

Abstract
Alcohol dehydrogenases are of high interest for stereoselective syntheses of chiral building blocks such as 1,2‐diols. As this class of enzymes requires nicotinamide cofactors, their application in biotechnological synthesis reactions is economically only feasible with appropriate cofactor regeneration. Therefore, a co‐substrate is oxidized to the respective co‐product that accumulates in equal concentration to the desired target product. Co‐product removal during the course of the reaction shifts the reaction towards formation of the target product and minimizes undesired side effects. Here we describe an atom efficient enzymatic cofactor regeneration system where the co‐product of the ADH is recycled as a substrate in another reaction set. A 2‐step enzymatic cascade consisting of a thiamine diphosphate (ThDP)‐dependent carboligase and an alcohol dehydrogenase is presented here as a model reaction. In the first step benzaldehyde and acetaldehyde react to a chiral 2‐hydroxy ketone, which is subsequently reduced by to a 1,2‐diol. By choice of an appropriate co‐substrate (here: benzyl alcohol) for the cofactor regeneration in the alcohol dehydrogenases (ADH)‐catalyzed step, the co‐product (here: benzaldehyde) can be used as a substrate for the carboligation step. Even without any addition of benzaldehyde in the first reaction step, this cascade design yielded 1,2‐diol concentrations of >100 mM with optical purities (ee, de) of up to 99%. Moreover, this approach overcomes the low benzaldehyde solubility in aqueous systems and optimizes the atom economy of the reaction by reduced waste production. The example presented here for the 2‐step recycling cascade of (1R,2R)‐1‐phenylpropane‐1,2‐diol can be applied for any set of enzymes, where the co‐products of one process step serve as substrates for a coupled reaction.
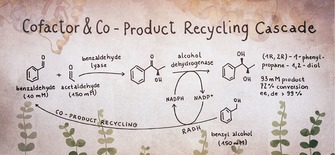
Keywords: Enzymatic cascade reaction; vicinal 1,2-diols; cofactor and co-product recycling cascade; alcohol dehydrogenase; carboligase; chiral 2-hydroxy ketone
Introduction
Chiral 1,2‐diols find broad application as building blocks for pharmaceuticals, agrochemicals, and chemical catalysts.1 NAD(P)H‐dependent alcohol dehydrogenases (ADH) can catalyze synthesis of 1,2‐diols from prochiral 2‐hydroxy ketones in high stereoselectivities, mild reaction conditions, and without toxic reagents1f, 3 compared to classical chemical methods such as Sharpless asymmetric dihydroxylation, epoxide hydrolysis and asymmetric aldol condensation.2 Various 1,2‐diols are accessible by an enzymatic 2‐step cascade starting with the carboligation of aldehydes catalyzed by thiamine diphosphate (ThDP)‐dependent enzymes towards different chiral 2‐hydroxy ketones.3c In the second step these 2‐hydroxy ketones are reduced by alcohol dehydrogenases (ADH) yielding vicinal chiral 1,2‐diols.1f, 1g, 3a Such alcohol dehydrogenases require expensive nicotinamide cofactors (NAD(P)H) to deliver the necessary reduction equivalents. Therefore in situ regeneration of such cofactors is decisive for economic feasibility of these biocatalytic processes.4 According to the classification of Chenault and Whitesides5 there are five general methods for redox cofactor regeneration including biological, chemical, electrochemical, photochemical, and enzymatic methods. Enzymatic cofactor regeneration methods are most frequently used4b, 6 and can be further subdivided into substrate‐coupled and enzyme‐coupled approaches. In enzyme‐coupled approaches two different enzymes are involved, one in the main reaction and the other catalyzes the cofactor regeneration reaction. In the substrate‐coupled approach, the main reaction and the cofactor regeneration is catalyzed by the same enzyme, but requires a co‐substrate and generates co‐products. This concept has the advantage of avoiding an additional enzyme therefore keeping production costs in industrial scale applications lower.7 Some biocatalytic processes implemented via substrate‐coupled cofactor regeneration require removal of the co‐product to shift the equilibrium or to avoid negative effects on enzyme activity and stability.4, 8
No matter if enzyme‐ or substrate‐coupled cofactor regeneration is chosen, the co‐substrate is usually used in excess to shift the equilibrium of the main reaction appropriately. In contrast to formate dehydrogenases, which generate CO2 as a co‐product by oxidation of formate and that CO2 leaves the liquid phase, the resulting co‐products produced by glucose dehydrogenases (D‐gluconate), and ADHs (e. g. acetone, acetaldehyde) accumulate in the reaction mixture and must be separated from the product either during the course of the reaction to shift the reaction equilibrium or during downstream processing. The use of “smart co‐substrates”, like 1,4‐butandiol or 1,6‐hexandiol enables the use of only a 0.5‐fold molar concentration relative to the main substrate.9 However, here also co‐products are produced, which increase the waste‐stream and must be separated during downstream processing. In so‐called recycling cascades, also known as “closed‐loop” or “self‐sufficient” cascades,10 oxidizing and reducing steps are directly coupled and neither co‐substrate nor further regenerating enzymes are required. This concept has the potential to significantly improve the atom economy and reduce the waste stream. However, this beneficial concept only functions if NAD(P)H‐based oxidation/reduction can be coupled red/ox‐neutral in a desired cascade setup.
To broaden the operational window of smart recycling cascades processes, we present a cascade where the cofactor NADPH is regenerated in a way that the co‐product is a substrate for a coupled reaction step. This recycling cascade was recently applied by our group in micro‐aqueous reaction systems using whole lyophilized cells.11 In this study here, we show its general application in buffered systems using cell free catalyst formulations. Further, we studied more deeply the reaction parameters necessary in such buffered reaction environment and identified limiting factors (such as kinetics and thermodynamic equilibrium) in order to get optimal conversion. We demonstrate this concept using a 2‐step cascade for the synthesis of (1R,2R)‐1‐phenylpropane‐1,2‐diol (PPD) (Scheme 1). Starting from benzaldehyde and acetaldehyde, the thiamine diphosphate (ThDP)‐dependent benzaldehyde lyase from Pseudomonas fluorescens (PfBAL) gives access to the 2‐hydroxy ketone (R)‐2‐hydroxy‐1‐phenylpropane‐1‐one, short (R)‐2‐HPP, with high regio‐ and enantioselectivity (ee > 99%).12 For the subsequent reduction to the respective 1,2‐diol (1R,2R)‐PPD the alcohol dehydrogenase from Ralstonia sp. (RADH) is well suited to reduce (R)‐2‐HPP with excellent diastereoselectivities and high activities (up to ∼360 U mg−1).3b
Scheme 1.
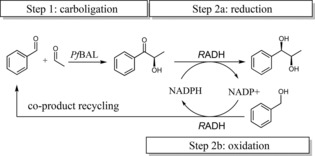
1‐Pot 2‐step co‐product recycling cascade for the synthesis of (1R,2R)‐1‐phenylpropane‐1,2‐diol. (1R,2R)‐1‐phenylpropane‐1,2‐diol (PPD) is formed by combining a carboligation step (1) and an oxidoreduction step (2a). By using benzyl alcohol as a co‐substrate for NADPH cofactor regeneration, the co‐product benzyl alcohol (step 2b) can be recycled as a substrate in the carboligation step (1). PfBAL=benzaldehyde lyase from Pseudomonas fluorescens, RADH=alcohol dehydrogenase from Ralstonia sp.
Results and Discussion
Based on previously optimized reaction conditions for both enzymes, PfBAL6a, 13 and RADH,3b, 14 we here focused on a feasibility study of the co‐product recycling concept towards chiral 1,2‐diols in buffered systems. Although in principle, benzyl alcohol (via benzaldehyde) and ethanol (via acetaldehyde) could theoretically be used as co‐substrates in this recycling cascade concept, we focused on benzyl alcohol due to the higher activity of RADH towards large cyclic and aromatic alcohols compared to small aliphatic substrates.3b, 14
A) Benzyl Alcohol for Cofactor Regeneration
Since the PfBAL‐catalyzed reaction was already well characterized in buffered reaction systems,12b, 13 the RADH reaction was initially optimized separately with the special focus on the application of benzyl alcohol as a co‐substrate. As demonstrated earlier, RADH exhibits different non‐overlapping pH‐optima for reduction (pH 6.0–9.5) and oxidation (pH 10.0–11.5).14 Furthermore, stability tests indicated that RADH is rather stable between pH 5.5 and pH 8.0 (half‐life: ∼60–70 h), whereas higher pH‐values induced progressive inactivation,14 making the application of RADH for the simultaneous oxidation of benzy alcohol and the reduction of 2‐HPP specifically challenging.
Based on these previous results, the RADH‐catalyzed reduction rac‐2‐HPP3b, 14 was tested between pH 8.0 to 9.0 (Figure 1‐A) for determining optimal reaction conditions for the cascade setup. The results indicate 1.7‐fold higher conversions of rac‐2‐HPP to the respective 1,2‐diols (1R,2R)‐ and (1R,2 S)‐PPD at pH 9.0 compared to pH 8.0 (35% for pH 8 and 59% for pH 9). The applied buffer species (TEA‐HCl or TRIS‐HCl) had no influence at pH 8.0, resulting in 35% conversion in both cases (Figure 1‐A). Thus, a concentration range of 10–250 mM benzyl alcohol was tested to convert 10 mM of rac 2‐HPP (Figure 1‐B). Higher concentrations were not investigated in order to avoid the formation of a two‐phase system due to the limited solubility of benzyl alcohol in aqueous media. Although RADH accepts both enantiomers of rac‐2‐HPP3b, 14 in none of these experiments conversion of rac‐2‐HPP exceeded 59% (Figure 1‐B). This could be partly due to the fact that the (R)‐enantiomer is 24‐fold faster converted compared to the (S)‐enantiomer.3b Additionally, deactivation of RADH under reaction conditions could occur. To test whether the RADH concentration was limiting the conversion, it was increased from 0.05 mg/mL to 0.30 mg/mL, which indeed resulted in almost full conversion (98.5%; Figure 1‐C).
Figure 1.
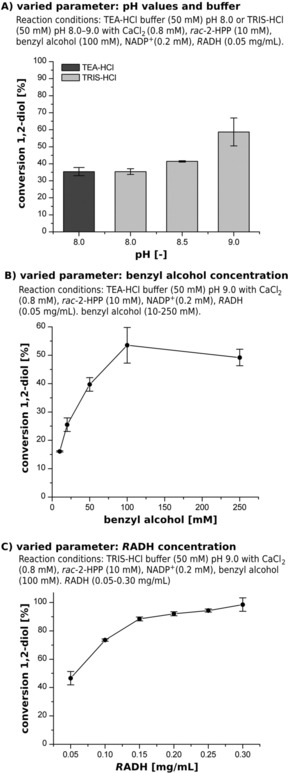
Reaction optimization for RADH‐catalyzed reduction of 2‐HPP reactions with benzyl alcohol as co‐substrate for cofactor regeneration. A) Identification of optimal pH for the oxidoreduction. B) Optimization of benzyl alcohol concentration. C) Optimization of the RADH concentration. All reactions were carried out in duplicates at 20 °C as endpoint‐determination for 24 h under constant shaking (500 rpm) in aliquots (1 mL each) in closed glass vials.
These results prove that benzyl alcohol is a suitable co‐substrate for cofactor regeneration in RADH‐catalyzed reductions for single step reactions of 2‐HPP to the 1,2‐diol.
B) 2‐Step Cofactor and Co‐Product Recycling Cascade
After identification of optimal reaction conditions for the RADH‐catalyzed reduction of HPP, the 1‐pot 2‐step cascade was investigated by combining the carboligation and the reduction step with in‐situ benzaldehyde removal (see Scheme 1). The cascade reaction was evaluated using two starting conditions (see Figure 2 and SI Figure S1), which differed in the initial concentrations of benzaldehyde. In route A the one‐pot recycling cascade was started with benzaldehyde (10 mM), acetaldehyde (150 mM), benzyl alcohol (120 mM) and NADP+ (in different concentrations). Small concentrations of benzaldehyde were added in this route to detect weather starting material of benzaldehyde is important to start the cascade reaction. In route B no benzaldehyde was added at all, so that the benzaldehyde (required for the first carboligation step) had to be completely generated by recycling through oxidation of benzyl alcohol. Both cascades were optimized with respect to pH and the concentrations of NADP+ and RADH, respectively (Figure 2).
Figure 2.
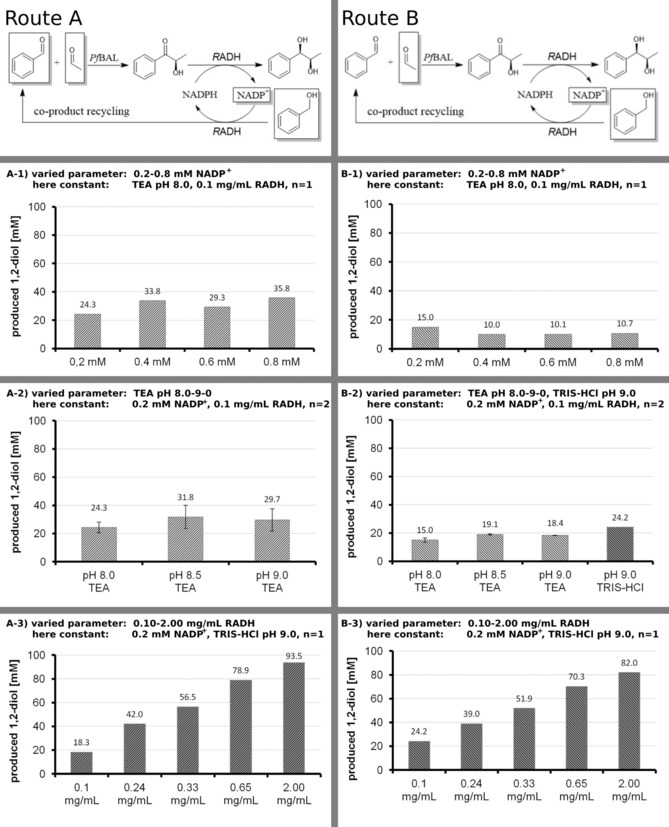
Optimization of the 1‐pot 2‐step cofactor and co‐product recycling cascade based on route A and route B. Constant parameter: 50 mM buffer, 0.8 mM CaCl2, 2.5 mM MgSO4, 0.15 mM ThDP, 0.05 mg/mL PfBAL. Varied parameter: pH: 8.0–9.0, NADP+: 0.2–0.8 mM, TEA or TRIS‐HCl and RADH: 0.1–2.0 mg/mL. Route A: benzaldehyde (10 mM), acetaldehyde (150 mM), benzyl alcohol (120 mM). Route B: acetaldehyde (150 mM), benzyl alcohol (120 mM). All reactions were carried out in a 1 mL at 20 °C with 150 rpm shaking and measured within a maximum of 240 h.
Conversions were calculated based on the total amount of aromatic substrate (benzaldehyde) and co‐substrate (benzyl alcohol), which finally gave the 1,2‐diol. Thus, 100% conversion refers to 130 mM 1,2‐diol for route A (10 mM benzaldehyde +120 mM benzyl alcohol) and 120 mM for route B (120 mM benzyl alcohol), respectively. As a consequence, 1,2‐diol concentrations greater than the initial benzaldehyde concentration (route A: 10 mM benzaldehyde; route B: 0 mM) are indicative for the successful co‐product (benzaldehyde) recycling in the first reaction step (catalyzed by PfBAL).
NADP+ Concentration
In order to identify the optimal cofactor concentration, NADP+ concentration was varied within a range of 0.2–0.8 mM while the other parameter for route A and route B were kept constant (see Figure 2). Whereas the conversions were about three‐fold higher following route A (see Figure 2; A‐1) compared to route B (see Figure 2; B‐1), the influence of the NADP+ concentration was almost negligible. In both cases, NADP+ concentrations >0.2 mM had no influence on amount of formed 1,2‐diol product (Figure 2; A‐1 and B‐1) or even on the reaction velocity (see SI Chapter 3 Figure S3–S5).
Optimization of pH and Buffer
As previously demonstrated, RADH shows different pH optima for the reduction and oxidation reaction, with pH 9.0 being a compromise for the simultaneous oxidation and reduction reactions.14 The challenging task for the co‐product recycling cascade was to combine these requirements with conditions that are also optimal for the PfBAL12b, 13 and the RADH14 enzymes in the 1‐pot 2‐step cascade. Thus, we investigated the cascade within a pH range of 8.0, 8.5 and 9.0 using TEA‐buffer. At pH 8.5–9.0, slightly higher 1,2‐diol values were observed in both routes A and B. Although the trend was not clear, higher pH values were seen as beneficial within the cascade due to the activity/stability profiles of both enzymes PfBAL and RADH.12b, 13, 14 Additionally, the TEA‐buffer (buffer range pH 7.0–8.3) was compared to TRIS‐HCl (buffer range pH 7.5–9.0) at pH 9.0 for route A. As slightly better results were obtained using TRIS buffer, which additionally has a higher buffer capacity at pH 9.0, this buffer was used for the subsequent studies.
RADH Concentration
To investigate a limitation caused by reaction velocity and/or enzyme stability, the RADH enzyme concentration was varied form 0.10 mg/mL to 2.00 mg/mL. Here, we found a strong influence of the RADH concentration on the amount of the formed 1,2‐diol product in both routes A and B (Figure 2; A‐3 and B‐3). The highest tested RADH concentration resulted in a final product concentration of 82.8 mM (Figure 2, A‐3) via route A and 75.7 mM via route B (Figure 2, B‐3).
Proof‐of‐Principle Successfully Demonstrated
In all shown cases of the optimization using route A (Figure 2; entry A‐1 and B‐1), higher 1,2‐diol concentrations than the initially added benzaldehyde concentration (10 mM benzaldehyde) were found. This clearly demonstrates that the recycling of the benzaldehyde formed as a co‐product via the RADH‐catalyzed oxidation of benzyl alcohol was successful in the first reaction step catalyzed by PfBAL (Figure 3). The highest value of 93.5 mM indicates that at least 83.5 mM 1,2‐diol was formed via co‐product recycling. Here an overall conversion (relatative to the benzaldehyde and benzyl alcohol) of 72% was reached. Moreover, even if no benzaldehyde was added initially (route B) the 1,2‐diol is formed to concentrations of 82.0 mM. This corresponds to 68% conversion of the benzyl alcohol.
Figure 3.
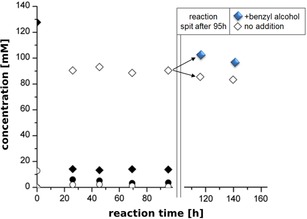
Conversion curve of the 2‐step recycling cascade with 3.75 mg mL−1 RADH concentration. Reaction conditions: TRIS‐HCl buffer (50 mM) supplemented with CaCl2 (0.8 mM), MgSO4 (2.5 mM), ThDP (0.15 mM), pH 9.0, NADP+ (0.8 mM), benzaldehyde (10 mM), acetaldehyde (150 mM), benzyl alcohol (120 mM), PfBAL (0.05 mg/mL), RADH (3.75 mg/mL). Symbols: ⧫ benzyl alcohol, ○benzaldehyde, • (R)‐2‐HPP, ◊1,2‐diol. Reactions were carried out at 20 °C with constant shaking (150 rpm). Samples were taken in defined time intervals. After 96 h the reaction solution was split and to one aliquot 50 mM fresh benzyl alcohol was added (⧫ 1,2‐diol) to another not (◊1,2‐diol) and the reaction further incubated at 20 °C with constant shaking (150 rpm).
Moreover, in all reactions with RADH concentrations <0.33 mg/mL, an optical purity of >97% (in some cases even >99%; see SI Chapter 3, Figure S3–S5) was reached for both route A and B. The optical impurities of <3% were caused by the isomers (1S,2R)‐ and (1R,2S)‐PPD. While the formation of the (1S,2R)‐isomer can be explained by the imperfect stereoselectivity of RADH, the formation of the (1R,2 S)‐diol is most likely caused by keto‐enol tautomerism of the 1,2‐diol. With higher RADH concentrations (>0.65 mg/mL) the optical purity dropped to 95–96%. In both routes (A, B) small amounts of benzoin (<1 mM) were formed as a side product of the PfBAL‐catalyzed carboligation step.
Limitations of the 1‐Pot 2‐Step Cascade
The maximal conversions obtained under optimized conditions did not exceed 72% (93.5 mM; route A), suggesting that full conversion might be hampered by an unfavourable thermodynamic equilibrium. Therefore, a rough estimation on the theoretical equilibrium constant was calculated (see SI, Chapter 4). According to these rough calculations, conversions up to 99% should be possible via route B starting from 120 mM benzyl alcohol and 150 mM acetaldehyde, whereas a significant lower theoretical total conversion of ∼90% was calculated for route A (SI Tab. S2). The difference in theoretical total conversion for route A (∼99%) and route B (∼90%) are based on the ratio of the aromatic substrates (benzyl alcohol+benzaldehyde) and acetaldehyde (150 mM). For route A the ratio is (130 mM/150 mM)=0.87 and for route B (120 mM/150 mM)=0.80.
Further increasing the RADH concentration to 3.75 mg mL−1 and NADP+ concentration to 0.8 mM did not result in full theoretical conversion. Here demonstrated for route A, 93.2 mM (Figure 3) 1,2‐diol was observed, corresponding to a conversion of 72%.
Considering kinetic limitations, we found that at this maximum conversion, the residual concentration of benzyl alcohol was 17 mM (Figure 3), which is in the range of the KM‐value of RADH for benzyl alcohol under the conditions tested (12.3±2.7 mM),14 Thus, the enzyme is not operating under Vmax conditions anymore when 72% conversion (route A) was reached. Indeed, the product concentration could be further increased to 102 mM by addition of further benzyl alcohol (50 mM) after 96 h (Figure 3; blue diamonds). Since thermodynamic parameter seems not to be the limiting factor, enzyme activity might hamper the reaction to go to theoretical maximal conversion of ∼90% (for route A).
To overcome such limitations in future, one option might be to continuously fed benzyl alcohol to the process to ensure maximal RADH activity in fed‐batch reactions. Another possibility would be to increase the operation window of both enzymes (PfBAL and RADH) with respect to their activity profile. In this part I of the paper we did not address the inactivation of the enzymes over time under process conditions, however this was considered in part II. Moreover, the addition of even higher enzyme loads is not beneficial for industrial application since the reaction process costs might be significantly increased. The results of part II of the publication17 indicate that an improvement of the enzyme stability for both PfBAL and RADH increases the conversion. Under similar conditions, the thermodynamically maximal conversion of ∼90% (for route A) was reached by maintaining high optical purities of >99% ee and >99% de.
Conclusion
We developed a step‐efficient recycling cascade for the highly stereo selective production of (1R,2R)‐1‐phenylpropane‐1,2‐diol starting from inexpensive benzaldehyde and acetaldehyde as substrates for the carboligation step and benzyl alcohol as co‐substrates for the cofactor regeneration step. To increase the atom‐efficiency, the co‐substrate (here: benzyl alcohol) was chosen such that the resulting co‐product (here: benzaldehyde) is used as a substrate in the carboligation step of the synthetic enzyme cascade. After optimization of the one‐pot 2‐step cascade, 93.5 mM (1R,2R)‐1‐phenylpropane‐1,2‐diol was produced starting with 10 mM benzaldehyde and 150 mM acetaldehyde by successful recycling of the co‐product benzyl alcohol (added initially: 120 mM) according to route A. Moreover, even without any addition of benzaldehyde 82 mM of the final product was formed with high stereoselectivity (>96–99% isomeric purity) in route B. By addition of 50 mM fresh benzyl alcohol, a product concentration of 102 mM was reached in this monophase batch reaction system. We could further demonstrate that the batch systems are limited by the activity and stability specifically of the RADH. An increase of the RADH concentration from 0.1 mg/mL to 2 mg/mL and keeping the concentration of PfBAL constant (0.05 mg/mL) resulted in an increase in conversion from about 15% to 72% (route A). At these highest conversion values, the benzyl alcohol concentration might cause kinetic limitations since the RADH is not vmax. Here, on the one hand a fed‐batch strategy might be useful to further increase the 1,2‐diol product concentration and to reach the theoretical conversion of ∼90% (route A) and >99% (route B). On the other hand, the activity and stability issue was solved in part II of the paper where ∼90% (for route A) was reached by maintaining high optical purities of >99% ee and >99% de.17
Our method describes a process strategy for more sustainable and eco‐friendly synthetic enzyme cascades employing nicotine amide red‐ox cofactor recycling without any waste production. This approach optimizes the atom economy of a reaction, reduces waste production and thus increases process economy can be applied for any set of enzymes, where the co‐products of one process step serve as substrates for another reaction step.
Experimental Section
Materials and Methods
Chemicals
All chemicals were of high analytical grade. Tris‐2‐(hydroxyethyl)amine (TEA) was purchased from Sigma (Steinheim, Germany), 2‐amino‐2‐hydroxymethylpropane‐1,3‐diol (TRIS) and calcium chloride (CaCl2 ⋅ 2 H2O) were from Merck (Darmstadt, Germany), benzyl alcohol and benzaldehyde from Sigma‐Aldrich (Steinheim, Germany), and acetaldehyde and magnesium sulphate (MgSO4) from Fluka (Steinheim, Germany). Cofactors: thiamine diphosphate (ThDP) and NADP+ were from AppliChem (Darmstadt, Germany) and Biomol (Hamburg, Germany), respectively. (R)‐2‐Hydroxy‐1‐phenylpropan‐1‐one ((R)‐2‐HPP)3a, 12a and rac‐2‐hydroxy‐1‐phenylpropan‐1‐one (rac‐2‐HPP)15 were synthesized as described in the literature.
Expression and Purification of the Recombinant Enzymes
Expression and purification of His‐tagged benzaldehyde lyase from Pseudomonas fluorescens (PfBAL) employing metal chelate affinity chromatography (Ni‐NTA column) was carried out as described elsewhere.12b Alcohol dehydrogenase from Ralstonia sp. (RADH) was expressed and purified by ion exchange chromatography (anion exchanger) as recently published.8
Determination of Protein Concentration
Protein concentration was determined by the Bradford method16 using bovine serum albumin as a standard (Sigma‐Aldrich, Steinheim, Germany).
A) Benzyl Alcohol for Cofactor Regeneration
Optimization of cofactor regeneration was carried out at a 1 mL scale in screwed glass vials equipped with a Teflon‐membrane in order to avoid evaporation of aldehydes. Reactions were conducted at 20 °C for 24 h with constant shaking of 500 rpm (Eppendorf thermomixer, Hamburg, Germany) and stopped by extraction with ethyl acetate (50% v v−1). All reactions were performed in duplicate. Conversions were determined using GC‐analysis based on a calibration with the formed product 1‐phenylpropane‐1,2‐diol. For instrumental analysis and origins of reference compounds see section Chiral analysis.
All reactions were conducted in TRIS‐HCl buffer (50 mM) supplemented with CaCl2 (0.8 mM), pH 9.0, if not stated otherwise. For biotransformations the following standard conditions were applied (if not stated otherwise): rac‐2‐HPP (10 mM), NADP+ (0.2 mM) and RADH (0.05 mg mL−1). To determine the optimal co‐substrate concentration benzyl alcohol (10 mM to 250 mM) was tested under standard conditions. To identify the optimal pH and buffer conditions, batch experiments were performed in TRIS‐HCl (50 mM, pH 8.0–9.0) and in TEA‐HCl (50 mM, pH 8.0) buffer systems, both supplemented with CaCl2 (0.8 mM). The optimal RADH concentration was tested by varying the enzyme concentration between 0.05–0.30 mg mL−1 under standard biotransformation conditions, using benzyl alcohol (100 mM) as co‐substrate.
B) 2‐Step cofactor and Co‐Product Recycling Cascade
Two different routes were studied differing in substrate supply strategy (Figure 2 and SI Figure S1). Depending on the mode, initial amounts of substrates and co‐substrates were varied. Route A: acetaldehyde (150 mM), benzaldehyde (10 mM), and benzyl alcohol (120 mM). Route B: acetaldehyde (150 mM) and benzyl alcohol (120 mM). All reaction were performed in 50 mM buffer, 0.8 mM CaCl2, 2.5 mM MgSO4, 0.15 mM ThDP, 0.05 mg/mL PfBAL. For NADP+ optimization, the NADP+ concentration was varied in the range of 0.2 mM to 0.8 mM. For pH optimization TEA‐HCl (pH 8.0–9.0) and TRIS‐HCl buffer systems (50 mM, pH 9.0) were tested. To find optimal RADH concentrations, catalyst loads from 0.10 mg mL−1 to 2.00 mg mL−1 were examined. For reaction details of the varied parameter see description Figure 2. All reactions were carried out at a 1 mL scale in screwed glass vessels equipped with a Teflon membrane at 20 °C under constant shaking (500 rpm). Samples (100 μL) were taken in defined time intervals for 240 h.
Determination of the Thermodynamic Equilibrium Constant
The equilibrium constant (Keq) for the carboligation reaction was calculated according to Formula 1:
| (1) |
The equilibrium constant for the oxidoreduction step was calculated according to Formula 2 and Formula 3:
| (2) |
| (3) |
Equilibrium constants were calculated using initial and final concentrations of all available compounds in the reaction vessel measured for the respective route. The theoretical overall conversions of the 2‐step recycling cascade were calculated with MATLAB (The MathWorks, Germany). For details concerning calculations and scripts see SI, Chapter 4.
Synthetic Cascade Reaction with 3.75 mg mL−1 RADH Concentration using Route A
Reactions were conducted under the following conditions: TRIS‐HCl buffer system (50 mM), pH 9.0, supplemented with CaCl2 (0.8 mM), MgSO4 (2.5 mM), ThDP (0.15 mM), benzyl alcohol (120 mM) and NADP+ (0.8 mM). Reactions were carried out at 20 °C with constant shaking (500 rpm) in closed glass vials. After 96 h additional fresh 50 mM benzyl alcohol was added to the reaction vessel. Samples were taken in defined time intervals and reaction progress was measured by GC and HPLC (see paragraph Chiral analysis).
Chiral Analysis – High Performance Liquid Chromatography (HPLC)
Concentrations of substrates (benzaldehyde and benzyl alcohol) and the intermediately formed (R)‐2‐HPP were determined by HPLC‐analysis. Therefore 10 μL sample (10‐times diluted in acetonitrile with 2‐methylbenzaldehyde as internal standard) was analyzed by chiral HPLC using a HiBar 250–4 LiChrosphere 100 RP‐8 (5 μm) column (Merck KGaA, Germany; flow: 1 mL min−1, temperature: 25 °C). As mobile phase, a gradient of 25–60% (v v−1) acetonitrile was used, mixed with ultrapure water (0–12 min: 25% (v v−1) acetonitrile, 12–13 min: gradient 25–60% (v v−1) acetonitrile, 13–20 min: 60% (v v−1) acetonitrile, 20–23 min: 60–25% (v v−1) acetonitrile, 23–25 min: 25% (v v−1) acetonitrile). Absorption was determined at 200 nm (benzyl alcohol) and 250 nm (2‐HPP and benzaldehyde) with the following retention times: Rt,(benzyl alcohol)=7.6 min, Rt,(2‐HPP)=10.5 min and Rt,(benzaldehyde)=16.2 min.
Chiral Analysis – Gas Chromatography (GC)
Final concentration and diastereomeric excess (de) of the cascade product (non‐derivatized stereoisomers of 1‐phenyl‐propane‐1,2‐diol) were determined via GC‐analysis. Therefore samples were extracted with ethyl acetate including 1‐dodecanol as internal standard. 5 μL of the obtained organic phase were analyzed by gas chromatography using a chiral CP‐Chirasil‐DEX CB (Varian, Germany) column (25 m×0.25 mm×0.25 μm) with a flame ionisation detector (FID) and hydrogen as carrier gas. The following GC‐program was applied: 140 °C injection temperature, isothermic run for 30 min. Retention times: Rt,(benzaldehyde)=3.1 min and Rt,(benzyl alcohol)=4.9 min. Retention times of the four stereoisomers of phenylpropan‐1,2‐diol: Rt,(1S,2S)=24.1 min, Rt,(1R,2R)=25.8 min, Rt,(1S,2R)=27.4 min, Rt,(1R,2S)=28.5 min. Detailed chemical syntheses of reference compounds and the assignment of absolute configuration are described elsewhere.3b
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Wolfgang Kroutil (University of Graz, Austria) for providing the radh gene and Dr. Christiane Claaßen for her help with the NMR data processing. This work was financed by the Marie Curie Initial Training Network “BIOTRAINS – a European biotechnology training network for the support of chemical manufacturing”, grant agreement no. 238531 and by the Helmholtz Association in frame of the W2/W3‐professor program (grant no. W2/W3‐110).
J. Kulig, T. Sehl, U. Mackfeld, W. Wiechert, M. Pohl, D. Rother, Adv. Synth. Catal. 2019, 361, 2607.
References
- 1.
- 1a. Kurina-Sanz M., Bisogno F. R., Lavandera I., Orden A. A., Gotor V., Adv. Synth. Catal. 2009, 351, 1842–1848; [Google Scholar]
- 1b. Agudo R., Roiban G.-D., Lonsdale R., Ilie A., Reetz M. T., J. Org. Chem. 2015, 80, 950–956; [DOI] [PubMed] [Google Scholar]
- 1c. Gala D., DiBenedetto D. J., Clark J. E., Murphy B. L., Schumacher D. P., Steinman M., Tetrahedron Lett. 1996, 37, 611–614; [Google Scholar]
- 1d. Wachtmeister J., Jakoblinnert A., Kulig J., Offermann H., Rother D., ChemCatChem 2014, 6, 1051–1058; [Google Scholar]
- 1e. Wachtmeister J., Jakoblinnert A., Rother D., Org. Process Res. Dev. 2016, 20, 1744–1753; [Google Scholar]
- 1f. Husain S. M., Stillger T., Dünkelmann P., Lödige M., Walter L., Breitling E., Pohl M., Bürchner M., Krossing I., Müller M., Romano D., Molinari F., Adv. Synth. Catal. 2011, 353, 2359–2362; [Google Scholar]
- 1g. Muschallik L., Molinnus D., Bongaerts J., Pohl M., Wagner T., Schöning M. J., Siegert P., Selmer T., J. Biotechnol. 2017, 258, 41–50; [DOI] [PubMed] [Google Scholar]
- 1h. Nestl B. M., Hammer S. C., Nebel B. A., Hauer B., Angew. Chem. Int. Ed. 2014, 53, 3070–3095. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Griffith J. C., Jones K. M., Picon S., Rawling M. J., Kariuki B. M., Campbell M., Tomkinson N. C. O., J. Am. Chem. Soc. 2010, 132, 14409–14411; [DOI] [PubMed] [Google Scholar]
- 2b. Mohan R. S., Gavardinas K., Kyere S., Whalen D. L., J. Org. Chem. 2000, 65, 1407–1413; [DOI] [PubMed] [Google Scholar]
- 2c. Ashikari Y., Nokami T., Yoshida J.-i., Org. Lett. 2012, 14, 938–941; [DOI] [PubMed] [Google Scholar]
- 2d. Notz W., List B., J. Am. Chem. Soc. 2000, 122, 7386–7387; [Google Scholar]
- 2e. Torres C. C., Campos C. H., Fierro J. L. G., Reyes P., Ruiz D., J. Mol. Catal. A 2014, 392, 321–328; [Google Scholar]
- 2f. Sharpless K. B., Amberg W., Bennani Y. L., Crispino G. A., Hartung J., Jeong K. S., Kwong H. L., Morikawa K., Wang Z. M., J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar]
- 3.
- 3a. Kihumbu D., Stillger T., Hummel W., Liese A., Tetrahedron: Asymmetry 2002, 13, 1069–1072; [Google Scholar]
- 3b. Kulig J., Simon R. C., Rose C. A., Husain S. M., Häckh M., Lüdeke S., Zeitler K., Kroutil W., Pohl M., Rother D., Catal. Sci. Technol. 2012, 2, 1580–1589; [Google Scholar]
- 3c. Pohl M., Dresen C., Beigi M., Müller M., and Acyloin Condensations Benzoin, in Enzyme Catalysis in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, doi:10.1002/9783527639861.ch22. [Google Scholar]
- 4.
- 4a. Kara S., Schrittwieser J. H., Hollmann F., Ansorge-Schumacher M. B., Appl. Microbiol. Biotechnol. 2014, 98, 1517–1529; [DOI] [PubMed] [Google Scholar]
- 4b. Weckbecker A., Gröger H., Hummel W., in Biosystems Engineering I: Creating Superior Biocatalysts (Eds.: C. Wittmann, R. Krull), Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 195–242; [Google Scholar]
- 4c. Josa-Culleré L., Lahdenperä A. S. K., Ribaucourt A., Höfler G. T., Gargiulo S., Liu Y.-Y., Xu J.-He, Cassidy J., Paradisi F., Opperman D. J., Hollmann F., Paul C. E., Catalysts 2019, 9, 207–218. [Google Scholar]
- 5. Chenault H. K., Whitesides G. M., Appl. Biochem. Biotechnol. 1987, 14, 147–197. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wichmann R., Vasic-Racki D., in Technology Transfer in Biotechnology: From lab to Industry to Production (Ed.: U. Kragl), Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 225–260; [Google Scholar]
- 6b. Hollmann F., Arends I. W. C. E., Buehler K., ChemCatChem 2010, 2, 762–782. [Google Scholar]
- 7.
- 7a. Bastos F. d. M., dos Santos A. G., Jones J., Oestreicher E. G., Pinto G. F., Paiva L. M. C., Biotechnol. Tech. 1999, 13, 661–664; [Google Scholar]
- 7b. Bastos F. M., França T. K., Machado G. D. C., Pinto G. F., Oestreicher E. G., Paiva L. M. C., J. Mol. Catal. B 2002, 19–20, 459–465; [Google Scholar]
- 7c. Röthig T. R., Kulbe K. D., Bückmann F., Carrea G., Biotechnol. Lett. 1990, 12, 353–356; [Google Scholar]
- 7d. Peters J., Zelinski T., Minuth T., Kula M.-R., Tetrahedron: Asymmetry 1993, 4, 1683–1692; [Google Scholar]
- 7e. Daußmann T., Rosen T. C., Dünkelmann P., Eng. Life Sci. 2006, 6, 125–129. [Google Scholar]
- 8.
- 8a. Julliard M., Le Petit J., Ritz P., Biotechnol. Bioeng. 1986, 28, 1774–1779; [DOI] [PubMed] [Google Scholar]
- 8b. Abu R., Woodley J. M., ChemCatChem 2015, 7, 3094–3105. [Google Scholar]
- 9.
- 9a. Kara S., Spickermann D., Schrittwieser J. H., Leggewie C., van Berkel W. J. H., Arends I. W. C. E., Hollmann F., Green Chem. 2013, 15, 330–335; [Google Scholar]
- 9b. Bornadel A., Hatti-Kaul R., Hollmann F., Kara S., ChemCatChem 2015, 7, 2442–2445. [Google Scholar]
- 10.
- 10a. Sehl T., Hailes H. C., Ward J. M., Wardenga R., von Lieres E., Offermann H., Westphal R., Pohl M., Rother D., Angew. Chem. Int. Ed. 2013, 52, 6772–6775; [DOI] [PubMed] [Google Scholar]
- 10b. Simon R. C., Richter N., Busto E., Kroutil W., ACS Catal. 2014, 4, 129–143; [Google Scholar]
- 10c. Wachtmeister J., Rother D., Curr. Opin. Biotechnol. 2016, 42, 169–177; [DOI] [PubMed] [Google Scholar]
- 10d. Schrittwieser J. H., Velikogne S., Hall M., Kroutil W., Chem. Rev. 2018, 118, 270–348; [DOI] [PubMed] [Google Scholar]
- 10e. Gupta P., Mahajan N., New J. Chem. 2018, 42, 12296–12327. [Google Scholar]
- 10f. Liu S., Zhang X., Liu F., Xu M., Yang T., Long M., Zhou J., Osire T., Yang S., Rao Z., ACS Synth. Biol. 2019, DOI: 10.1021/acssynbio.8b00364 [DOI] [PubMed] [Google Scholar]
- 11. Oeggl R., Maßmann T., Jupke A., Rother D., ACS Sustainable Chem. Eng. 2018, 6, 11819–11826. [Google Scholar]
- 12.
- 12a. Demir A. S., Pohl M., Janzen E., Müller M., J. Chem. Soc. Perkin Trans. 1 2001, 633–635; [Google Scholar]
- 12b. Janzen E., Müller M., Kolter-Jung D., Kneen M. M., McLeish M. J., Pohl M., Bioorg. Chem. 2006, 34, 345–361. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. de María P. Dominguez, Stillger T., Pohl M., Wallert S., Drauz K., Gröger H., Trauthwein H., Liese A., J. Mol. Catal. B 2006, 38, 43–47; [Google Scholar]
- 13b. Stillger T., Pohl M., Wandrey C., Liese A., Org. Process Res. Dev. 2006, 10, 1172–1177. [Google Scholar]
- 14. Kulig J., Frese A., Kroutil W., Pohl M., Rother D., Biotechnol. Bioeng. 2013, 110, 1838–1848. [DOI] [PubMed] [Google Scholar]
- 15. Chen C., Feng X., Zhang G., Zhao Q., Huang G., Chem. Informationsdienst 2009, 40. [Google Scholar]
- 16. Bradford M. M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 17. Jäger V. D., Piqueray M., Seide S., Pohl M., Wiechert W., Jaeger K.-E., Krauss U., Adv. Synth. Catal. 2019, 361, DOI: 10.1002/adsc.201900189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary