

Abstract
Multi‐step biocatalytic reactions have gained increasing importance in recent years because the combination of different enzymes enables the synthesis of a broad variety of industrially relevant products. However, the more enzymes combined, the more crucial it is to avoid cross‐reactivity in these cascade reactions and thus achieve high product yields and high purities. The selective control of enzyme activity, i.e., remote on‐/off‐switching of enzymes, might be a suitable tool to avoid the formation of unwanted by‐products in multi‐enzyme reactions. This review compiles a range of methods that are known to modulate enzyme activity in a stimulus‐responsive manner. It focuses predominantly on in vitro systems and is subdivided into reversible and irreversible enzyme activity control. Furthermore, a discussion section provides indications as to which factors should be considered when designing and choosing activity control systems for biocatalysis. Finally, an outlook is given regarding the future prospects of the field.
Keywords: activity regulation, biocatalysis, enzyme catalysis, on-/off-switching, stimuli
1. Introduction
Enzymatic reactions intrinsically offer several advantages, making them ideal alternatives to classical synthesis strategies: Enzymes typically exhibit high substrate specificities, both regio‐ and stereoselectivities,1 circumventing the problem of expensive and time‐consuming isolation of by‐products and intermediates.2 Enzymatic reactions typically take place under environmentally benign reaction conditions;1c, 3 toxic auxiliaries can (mostly) be prevented.1c, 3 Furthermore, enzymes are intrinsically biodegradable, thus increasing the eco‐efficiency of enzymatic reactions.4
Due to ongoing research, a steadily increasing diversity of enzymes is available, enabling chemical reactions that are extremely challenging with classical synthesis approaches,5 as well as access to a broad and steadily increasing variety of products.6 The accessible range of products for biocatalysis is extended even further by combining different enzymes into complex multi‐step reaction cascades.7 The combination of enzymes in cascades has attracted increasing attention over the last decade, as can be seen by the increasing number of articles published in this field (a small collection can be found under ref.8).
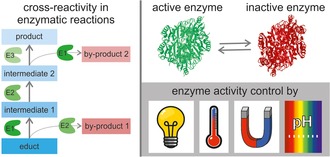
An issue that has only recently emerged in the field of multi‐step biocatalysis is cross‐reactivity.7, 9 Cross‐reactivity, in the sense this review, occurs when one enzyme is not only able to accept the targeted substrate, but also the substrates/intermediates/products of the other enzymatic reactions, which take place in the cascade. This issue is closely related to enzyme substrate promiscuity.10 Many enzymes used in biocatalysis are even intentionally designed to show substrate promiscuity in order to enable the synthesis of a broader variety of products with the same enzyme but different, closely related substrates.10 For enzymatic cascade reactions, not only does product diversity increase with each additional enzymatic step, but also does the risk of unspecific substrate uptake, leading to by‐product formation.7b As enzyme cascade reactions show the immense potential for accessing almost any desired product through the combination of enzymes run in a sequence under the same (or at least similar) reaction conditions, avoiding cross‐reactivity becomes an increasingly crucial factor to combine high product yields with high purities.
Technical solutions to avoid undesired by‐product formation include the catalyst removal after each reaction step8h or the spatio‐temporal separation of the reaction steps, for example, via enzyme retention in reaction modules.11 Recent reviews addressing these approaches can be found in refs.7b, 12 However, an alternative solution is the dynamic regulation of enzyme activity by controlled on‐/off‐switching of the catalyst.12b, 13 Specifically, if an enzyme could be switched on and off remotely and on demand during a one‐pot enzymatic cascade reaction, unspecific substrate uptake could be prevented, since the enzyme would only be active within the desired period. During this time, only its targeted substrate would be available for conversion. A precise in situ adjustment of enzyme activity could then prevent cross‐reactivity in one‐pot systems.
External stimuli such as light have long been known to be suitable to change the enzyme activity of “switchable” enzymes. For example, irradiation‐induced enzyme activity changes were reported as early as the 1970s for a number of enzymes modified with photo‐isomerizable units.14 A possible application of in situ activity control by enzyme immobilization in thermosensitive hydrogels was proposed in the 1980s to enable feedback reaction control in future.15 Despite that, there are still only a few reports of enzymatic activity being successfully controlled in situ, and even fewer providing a description of methods with industrially relevant reactions.
This review aims to give an overview of different concepts that have been used to regulate enzymatic activity. We furthermore highlight future perspectives for the field of multi‐step cascades with regard to the orthogonal control of different enzymes in cascade reactions. Rather than giving a complete overview of the literature from the last few decades, this review focuses on the general strategies underlying activity control. Free (purified) enzymes are primarily targeted as catalysts, while some of the methods applied for activity control have been proven to work in whole cells as well. Furthermore, the review only addresses the direct enzyme activity regulation, aspects such as the regulation of gene expression are not covered.
To this end, an overview of the different stimuli that can be used for remote control is given. The methods for enzyme activity control are then compiled in two sections: methods for reversible control and methods for irreversible control. Reversible control covers concepts in which enzyme activity can be switched on/off multiple times in a row, while irreversible control allows the enzyme to be switched on or off only once. A range of factors that are generally important when designing enzyme activity control systems are compiled, and a future perspective for enzyme activity control in one‐ and multi‐step reaction cascades is proposed.
2. Methods for the Regulation of Enzyme Activity
The control of enzyme activity has played a role in the research community for roughly half a century. Especially during the last few years, an increasing number of different approaches have been published, aiming to control enzyme activity remotely in various systems.13, 16
To achieve controlled on‐/off‐switching of enzymes, different stimuli can be applied as “switches”. The available stimuli are often subdivided into internal, environmental and external stimuli.17 Suitable stimuli for controlling enzymatic activity should generally enable reactions to be controlled remotely, and should therefore be easily adjustable during the reaction by the experimenter. This prerequisite excludes internal stimuli in almost all cases. Frequently applied environmental and external stimuli are temperature, pH, light, magnetic and electric fields, and ultrasound.17, 18
The environmental stimuli temperature and pH value often show a direct effect on the enzyme activity, even without an engineered switch.13, 19 When using these stimuli in enzyme activity control, the inherent response of the enzyme itself has to be considered carefully, as these inherent changes and “switch‐dependent” changes go hand in hand and are frequently difficult to distinguish sufficiently. External stimuli such as light, magnetic field and electric field offer the possibility of remote on‐demand control in real time.17 Light is especially attractive as an external stimulus, as it allows an extremely high temporal and spatial control.20
2.1. Methods for Reversible Control
This chapter describes the most common generic approaches to the reversible control over enzyme activity as compiled in Table 1 and depicted in Figure 1 to Figure 5. Reversible control in the context of this review means that the catalyst can be activated and deactivated multiple times. Reversible control is generally possible with all of the above‐mentioned environmental and external stimuli. But, light, temperature and magnetic fields are most commonly applied. Under certain conditions, all of the described methods can be conferrable to enzymes other than the examples given in this review. Where known, the conditions for applying the methods to other enzymes are given in the respective subsections.
Table 1.
Overview of different methods for reversible control of enzyme activity, and the respective used stimuli.
| Methods | Stimulus | Enzyme | ||
|---|---|---|---|---|
| Changed accessibility of the active site | ||||
| Reversible | ||||
| inhibitor molecule approach | light | thrombin;21a α‐chymotrypsin;60 papain;60d subtilisin;60d glycogen synthase;61 glycogen phosphorylase;61 phosphoribosyl‐isomerase A;62 NADH‐ubiquinone oxidoreductase;63 tyrosinase;64 carbonic anhydrase65 |
||
| cover slip approach (sometimes called molecular gate approach) | light | endoglucanase 12A;21b carbonic anhydrase;21e restriction endonuclease SsoII;21f heparinase III21d | ||
| temperature | endoglucanase 12A;21c restriction endonuclease SsoII66 | |||
| formation of reactive nano‐compartments | light | horseradish peroxidase22a | ||
| magnetic field | papain;22b amyloglucosidase;67 glucose oxidase;67 horseradish peroxidase67 | |||
| Conformational changes in the enzyme | ||||
| random modification | light | α‐amylase;14a–14c β‐amylase;14b urease;14b, 24a α‐chymotrypsin;14b, 24b β‐glucosidase;14b aldolase;14d papain;24c horseradish peroxidase68 | ||
| site‐selective modification | light | kinesin ATPase;23d restriction endonuclease PvuII;23c restriction endonuclease BamHI;69 lipase B;23b lipase;23a ribonuclease S;70 horseradish peroxidase71 | ||
| interaction with matrix | light | lysozyme;26 (bovine serum albumin25) | ||
| Immobilization in stimulus‐responsive hydrogels | ||||
| light | urease;27h microperoxidase‐11;72 α‐chymotrypsin73 | |||
| temperature | asparaginase;15 β‐galactosidase;27a, 27e catalase;27b trypsin;27c laccase;27d lipase B;27f urease27g | |||
| Change in local temperature | ||||
| immobilization on the surface of magnetic particles | alternating magnetic field | α‐amylase;30a, 31 l‐aspartate oxidase;31 T4 DNA ligase;30b l‐2‐HADST dehalogenase74 | ||
| radio‐frequency field | thermolysin;35 carbonic anhydrase75 | |||
| immobilization on the surface of plasmonic gold nanoparticles | light | glucokinase;32 laccase;76 horseradish peroxidase77 | ||
| combination with organic nanoparticles in solution | light | α‐amylase13, 33 | ||
| combination with carbon nanotubes | light | cyclodextrin glycosyltransferase34 | ||
| combination of stimulus‐sensitive hydrogels with magnetic particles for immobilization | alternating magnetic field | invertase;36a β‐galactosidase;36b trypsin37 | ||
Figure 1.
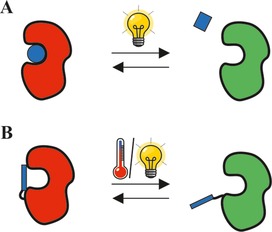
Reversible blocking of the active site of an enzyme with A: a stimulus‐responsive inhibitor molecule (blue circle and square, respectively); B: a stimulus‐sensitive residue (blue rectangle) acting as reversible cover slip. Red enzyme=inactive form; green enzyme=active form.
Figure 5.

Immobilization of enzymes on the surface of magnetic particles (or gold particles respectively) and reversible changes in the temperature of the particles as well as the direct environment upon application of an external alternating magnetic field or irradiation respectively. Red enzyme=inactive form; green enzyme=active form.
2.1.1. Changed Accessibility of the Active Site for the Substrate
The first approach in this chapter deals with a changed accessibility of the active site, prohibiting the substrate from entering. This approach has been described for free enzymes in solution by blocking the active site21 and for immobilized enzymes by formation of reactive nano‐compartments.22
a) Blocking the active site: To the best of our knowledge the different approaches published so far for free enzymes are as follows (Figure 1): blocking the active site using a stimulus‐sensitive inhibitor molecule which can be released on demand (Figure 1A; inhibitor molecule approach),21a or covalently tethering a stimulus‐sensitive residue to the enzyme which acts as a kind of lid, covering the active site on demand (Figure 1B; cover slip approach/molecular gate approach).21b–21f
In the first case, the inhibitor molecule changes its conformation in response to a stimulus, mostly light, but can only bind to the active site of the enzyme in one of the two possible conformations.21a Tian et al. showed that the activity of thrombin can be reversibly turned on and off by light‐induced cis‐trans isomerization of an azobenzene molecule (Razo) interacting with a telomere‐DNA‐based thrombin inhibitor (Itelo).21a Still, the overall activity of the thrombin was almost halved after 12 cycles of UV/Vis irradiation.21a
Since inhibitor molecules typically only work for one specific or a narrow range of enzymes, new inhibitors have to be found for each new or other enzyme. Although this process is quite laborious, it appears to be manageable, as shown by the variety of enzymes for which this approach has already proven to be suitable (see Table 1). However, for industrial biocatalysis, the need to separate the abundant enzyme inhibitor and product at the end of the reaction has to be considered.
In the second case, the conformation of a switchable residue is changed upon stimulation, meaning it covers the active site and thus prevents the substrate from entering the active site.21b–21f Shimoboji et al. applied a site‐selective modification of endoglucanase 12A mutants with thermo‐switchable21c and light‐switchable21b polymeric residues (N,N‐dimethylacrylamide copolymerized with N‐4‐phenylazo‐phenylacrylamide, or 4‐phenylazo‐phenylacrylamide) to gain remote control over the hydrolysis of cellulose (derivatives). Depending on the size of the substrate and the polymer chain, an almost complete, but reversible, shutdown of the activity was achieved.21b, 21c Similar outcomes were realized most recently for a heparinise III mutant as well.21d
If suitable knowledge on the 3D conformation of the enzyme and the active site is available together with know‐how in protein engineering and materials science, the method will be universally conferrable to most enzymes in the author's opinion. A prerequisite for this is the site‐selective incorporation of stimulus‐sensitive residues next to the active site, which can then cover the entire active site upon stimulation. The size and location of the tethered residue are therefore crucial for successful activity control, as described above.
b) Formation of reactive nano‐compartments: For immobilized enzymes, changes in the accessibility of the active site were achieved, for example, by enzyme and substrate immobilization onto separate superparamagnetic nanoparticles grafted with polymers (Figure 2).22b Without application of an external magnetic field, the particles are dispersed in the solution, and enzyme and substrate are spatially separated but remain in the same reaction pot.22b The application of an external magnetic field results in the formation of biocatalytic nano‐compartments between the enzyme and substrate particles, whereas if the magnetic field is removed, the nano‐compartments disassembled again and the reaction of the immobilized papain was stopped.22b
Figure 2.
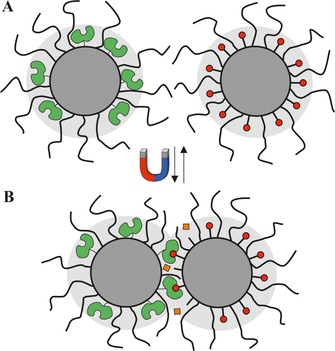
Reversible formation of reactive nano‐compartments upon application of a magnetic field. A: inactive magnetic particles with immobilized enzyme (green) and substrate (red circles) on separate particles; B: clustering of particles and formation of reactive nano‐compartments. Due to their close proximity, the enzyme is now able to convert the substrate (red circles) on the other particle into a product (orange squares). The light grey area represents the exclusion volume without application of a magnetic field.
Another approach made use of encapsulating horseradish peroxidase in light‐switchable polymersomes.22a The polymersome membrane was engineered to change from impermeable to permeable for the substrate upon isomerization of a photo‐switchable spiropyran (donor‐acceptor Stenhouse adduct) with visible light.22a Selective on‐ and off‐switching of the reaction was thus achieved, as the substrate is only able to enter the polymersome in one membrane conformation.
The formation of reactive nano‐compartments might be a generic method for a multitude of enzymes. However, for industrial biocatalysis the approaches published so far appear to be rather specialized. In these cases, formation of reactive nano‐compartments and the aim to achieve high product concentrations currently preclude each other.
2.1.2. Conformational Changes in the Enzyme
Conformational changes in enzymes and proteins (Figure 3) have been achieved through site‐selective23 and random incorporation14, 24 of photo‐switchable residues into the enzyme, or by interactions with a stimulus‐sensitive matrix, for example, light‐switchable surfactants.25 In the first case, photo‐switchable moieties are incorporated into the central enzyme structure, more specifically in regions that are important for the 3D‐structure of the active site of the enzyme. Upon light‐induced isomerization of the moieties, the enzyme conformation is altered. This change in the conformation of the enzyme results in either the substrate no longer fitting into the active site, or the (partial) prevention of the function of the active site.14a–14c, 23, 24, 25
Figure 3.
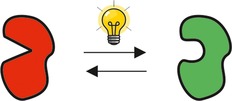
Reversible conformational change of an enzyme due to an external stimulus. Red enzyme=inactive form; green enzyme=active form.
The site‐selective incorporation of photo‐switchable moieties has been reported, for example, for azobenzene derivatives tethering to kinesin ATPase mutants.23d The ATPase showed reversible alterations in its activity by a factor of two upon light‐induced isomerization of the azobenzenes.23d Mutants of the restriction endonuclease PvuII, which were modified with a bifunctional azobenzene derivative, showed an activity change up to a maximum factor of 16 upon irradiation and thus cis‐trans isomerization.23c Lipase B was modified with azobenzene as well, and showed an 8‐ to 52‐fold activity increase compared to the unmodulated enzyme, depending on the cross‐linked amino acid residues.23b
In all the conducted studies, the (site‐directed) positioning of the photo‐switchable moiety was identified as being crucial to achieving the desired modulation of activity.23 The random incorporation of photo‐switchable residues, which was mostly performed at the beginning, is therefore no longer that common. Changes in enzyme activity through the random incorporation of photo‐switchable moieties were achieved in earlier days, for example, for α‐amylase,14a–14c β‐amylase,14b urease,14b, 24a α‐chymotrypsin,14b, 24b β‐glucosidase,14b aldolase14d and papain24c by incorporating spiropyran14a–14c, 24a or azobenzene14d, 24b, 24c moieties.
Conformational changes due to interactions with a stimulus‐sensitive matrix were achieved for lysozyme26 and the protein bovine serum albumin25 by applying surfactants with an azobenzene moiety. The photo‐switchable surfactants show a cis‐trans isomerization upon irradiation, leading to distinct changes in their 3D‐structure and, therefore, changed interactions with the protein as well as a change in protein conformation.25, 26
2.1.3. Immobilization in Stimulus‐Sensitive Hydrogels
The immobilization of enzymes in stimulus‐sensitive hydrogels, hydrogel coatings or hydrogel particles is another generic approach.15, 27 As illustrated in Figure 4, a change in enzyme activity is achieved by changing the swelling degree of the gel. The reasons behind the changes in the enzyme activity upon deswelling are most likely a combination of the following factors: a changed accessibility of the active site for the substrate, a changed enzyme conformation and moving flexibility due to the packing of enzyme into the hydrogel meshes, a change in enzyme environment (hydrophilicity, presence of charged groups), and a change in substrate/product diffusion due to differences in water content. The factor with the biggest impact most likely varies with the applied material.
Figure 4.
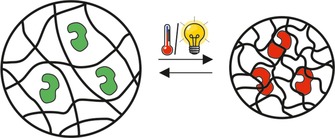
Immobilization of enzymes in stimulus‐sensitive hydrogels, which reversibly change their degree of swelling upon stimulation. Red enzyme=inactive form; green enzyme=active form.
Polymers showing a decrease in the swelling degree upon an increase of temperature above a certain value, referred to as lower critical solution temperature (LCST), are most commonly used for enzyme immobilization and activity control.15, 27a–27g The LCST for N‐isopropyl‐acrylamide‐based materials is typically between 32 °C15, 27b, 27e–27g and 40 °C.27a The decrease in the swelling degree upon an increase of temperature above the LCST can either be an abrupt or a gradual process, depending on the material used.28
A change in enzyme activity upon stimulation has so far been reported for a broad variety of enzymes, for example, asparaginase,15 β‐galactosidase,27a, 27e catalase,27b trypsin,27c laccase,27d lipase B27f and urease,27g in LCST hydrogels such as N‐isopropylacrylamide,27d–27f N‐isopropylacrylamide‐acrylamide copolymers,15, 27a, 27c isopropylacrylamide‐hydroxymethacrylate copolymers27b and N‐isopropylacrylamide‐poly(ethylene glycol) methacrylate copolymers.27g
Yet, the change in activity of the immobilized enzymes varied immensely. A number of studies reported an increase in activity despite hydrogel shrinking27e, 27f (in contrast to the activation direction shown in Figure 4); while some studies reported almost no change in activity upon shrinking.15 Other studies showed a roughly 3‐ to 5‐fold reduced activity in the shrunken state compared to the swollen state.27c, 27f, 27g Furthermore, some published studies describe a complete shutdown of enzyme activity in the shrunken gel, while the enzyme remained active in the free form at the same temperature.15, 27a Even within the same study, there were major differences in enzyme activity upon the shrinking of the gel when different gel compositions were used.15, 27f
One factor for this considerable variation in enzyme activity change could be that it is so far difficult to predict the range in which parameters such as the initial swelling degree, the resulting swelling degree after deswelling, and the mesh sizes of the gels have to be adjusted to achieve the desired effect on enzyme activity. The impact of meshes on enzyme activity is evident, but their intensity is highly dependent on the material, reaction environment, and type of enzyme selected.
Another factor behind the considerable variation in enzyme activity change might be the conflicting impact of temperature on both enzyme activity and hydrogel swelling. Enzyme activity is typically increased proportionally with temperature within a certain range,13, 19a while swelling degree is decreased in LCST materials with increasing temperature, thus decreasing the enzyme activity typically.15, 27a–27g An alternative approach could be the immobilization in hydrogel‐forming polymers exhibiting an increase in swelling degree upon an increase of temperature, referred to as upper critical solution temperature (UCST).29 However, to the best of our knowledge, the immobilization of enzymes in UCST hydrogels to modulate their activity has not been investigated so far.
Since it is difficult to predict the outcome of enzyme activity change upon application of the stimulus, it is almost impossible to rationally design enzyme activity control by immobilization in stimulus‐sensitive hydrogels. Indeed, further knowledge of the essential factors involved might enable a more universal use of this method in future.
2.1.4. Changes in Local Temperature
Activity control via a change in the (micro) temperature by means of enzyme immobilization on magnetic nanoparticles (ferromagnetic30 or superparamagnetic31) or plasmonic gold nanoparticles32 are other potential methods for enzyme activity control. For immobilization on magnetic particles, the application/removal of an external alternating magnetic field leads to subsequent changes in the micro temperature around the magnetic beads; for immobilization on plasmonic gold nanoparticles irradiation leads to changes in the micro temperature (Figure 5).
Suzuki et al. and Armenia et al. showed that the micro temperatures around the superparamagnetic particles could be varied between 25 °C and 90 °C, aiming for optimal reaction conditions for thermophilic enzymes, while the reaction medium temperature remained almost constant.30a, 31 To demonstrate the feasibility of the system for activating thermophilic enzymes, Armenia et al. conjugated α‐amylase from Bacillus licheniformis and l‐aspartate oxidase from Solfolobus tokodaii to iron oxide nanoparticles.31 By applying an external alternating magnetic field, they successfully, selectively, and remotely activated the enzymes with an increase in micro temperature.31
For immobilization on plasmonic gold nanoparticles, the application of light as an external stimulus leads to a subsequent increase in the microtemperature, with this photo‐thermal response being tunable with nanoparticle size and geometry.32 In this manner, the activity of glucokinase immobilized on plasmonic gold nanorods was increased by irradiation with near infrared light by a factor of approximately 1.5 compared to a sample without nanorods.32
Further approaches in which changes in (local) temperature affect enzymatic activity include the laser‐induced heating of polymeric nanoparticles (e.g., polydopamine‐polylactic acid nanoparticles for laser‐induced heating of α‐amylase/starch solutions13, 33), or graphene materials (carbon nanotubes for laser‐induced heating of cyclodextrin glycosyltransferase/starch solutions34). The use of radio‐frequency fields to heat enzymes immobilized on magnetic particles was described as well.35
In addition, techniques are described which combine magnetic particles with LCST polymers by (i) incorporating magnetic particles into hydrogels together with an enzyme,36 and (ii) immobilizing an enzyme in the polymeric shell of magnetic particles.37 Deswelling of the hydrogel material was achieved by applying an alternating magnetic field, resulting in the heating of the magnetic particles.36 Thereby, a modulation of enzyme activity was demonstrated for invertase,36a β‐galactosidase36b and trypsin,37 while a copolymer of N‐isopropylacrylamide and acrylamide served as hydrogel material in both cases.
In the author's opinion, approaches applying magnetic nanoparticles together with a magnetic field as a stimulus, or plasmonic gold nanoparticles together with light as a stimulus, are particularly promising, not only for controlling the activity of thermophilic enzymes. As shown in the depicted examples, enzyme immobilization on nanoparticles is already well established for a broad variety of systems, and enzymes generally show distinct activity changes upon an increase of temperature. As long as the heating of particles can be precisely controlled, optimal reaction temperatures can be achieved for all enzymes. The combination of different particles, responding to different stimuli or, for example, different irradiation wavelengths, could even enable the orthogonal control of different enzymes in one pot in future.
2.2. Methods for Irreversible Control
The term irreversible control refers to when a catalyst is switched, but the process cannot be reversed. Irreversible control of enzyme activity can generally be achieved either by selective in situ on‐switching of the enzyme, or by selective in situ off‐switching of the enzyme. In the following, methods for irreversible on‐ and off‐switching are compiled (Table 2). Notably, irreversible on‐switching of enzymes can generally be achieved with most established methods for stimulus‐induced release, for example immobilization in pH‐, temperature‐, or light‐sensitive hydrogels, micelles, polymersomes, or capsules. A precondition for using these “controlled release” approaches is that the enzyme exhibits no or low activity in the non‐released state. In the following, methods are referred to as “controlled release” approaches if they fulfil this basic requirement and start the release of the enzyme on demand by applying a stimulus. For a more detailed overview of controlled release methods in general, the reader is referred to the relevant publications.17, 18, 20b, 20c, 38
Table 2.
Overview of different methods for irreversible control of enzyme activity, and the respective used stimuli.
| Methods | Stimulus | Enzyme | ||
|---|---|---|---|---|
| Irreversible on | “Controlled release” approaches | |||
| immobilization in stimulus degradable materials=>(partial) degradation of the carrier upon stimulation | light | trypsin;39 chymotrypsin;39, 78 elastase;39, 78 γ‐glutamyltransferase;78 papain;78 thermolysin78 | ||
| immobilization in stimulus‐responsive hollow carriers=>(ir‐)reversible opening of pores in the carrier upon stimulation | light | lysozyme;40b alkaline phosphatase;40a β‐galactosidase41 | ||
| temperature | horseradish peroxidase79 | |||
| Uncaging | ||||
| light | lysozyme;44a restriction endonuclease MunI;42a, 42b restriction endonuclease PVuII;42a restriction endonuclease Bse634I;42b restriction endonuclease BamHI;42c T7 RNA polymerase;44b protein kinase A;43 ribonuclease S;80 caspase‐3;81 cAMP‐dependent protein kinase;82 Taq polymerase;83 Cre recombinase;84 butyrylcholinesterase;85 α‐chymotrypsin86 | |||
| Laser‐induced pH jump | ||||
| light | acid phosphatase type IV‐S.45 | |||
| Irreversible off | ||||
| Change in local temperature | ||||
| immobilization on the surface of gold nanoparticles | light | horseradish peroxidase;47a alkaline phosphatase;47b chymotrypsin;47b α‐chymotrypsin47c | ||
| Localized generation of free radicals | ||||
| light | alkaline phosphatase;49 β‐galactosidase;48a, 48f, 49, 50 acetylcholinesterase;49 thrombin;87 glutathione‐S‐transferase88 | |||
2.2.1. On‐Switching by Controlled‐Release Approaches
The first “controlled release”, or irreversible on‐switching method applied for biocatalysis is the encapsulation of a biocatalyst into a photodegradable hydrogel. The on‐switching of trypsin, chymotrypsin and elastase was achieved by irradiating (365 nm) a polypropyleniminetetramine acrylate‐based hydrogel with incorporated photo‐cleavable ortho‐nitrobenzyl moieties.39 Upon irradiation, the cross‐linking points are cleaved, leading to the disintegration of the hydrogel network structure, the release, and thereby activation of the enzyme as depicted in Figure 6.39
Figure 6.
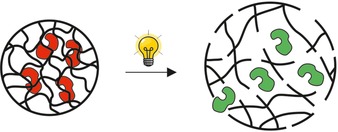
Immobilization of enzymes in stimulus‐degradable hydrogels, and controlled release upon stimulation. Red enzyme=inactive form; green enzyme=active form.
The second method is a controlled release through the formation of pores in hollow carrier materials, for example, utilizing the photo‐thermal effect.40 Here, one example is the immobilization of enzymes in hollow and porous gold nanoparticles coated with an LCST polymer.40 If these particles are irradiated, thermal energy is set free leading to the collapse of the LCST polymer coating and enabling the enzyme to diffuse through the now open pores of the carrier.40 In this case, the opening and closing of the pores upon irradiation is reversible. However, as the enzyme diffuses out of the carrier upon the opening of the pores, the on‐switching of enzyme activity is irreversible. A controlled on‐switching with this method was shown, for example, with lysozyme40b and alkaline phosphatase.40a
Other methods use the thermal energy set free upon irradiation for a polymerization of the polymer coating, which thus irreversibly forms open pores.41 Controlled release methods can generally be applied to other enzymes, provided that the enzyme exhibits no or low activity in the non‐released state as indicated above.
2.2.2. On‐Switching by Uncaging
On‐switching by uncaging is a common method, especially in drug delivery applications, where an inactive pro‐drug is converted into the active agent, for example, by a stimulus‐induced cleavage of a tethered residue. The uncaging method has been applied for proteins and enzymes as well, however, mainly in the context of controlling cellular processes, for example, by controlling restriction enzymes.42
For uncaging, the enzyme is typically modified with a photo‐cleavable residue, for example, phenacyl43 or ortho‐nitrobenzyl moieties.42a, 44 This residue works as an enzyme inactivator, meaning that as long as the residue is bound to the enzyme, the enzyme exhibits no or low activity.42a, 43, 44 Once the residue is cleaved upon stimulation, typically irradiation, the enzyme activity is restored.42a, 42b, 43, 44 The mechanism of uncaging an enzyme is schematically shown in Figure 7.
Figure 7.
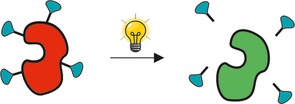
Uncaging of enzymes upon a stimulus. The deactivating residues (turquiose darts) are cleaved upon a stimulus, thereby activating the enzyme. Red enzyme=inactive form; green enzyme=active form.
Similar to controlled release methods, uncaging is generally conferrable to other enzymes provided that the enzyme does not exhibit significant activity in the caged state, and regains the majority of activity after uncaging.
2.2.3. On‐Switching by a Laser‐Induced pH Jump
Another approach for irreversible on‐switching introduced by Kohse et al. is based on enzyme activation in response to a remotely induced pH change.45 A remote change in pH can be achieved, for example, using a photoacid generator, that is, a compound that undergoes a reaction or dissociation upon irradiation with one final product being an acid.46 For example, a laser‐induced pH jump was employed to activate acid phosphatase type IV‐S.45 Upon irradiation, the photoacid generator derivatives based on 2‐nitrobenzaldehyde were converted to 2‐nitrosobenzoic acid, thus releasing one acid proton per molecule. This proton release induced a pH jump from 8 to 5, and an enzyme activity increase from 3% to 78%.45
On‐switching of enzyme activity via a change in the pH value is only applicable if the enzyme exhibits a distinct and significant change in activity dependent on the pH, combined with a high stability within a broad range of pH values.
2.2.4. Off‐Switching by Changes in Local Temperature
In line with the method of local temperature change for the reversible control over enzyme activity (Section 2.1.4, Figure 5), this method has been applied for irreversible off‐switching of enzymes as well.47 For example, horseradish peroxidase was immobilized on polyethylene glycol‐coated gold nanorods via the biotin‐avidin interaction, and selectively switched off by photo‐thermal inactivation upon heating of the nanorods with irradiation.47a
As all enzymes are denatured at a certain temperature, the method is universally conferrable to other enzymes, in the author's opinion on condition that suitably high temperatures are achieved with the nanoparticles.
2.2.5. Off‐Switching by Localized Generation of Free Radicals
Another method for the irreversible off‐switching of enzymatic activity is the use of photochromic groups that generate free radicals, mostly reactive oxygen species (ROS), for example, hydroxyl or oxygen radicals, upon irradiation with a specific wavelength.48 As depicted in Figure 8, ROS are formed upon irradiation, leading to irreversible changes in the protein structures, and thus destroying their function.49 ROS are sufficiently short‐lived to only inactivate the targeted molecules, if well‐selected, thus minimizing damage to the surrounding area.48d, 48f, 48g, 50 Activity control is most likely achievable for all enzymes that react sensitively to the presence of radicals.
Figure 8.

Irreversible deactivation of enzymes by light‐induced formation of reactive oxygen species (stars) from a chromophore or photosensitizer (orange circle) directly bound to the enzyme. Red enzyme=inactive form; green enzyme=active form.
This technique of inactivation by generating ROS is known by a number of different names, all terming similar approaches: chromophore‐assisted light inactivation (CALI),48c, 48f, 48g, 51 chromophore‐assisted laser inactivation (also CALI),48a, 48d, 49 or fluorophore‐assisted light inactivation (FALI).48e, 52 Chromophores,49 fluorophores48e, 52 or phototoxic proteins (photosensitizers)48c are fused to the target structure on a genetic level or via affinity binding.
Radical‐generating photochromic groups have been used for protein deactivation in various fields, with a particular focus on medical applications including photodynamic therapy.48b They have, furthermore, been used to regulate gene expression,16a to gain a better understanding of cell surface phenomena,49 and for the light‐driven killing of bacterial cells.53 In terms of enzyme inactivation, Jay et al. conjugated alkaline phosphatase, β‐galactosidase, and acetylcholinesterase to malachite green via the biotin‐streptavidin interaction and observed a controlled inactivation of these enzymes upon irradiation.49 Similar results were obtained by Takemoto et al. using eosin as a chromophore for the deactivation of β‐galactosidase.48f
3. Factors to Consider when Designing and Choosing Systems for Enzyme Activity Regulation
To achieve enzymatic activity control by environmental and external stimuli (e.g., temperature change, irradiation, or magnetic field) as described in Chapter 2, several factors have to be considered. This chapter aims to provide an overview of the factors to consider when designing systems for enzyme activity regulation or when choosing the best available system for a specific application (see also Scheme 1).
Scheme 1.
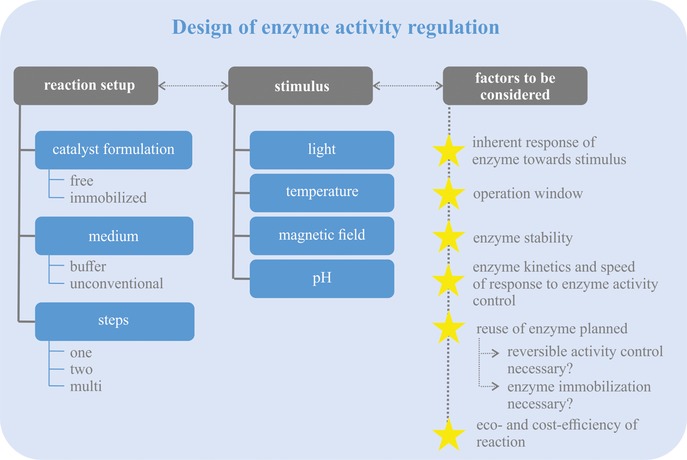
Factors to consider when designing and choosing systems for enzyme activity regulation.
Firstly, (some) enzymes inherently show an activity change when treated with the above‐mentioned stimuli even without an engineered “switch”. For example, enzymes respond with an increase in reaction speed to an increase in temperature.13, 19a They further show a distinct pH optimum for the catalyzed reaction.19a Therefore, the inherent response of the enzyme to the stimulus has to be considered when designing the reaction control. It is generally desirable that stimulus‐induced changes in enzyme activity and the inherent response of the enzyme to the stimulus do not work against each other, since this can limit the range in which enzyme activity can be adjusted (cf. Section 2.1.3). Furthermore, the point of inherent response of the enzyme to a stimulus applies to enzyme stability as well. The enzyme has to be stable under the chosen reaction conditions, and stable upon application of the stimulus, at least for reversible control methods.
A second point to be considered is the reversibility of activity control. Concepts enabling a repetitive, reversible on‐/off‐switching are especially attractive for most applications as these systems offer the general possibility of reusing the enzyme multiple times, for example, for repetitive batch processes. In contrast, irreversible off‐switching, for example, by temperature‐assisted denaturation,47a would destroy the biocatalyst and make a reuse impossible. This reduces the economic and ecological efficiency of the process,54 as new enzyme has to be produced for each reaction batch. In some cases, for example, for enzyme activity regulation in whole cells, many of the reversible methods might not be applicable. Then, irreversible control options (e.g., CALI) represent good alternatives.
A third aspect to consider is the interplay between enzyme kinetics and promptness of reaction control. To enable precise adjustments of enzyme activity, the response to the stimulus should ideally be faster, or at least similarly fast, compared to the enzymatic reaction. Still, for some applications also the gradual adjustment of enzyme activity might be suitable, and applicable if the reaction speed of the enzyme is faster than the reaction control.
A fourth point of consideration is whether immobilization makes sense, or is even mandatory, for a specific application. Undoubtedly, there are various opinions, publications, reviews, and books concerning immobilization in general as well as its advantages and disadvantages (see, for example, ref.55). In general, immobilization facilitates the reuse of a biocatalyst,55b–55d for example, magnetic beads can be easily removed from the reaction and reused multiple times.56 Furthermore, enzyme immobilization might increase enzyme stability,55b–55d for example, against higher temperatures or pH changes,56b, 57 and, in selected cases, can even enable the use of enzymes in organic solvents or neat substrate systems as well.54, 57b, 58 However, immobilization might also negatively affect enzyme activity,58a, 59 or can furthermore lead to mass transport limitations.55d Additionally, immobilization is a cost‐driving factor for many applications in biocatalysis.54, 55d The advantages and disadvantages of immobilization have to be balanced against each other in the end.
For multi‐step reactions, in which more than one of the enzymes exhibits cross‐reactivity, suitable methods have to be chosen that enable orthogonal control over several enzymes or even each single enzyme. Indeed, studies utilizing this approach have to the best of our knowledge not been published so far.
To date, light, temperature, and magnetic fields have most commonly served as stimuli for enzyme activity control. Since a broad variety of methods for activity control has already been published, choosing the best method for each application might be a challenge. In general, we would strongly advice researchers to consider the above‐mentioned factors: whether there is an inherent response of the enzyme to the applied stimulus, whether the aim is to reuse the enzyme and the reaction must be reversible, whether the reaction set‐up would benefit from immobilizing the enzyme, and, lastly, whether multiple enzymes exhibit cross‐reactivity and therefore require orthogonal control of all enzymes involved in the reaction.
4. Future Prospects
In future, selective in situ remote control will bring biocatalysis a significant step closer to mimicking physiological control mechanisms. It will thus enable sophisticated multi‐step reaction cascades with high yields, while minimizing side reactions. In particular, concepts enabling orthogonal and reversible control over the activity of different enzymes, meaning they are able to control the activity of each enzyme in the reaction independently from each other, will have to be further developed. Although this is currently not possible, to the best of our knowledge, the methods compiled in this review have the potential of achieving this objective in future. For example, Armenia et al. reported the first steps towards reaching this goal in their study on the selective heating of enzymes immobilized on magnetic particles with an external alternating magnetic field.31 They demonstrated how the activity of the immobilized thermophilic enzymes was controlled without impeding the activity of a non‐thermophilic enzyme in the continuous phase.31
Furthermore, knowledge of a detailed kinetic reaction would help in selecting the right time points at which a distinct cascade step reaches its peak in order to then selectively switch on the next step and/or inactivate the previous one. Suitable on‐line analytical systems enabling feedback control of the reaction might therefore be of immense help. With progress being made with in‐line infrared spectroscopy and on‐line nuclear magnetic resonance spectroscopy using benchtop devices, there is the potential for an optimal regulation of the overall process, for example, regarding conversions or specific space‐time yields for industrial applications. Even a self‐controlled regulation of each step seems possible in the near future if in‐line analytics are sensitive enough to determine the substrate, intermediate, and product concentrations reliably. This would enable automated on‐/off‐switching of the stimulus when a defined minimum/maximum is reached.
Biographical Information
Christiane Claaßen received her M.Sc. (2014) degree from the Niederrhein University of Applied Sciences and her Ph.D. from the University of Stuttgart in 2018. She is currently conducting research under the guidance of Prof. Dr. Dörte Rother at Forschungszentrum Jülich GmbH. Her research focuses on the implementation of nuclear magnetic resonance spectroscopy as an on‐line analytics system for the biocatalytic multi‐step synthesis of diversely substituted tetrahydroisoquinolines.

Biographical Information
Tim Gerlach received his M.Sc. (2018) from RWTH Aachen University and is currently undertaking his Ph.D. studies in the same group at Forschungszentrum Jülich GmbH and RWTH Aachen University. His research is focused on establishing a toolbox with various photoactive fusion enzymes and their application in multi‐enzyme cascades.

Biographical Information
Dörte Rother received her Diplom degree in biology with a focus on biotechnology from RWTH Aachen University in 2003 and her Ph.D. from Heinrich‐Heine University Düsseldorf in 2008. Thanks to a Helmholtz young investigator group scholarship in 2012, she was able to begin her independent research on modular enzyme cascades at the Institute of Bio‐ and Geosciences – Biotechnology (IBG‐1), Forschungszentrum Jülich GmbH. Since then, her group has been working on an integrated engineering approach, combining enzyme engineering, reaction optimization, and process design to produce active pharmaceutical compounds in an economically and ecologically efficient way. For this work, she received the DECHEMA Prize 2018. She is currently head of the biocatalysis group at Forschungszentrum Jülich GmbH and since June 2018 a full professor for synthetic enzyme cascades at RWTH Aachen University. Supported by a Starting Grant from the European Research Council, one current research target is the regulation of enzyme activity using light and other stimuli (LightCas project), as well as, in the near future, by magnetic field as part of the FET Open project HOTZYMES.

Acknowledgements
The authors would like to thank Clare Dibble (IBG‐1, Forschungszentrum Jülich) and Reinhard Oeggl (IBG‐1, Forschungszentrum Jülich) for their support in proofreading the manuscript. The authors received funding from the European Research Council (ERC) as part of the European Union's Horizon 2020 research and innovation programme (grant agreement No 757320) within the scope of the LightCas (light‐controlled synthetic enzyme cascades) project.
C. Claaßen, T. Gerlach, D. Rother, Adv. Synth. Catal. 2019, 361, 2387.
References
- 1.
- 1a. Beigi M., Gauchenova E., Walter L., Waltzer S., Bonina F., Stillger T., Rother D., Pohl M., Müller M., Chem. Eur. J. 2016, 22, 13999–14005; [DOI] [PubMed] [Google Scholar]
- 1b. Turner N. J., Humphreys L., Biocatalysis in Organic Synthesis: The Retrosynthesis Approach, Royal Society of Chemistry, 2018; [Google Scholar]
- 1c. Ghaffari-Moghaddam M., Eslahi H., Aydin Y. A., Saloglu D., J. Biol. Methods 2015, 2, e25. [Google Scholar]
- 2. Lopez-Gallego F., Schmidt-Dannert C., Curr. Opin. Chem. Biol. 2010, 14, 174–183. [DOI] [PubMed] [Google Scholar]
- 3. Shoda S.-I., Uyama H., Kadokawa J.-I., Kimura S., Kobayashi S., Chem. Rev. 2016, 116, 2307–2413. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Bruggink A., Schoevaart R., Kieboom T., Org. Process Res. Dev. 2003, 7, 622–640; [Google Scholar]
- 4b. Mayer S. F., Kroutil W., Faber K., Chem. Soc. Rev. 2001, 30, 332–339; [Google Scholar]
- 4c. Schrittwieser J. H., Sattler J., Resch V., Mutti F. G., Kroutil W., Curr. Opin. Chem. Biol. 2011, 15, 249–256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Sheldon R. A., Green Chem. 2007, 9, 1273–1283; [Google Scholar]
- 4e. Sheldon R. A., Chem. Commun. 2008, 3352–3365; [DOI] [PubMed] [Google Scholar]
- 4f. Oeggl R., Maßmann T., Jupke A., Rother D., ACS Sustainable Chem. Eng. 2018, 6, 11819–11826. [Google Scholar]
- 5.
- 5a. Hailes H. C., Rother D., Müller M., Westphal R., Ward J. M., Pleiss J., Vogel C., Pohl M., FEBS J. 2013, 280, 6374–6394; [DOI] [PubMed] [Google Scholar]
- 5b. Pavlidis I. V., Weiß M. S., Genz M., Spurr P., Hanlon S. P., Wirz B., Iding H., Bornscheuer U. T., Nat. Chem. 2016, 8, 1076; [DOI] [PubMed] [Google Scholar]
- 5c. Westphal R., Vogel C., Schmitz C., Pleiss J., Müller M., Pohl M., Rother D., Angew. Chem. 2014, 126, 9530–9533; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 9376–9379; [DOI] [PubMed] [Google Scholar]
- 5d. Payer S. E., Schrittwieser J. H., Grischek B., Simon R. C., Kroutil W., Adv. Synth. Catal. 2016, 358, 444–451; [Google Scholar]
- 5e. Zhang W., Fernández-Fueyo E., Ni Y., van Schie M., Gacs J., Renirie R., Wever R., Mutti F. G., Rother D., Alcalde M., Hollmann F., Nat. Catal. 2018, 1, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Bommarius A. S., Riebel-Bommarius B. R., Biocatalysis: Fundamentals and Applications, Wiley-VCH, Weinheim, 2004; [Google Scholar]
- 6b. Faber K., Biotransformations in organic chemistry: a textbook, 6 th edn., Springer Verlag, Heidelberg, 2011; [Google Scholar]
- 6c. Reetz M. T., J. Am. Chem. Soc. 2013, 135, 12480–12496. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Schrittwieser J. H., Velikogne S., Hall M., Kroutil W., Chem. Rev. 2018, 118, 270–348; [DOI] [PubMed] [Google Scholar]
- 7b. Sperl J. M., Sieber V., ACS Catal. 2018, 8, 2385–2396. [Google Scholar]
- 8.
- 8a. France S. P., Hussain S., Hill A. M., Hepworth L. J., Howard R. M., Mulholland K. R., Flitsch S. L., Turner N. J., ACS Catal. 2016, 6, 3753–3759; [Google Scholar]
- 8b. Jakoblinnert A., Rother D., Green Chem. 2014, 16, 3472–3482; [Google Scholar]
- 8c. Oroz-Guinea I., García-Junceda E., Curr. Opin. Chem. Biol. 2013, 17, 236–249; [DOI] [PubMed] [Google Scholar]
- 8d. Ricca E., Brucher B., Schrittwieser J. H., Adv. Synth. Catal. 2011, 353, 2239–2262; [Google Scholar]
- 8e. Santacoloma P. A., Sin G., Gernaey K. V., Woodley J. M., Org. Process Res. Dev. 2011, 15, 203–212; [Google Scholar]
- 8f. Sehl T., Hailes H. C., Ward J. M., Wardenga R., von Lieres E., Offermann H., Westphal R., Pohl M., Rother D., Angew. Chem. 2013, 125, 6904–6908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013, 52, 6772–6775; [DOI] [PubMed] [Google Scholar]
- 8g. Sehl T., Hailes H. C., Ward J. M., Menyes U., Pohl M., Rother D., Green Chem. 2014, 16, 3341–3348; [Google Scholar]
- 8h. Erdmann V., Lichman B. R., Zhao J., Simon R. C., Kroutil W., Ward J. M., Hailes H. C., Rother D., Angew. Chem. 2017, 129, 12677–12681; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2017, 56, 12503–12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. France S. P., Hepworth L. J., Turner N. J., Flitsch S. L., ACS Catal. 2017, 7, 710–724. [Google Scholar]
- 10. Hult K., Berglund P., Trends Biotechnol. 2007, 25, 231–238. [DOI] [PubMed] [Google Scholar]
- 11. Brahma A., Musio B., Ismayilova U., Nikbin N., Kamptmann S. B., Siegert P., Jeromin G. E., Ley S. V., Pohl M., Synlett 2016, 27, 262–266. [Google Scholar]
- 12.
- 12a. Quin M. B., Wallin K. K., Zhang G., Schmidt-Dannert C., Org. Biomol. Chem. 2017, 15, 4260–4271; [DOI] [PubMed] [Google Scholar]
- 12b. Schmidt-Dannert C., Lopez-Gallego F., Microb. Biotechnol. 2016, 9, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao Y., Wang Y., ChemCatChem 2016, 8, 2740–2747. [Google Scholar]
- 14.
- 14a. Aizawa M., Namba K., Suzuki S., Arch. Biochem. Biophys. 1977, 180, 41–48; [DOI] [PubMed] [Google Scholar]
- 14b. Aizawa M., Namba K., Suzuki S., Arch. Biochem. Biophys. 1977, 182, 305–310; [DOI] [PubMed] [Google Scholar]
- 14c. Kenryo N., Shuichi S., Chem. Lett. 1975, 4, 947–950; [Google Scholar]
- 14d. Montagnoli G., Montt S., Nannicini L., Giovannitti M. P., Ristori M. G., Photochem. Photobiol. 1978, 27, 43–49. [DOI] [PubMed] [Google Scholar]
- 15. Dong L. C., Hoffman A. S., J. Control. Release 1986, 4, 223–227. [Google Scholar]
- 16.
- 16a. Mayer G., Heckel A., Angew. Chem. 2006, 118, 5020–5042; [Google Scholar]; Angew. Chem. Int. Ed. 2006, 45, 4900–4921; [DOI] [PubMed] [Google Scholar]
- 16b. Willner I., Rubin S., Angew. Chem. 1996, 108, 419–439; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1996, 35, 367–385; [Google Scholar]
- 16c. Szymański W., Beierle J. M., Kistemaker H. A. V., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178. [DOI] [PubMed] [Google Scholar]
- 17. Deshayes S., Kasko A. M., J. Polym. Sci. Part A: Polym. Chem. 2013, 51, 3531–3566. [Google Scholar]
- 18. Qiu Y., Park K., Adv. Drug Delivery Rev. 2001, 53, 321–339. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Kulig J., Frese A., Kroutil W., Pohl M., Rother D., Biotechnol. Bioeng. 2013, 110, 1838–1848; [DOI] [PubMed] [Google Scholar]
- 19b. Gocke D., Graf T., Brosi H., Frindi-Wosch I., Walter L., Müller M., Pohl M., J. Mol. Catal. B: Enzym. 2009, 61, 30–35. [Google Scholar]
- 20.
- 20a. Willner I., Rubin S., Angew. Chem. 1996, 108, 419–439; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1996, 35, 367–385; [Google Scholar]
- 20b. Tomatsu I., Peng K., Kros A., Adv. Drug Delivery Rev. 2011, 63, 1257–1266; [DOI] [PubMed] [Google Scholar]
- 20c. Lu Y., Sun W. J., Gu Z., J. Control. Release 2014, 194, 1–19; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Barhoumi A., Liu Q., Kohane D. S., J. Control. Release 2015, 219, 31–42. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Tian T., Song Y., Wang J., Fu B., He Z., Xu X., Li A., Zhou X., Wang S., Zhou X., J. Am. Chem. Soc. 2016, 138, 955–961; [DOI] [PubMed] [Google Scholar]
- 21b. Shimoboji T., Larenas E., Fowler T., Kulkarni S., Hoffman A. S., Stayton P. S., Proc. Natl. Acad. Sci. USA 2002, 99, 16592–16596; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Shimoboji T., Larenas E., Fowler T., Hoffman A. S., Stayton P. S., Bioconjugate Chem. 2003, 14, 517–525; [DOI] [PubMed] [Google Scholar]
- 21d. Gu Y., Wu X., Liu H., Pan Q., Chen Y., Org. Lett. 2018, 20, 48–51; [DOI] [PubMed] [Google Scholar]
- 21e. Harvey J. H., Trauner D., ChemBioChem 2008, 9, 191–193; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21f. Hien L. T., Zatsepin T. S., Schierling B., Volkov E. M., Wende W., Pingoud A., Kubareva E. A., Oretskaya T. S., Bioconjugate Chem. 2011, 22, 1366–1373. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Rifaie-Graham O., Ulrich S., Galensowske N. F. B., Balog S., Chami M., Rentsch D., Hemmer J. R., Read deAlaniz J., Boesel L. F., Bruns N., J. Am. Chem. Soc. 2018, 140, 8027–8036; [DOI] [PubMed] [Google Scholar]
- 22b. Zakharchenko A., Guz N., Laradji A. M., Katz E., Minko S., Nat. Catal. 2018, 1, 73–81. [Google Scholar]
- 23.
- 23a. Poloni C., Szymanski W., Feringa B. L., Chem. Commun. 2014, 50, 12645–12648; [DOI] [PubMed] [Google Scholar]
- 23b. Agarwal P. K., Schultz C., Kalivretenos A., Ghosh B., Broedel S. E., J. Phys. Chem. Lett. 2012, 3, 1142–1146; [Google Scholar]
- 23c. Schierling B., Noel A. J., Wende W., Hien L. T., Volkov E., Kubareva E., Oretskaya T., Kokkinidis M., Rompp A., Spengler B., Pingoud A., Proc. Natl. Acad. Sci. USA 2010, 107, 1361–1366; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23d. Yamada M. D., Nakajima Y., Maeda H., Maruta S., J. Biochem. 2007, 142, 691–698. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Karube I., Nakamoto Y., Namba K., Suzuki S., Biochim. Biophys. Acta 1976, 429, 975–981; [DOI] [PubMed] [Google Scholar]
- 24b. Erlanger B. F., Wassermann N. H., Cooper A. G., Monk R. J., Eur. J. Biochem. 1976, 61, 287–295; [DOI] [PubMed] [Google Scholar]
- 24c. Willner I., Rubin S., Riklin A., J. Am. Chem. Soc. 1991, 113, 3321–3325. [Google Scholar]
- 25.
- 25a. Lee C. T. J., Smith K. A., Hatton T. A., Biochemistry 2005, 44, 524–536; [DOI] [PubMed] [Google Scholar]
- 25b. Wang S.-C., Lee C. T. J., J. Phys. Chem. B 2006, 110, 16117–16123. [DOI] [PubMed] [Google Scholar]
- 26. Wang S.-C., Lee C. T. J., Biochemistry 2007, 46, 14557–14566. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Park T. G., Hoffman A. S., J. Biomed. Mater. Res. 1990, 24, 21–38; [DOI] [PubMed] [Google Scholar]
- 27b. Arıca M. Y., Öktem H. A., Öktem Z., Tuncel S. A., Polym. Int. 1999, 48, 879–884; [Google Scholar]
- 27c. Harada A., Johnin K., Kawamura A., Kono K., J. Polym. Sci. Part A: Polym. Chem. 2007, 45, 5942–5948; [Google Scholar]
- 27d. Klis M., Karbarz M., Stojek Z., Rogalski J., Bilewicz R., J. Phys. Chem. B 2009, 113, 6062–6067; [DOI] [PubMed] [Google Scholar]
- 27e. Welsch N., Wittemann A., Ballauff M., J. Phys. Chem. B 2009, 113, 16039–16045; [DOI] [PubMed] [Google Scholar]
- 27f. Gawlitza K., Wu C., Georgieva R., Ansorge-Schumacher M., von Klitzing R., Z. Phys. Chem. (Muenchen Ger.) 2012, 226, 749; [DOI] [PubMed] [Google Scholar]
- 27g. Bayramoglu G., Arica M. Y., Bioproc. Biosyst. Eng. 2014, 37, 235–243; [DOI] [PubMed] [Google Scholar]
- 27h. Karube I., Nakamoto Y., Suzuki S., Biochim. Biophys. Acta 1976, 445, 774–779. [DOI] [PubMed] [Google Scholar]
- 28. Teotia A. K., Sami H., Kumar A., in: Switchable and Responsive Surfaces and Materials for Biomedical Applications, (Ed.: Z. Zhang), Woodhead Publishing, Oxford, 2015, pp 3–43. [Google Scholar]
- 29.
- 29a. Aoki T., Kawashima M., Katono H., Sanui K., Ogata N., Okano T., Sakurai Y., Macromolecules 1994, 27, 947–952; [Google Scholar]
- 29b. Seuring J., Agarwal S., Macromolecules 2012, 45, 3910–3918; [Google Scholar]
- 29c. Seuring J., Agarwal S., ACS Macro Lett. 2013, 2, 597–600. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Suzuki M., Aki A., Mizuki T., Maekawa T., Usami R., Morimoto H., PLoS One 2015, 10, e0127673; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Suzuki M., Hayashi H., Mizuki T., Maekawa T., Morimoto H., Biochem. Biophys. Rep. 2016, 8, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Armenia I., Grazú Bonavia M. V., De Matteis L., Ivanchenko P., Martra G., Gornati R., de La Fuente J. M., Bernardini G., J. Colloid Interface Sci. 2019, 537, 615–628. [DOI] [PubMed] [Google Scholar]
- 32. Blankschien M. D., Pretzer L. A., Huschka R., Halas N. J., Gonzalez R., Wong M. S., ACS Nano 2013, 7, 654–663. [DOI] [PubMed] [Google Scholar]
- 33. Cao Y., Wang Z., Liao S., Wang J., Wang Y., Chem. Eur. J. 2016, 22, 1152–1158. [DOI] [PubMed] [Google Scholar]
- 34. Miyako E., Nagata H., Hirano K., Hirotsu T., Lab Chip 2009, 9, 788–794. [DOI] [PubMed] [Google Scholar]
- 35.C. B. Collins, C. J. Ackerson, in: SPIE BiOS, Vol. 10507, SPIE, 2018, p 6.
- 36.
- 36a. Takahashi F., Sakai Y., Mizutani Y., J. Ferment. Bioeng. 1997, 83, 152–156; [Google Scholar]
- 36b. Kato N., Oishi A., Takahashi F., Mater. Sci. Eng. C 1998, 6, 291–296. [Google Scholar]
- 37. Marten G. U., Gelbrich T., Schmidt A. M., Beilstein J. Org. Chem. 2010, 6, 922–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koetting M. C., Peters J. T., Steichen S. D., Peppas N. A., Mater. Sci. Eng. R 2015, 93, 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murayama S., Ishizuka F., Takagi K., Inoda H., Sano A., Santa T., Kato M., Anal. Chem. 2012, 84, 1374–1379. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Shi P., Ju E., Ren J., Qu X., Adv. Funct. Mater. 2014, 24, 826–834; [Google Scholar]
- 40b. Yavuz M. S., Cheng Y., Chen J., Cobley C. M., Zhang Q., Rycenga M., Xie J., Kim C., Song K. H., Schwartz A. G., Wang L. V., Xia Y., Nat. Mater. 2009, 8, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hindley J. W., Elani Y., McGilvery C. M., Ali S., Bevan C. L., Law R. V., Ces O., Nat. Commun. 2018, 9, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Silanskas A., Foss M., Wende W., Urbanke C., Lagunavicius A., Pingoud A., Siksnys V., Biochemistry 2011, 50, 2800–2807; [DOI] [PubMed] [Google Scholar]
- 42b. Silanskas A., Zaremba M., Sasnauskas G., Siksnys V., Bioconjugate Chem. 2012, 23, 203–211; [DOI] [PubMed] [Google Scholar]
- 42c. Endo M., Nakayama K., Majima T., J. Org. Chem. 2004, 69, 4292–4298. [DOI] [PubMed] [Google Scholar]
- 43. Zou K., Cheley S., Givens R. S., Bayley H., J. Am. Chem. Soc. 2002, 124, 8220–8229. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Takamori S., Yamaguchi S., Ohashi N., Nagamune T., Chem. Commun. 2013, 49, 3013–3015; [DOI] [PubMed] [Google Scholar]
- 44b. Chou C., Young D. D., Deiters A., ChemBioChem 2010, 11, 972–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.
- 45a. Kohse S., Neubauer A., Lochbrunner S., Kragl U., ChemCatChem 2014, 6, 3511–3517; [Google Scholar]
- 45b. Kohse S., Neubauer A., Pazidis A., Lochbrunner S., Kragl U., J. Am. Chem. Soc. 2013, 135, 9407–9411. [DOI] [PubMed] [Google Scholar]
- 46. Martin C. J., Rapenne G., Nakashima T., Kawai T., J. Photochem. Photobiol. C 2018, 34, 41–51. [Google Scholar]
- 47.
- 47a. Thompson S. A., Paterson S., Azab M. M. M., Wark A. W., de La Rica R., Small 2017, 13, 1603195; [DOI] [PubMed] [Google Scholar]
- 47b.G. Huettmann, B. Radt, J. Serbin, R. Birngruber, in: Proc. SPIE, Vol. 5142, SPIE, 2003, p 8;
- 47c. Kang P., Chen Z., Nielsen S. O., Hoyt K., D′Arcy S., Gassensmith J. J., Qin Z., Small 2017, 13, 1700841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.
- 48a. Liao J. C., Roider J., Jay D. G., Proc. Natl. Acad. Sci. USA 1994, 91, 2659–2663; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48b. Abrahamse H., Hamblin M. R., Biochem. J. 2016, 473, 347–364; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48c. Bulina M. E., Lukyanov K. A., Britanova O. V., Onichtchouk D., Lukyanov S., Chudakov D. M., Nat. Protoc. 2006, 1, 947; [DOI] [PubMed] [Google Scholar]
- 48d. Jacobson K., Rajfur Z., Vitriol E., Hahn K., Trends Cell Biol. 2008, 18, 443–450; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48e. Morton R. A., Luo G., Davis M. I., Hales T. G., Lovinger D. M., Mol. Cell. Neurosci. 2011, 47, 79–92; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48f. Takemoto K., Matsuda T., McDougall M., Klaubert D. H., Hasegawa A., Los G. V., Wood K. V., Miyawaki A., Nagai T., ACS Chem. Biol. 2011, 6, 401–406; [DOI] [PubMed] [Google Scholar]
- 48g. Tour O., Meijer R. M., Zacharias D. A., Adams S. R., Tsien R. Y., Nat. Biotechnol. 2003, 21, 1505–1508. [DOI] [PubMed] [Google Scholar]
- 49. Jay D. G., Proc. Natl. Acad. Sci. USA 1988, 85, 5454–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Linden K. G., Liao J. C., Jay D. G., Biophys. J. 1992, 61, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Surrey T., Elowitz M. B., Wolf P. E., Yang F., Nedelec F., Shokat K., Leibler S., Proc. Natl. Acad. Sci. USA 1998, 95, 4293–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beck S., Sakurai T., Eustace B. K., Beste G., Schier R., Rudert F., Jay D. G., Proteomics 2002, 2, 247–255. [DOI] [PubMed] [Google Scholar]
- 53. Endres S., Wingen M., Torra J., Ruiz-González R., Polen T., Bosio G., Bitzenhofer N. L., Hilgers F., Gensch T., Nonell S., Jaeger K.-E., Drepper T., Sci. Rep. 2018, 8, 15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tufvesson P., Lima-Ramos J., Nordblad M., Woodley J. M., Org. Process Res. Dev. 2011, 15, 266–274. [Google Scholar]
- 55.
- 55a. Sheldon R. A., van Pelt S., Chem. Soc. Rev. 2013, 42, 6223–6235; [DOI] [PubMed] [Google Scholar]
- 55b. Guisan J. M., in: Immobilization of Enzymes and Cells, 3 rd edn., (Ed.: J. M. Guisan), Humana Press, Totowa, NJ, 2013, pp 1–13; [Google Scholar]
- 55c. DiCosimo R., McAuliffe J., Poulose A. J., Bohlmann G., Chem. Soc. Rev. 2013, 42, 6437–6474; [DOI] [PubMed] [Google Scholar]
- 55d. Liese A., Hilterhaus L., Chem. Soc. Rev. 2013, 42, 6236–6249; [DOI] [PubMed] [Google Scholar]
- 55e. Datta S., Christena L. R., Rajaram Y. R. S., 3 Biotech. 2013, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.
- 56a. Goldberg K., Krueger A., Meinhardt T., Kroutil W., Mautner B., Liese A., Tetrahedron: Asymmetry 2008, 19, 1171–1173; [Google Scholar]
- 56b. Wang F., Guo C., Liu H.-Z., Liu C.-Z., J. Mol. Catal. B: Enzym. 2007, 48, 1–7. [Google Scholar]
- 57.
- 57a. Mateo C., Palomo J. M., Fernandez-Lorente G., Guisan J. M., Fernandez-Lafuente R., Enzyme Microb. Technol. 2007, 40, 1451–1463; [Google Scholar]
- 57b. Guzik U., Hupert-Kocurek K., Wojcieszynska D., Molecules 2014, 19, 8995–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.
- 58a. Döbber J., Pohl M., Ley S. V., Musio B., React. Chem. Eng. 2018, 3, 8–12; [Google Scholar]
- 58b. Okrob D., Paravidino M., Orru R. V. A., Wiechert W., Hanefeld U., Pohl M., Adv. Synth. Catal. 2011, 353, 2399–2408; [Google Scholar]
- 58c. Hischer T., Gocke D., Fernández M., Hoyos P., Alcántara A. R., Sinisterra J. V., Hartmeier W., Ansorge-Schumacher M. B., Tetrahedron 2005, 61, 7378–7383. [Google Scholar]
- 59. Döbber J., Gerlach T., Offermann H., Rother D., Pohl M., Green Chem. 2018, 20, 544–552. [Google Scholar]
- 60.
- 60a. Bagchi D., Ghosh A., Singh P., Dutta S., Polley N., Althagafi I. I., Jassas R. S., Ahmed S. A., Pal S. K., Sci. Rep. 2016, 6, 34399; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60b. Pearson D., Abell A. D., Org. Biomol. Chem. 2006, 4, 3618–3625; [DOI] [PubMed] [Google Scholar]
- 60c. Pearson D., Alexander N., Abell A. D., Chem. Eur. J. 2008, 14, 7358–7365; [DOI] [PubMed] [Google Scholar]
- 60d. Westmark P. R., Kelly J. P., Smith B. D., J. Am. Chem. Soc. 1993, 115, 3416–3419; [Google Scholar]
- 60e. Harvey A. J., Abell A. D., Bioorg. Med. Chem. Lett. 2001, 11, 2441–2444; [DOI] [PubMed] [Google Scholar]
- 60f. Harvey A. J., Abell A. D., Tetrahedron 2000, 56, 9763–9771. [Google Scholar]
- 61. Diaz-Lobo M., Garcia-Amoros J., Fita I., Velasco D., Guinovart J. J., Ferrer J. C., Org. Biomol. Chem. 2015, 13, 7282–7288. [DOI] [PubMed] [Google Scholar]
- 62. Reisinger B., Kuzmanovic N., Löffler P., Merkl R., König B., Sterner R., Angew. Chem. 2014, 126, 606–609; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 595–598. [DOI] [PubMed] [Google Scholar]
- 63. Fujita D., Murai M., Nishioka T., Miyoshi H., Biochemistry 2006, 45, 6581–6586. [DOI] [PubMed] [Google Scholar]
- 64. Komori K., Yatagai K., Tatsuma T., J. Biotechnol. 2004, 108, 11–16. [DOI] [PubMed] [Google Scholar]
- 65.
- 65a. Vomasta D., Hogner C., Branda N. R., Konig B., Angew. Chem. 2008, 120, 7756–7759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008, 47, 7644–7647; [DOI] [PubMed] [Google Scholar]
- 65b. Vomasta D., Innocenti A., Konig B., Supuran C. T., Bioorg. Med. Chem. Lett. 2009, 19, 1283–1286. [DOI] [PubMed] [Google Scholar]
- 66. Abrosimova L. A., Monakhova M. V., Migur A. Y., Wolfgang W., Pingoud A., Kubareva E. A., Oretskaya T. S., IUBMB Life 2013, 65, 1012–1016. [DOI] [PubMed] [Google Scholar]
- 67. Filipov Y., Zakharchenko A., Minko S., Katz E., ChemPhysChem 2018, 19, 3035–3043. [DOI] [PubMed] [Google Scholar]
- 68. Weston D. G., Kirkham J., Cullen D. C., Biochim. Biophys. Acta 1999, 1428, 463–467. [DOI] [PubMed] [Google Scholar]
- 69.
- 69a. Nakayama K., Endo M., Majima T., Bioconjugate Chem. 2005, 16, 1360–1366; [DOI] [PubMed] [Google Scholar]
- 69b. Nakayama K., Endo M., Majima T., Chem. Commun. 2004, 2386–2387. [DOI] [PubMed] [Google Scholar]
- 70.
- 70a. James D. A., Burns D. C., Woolley G. A., Protein Eng. 2001, 14, 983–991; [DOI] [PubMed] [Google Scholar]
- 70b. Liu D., Karanicolas J., Yu C., Zhang Z., Woolley G. A., Bioorg. Med. Chem. Lett. 1997, 7, 2677–2680. [Google Scholar]
- 71. Muranaka N., Hohsaka T., Sisido M., FEBS Lett. 2002, 510, 10–12. [DOI] [PubMed] [Google Scholar]
- 72. Dubner M., Cadarso V. J., Gevrek T. N., Sanyal A., Spencer N. D., Padeste C., ACS Appl. Mater. Interfaces 2017, 9, 9245–9249. [DOI] [PubMed] [Google Scholar]
- 73.
- 73a. Willner I., Rubin S., Shatzmiller R., Zor T., J. Am. Chem. Soc. 1993, 115, 8690–8694; [Google Scholar]
- 73b. Willner I., Rubin S., Zor T., J. Am. Chem. Soc. 1991, 113, 4013–4014. [Google Scholar]
- 74. Knecht L. D., Ali N., Wei Y., Hilt J. Z., Daunert S., ACS Nano 2012, 6, 9079–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Andreeva Y. I., Drozdov A. S., Avnir D., Vinogradov V. V., ACS Biomater. Sci. Eng. 2018, 4, 3962–3967. [DOI] [PubMed] [Google Scholar]
- 76. Guo S., Li H., Liu J., Yang Y., Kong W., Qiao S., Huang H., Liu Y., Kang Z., ACS Appl. Mater. Interfaces 2015, 7, 20937–20944. [DOI] [PubMed] [Google Scholar]
- 77.
- 77a. Tadepalli S., Yim J., Cao S., Wang Z., Naik R. R., Singamaneni S., Small 2018, 14, 1702382; [DOI] [PubMed] [Google Scholar]
- 77b. Tadepalli S., Yim J., Madireddi K., Luan J., Naik R. R., Singamaneni S., Chem. Mater. 2017, 29, 6308–6314. [Google Scholar]
- 78. Murayama S., Kato M., Anal. Chem. 2010, 82, 2186–2191. [DOI] [PubMed] [Google Scholar]
- 79. Juul S., Iacovelli F., Falconi M., Kragh S. L., Christensen B., Frøhlich R., Franch O., Kristoffersen E. L., Stougaard M., Leong K. W., Ho Y.-P., Sørensen E. S., Birkedal V., Desideri A., Knudsen B. R., ACS Nano 2013, 7, 9724–9734. [DOI] [PubMed] [Google Scholar]
- 80. Hiraoka T., Hamachi I., Bioorg. Med. Chem. Lett. 2003, 13, 13–15. [DOI] [PubMed] [Google Scholar]
- 81. Endo M., Nakayama K., Kaida Y., Majima T., Angew. Chem. 2004, 116, 5761–5763; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004, 43, 5643–5645. [DOI] [PubMed] [Google Scholar]
- 82. Chang C.-y., Fernandez T., Panchal R., Bayley H., J. Am. Chem. Soc. 1998, 120, 7661–7662. [Google Scholar]
- 83. Chou C., Young D. D., Deiters A., Angew. Chem. 2009, 121, 6064–6067; [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 5950–5953. [DOI] [PubMed] [Google Scholar]
- 84. Edwards W. F., Young D. D., Deiters A., ACS Chem. Biol. 2009, 4, 441–445. [DOI] [PubMed] [Google Scholar]
- 85. Loudwig S., Nicolet Y., Masson P., Fontecilla-Camps J. C., Bon S., Nachon F., Goeldner M., ChemBioChem 2003, 4, 762–767. [DOI] [PubMed] [Google Scholar]
- 86. Thompson S., Fawcett M. C., Pulman L. B., Self C. H., Photochem. Photobiol. Sci. 2006, 5, 326–330. [DOI] [PubMed] [Google Scholar]
- 87. Fang W., Liu S., Tan C., Li A., Tan Y., Jiang Y., Anal. Methods 2018, 10, 2205–2210. [Google Scholar]
- 88. McLean M. A., Rajfur Z., Chen Z., Humphrey D., Yang B., Sligar S. G., Jacobson K., Anal. Chem. 2009, 81, 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]