

Abstract
Antibody–drug conjugates (ADCs) are a growing class of therapeutics that harness the specificity of antibodies and the cell‐killing potency of small‐molecule drugs. Beyond cytotoxics, there are few examples of the application of an ADC approach to difficult drug discovery targets. Here, we present the initial development of a non‐internalising ADC, with a view to selectively inhibiting an extracellular protein. Employing the wellinvestigated matrix metalloproteinase‐9 (MMP‐9) as our model, we adapted a broad‐spectrum, nonselective MMP inhibitor for conjugation and linked this to a MMP‐9‐targeting antibody. The resulting ADC fully inhibits MMP‐9, and ELISA results suggest antibody targeting can direct a nonselective inhibitor.
Keywords: antibodies, antibody–drug conjugates, inhibitors, MMP-9
Current clinical antibody–drug conjugates (ADCs) combine the high specificity and long circulating half‐life of an antibody with the cell‐killing potency of a small‐molecule payload to generate a targeted chemotherapeutic.1 Designed to improve the therapeutic index of cytotoxic agents, two distinct mechanistic classes of payload have most often been conjugated to monoclonal antibodies; microtubule inhibitors (e.g., maytansines, auristatins) and DNA‐modifying agents (e.g., calcheamicins, PBD dimers, duocarmycins).2, 3, 4, 5, 6 Currently, there are four ADCs on the market and over 65 in clinical evaluation; however, there are limited examples of ADCs employing non‐cytotoxic small molecules.7, 8, 9, 10, 11, 12, 13, 14 As part of a growing interest in the field of targeted delivery of small molecules, we began to explore a novel application of the ADC approach to the selective inhibition of an extracellular protein. Using matrix metalloproteinase‐9 (MMP‐9, also known as gelatinase B) as our model, we conjugated a broad‐spectrum MMP inhibitor to a selective MMP‐9 antibody.
MMPs are a family of structurally related zinc‐binding proteolytic enzymes.15 Individual MMPs are promising drug targets; many diseases, including cancer, inflammation, and vascular disease, are associated with altered MMP expression and aberrant proteolysis.16, 17, 18, 19 Significant drug discovery effort has been invested into generating small‐molecule MMP inhibitors that target the active‐site zinc in the catalytic domain. Despite the investigation of more than 50 of these inhibitors in clinical trials, efforts with first‐generation compounds were hampered either by dose‐limiting toxicity or insufficient clinical benefit.20 One explanation for this failure is the high degree of sequence and structural similarity in the catalytic domain of MMPs, which results in broad‐spectrum, nonspecific inhibitors, although more selective next‐generation compounds are now beginning to appear.18 Monoclonal antibodies selective for specific MMPs have been successfully generated.21 However, as these targeting antibodies interact with surface loops rather than the active site, they often lack sufficient functional potency.
We describe herein a new approach towards the selective inhibition of MMPs, specifically MMP‐9, through combining the specificity and high affinity of an antibody with the potency of a small‐molecule inhibitor. MMP‐9 shows particular promise as a therapeutic target, having been associated with a number of pathological processes that contribute to tumorigenesis, metastasis and chronic inflammation.22, 23 As a result, MMP‐9 is perhaps the best investigated of the MMPs and thus provides a valuable model to begin exploring the application of an ADC to retarget a nonselective inhibitor.
To investigate our ADC approach, we needed to adapt a broad‐spectrum MMP inhibitor for conjugation. Many MMP inhibitors are hydroxamate based; the hydroxamic acid motif coordinates the active‐site zinc ion in a bidentate fashion to generate inhibitors with high affinity but poor MMP selectivity.24 One such inhibitor is CGS27023A (Figure 1 A), which was originally discovered as an orally active MMP‐3 inhibitor. It was soon demonstrated to be a potent inhibitor of many MMP family members, including MMPs 9 and 2.25, 26 The crystal structure of CGS27023A in complex with the MMP‐12 catalytic domain has been successfully resolved.26 This reveals the pyridine ring of the inhibitor to be relatively solvent exposed, thus potentially providing a site for linker derivatisation. In fact, there is literature precedent for the PEGylation of CGS27023A through a benzyl derivative.27 Consequently, we opted to design and synthesise CGS27023A–linker derivatives for conjugation to a monoclonal antibody.
Figure 1.
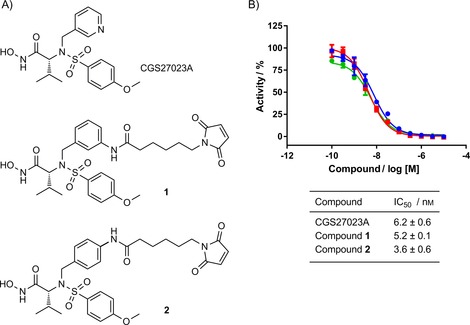
CGS27023A and its derivatives inhibit MMP‐9 activity. A) Structure of CGS27023A and two linker derivatives 1 and 2. B) Inhibition of human catalytic MMP‐9 activity by •: CGS27023A, ▪: 1 and •: 2 in the SensoLyte fluorometric assay. Data were normalised to the MMP‐9‐alone control and are expressed as means±SEM for three independent experiments.
To install a linker for conjugation, we replaced the pyridine ring of CGS27023A with an aniline, exploring both the 3‐ and 4‐positions initially to determine if there was an impact on inhibitor potency (Schemes S1 and S2 in the Supporting Information). Both anilines were subsequently coupled to maleimide hexanoic acid to provide a handle for cysteine conjugation, thus generating linker derivatives 1 and 2 (Figure 1 A). CGS27023A and linker compounds 1 and 2 were tested in a fluorometric biochemical assay against MMP‐9 by using the SensoLyte 520 MMP‐9 assay kit (AnaSpec). This indicated no loss in potency at MMP‐9 (Figure 1 B). Furthermore, 1 and 2 displayed potent inhibition of the most closely related protease, MMP‐2 (also known as gelatinase A), thereby confirming that the broad‐spectrum behaviour of this compound was maintained (Figure S1). We chose to move forward with CGS27023A derivative 2 as more material was available.
In addition to adapting a broad‐spectrum inhibitor for conjugation, we required an antibody that specifically binds to MMP‐9 with high affinity. Ideally this antibody would target an epitope near to, but distinct from, the zinc‐binding site, thereby leaving the site free for occupation by a small‐molecule inhibitor. One of the earliest anti‐MMP‐9 antibodies reported was REGA‐3G12, a mouse monoclonal generated by hybridoma technology against the human MMP‐9 catalytic domain.28 This antibody is reported to be highly selective for MMP‐9 and to recognise Trp116–Lys214, a region of the catalytic domain separate from the zinc‐binding site.29 This suggested that REGA‐3G12 might be suitable for selective delivery of a small‐molecule inhibitor to MMP‐9.
In view of our intention to conjugate REGA‐3G12 to a small molecule through its interchain disulfides, we generated the antibody in chimeric IgG1 format, thus providing a maximum of eight sites for conjugation as opposed to the ten found in mouse IgG1. Transient expression of REGA‐3G12 in Expi293F cells and purification by protein A and size‐exclusion chromatography resulted in a final yield of 51 mg L−1. Analysis of the antibody by SDS‐PAGE demonstrated it to be pure (>95 %), and high‐resolution Orbitrap mass spectrometry enabled sequence matching to the observed molecular weight (Figure S2). Next, it was imperative to determine the affinity of REGA‐3G12 for MMP‐9, as the principle of our retargeting ADC approach relies upon strong antibody binding driving selectivity.
We measured the affinity (K D) of REGA‐3G12 for the human MMP‐9 catalytic domain by using biolayer interferometry (BLI). Briefly, the antibody was captured on protein G sensors, and its binding to human catalytic MMP‐9 was analysed by using a twofold dilution series with a top concentration of 12.5 nm. REGA‐3G12 was found to have very high affinity (K D=97.4 pm), displaying the ideal antibody‐binding kinetics of a fast on‐rate and slow off‐rate (Figures 2 A and S3). To confirm the selectivity of REGA‐3G12, we tested binding to MMP‐2, which shares 78 % sequence identity in the catalytic domain. Pleasingly, no binding to MMP‐2 was observed by BLI or ELISA (Figures 2 B and S4).
Figure 2.
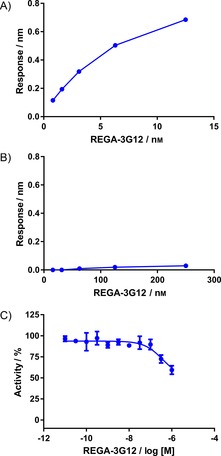
REGA‐3G12 specifically binds MMP‐9 and displays weak inhibitory activity A) BLI‐derived K D (97.4±1.0 pm) determined for REGA‐3G12 binding to the human MMP‐9 catalytic domain. For the BLI curves and K D fitting curves, see Figure S3. B) BLI analysis of REGA‐3G12 with human MMP‐2. C) Inhibition of human catalytic MMP‐9 activity by REGA‐3G12 in the SensoLyte fluorometric assay; IC50=265±63.8 nm. Data were normalised to the MMP‐9‐alone controls and are expressed as means±SEM for three independent experiments.
Having confirmed the binding selectivity of REGA‐3G12 for MMP‐9, it was next tested for activity in the fluorometric assay. Despite the high affinity of REGA‐3G12, it displayed very weak inhibition of MMP‐9 activity (Figure 2 C). Consequently, potency changes after small‐molecule inhibitor conjugation could be readily observed.
Prior to generating antibody–drug conjugates, we confirmed whether the CGS27023A payload could inhibit MMP‐9 in the presence of REGA‐3G12. We compared the inhibition of MMP‐9 by REGA‐3G12, small molecule 2 and a mixed sample mimicking an ADC with a drug‐to‐antibody ratio (DAR) of 4 (Figure 3). In the fluorometric assay, REGA‐3G12 alone behaved as a weak, partial inhibitor of MMP‐9, as observed previously. When mixed with 2 as a unconjugated DAR=4 mimic, full inhibition of MMP‐9 was achieved and the potency of the small molecule was well maintained (IC50=7.8±1.2 nm). This confirmed the possibility that REGA‐3G12 and 2 could bind simultaneously at MMP‐9.
Figure 3.
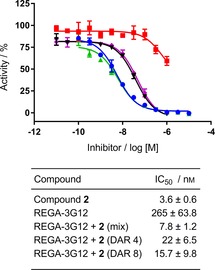
REGA‐3G12 and 2 can inhibit MMP‐9 activity simultaneously. Complete inhibition of MMP‐9 activity is seen when REGA‐3G12 is conjugated to 2. Inhibition of human catalytic MMP‐9 activity by ▪: REGA‐3G12, •: compound 2, ▴: REGA‐3G12+2 mixture (an unconjugated DAR 4 mimic) and REGA‐3G12+2 conjugates with a DAR of ▾: 4 and ⧫: 8 in the SensoLyte fluorometric assay. Data were normalised to the MMP‐9 alone control and are expressed as means±SEM for three independent experiments.
Having successfully demonstrated the simultaneous binding of antibody and small molecule, we constructed the ADC. For conjugation, REGA‐3G12 was immobilised on protein A beads and reduced with the mild reducing agent tris(2‐carboxyethyl)phosphine hydrochloride (TCEP). Following simultaneous removal of TCEP and buffer exchange to give conditions suitable for maintaining the reduced antibody (PBS, 5 mm EDTA, pH 7.4), 4.4 or 8.8 equivalents of 2 were added to yield conjugates of average DARs of 4 and 8, respectively, as determined by LCMS (Figures S5 and S6). Solid‐phase conjugation was found to be necessary to completely remove all of the potent, unconjugated small molecule by using repeated PBS+10 % DMA washes. Alternative purification methods usually employed after solution‐phase conjugation (e.g., gel filtration, ultracentrifugation) were found to be inadequate. The conjugates were subsequently tested by ELISA, which confirmed that binding of the antibody was maintained after conjugation (Figure S7).
Next, we examined the activity of the ADCs in the fluorometric assay. Both DAR=4 and DAR=8 ADCs fully inhibited MMP‐9 with an acceptable level of potency, and no significant change in potency was observed upon increasing the DAR from 4 to 8 (IC50=22±6.5 and 15.7±9.8 nm, respectively; Figure 3). This result provided excellent promise for our ADC approach, demonstrating that both antibody and small molecule could maintain binding to MMP‐9 as part of an ADC.
This ADC approach to selective inhibition is driven by the theory that superior antibody‐binding kinetics would drive selectivity. To examine this, we developed an ELISA to indicate whether ADC binding is driven by the antibody or the small molecule. For this, two ADCs were compared; REGA‐3G12 and an isotype control hIgG1, both conjugated to 2.
First, 384‐well plates were coated with human catalytic MMP‐9 or the closely related human MMP‐2, and conjugates of either REGA‐3G12 or nonbinding isotype control were added. Subsequent wash‐off and incubation with anti‐human Fc horseradish peroxidase would therefore only lead to a signal if either ADC was still present, thus demonstrating whether the broad‐spectrum small‐molecule inhibitor alone could drive binding of an ADC at either MMP‐9 or MMP‐2. At MMP‐9, binding curves were observed only for REGA‐3G12 alone and its conjugate with 2 (Figure 4 A). However, no signal was observed for the isotype control ADC. Furthermore, at MMP‐2 no read out was observed for either ADC. This result suggests that antibody‐binding kinetics could drive ADC selectivity, as no binding of our REGA‐3G12 ADC was observed at MMP‐2 (Figure 4 B). Following this result, we endeavoured to compare the impact of conjugation on the binding affinity of compound 2 at MMP‐9 and ‐2 by using surface plasmon resonance (SPR). Unfortunately, these efforts were without success. We were unable to determine the binding affinity of compound 2, and, to the best of our knowledge, no binding affinity for CGS27023A is reported in the literature, neither are conditions for SPR analysis of inhibitors at MMP‐9 or ‐2.
Figure 4.
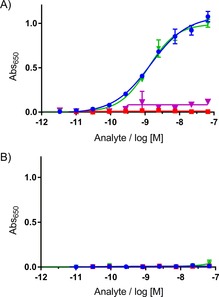
ELISA demonstrates ADC selectivity for MMP‐9. ELISA against A) human catalytic MMP‐9 and B) human MMP‐2; •: REGA‐3G12, ▪: hlgG1 control, ▴: REGA‐3G12+2 (DAR=4) and ▴: hlgG1+2 (DAR=4). See Figure S8 for all ELISA controls.
In summary, we have presented the first example of an ADC targeting an extracellular protein in which both the antibody and small molecule bind to the same target, with the aim of imparting selectivity to a nonselective, broad‐spectrum inhibitor. This ADC was designed for MMP‐9, an attractive therapeutic target for which the progress of small‐molecule inhibitors has previously been hampered by lack of selectivity. The anti‐MMP‐9 antibody REGA‐3G12 was identified and successfully demonstrated to fit the binding selectivity and kinetics requirements for our ADC model. The nonselective MMP inhibitor CGS27023A was effectively adapted for cysteine conjugation to an antibody with maintained potency at MMP‐9 and the closely related MMP‐2. The resulting ADC displayed potent inhibition of MMP‐9, and we successfully developed an ELISA that encouragingly suggested that selectivity for MMP‐9 over MMP‐2 in vitro is possible. This also validated the hypothesis that a suitable antibody can drive the binding of an ADC and provide a basis for retargeting a nonselective small‐molecule inhibitor. This early work therefore suggests that such an ADC approach could provide a selectivity solution for difficult drug‐discovery targets. We shall report on the further development of this ADC approach to additional targets in due course.
Experimental Section
Details of all synthetic procedures, characterisation data for new compounds, protein expression and characterisation, conjugation and assay protocols are given in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
E. A. Love, A. Sattikar, H. Cook, K. Gillen, J. M. Large, S. Patel, D. Matthews, A. Merritt, ChemBioChem 2019, 20, 754.
Contributor Information
Dr. Elizabeth A. Love, Email: elizabeth.love@lifearc.org.
Afrah Sattikar, Email: afrah.sattikar@lifearc.org.
References
- 1. Polakis P., Pharmacol. Rev. 2016, 68, 3–19. [DOI] [PubMed] [Google Scholar]
- 2. Lopus M., Oroudjev E., Wilson L., Wilhelm S., Widdison W., Chari R., Jordan M. A., Mol. Cancer Ther. 2010, 9, 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Francisco J. A., Cerveny C. G., Meyer D. L., Mixan B. J., Klussman K., Chace D. F., Rejniak S. X., Gordon K. A., DeBlanc R., Toki B. E., Law C. L., Doronina S. O., Siegall C. B., Senter P. D., Wahl A. F., Blood 2003, 102, 1458–1465. [DOI] [PubMed] [Google Scholar]
- 4. Hamann P. R., Hinman L. M., Hollander I., Beyer C. F., Lindh D., Holcomb R., Hallett W., Tsou H. R., Upeslacis J., Shochat D., Mountain A., Flowers D. A., Bernstein I., Bioconjugate Chem. 2002, 13, 47–58. [DOI] [PubMed] [Google Scholar]
- 5. Mantaj J., Jackson P. J., Rahman K. M., Thurston D. E., Angew. Chem. Int. Ed. 2017, 56, 462–488; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 474–502. [Google Scholar]
- 6. Dokter W., Ubink R., van der Lee M., van der Vleuten M., van Achterberg T., Jacobs D., Loosveld E., van den Dobbelsteen D., Egging D., Mattaar E., Groothuis P., Beusker P., Coumans R., Elgersma R., Menge W., Joosten J., Spijker H., Huijbregts T., de Groot V., Eppink M., de Roo G., Verheijden G., Timmers M., Mol. Cancer Ther. 2014, 13, 2618–2629. [DOI] [PubMed] [Google Scholar]
- 7. Beck A., Goetsch L., Dumontet C., Corvaia N., Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- 8. Graversen J. H., Svendsen P., Dagnaes-Hansen F., Dal J., Anton G., Etzerodt A., Petersen M. D., Christensen P. A., Moller H. J., Moestrup S. K., Mol. Ther. 2012, 20, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang R. E., Liu T., Wang Y., Cao Y., Du J., Luo X., Deshmukh V., Kim C. H., Lawson B. R., Tremblay M. S., Young T. S., Kazane S. A., Wang F., Schultz P. G., J. Am. Chem. Soc. 2015, 137, 3229–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lehar S. M., Pillow T., Xu M., Staben L., Kajihara K. K., Vandlen R., DePalatis L., Raab H., Hazenbos W. L., Morisaki J. H., Kim J., Park S., Darwish M., Lee B. C., Hernandez H., Loyet K. M., Lupardus P., Fong R., Yan D., Chalouni C., Luis E., Khalfin Y., Plise E., Cheong J., Lyssikatos J. P., Strandh M., Koefoed K., Andersen P. S., Flygare J. A., Wah Tan M., Brown E. J., Mariathasan S., Nature 2015, 527, 323–328. [DOI] [PubMed] [Google Scholar]
- 11. Kern J. C., Dooney D., Zhang R., Liang L., Brandish P. E., Cheng M., Feng G., Beck A., Bresson D., Firdos J., Gately D., Knudsen N., Manibusan A., Sun Y., Garbaccio R. M., Bioconjugate Chem. 2016, 27, 2081–2088. [DOI] [PubMed] [Google Scholar]
- 12. Kern J. C., Cancilla M., Dooney D., Kwasnjuk K., Zhang R., Beaumont M., Figueroa I., Hsieh S., Liang L., Tomazela D., Zhang J., Brandish P. E., Palmieri A., Stivers P., Cheng M., Feng G., Geda P., Shah S., Beck A., Bresson D., Firdos J., Gately D., Knudsen N., Manibusan A., Schultz P. G., Sun Y., Garbaccio R. M., J. Am. Chem. Soc. 2016, 138, 1430–1445. [DOI] [PubMed] [Google Scholar]
- 13. Lerchen H.-G., Wittrock S., Stelte-Ludwig B., Sommer A., Berndt S., Griebenow N., Rebstock A. S., Johannes S., Cancho-Grande Y., Mahlert C., Greven S., Terjung C., Angew. Chem. Int. Ed. 2018, 57, 15243–15247; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15463–15647. [Google Scholar]
- 14. Cini E., Faltoni V., Petricci E., Taddei M., Salvini L., Giannini G., Vesci L., Milazzo F. M., Anastasi A. M., Battistuzzi G., De Santis R., Chem. Sci. 2018, 9, 6490–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagase H., Visse R., Murphy G., Cardiovasc. Res. 2006, 69, 562–573. [DOI] [PubMed] [Google Scholar]
- 16. Sbardella D., Fasciglione G. F., Gioia M., Ciaccio C., Tundo G. R., Marini S., Coletta M., Mol. Aspects Med. 2012, 33, 119–208. [DOI] [PubMed] [Google Scholar]
- 17. Cathcart J., Pulkoski-Gross A., Cao J., Genes Dis. 2015, 2, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nissinen L., Kahari V. M., Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2571–2580. [DOI] [PubMed] [Google Scholar]
- 19. Lindsey M. L., Zamilpa R., Cardiovasc. Ther. 2012, 30, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skiles J. W., Gonnella N. C., Jeng A. Y., Curr. Med. Chem. 2004, 11, 2911–2977. [DOI] [PubMed] [Google Scholar]
- 21. Fields G. B., Matrix Biol. 2015, 44–46, 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vandooren J., Van den Steen P. E., Opdenakker G., Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [DOI] [PubMed] [Google Scholar]
- 23. Ram M., Sherer Y., Shoenfeld Y., J. Clin. Immunol. 2006, 26, 299–307. [DOI] [PubMed] [Google Scholar]
- 24. Hu J., Van den Steen P. E., Sang Q. X., Opdenakker G., Nat. Rev. Drug Discovery 2007, 6, 480–498. [DOI] [PubMed] [Google Scholar]
- 25. MacPherson L. J., Bayburt E. K., Capparelli M. P., Carroll B. J., Goldstein R., Justice M. R., Zhu L., Hu S., Melton R. A., Fryer L., Goldberg R. L., Doughty J. R., Spirito S., Blancuzzi V., Wilson D., O′Byrne E. M., Ganu V., Parker D. T., J. Med. Chem. 1997, 40, 2525–2532. [DOI] [PubMed] [Google Scholar]
- 26. Nar H., Werle K., Bauer M. M., Dollinger H., Jung B., J. Mol. Biol. 2001, 312, 743–751. [DOI] [PubMed] [Google Scholar]
- 27. Faust A., Waschkau B., Waldeck J., Holtke C., Breyholz H. J., Wagner S., Kopka K., Schober O., Heindel W., Schafers M., Bremer C., Bioconjugate Chem. 2009, 20, 904–912. [DOI] [PubMed] [Google Scholar]
- 28. Paemen L., Martens E., Masure S., Opdenakker G., Eur. J. Biochem. 1995, 234, 759–765. [DOI] [PubMed] [Google Scholar]
- 29. Martens E., Leyssen A., Van Aelst I., Fiten P., Piccard H., Hu J., Descamps F. J., Van den Steen P. E., Proost P., Van Damme J., Liuzzi G. M., Riccio P., Polverini E., Opdenakker G., Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 178–186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary