degradation and transcription (Figure S5). The assay revealed deeper insight into the differences in the stressed and unstressed cells. The transcription and/or degradation rate of tRNA seemed to be faster in unstressed cells than in stressed cells in the first 3 h after medium exchange. This indicated that there was no massive degradation of tRNAs in the first 3 h of recovery. After that, the ratio flipped, and we found that the original nucleosides were more diluted in the stressed cells than in the unstressed cells. This was potentially caused by faster transcription in the stressed cells or by excessive tRNA degradation of damaged tRNA in the stressed cells after 3 h.
Figure 5.
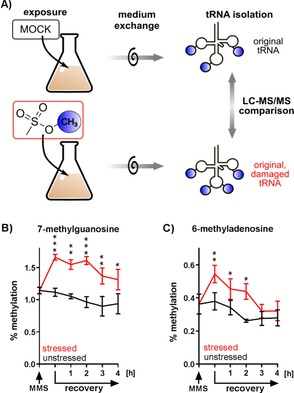
Pulse‐chase NAIL‐MS study of MOCK‐ and MMS‐incubated bacterial cultures. A) Principle of the study for comparison of the percent methylation in original tRNAs. original tRNAs: tRNAs present during MOCK incubation; original, damaged tRNAs: tRNAs present during the MMS exposure. B) 7‐Methylguanosine abundance in stressed (red) and unstressed (black) tRNAs. C) 6‐Methyladenosine abundance in stressed (red) and unstressed (black) tRNAs. Error bars represent the standard error of three biological replicates. P values of student t‐test for significance between stressed and unstressed sample at the same timepoint are indicated as * p<0.05, ** p<0.005, *** p<0.001.
We could thus exclude tRNA degradation as the main pathway of tRNA damage repair in the first 3 h after MMS removal. Figure 5 B, C shows the results of the methylation content in the original tRNA from stressed (red) and unstressed (black) bacteria.
The abundance of m7G before MMS exposure was around 1.1 %. After MMS exposure, the m7G abundance rose to 1.6 %, whereas the unstressed cells remained at around 1.1 % methylation. The difference in m7G abundance in the stressed and unstressed cells was statistically significant throughout the observed timeframe (Figure 5 B). After recovery for 3 h, the abundance of m7G decreased in the stressed bacteria, but it stayed elevated compared to the unstressed bacteria. This is in stark contrast to the results we saw for m6A. In Figure 5 C, we observed an initial increase in the abundance of m6A, followed by a rapid decrease in the first 3 h of recovery. In fact, after the 3 h recovery phase, we did not see any statistically significant difference in the abundance of m6A in the stressed and unstressed bacterial tRNAs. As we excluded tRNA degradation as a tRNA repair mechanism, the fast return of m6A to the starting abundance hinted towards a demethylation process of m6A. For m7G, we did not have any such indication, as the abundance of m7G stayed elevated in the stressed bacterial tRNAs. Future experiments will deepen our understanding of this intriguing finding.
We also analyzed other modified nucleosides in the bacterial tRNAs. Unlike m7G and m6A, we did not observe any difference in the abundance of other modified nucleosides upon comparing the tRNAs from stressed and unstressed cells. This was more proof that the overall compositions of the tRNA pools of the stressed and unstressed bacteria were comparable. We observed a minor decrease in the abundances of m1G and m5U in the original tRNAs from both stressed and unstressed bacteria (Figure S6). This decrease was also seen in the abundances of m7G and m6A in unstressed cells (Figure 5 B, C). This was most likely growth‐phase dependent, as already described for Saccharomyces cerevisiae by using NAIL‐MS.11
Conclusion
In the presented work, we systematically defined the main target sites for methylation within ribonucleosides in vitro and in vivo. Using MMS as an electrophile, we found position 7 of guanosine to be the main target in vitro. Position 1 of adenosine was found to be similarly reactive. Furthermore, position 3 of cytidine and uridine were prominent targets for methylation. As expected, we observed no methylation of non‐nucleophilic positions such as C5 in cytidine and the 2′‐OH of the ribose moiety.
Although 7‐methyl‐2′‐deoxyguanosine was described as a damage product of DNA methylation, m7G in RNA has not yet been observed as a damage in vivo.9 In fact, m1A was described to be the main methylation product of MMS damage in bacterial RNA. The detection of m7G as a damage product was impossible in previous studies, as m7G is also formed enzymatically and, thus, damage and enzymatic m7G could not be distinguished. We developed a NAIL‐MS‐based methylome discrimination assay that found m7G to be a major damage product of bacterial tRNAs. After 60 minutes′ exposure of E. coli to MMS, we found that about 0.5 % of all guanosines and adenosines were methylated. The results of our in vivo methylome discrimination assay nicely reflected our in vitro results in terms of these major damage products. For the minor methylation products m3U and m3C, we observed less formation in vivo than in the nucleoside‐based in vitro assay. We assumed that in vivo base pairing protected the N3 position of these nucleosides from the reaction with MMS. We also found a damage product that we did not expect from our in vitro studies, namely, m6A. Although in vitro the N1 of adenosine was the most reactive center, it was blocked in vivo because of base pairing, and thus, the N6 position became an easily accessible nucleophile for reaction with MMS. Similar to m7G, m6A is a damage product that was previously masked by the abundance of enzymatically produced m6A in bacterial tRNA.
With the adapted pulse‐chase NAIL‐MS assay, we followed the fate of the MMS‐damaged tRNAs over time. For m1A, repair by active demethylation was reported in the past.8 We also found repair of m1A by demethylation in vivo. However, the decrease that we found in the m1A abundance was slower than that in the 2004 publication. This discrepancy was explained by the fact that we used regular E. coli bacteria, whereas the study in 2004 used bacteria with an induced AlkB demethylase. AlkB is not constitutively expressed, but sublethal doses of MMS lead to its expression within 2 h and a plateau after 4 h of continuous MMS exposure.9 Thus, they observed immediate repair, whereas we saw no repair in the early recovery phase but only at later time points after MMS induced expression of AlkB.
In addition to the repair of m1A, we clearly saw the repair of m3C in vivo. To our knowledge, this is the first report of m3C repair in vivo upon alkylation stress. Intriguingly, the repair of m3C was more efficient than the repair of m1A. One factor might have been the relative low abundance of m3C in the damaged tRNAs (less than 0.01 %) relative to that of m1A with approximately 0.5 % (compare Figures 3 B and 4 B). It is also possible that m3C is a better substrate for the demethylase than m1A. Although we assumed that AlkB, as previously reported, repaired m1A and m3C damage, in this study we focused on the chemical fate of the tRNA and not on the biological players of repair. Currently, AlkB is the only known demethylase in E. coli, but future studies will reveal its substrate specificity.
Specifically, the results we received for m6A indicated potential repair of damage‐derived m6A nucleosides in bacterial tRNAs. Here, a systematic screen for potential demethylases in E. coli might reveal the biological impact of our observation.
The combination of nucleic acid isotope labeling with mass spectrometry is a powerful tool to study damage and repair of RNA. With NAIL‐MS, we uncovered in vivo two MMS damage products that could previously not be detected in RNA. Furthermore, we followed the fate of the damaged RNA and observe its repair mechanisms in vivo.
Experimental Section
Salts, reagents, isotopes, and nucleosides: All salts were obtained from Sigma–Aldrich (Munich, Germany) at molecular biology grade unless stated otherwise. The isotopically labeled compounds 15NH4Cl (>98 atom %) and [D3]‐l‐methionine (98 atom %) were also obtained from Sigma–Aldrich. All solutions and buffers were made with water from a Millipore device (Milli‐Q, Merck). The nucleosides adenosine, cytidine, guanosine, uridine, and N2‐methylguanosine (m2G) were obtained from Sigma–Aldrich. 1‐Methyladenosine (m1A), 2‐methyladenosine (m2A), N3‐methylcytidine (m3C), N6‐methyladenosine (m6A), 7‐methylguanosine (m7G), 5‐methylcytidine (m5C), 5‐methyluridine (m5U), 2′‐O‐methylcytidine (Cm), 2′‐O‐methylguanosine (Gm), 1‐methylguanosine (m1G), and 3‐methyluridine (m3U) were obtained from Carbosynth (Newbury, UK).
In vitro methylation of nucleosides: For the in vitro methylation assay, a canonical stock solution was prepared containing 1 mm adenosine, guanosine, cytidine, and uridine, respectively. The stock solution (10 μL) was incubated with potassium phosphate buffer (10 μL, 50 mm, pH 7) and methyl methanesulfonate (MMS; 0.1 mm, diluted in water, 10 μL) at 37 °C for 1 h. Following incubation, potassium phosphate buffer (60 μL, 50 mm, pH 9) was added before the reaction mixture was quenched with freshly prepared dithiothreitol (10 μL, 0.01 m). The same procedure was used to conduct the assay with MMS concentrations of 0.5, 1, 5, 10, 100 mm as well as a water control. The reactions were stopped by the addition of DTT in a hundredfold excess. As part of the sample preparation, the sample solution (10 μL) was diluted with water (990 μL). An aliquot (18 μL) of the dilution was transferred into a vial and was combined with a yeast‐derived, stable isotope labeled internal standard (SIL‐IS; 2 μL).5
In vivo methylation
Strains: A single‐colony lysogeny broth (LB) plate of E. coli BW25113 was prepared, and for each experiment, a single colony was picked. The LD50 of MMS for this strain at OD600 values of 0.1 and 1.0 was determined to be 20 mm by colony counting after exposure to various MMS concentrations for 60 min.
Media for isotope labeling: Minimal medium M9 was used with and without the indicated isotopes. Unlabeled M9 was prepared by mixing a 10× M9 stock solution with glucose, MgCl2, Na2SO4, and CaCl2. For unlabeled 10× M9 stock solution, Na2HPO4 (68 g L−1), KH2PO4 (30 g L−1), NaCl (2.5 g L−1), and NH4Cl (10 g L−1) were mixed and autoclaved. For 15N‐labeled 10× M9 stock solution, Na2HPO4 (68 g L−1), KH2PO4 (30 g L−1), NaCl (2.5 g L−1), and 15NH4Cl (10 g L−1) were mixed and autoclaved. MgCl2 (0.1 m), CalCl2 (0.1 m), Na2SO4 (0.1 m), and 20 % (w/w) glucose were prepared by sterile filtration. For a 5 mL M9 preculture (or 50 mL exposure culture, respectively), 500 μL (5 mL) M9 stock solution was mixed with 100 μL (1 mL) glucose, 100 μL (1 mL) MgCl2, 100 μL (1 mL) Na2SO4, and 5 μL (50 μL) CaCl2. For 15N‐labeled cultures, the 15N‐10× M9 stock solution was used. For CD3‐methylome labeling, 200 μL CD3‐methionine (stock 5 g L−1) was added to 5 mL of culture volume.
RNA isolation and tRNA purification: The bacteria culture was centrifuged at 1200 g for 5 min. The supernatant was discarded, and the cell pellet was suspended in 1 mL TRI reagent (Sigma–Aldrich) per 5 mL bacteria culture, and the total RNA was isolated according to the supplier's manual.
For purification of tRNA from total RNA, size‐exclusion chromatography (SEC)14 was used with an Agilent 1100 HPLC system with an AdvanceBio column, 300 Å pore size, 2.7 μm particle size, 7.8×300 mm (Agilent, Waldbronn, Germany) at 40 °C. For elution, an isocratic flow of 1 mL min−1 0.1 m ammonium acetate buffer was used. Eluting RNA was detected at λ=254 nm with a diode‐array detector. The tRNA fraction was collected, and the solvent was evaporated (GeneVac, EZ‐2 PLUS, Ipswich, UK) to a volume of about 100 μL before ethanol precipitation. The tRNA was resuspended in water (30 μL).
Distinguishing enzymatic methylation from damage methylation: A single colony was picked and inoculated in CD3‐labeled M9 medium (5 mL) at 37 °C and 250 rpm shaking overnight. From this culture, a bacterial solution with an OD600 of 0.1 was prepared (5 mL, CD3‐labeled M9) by dilution. The culture was allowed to transit from stationary into early log phase by shaking at 37 °C for 60 min with 250 rpm in a shaking incubator. Methyl methanesulfonate (MMS, 99 % solution) was added to a final MMS concentration of 20 mm (8.5 μL of MMS stock solution for a 5 mL exposure culture). MMS was distributed evenly in the culture by inverting the tube several times. After 60 min of exposure to MMS at 37 °C and 250 rpm, the RNA was isolated. As a control, water was added instead of MMS.
Pulse‐chase NAIL‐MS: A single colony was picked and grown in unlabeled M9 medium (5 mL) overnight. From this preculture, a 50 mL culture was prepared in unlabeled M9 medium and grown overnight. Unlabeled bacteria solution (120 mL, OD600=1.0) was prepared by adding the appropriate amount of overnight culture to fresh, unlabeled M9 medium. After 60 min growth (37 °C, 250 rpm), the first aliquot (7 mL) was taken for RNA isolation. The remaining culture was equally split into two flasks of 56.5 mL each. One was exposed to MMS stock solution (95.7 μL) or water and was inverted before both cultures were left to grow for 60 min at 37 °C and with 250 rpm shaking. After 60 min exposure, an aliquot (7 mL) was drawn from each culture, and the RNA was isolated. The remaining bacteria were centrifuged (1200 g, 5 min), and the MMS/MOCK‐containing supernatants were discarded. The bacteria pellets were washed with 15N CD3‐methionine labeled M9 medium (5 mL), and each was suspended in fresh CD3/15N M9 medium (50 mL). For recovery, the bacteria were grown at 37 °C, 250 rpm, and after 1, 2, 3, 4, and 10 h, aliquots (7 mL) were drawn for RNA isolation.
tRNA digestion for mass spectrometry: tRNA (100 ng) in aqueous digestion mix (30 μL) was digested to single nucleosides by using alkaline phosphatase (0.2 U, Sigma–Aldrich), phosphodiesterase I (0.02 U, VWR, Radnor, Pennsylvania, USA), and benzonase (0.2 U, Sigma–Aldrich) in Tris (pH 8, 5 mm) and MgCl2 (1 mm) containing buffer. Furthermore, tetrahydrouridine (THU, 0.5 μg from Merck), butylated hydroxytoluene (BHT,1 μm, Sigma–Aldrich), and pentostatin (0.1 μg Sigma–Aldrich) were added to avoid deamination and oxidation of the nucleosides.15 The mixture was incubated with the RNA for 2 h at 37 °C and was filtered through 96‐well filter plates (AcroPrep Advance 350 10 K Omega, PALL Corporation, New York, USA) at 3000 g and 4 °C for 30 min. The filtrate was combined with E. coli SILIS 10:1 (stable isotope labeled internal standard16) and was measured with a triple quadrupole mass spectrometer.
LC‐MS instruments and methods
Triple quadrupole instrument no. 1: Agilent 1290 LC equipped with a variable wavelength detector (VWD) combined with an Agilent G6490A Triple Quad system with electrospray ionization (ESI‐MS, Agilent Jetstream). The used parameters for this instrument were as follows: 250 V fragmentor voltage, cell accelerator voltage of 5 V, N2 gas temperature of 150 °C and N2 gas flow of 15 L min−1, sheath gas (N2) temperature of 275 °C with a flow of 11 L min−1, capillary voltage of 2500 V, nozzle voltage of 500 V, nebulizer pressure of 30 psi, and positive‐ion mode. The instrument was operated in dynamic MRM mode, and the individual mass spectrometric parameters for the nucleosides are given in Table S2.
Triple quadrupole instrument no. 2: Agilent 1290 Infinity II equipped with a diode‐array detector (DAD) combined with an Agilent Technologies G6470A Triple Quad system and electrospray ionization (ESI‐MS, Agilent Jetstream). Operating parameters: positive‐ion mode, skimmer voltage of 15 V, cell accelerator voltage of 5 V, N2 gas temperature of 230 °C and N2 gas flow of 6 L min−1, sheath gas (N2) temperature of 400 °C with a flow of 12 L min−1, capillary voltage of 2500 V, nozzle voltage of 0 V, and nebulizer at 40 psi. The instrument was operated in dynamic MRM mode, and the individual mass spectrometric parameters for the nucleosides are given in Tables S1 and S3.
Chromatography no. 1: For the in vitro assay, we used an RP‐18 column (Synergi, 2.5 μm Fusion‐RP C18 100 Å, 100×2 mm; Phenomenex) at 35 °C and a flow rate of 0.35 mL min−1 in combination with a binary mobile phase of 5 mm NH4OAc aqueous buffer A, brought to pH 5.6 with glacial acetic acid, and an organic buffer B of pure acetonitrile (Roth, LC‐MS grade, purity 95.95). The gradient started at 100 % solvent A for 1 min, followed by an increase to 10 % over 4 min. At minute 5, solvent B was increased to 40 % and was maintained for 1 min before returning to 100 % solvent A and a 2.5 min re‐equilibration period.
Chromatography no. 2: A Core‐Shell Technology separation column (Phenomenex, Torrance, CA, USA; Kinetex 1.7 μm EVO C18 100 Å, 150×2.1 mm) at 35 °C and a flow rate of 0.35 mL min−1 were used for the in vivo experiments. Solvents A and B were identical to the solvents used for chromatography no. 1. The gradient started at 100 % solvent A, followed by an increase to 10 % over 10 min. From 10 to 15 min, solvent B was increased to 45 % and was maintained for 3 min before returning to 100 % solvent A and a 3 min re‐equilibration period.
Calibration and equations: For calibration, synthetic nucleosides were weighed and dissolved to a stock concentration of 1–10 mm. In vitro calibration solutions: ranging from 100 fmol to 1 nmol with respect to canonical nucleosides and 100 amol to 1 pmol in terms of modified nucleosides. In vivo calibration solutions: ranging from 0.3 to 500 pmol for each canonical nucleoside and from 0.3 to 500 fmol for each modified nucleoside. The calibration solutions were mixed with the same SILIS as the corresponding samples and were analyzed with the same methods. The value of each integrated peak area of the nucleosides was divided through the respective SILIS area. The linear regression for each nucleoside's normalized signal/concentration plot gave the relative response factor for nucleosides (rRFN).16 The sample data were analyzed by the Quantitative and Qualitative MassHunter Software from Agilent. The areas of the MRM signals were integrated for each modification and their isotope derivatives. The area value was divided through the respective SILIS area value and was divided through the rRFN value from the respective calibration to receive the absolute amount of the modification or canonical. Finally, the absolute amounts of the modifications were referenced to the absolute amounts of the precursor canonical. In the case of the pulse‐chase experiment, the different isotopomers were referenced to their respective similarly labeled canonicals, so that original modifications were referenced to original canonicals and new modifications were referenced to new canonicals. See Equations (1), (2) for m7G as an example:
| (1) |
| (2) |
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Kellner laboratory thanks Thomas Carell and his group for instrument time (HRMS) and valuable discussions. This project was funded by the Verband der Chemischen Industrie (VCI) and the Deutsche Forschungsgemeinschaft (DFG; CIPSM and SFB 1309). V.F.R. is funded by the VCI.
V. F. Reichle, V. Weber, S. Kellner, ChemBioChem 2018, 19, 2575.
References
- 1. Boccaletto P., Machnicka M. A., Purta E., Piatkowski P., Baginski B., Wirecki T. K., de Crecy-Lagard V., Ross R., Limbach P. A., Kotter A., Helm M., Bujnicki J. M., Nucleic Acids Res. 2018, 46, D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.B. Singer, D. Grunberger, Molecular Biology of Mutagens and Carcinogens, Plenum, New York, 1983.
- 3.
- 3a. Kellner S., Seidu-Larry S., Burhenne J., Motorin Y., Helm M., Nucleic Acids Res. 2011, 39, 7348–7360; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Schmid K., Adobes-Vidal M., Helm M., Bioconjugate Chem. 2017, 28, 1123–1134. [DOI] [PubMed] [Google Scholar]
- 4. Kellner S., Kollar L. B., Ochel A., Ghate M., Helm M., PLoS One 2013, 8, e67945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heiss M., Kellner S., RNA Biol. 2017, 14, 1166–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Fedeles B. I., Singh V., Delaney J. C., Li D., Essigmann J. M., J. Biol. Chem. 2015, 290, 20734–20742; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Yi C., He C., Cold Spring Harbor Perspect. Biol. 2013, 5, a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aas P. A., Otterlei M., Falnes P. O., Vagbo C. B., Skorpen F., Akbari M., Sundheim O., Bjoras M., Slupphaug G., Seeberg E., Krokan H. E., Nature 2003, 421, 859–863. [DOI] [PubMed] [Google Scholar]
- 8. Ougland R., Zhang C. M., Liiv A., Johansen R. F., Seeberg E., Hou Y. M., Remme J., Falnes P. O., Mol. Cell 2004, 16, 107–116. [DOI] [PubMed] [Google Scholar]
- 9. Vagbo C. B., Svaasand E. K., Aas P. A., Krokan H. E., DNA Repair 2013, 12, 188–195. [DOI] [PubMed] [Google Scholar]
- 10. Kellner S., DeMott M. S., Cheng C. P., Russell B. S., Cao B., You D., Dedon P. C., Nat. Chem. Biol. 2017, 13, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heiss M., Reichle V. F., Kellner S., RNA Biol. 2017, 14, 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chujo T., Suzuki T., RNA 2012, 18, 2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan C. T., Dyavaiah M., DeMott M. S., Taghizadeh K., Dedon P. C., Begley T. J., PLoS Genet. 2010, 6, e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chionh Y. H., Ho C. H., Pruksakorn D., Ramesh Babu I., Ng C. S., Hia F., McBee M. E., Su D., Pang Y. L., Gu C., Dong H., Prestwich E. G., Shi P. Y., Preiser P. R., Alonso S., Dedon P. C., Nucleic Acids Res. 2013, 41, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai W. M., Chionh Y. H., Hia F., Gu C., Kellner S., McBee M. E., Ng C. S., Pang Y. L., Prestwich E. G., Lim K. S., Babu I. R., Begley T. J., Dedon P. C., Methods Enzymol. 2015, 560, 29–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kellner S., Ochel A., Thuring K., Spenkuch F., Neumann J., Sharma S., Entian K. D., Schneider D., Helm M., Nucleic Acids Res. 2014, 42, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary