

Abstract
Key features of acute pancreatitis include excess cellular Ca2+ entry driven by Ca2+ depletion from the endoplasmic reticulum (ER) and subsequent activation of store-operated Ca2+ entry (SOCE) channels in the plasma membrane. In several cell types including pancreatic acinar, stellate cells (PaSC) and immune cells, SOCE is mediated via channels composed primarily of Orai1 and stromal interaction molecule 1 (STIM1). CM4620, a selective Orai1 inhibitor, prevents Ca2+ entry in acinar cells. This study investigates the effects of CM4620 in preventing or reducing acute pancreatitis features and severity. We tested the effects of CM4620 on SOCE, trypsinogen activation, acinar cell death, activation of NFAT and NF-κB, and inflammatory responses in ex-vivo and in vivo rodent models of acute pancreatitis and human pancreatic acini. We also examined whether CM4620 inhibited cytokine release in immune cells, fibro-inflammatory responses in PaSC, and oxidative burst in neutrophils, all cell types participating in pancreatitis. CM4620 administration to rats by IV infusion starting 30 min after induction of pancreatitis significantly diminished pancreatitis features including pancreatic edema, acinar cell vacuolization, intrapancreatic trypsin activity, cell death signaling and acinar cell death. CM4620 also decreased myeloperoxidase activity and inflammatory cytokine expression in pancreas and lung tissues, fMLF peptide-induced oxidative burst in human neutrophils, and cytokine production in human peripheral blood mononuclear cells (PBMC) and rodent PaSC, indicating that Orai1/STIM1 channels participate in the inflammatory responses of these cell types during acute pancreatitis. These findings support pathologic Ca2+ entry-mediated cell death and proinflammatory signaling as central mechanisms in acute pancreatitis pathobiology.
Keywords: Orai channels, SOCE, acute pancreatitis, acinar cell
Introduction
Ca2+ is a ubiquitous intracellular messenger involved in a plethora of cellular processes including protein secretion and exocytosis, endoplasmic reticulum (ER) and mitochondrial function, autophagy, cell growth and proliferation, and regulation of cell death and inflammation (Berridge, et al., 2000). Many physiological processes are linked to transient, limited elevations of Ca2+ concentrations in the cytosolic compartment ([Ca2+]C), while sustained elevated [Ca2+]C can lead to cellular pathology. In pancreatic acinar cells, physiologic neurohumoral stimulation causes relatively small amounts of Ca2+ to be released into the cytoplasm from ER stores, typically in an oscillatory pattern; these limited increases in [Ca2+]C mediate secretion of stored digestive enzymes and other cellular processes (Gerasimenko, et al., 2014a). In contrast, supra-physiological stimulation induces more profound discharge of ER Ca2+ stores, producing a Ca2+ depleted state of the stores and activation of plasma membrane store-operated Ca2+ entry (SOCE) channels, enabling Ca2+ to enter the cytoplasm from which the stores may be refilled (Pandol, et al., 1987, Muallem, et al., 1988a, b, Pandol, et al., 1994, Voronina, et al., 2004, Prakriya, et al., 2006, Sutton, et al., 2008, Kim, et al., 2009, Gerasimenko, et al., 2013, Gerasimenko, et al., 2014b, Wen, et al., 2015). Importantly, pancreatitis stimuli that cause sustained and marked ER Ca2+ depletion lead to massive Ca2+ influx via SOCE channels and sustained increases in [Ca2+]c, overloading cellular capacity to buffer Ca2+. Examples of such pathologic stimuli include hyperstimulation with cholecystokinin (CCK) or CCK analogues such as cerulein, as well as high concentrations of bile salts and ethanol metabolites (Criddle, et al., 2004, Criddle, et al., 2007, Mukherjee, et al., 2008, Gukovsky, et al., 2011). Such disturbances in Ca2+ mobilization and abnormally sustained high [Ca2+]c are associated with mitochondrial Ca2+ overload, abnormal intracellular trypsinogen activation, acinar cell death and pancreatic inflammation, all manifestations of acute pancreatitis (AP) (Shalbueva, et al., 2013, Mukherjee, et al., 2015).
Several years ago, SOCE channels were associated with Ca2+ release-activated Ca2+ (CRAC) currents. CRAC currents or channels were found in several cell types, but the molecular mechanisms underlying them remained elusive until the identification of Orai proteins as the pore-forming unit of CRAC channels in the plasma membrane and STIM1 as the main ER calcium sensor regulating activation of the Orai channels (Liou, et al., 2005, Roos, et al., 2005, Vig, et al., 2006, Zhang, et al., 2006). Orai proteins, the best characterized being Orai1 and Orai2, reside in the plasma membrane. Upon marked decrease of Ca2+ levels in the ER lumen, STIM1 activates and oligomerizes, with subsequent STIM-Orai engagement and activation of Orai1 channel initiating Ca2+ entry (Hooper and Soboloff, 2015). These channels are not voltage dependent, display high selectivity for Ca2+ and are modulated by intracellular Ca2+ (Hogan and Rao, 2015, Hooper and Soboloff, 2015). Orai1 and STIM1 are highly expressed in immune cells and pancreatic acinar cells (Zhang, et al., 2006, Cahalan, et al., 2007, Lur, et al., 2009, Hong, et al., 2011, Dingsdale, et al., 2012). More recently, CRAC channels were pharmacologically identified in pancreatic stellate cells (PaSC) (Gryshchenko, et al., 2016). All three of these cell types are believed to be involved in the mechanisms of initiation and progression of acute and recurrent acute pancreatitis.
Recent studies have shown that the small molecule Orai1 inhibitor, GSK-7675A and CM-128 (aka CM4620)(Stauderman, 2018), attenuate cell death responses to toxic stimuli such as bile acids in pancreatic acinar cells and ameliorate AP severity in experimental mouse models (Gerasimenko, et al., 2013, Gerasimenko, et al., 2014a, Voronina, et al., 2015, Wen, et al., 2015). Thus, Orai1 inhibitors have emerged as potential therapeutic agents for pancreatitis and as useful agents to investigate details of the pathologic events downstream of SOCE that lead to pancreatitis. In the present study, we sought to examine the efficacy of CM4620 ex vivo and then, administered by a clinically relevant route, to reduce acinar cell and pancreatic damage in various models of pancreatitis, and to further explore inflammatory pathways associated with SOCE in acinar cells, immune cells and PaSC.
Methods
Reagents
CM4620 was obtained from CalciMedica (La Jolla, CA). All salts, DMSO, N-Formyl-Met-Leu-Phe (fMLF peptide, #F3506), Phorbol 12-Myristate 13-Acetate (PMA; #P1585), Propidium iodide (PI, #81845), Dexamethasone (#D2915) and Lipopolysaccharide (LPS, from E. Coli, serotype 024:B6, #L8274) were obtained from Sigma-Aldrich (Saint Louis, MO). Cholecystokinin octapeptide (CCK; #1166) was purchased from Tocris (Bio-Techne Corporation; Minneapolis, MN); and Carbachol (CCh, #212385) from MilliporeSigma (Burlington, MA); Taurolithocholic acid 3-sulfate (TLCS, #T009115) from Toronto Research Chemicals (ON, Canada); and Fura-2 acetoxymethyl ester (fura-2 AM; #F1201) from ThermoFisher Scientific (Waltham, MA).
Pancreas tissue digestion for acinar cell isolation was performed using collagenase (#LS00–5273; CLSPA; Worthington Biochemical Corporation, Lakewood, NJ), bovine serum albumin fraction V (#03116956001; Roche Diagnostic, Indianapolis, IN), and soybean trypsin inhibitor (T9003; Sigma-Aldrich). Cell culture reagents include: 199 medium (for culturing primary acinar cells; #12340–030), F-12K medium (for culturing AR42J cells; #30–2004); DMEM/F12 medium (for culturing pancreatic stellate cells; #11330–032); L-Glutamine (#25030–081) and trypsin- EDTA (#25200056) from ThermoFisher Scientific (Waltham, MA); antibiotics/antimycotics (1% Penicillin-Streptomycin; #25030–081) and fetal bovine serum (FBS; #FB11) from Omega Scientific (Tarzana, CA); dexamethasone (#D2915) from Sigma-Aldrich.
The following antibodies were used for Western blotting analysis: cleaved Caspase-3 (#9661); CHOP (#5554); PARP (#9542), ERK1/2 (p44/42 MAPK; #9102), and corresponding HRP-linked secondary antibodies were from Cell Signaling Technology (Danvers, MA). SuperSignal™ West Pico (or Femto). Chemiluminescent Substrate reagents (#34080 and 34094) were from ThermoFisher Scientific. All chemicals and kits were used according to the manufacturer’s recommendations, unless otherwise indicated.
Rat acute cerulein pancreatitis model with CM4620 intravenous administration
Male rats (250–290 g) with established chronic in-dwelling jugular vein catheters were purchased from Envigo (Placentia, CA). Surgical protocols regarding jugular vein catheterization procedures and post-operative treatments are available at the Envigo website https://www.envigo.com/products-services/research-models-services/services/surgical-services/north-america-surgical-services/rat-catheterizations-options/. Rats were individually housed and maintained with a 12-h light-dark cycle with ad libitum intake of standard rat chow and sterilized tap water. Animal care and use followed National Institutes of Health’s Guide for the Care and Use of Laboratory Animals guidelines, and the Institutional Animal Care and Use Committee of the Cedars-Sinai Medical Center (CSMC) approved the protocol for this study (IACUC005207). Rats were acclimated to the CSMC environment for 1 week before experiments, and catheter patency was reestablished three days prior to and checked again immediately before the experiments. After 1-week acclimation, rats were randomly assigned to the following treatment groups: (1) saline (n = 4); (2) saline + 5mg/kg CM4620 (n = 4); (3) saline + 10mg/kg CM4620 (n = 4); (4) saline + 20mg/kg CM4620 (n = 4); (5) Cerulein (50μ/kg) (n = 12); (6) Cerulein + 5mg/kg CM4620 (n = 4); (7) Cerulein + 10mg/kg CM4620 (n = 4), and (8) Cerulein + 20mg/kg CM4620 (n = 4). CM4620 was prepared as an emulsion for intravenous administration using a vehicle admixture proprietary to CalciMedica; an identical placebo emulsion without CM4620 was also prepared as a control. Different dose concentrations of CM4620 were prepared by diluting the CM4620 emulsion with placebo emulsion to maintain the same dose volume. The cerulein AP model was carried out according to the schedules shown in Results. Cerulein (50 μg/kg; 4 hourly injections) or saline control were administered intraperitoneally (IP), with continuous intravenous (IV) 4-hour infusion of CM4620 (at 20, 10 or 5 mg/kg; 2.5 ml/kg/h) or placebo control initiated 30 min after the first cerulein or saline injection (therapeutic approach) or 2 h before the first cerulein or saline injection (preventive approach).
Animals were euthanized by CO2 inhalation followed by pneumothorax. This euthanasia procedure is in compliance with the current American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. Rats were euthanized 30 min (therapeutic approach) or 2 h after the IV infusion (preventive approach) and immediately blood samples were collected from the inferior vena cava and later processed for measurements of amylase and lipase activity (determined by Antech Diagnostics; Fountain Valley, CA). Pancreata and lung sections were removed, weighed and immediately frozen in liquid nitrogen and store at −80°C for subsequent studies, preserved in RNAlater (#AM7024; Thermofisher Scientific) for qPCR analysis, or fixed in 10 % buffered formalin phosphate for histological analysis.
Mouse acute cerulein pancreatitis model with CM4620 intraperitoneal administration
C57Bl/6 male mice (7-week-old) were purchased from Envigo (Placentia, CA). Animals were housed in groups of 3–5 mice per cage and maintained with a 12-h light-dark cycle with ad libitum intake of standard mouse chow and sterilized tap water. Animal care and use followed National Institutes of Health’s Guide for the Care and Use of Laboratory Animals guidelines, and the Institutional Animal Care and Use Committee of the Cedars-Sinai Medical Center (CSMC) approved the protocol for this study (IACUC007211). Mice were acclimated to the CSMC environment for 1 week before experiments and then randomly assigned to the following treatment groups: (1) saline (n = 3); (2) saline + 20mg/kg CM4620 (n = 3); (3) Cerulein (50μ/kg) (n = 4); (6) Cerulein + 20mg/kg CM4620 (n = 4). Mice were then subjected to an episode of cerulein AP as previously described (Lugea, et al., 2006). Briefly, 7 hourly IP injections of 50 μg/kg cerulein (or saline as control) were administered to mice; CM4620 (20 mg/kg) was coadministered IP at two time-points, i.e., 30 min before the first and fourth cerulein (or saline) injections; thus, the total dose was 40 mg/kg. The vehicle for CM4620 in this case was comprised of 75% polyethylene glycol (MW=400), 5% 2-hydroxypropyl-β-cyclodextrin and 20% water. Animals were euthanized by CO2 inhalation followed by cervical dislocation (following recommendations of the current American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals) 1 h after the last cerulein/saline injection, and blood and pancreas tissues were collected for analysis. Measurements included blood amylase and lipase and pancreas trypsin activity (see below).
Histological Analysis
Histological analysis was performed in formalin-fixed, paraffin embedded rodent pancreatic sections. Five μm sections were stained with H&E, and blinded scoring was performed for assessment of parenchymal structure, edema, acinar cell vacuolization and necrosis, and inflammatory cell infiltration as described elsewhere (Lugea, et al., 2006). Scoring system was as follows: Edema: “absent” (0), “low” (1), “moderate” (2) or “extensive” (3); Necrosis and Cell death: “absent” (0), “low” (1), “moderate” (2) or “extensive” (3); Vacuolization, “absent” (0), “few acinar cells” (1), “moderate number of cells” (2) or “high number of cells” (3); Inflammatory cell infiltration: “absent” (0), “low” (1), “moderate” (2) or “extensive” (3). A combined total score for pancreatitis severity comprised the sum of the individual scores.
Isolation of mouse, rat and human pancreatic acini
Dispersed pancreatic acini were isolated from mouse or rat pancreas and cultured as described previously (Lugea, et al., 2017a). Briefly, the pancreas was digested with CLSPA collagenase and the resulting acini preparation was suspended in Medium 199 supplemented with 50 μg/mL soybean trypsin inhibitor. After 20 min of stabilization at room temperate, cells were transferred to fresh medium and cultured for up to 4 h in a cell incubator at 37°C in a 5% CO2 air-humidified atmosphere.
Primary human pancreatic acini were obtained from pancreatic cadaveric tissues from organ donors as we recently reported (Lugea, et al., 2017b). Briefly, pancreata were digested at City of Hope (Duarte, CA) to separate islets from ductal-acinar trees as previously described (Qi, et al., 2015). The resulting acini were transferred to Cedars-Sinai Medical Center facilities (Los Angeles, CA) and cultured as indicated above for rodent pancreatic acini. The study was performed in accordance with regulations and protocols approved by the Institutional Review Boards of the Beckman Research Institute of the City of Hope and the Cedars-Sinai Medical Center (IRB Pro00032114). Rodent and human acinar cells were preincubated for up to 1 h with CM4620 or DMSO vehicle (as control) and then incubated with different concentrations of acinar cell stimuli as indicated in the Results section.
Ca2+ measurements in freshly isolated pancreatic acini
Shortly after isolation, pancreatic acini were attached to polylysine/collagen-coated glass cover slips for 30 min and rested in a 37°C culture incubator for 30 min in culture media, then washed briefly (5 min) in Ca2+-free Hanks-balanced saline solution (HBSS), supplemented with 1 mg/ml BSA, loaded with fura-2 acetoxymethyl ester (fura-2/AM), at a final concentration of 5 μM for 1 h in the dark at RT. Cover slips were then washed and mounted with HBSS buffer and incubated at 37°C for 30 min in the presence or absence of CM4620. To obtain Ca2+ imaging data, cover slips were mounted in imaging rings and imaging performed using a DeltaRam V system (Photon Technology Incorporated, Birmingham, NJ). Fura-2 fluorescence intensity ratio was obtained at 510 nm with excitation wavelengths switching between 340 and 380 nm every 0.5 s. Aggregate data were obtained by similar methods (Lugea, et al., 2017b), except using acini in suspension in a stirred cuvette with HBSS with or without Ca2+ or other additions as indicated in the Results section. These Ca2+ measurements were carried out using a Shimadzu RF5301 PC spectrofluorometer (Shimadzu Corp, Tokyo, Japan).
Propidium iodide (PI) uptake
Cell death was determined in primary pancreatic acini by PI uptake. Briefly, cells were treated with the experimental reagents for 3 h and then cells were labeled with PI (2 μg/ml medium) for 10 min, washed with PBS to remove excess of PI, and lysed in RIPA buffer (50 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 1% Triton X-100, 0.5 % deoxycholic acid, 0.1 % SDS, supplemented with protease and phosphatase inhibitors (Complete™ ULTRA and PhosSTOP™, Sigma-Aldrich). PI fluorescence was measured by fluorometry at 535/617 nm, and values normalized to those of total protein concentration in cell lysates. PI uptake in damaged acinar cells was further confirmed by assessing nuclear PI staining (indicative of compromised plasma membrane) in live acinar cells labeled with 2 μg/ml propidium iodide (PI) and 0.5 μg/ml Hoechst 33342 in PBS using a Nikon Eclipse TE2000 inverted microscope (data not shown).
Trypsin Activity
Trypsin activity in cell or tissue homogenates was measured as described (Lugea, et al., 2017b). Briefly, the tissue was homogenized on ice-cold buffer containing 5 mmol/L MES, 1 mmol/L MgSO4 and 250 mmol/L sucrose (pH 6.5), and assayed at 37°C for 300 sec in buffer containing 50 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, 1 mmol/L CaCl2 and 0.1 mg/ml BSA, and Boc-Gln-Ala-Arg-AMC as specific fluorogenic substrate for trypsin (440 nm, emission, and 380 nm, excitation). Trypsin activity was estimated using a standard curve for purified trypsin (Sigma Chemical, St. Louis, MO).
Myeloperoxidase Activity
Neutrophil infiltration in pancreas and lung tissues was assessed by measuring myeloperoxidase (MPO) activity as previously described (Lugea, et al., 2017b). Tissue samples were homogenized in 20 mmol/L potassium phosphate buffer (pH 7.4) containing 1 mmol/L EDTA, and thereafter resuspended in 50 mmol/L potassium phosphate buffer (pH 6) containing 0.5 % hexadecyltrimethyl ammonium bromide. The samples were then processed by three freeze-thaw cycles, sonicated at 60°C for 2 h, and centrifuged for 20 min at 14,000 g. MPO activity was measured in aliquots of the supernatants mixed with 3,3,5,5-tetramethylbenzidine as substrate for MPO (final concentration 1.6 mmol/L) diluted in 80 mmol/L potassium phosphate buffer (pH 5.4) containing freshly added H2O2 (final concentration 3 mmol/L). Absorbance at 620 nm was measured for 3 min at 30 sec intervals. MPO activity was estimated by comparison of values to those obtained with known concentrations of human MPO (Sigma Chemical, St. Louis, MO) and expressed as activity units per tissue weight.
Western blot analysis
Pancreatic tissues were homogenized in RIPA buffer prepared as described above. Equal amounts of proteins were loaded onto Novex® 4–20% Tris-Glycine Gels (ThermoFisher Scientific), and proteins were electrophoretically transferred onto nitrocellulose membranes using Trans-Blot® Turbo™ Transfer Pack and Trans-Blot® Turbo™ Transfer System (Bio-Rad, Hercules, CA). Membranes were incubated with primary and secondary antibodies; proteins were detected with chemiluminescence reagents using PXi6 Touch Imaging System (Syngene, Cambridge, UK).
RNA analysis by qPCR
RNA was prepared from pancreas and other tissues using Trizol (TRI Reagent®, #TR 118; MRC, Cincinnati, OH) extracted with chloroform and precipitated from the aqueous fraction with 2-propanol according to manufacturer’s instructions. This crude RNA was further purified using the RNeasy® Plus Mini Kit (#74034; Qiagen, Germantown, MD) per manufacturer’s instructions. The extracted RNA samples were measured using a spectrophotometer (Nanodrop 2000, Thermo Fisher Scientific, Waltham, MA). Reverse transcription was performed with the iScript Reverse Transcription Supermix (#170–8840; Bio-Rad) using 1–2 μg of total RNA, and the synthesized cDNA samples were used as templates for quantitative real-time PCR (qPCR) analysis. All reactions were performed using the Bio-Rad CFX Connect™ Real-Time PCR Detection System and the amplifications were done with the iTaq™ Universal SYBR® Green Supermix (Bio-Rad). The qPCR specific oligonucleotide primers used are listed in Table 1. Relative transcript levels were calculated using the comparative 2-ΔΔCt method and normalized to the housekeeping gene, 18S rRNA. Relative mRNA levels were converted to fold increases by comparison with control values, as shown in the figures.
Table 1.
List of primer sequences for qPCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| 18S rRNA | 5’-AGTCCCTGCCCTTTGTACACA-3’ | 5’-CGATCCGAGGGCCTCACTA-3’ |
| rChop | 5’-CCCCAGGAAACGAAGAGGAAG-3’ | 5’-TCAGTCAGCCAAGCTAGGGA-3’ |
| rIl6 | 5’-AGAGACTTCCAGCCAGTTGC-3’ | 5’-AGTCTCCTCTCCGGACTTGT-3’ |
| rMpo | 5’-ATCTTCCCTGTGACCCCCAT-3’ | 5’-TGGACAGGGGGATGGATCAG-3’ |
| rOrai1 | 5’-GATGAGCCTCAACGAGCACT-3’ | 5’-GACTTCCACCATCGCTACCA-3’ |
| rOrai2 | 5’-GTGGGTCTCATCTTCGTGGT-3’ | 5’-CCACCTGTAGGCTTCTCTCG-3’ |
| rStim1 | 5’-ATGCCAATGGTGATGTGGAT-3’ | 5’-CCATGGAAGGTGCTGTGTTT-3’ |
| rTnfa | 5’-GGCTTTCGGAACTCACTGGA-3’ | 5’-GGGAACAGTCTGGGAAGCTC-3’ |
| mActa2 | 5’-GTTCAGTGGTGCCTCTGTCA-3’ | 5’-ACTGGGACGACATGGAAAAG-3’ |
| mCcl2 | 5’-GCCCTAAGGTCTTCAGCACCTT-3’ | 5’-TGCTTGAGGTGGTTGTGGAA-3’ |
| mCol1A1 | 5’-TAGGCCATTGTGTATGCAGC-3’ | 5’-ACATGTTCAGCTTTGTGGACC-3’ |
| mIl4 | 5’-AGCTATTGATGGGTCTCAAC-3’ | 5’-CTGTGACCTCGTTCAAAATG-3’ |
| mIl6 | 5’-CGTGGAAATGAGAAAAGAGTTGTG-3’ | 5’-CCAGTTTGGTAGCATCCATCATTTCT-3’ |
| mOrai1 | 5’-CAGGGTTGCT CAT CGTCTTT-3’ | 5’-ACCGAGTTGAGGTTGTGGAC-3’ |
| mOrai2 | 5’-CTGCTCATCAGCACTTGCAT-3’ | 5’-GGAACTTGATCCAGCAGAGC-3’ |
| mStim1 | 5’-CCTCTCTTGACTCGGCATAATC-3’ | 5’-CTTAGAGTAACGGTTCTGGATATAG-3’ |
| mSerpine1 | 5’-ATCCTGCCTAAGTTCTCTCTG-3’ | 5’-ATTGTCTCTGTCGGGTTGTG-3’ |
| mTgfb1 | 5’-CAACCCAGGTCCTTCCTAAA-3’ | 5’-GGAGAGCCCTGGATACCAAC-3’ |
| mTimp1 | 5’-AGGTGGTCTCGTTGATTCGT-3’ | 5’-GTAAGGCCTGTAGCTGTGCC-3’ |
NFAT and NF-κB gene reporter assays in mouse pancreatic acini and AR42J cells
NFAT-luciferase and NF-κB-luciferase adenoviruses constructs were constructed as described previously (Muili, et al., 2013b). Freshly isolated mouse pancreatic acini or AR42J rat acinar cells (ATCC® CRL1492™; ATCC, Rockville, MD, cultured and treated with 100 nM dexamethasone for 72 h) as described (Lugea, et al., 2017a). were acutely infected with adenovirus encoding Ad-NFAT-luciferase gene expression reporter construct for 30 min, then stimulated for 5 h with CCK (100 nM) in the presence or absence of 3 μM CM4620. At the end of the incubation period, NFAT-luciferase activity was measured using a Luciferase assay system (# E1500, Promega, Madison, WI) following manufacturer’s instructions. Briefly, cells were spun at 350 g for 5 min, washed with PBS, and lysed using proprietary lysis buffer. Samples were vortex mixed, spun at 12,000 g for 2 min and the supernatant assayed to measure luciferase activity using a SpectraMax M3 plate reader (Molecular Devices, San Jose, CA). Luminescence values were normalized to total protein in the cell lysates. In additional studies, AR42J cells were acutely infected with Ad-NF-κB-luciferase 20 h prior to treatment with 100 nM CCK in the presence or absence of 3 μM CM4620. NF-κB-luciferase activity was assayed using the same method described for NFAT-luciferase. Of note, primary mouse acini were not used in these studies because infection with the Ad-NF-kB luciferase construct was not consistent in these cells.
Cytokine release in human peripheral blood mononuclear cells (PBMC)
To determine the inhibitory effect of CM4620 on cytokine release, human PBMC were stimulated for 48 h with plate-bound anti-CD3/anti-CD28 in media containing 10% FBS in the presence or absence of different concentrations of CM4620. Levels of selected cytokines released to the conditioned media were measured by Luminex (EMD Millipore, St. Charles, MO).
Luminescence detection of superoxide anion produced by neutrophil respiratory burst
Human white blood cells were obtained from healthy platelet donors via leukoreductive apheresis at CSMC. Freshly obtained leukophoresis filtration devices were brought to the laboratory and the contents washed briefly by combining 1:1 with RPMI 1640 culture medium and pelleting intact cells by centrifugation at 500 × g for 5 min. Cell pellets were then resuspended in a residual amount of supernatant by tapping the tubes, and the red blood cells were selectively lysed on ice by adding 10 ml Milli-Q deionized water for 30 sec, followed by 40 ml of 0.2% NaCl solution and intact cells recovered by centrifugation at 400 × g for 5 min (this was repeated three times) to obtain a white cell pellet enriched in neutrophils. Aliquots of these cells were then pretreated with DMSO (as control) or 5 μM CM4620 for 30 min in RPMI 1640 culture medium, then transferred to 96-well plates and either left unstimulated or stimulated for 4 h with either 0.5 μM fMLF (induces oxidative burst in neutrophils) or 0.5 μM PMA (an activator of protein kinase C and a potent stimulator of neutrophil NADPH oxidase; used as positive control). Superoxide assays were performed using the chemiluminescence LumiMax (R) superoxide anion measurement kit (#204525, Agilent Technologies, La Jolla, CA) according to the manufacturer’s instructions. Chemiluminescence was measured in a SpectraMax M3 plate reader (Molecular Devices, San Jose, CA) at 37°C. Specific regulatory approval for the use of human lymphocytes was obtained in the CSMC IRB Study, Pro00036882. Informed consent was obtained for the use of all human samples in accordance with the Helsinki Declaration on Ethical Principles for Medical Research Involving Human Subjects.
Studies using mouse pancreatic stellate cells
Immortalized mouse pancreatic stellate cells (PaSC) were used in these studies as previously described (Su, et al., 2016). These immortalized PaSC have the characteristics of activated mouse PaSC (Omary, et al., 2007, Feig, et al., 2012). To determine the effects of CM4620 on fibro-inflammatory responses, cells cultured in DMEM media containing 10 mM glucose and 0.5% FBS were pretreated for 1 h with CM4620 and then stimulated for 3 h with LPS (1 μg/ml). Expression levels of selected genes involved in fibro-inflammatory PaSC responses were determined by qPCR.
Statistics
All experiments were performed in triplicate unless otherwise stated. Data are presented as mean ± SD or mean ± SEM. Data were subjected to analysis of variance (ANOVA) followed by Tukey’s post-hoc test, and two tailed Student t-test for comparison between 2 groups. The ANCOVA model was also used to determine joint effects of cerulein and different doses of CM4620 in the therapeutic rat cerulein pancreatitis model. P values <0.05 were considered significant.
Results
CM4620 efficiently blocks SOCE in primary pancreatic acini
Previous studies have examined the ability of the Orai1 inhibitors GSK-7975A, which can be delivered in vivo in its prodrug form GSK-6288B, or the CalciMedica agent, CM128 (also known as CM4620), a novel, selective inhibitor of Orai1 channels, to inhibit SOCE in acinar cells (Gerasimenko, et al., 2013, Voronina, et al., 2015, Wen, et al., 2015). The present study focused on CM4620 to confirm and extend previous results. For initial studies we used isolated rodent and human pancreatic acini. We first confirmed the ability of CM4620 to block SOCE in freshly isolated mouse pancreatic acini, as first reported by Wen et al. (Wen, et al., 2015). For these studies, fura-2 loaded acinar cells were treated with 10 nM cerulein (Cer) to mobilize Ca2+, and cytosolic Ca2+ concentrations ([Ca2+]c) were measured by Ca2+ fluorescence imaging as described in Methods. In the absence of extracellular Ca2+, Cer produced signaling from the CCK receptor to induce depletion of the ER Ca2+ pools. As evidence of the depleted state of the ER Ca2+ pools at this time, the addition of extracellular Ca2+ to an approximately normal physiologic level (1.8 mM) gave rise to an abrupt rise in [Ca2+ ]c (Figure 1A). As further evidence that this [Ca2+]c rise was dependent on extracellular Ca2+ influx, removal of the extracellular Ca2+ from the perfusion chamber caused a rapid drop in the [Ca2+]c. The [Ca2+]c pattern presented in (Figure 1A, upper trace) represents classic SOCE. In the lower trace, fura-2-loaded acinar cells were incubated first with Cer and 10 μM CM4620 in the absence of extracellular Ca2+, whereupon the Cer again caused release of the intracellular Ca2+ stores (not shown). However, in this case upon addition of 1.8 mM extracellular Ca2+, there was a major blockade of the Ca2+ entry response. This result demonstrates the remarkable efficacy of CM4620 to block SOCE triggered within acinar cells by activation of a surface receptor. Aggregated data from multiple experiments of this type are shown as a bar graph in Figure 1B.
Figure 1. CM4620 blocks store-operated Ca2+ entry (SOCE) in pancreatic acinar cells.
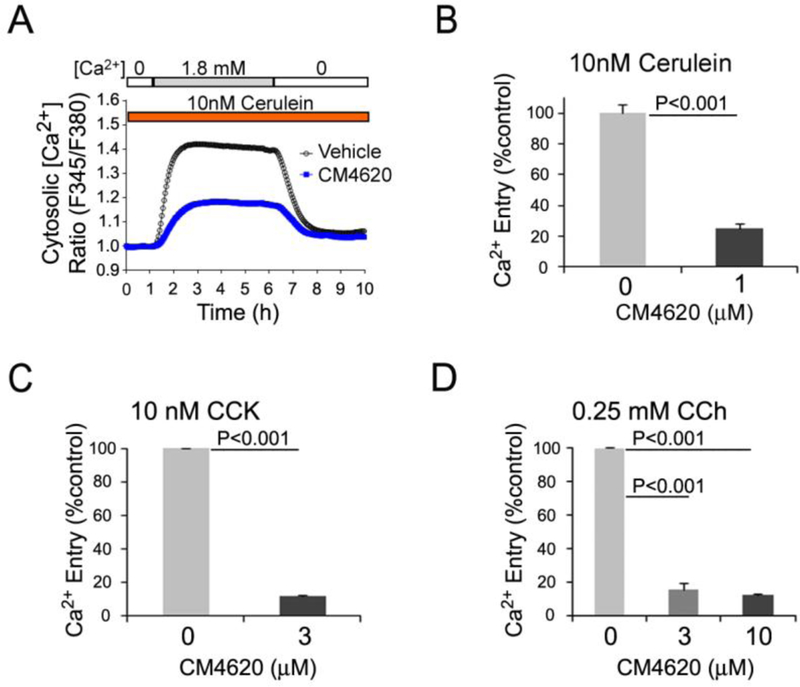
(A and B) Calcium levels in fura-2 loaded mouse pancreatic acini were monitored by fluorescence imaging microscopy. Acini were pretreated for 30 min with or without CM4620 at the indicated concentrations and then treated with Ca2+ mobilizing agents to induce Ca2+ release from ER stores. A, CM4620-induced blockade of SOCE in mouse acini stimulated with cerulein. Graph illustrates a representative trace showing the absence and presence of SOCE by fura-2 ratio (proportional to [Ca2+]c) in conditions of 0 mM (0 Ca) or 1.8 mM (1.8 Ca) Ca2+ in the extracellular media, respectively. CM4620 attenuated SOCE, indicating that Orai/STIM1 channels mediate SOCE in mouse acini. (B) Data obtained from multiple experiments similar to that shown in A show that cerulein-induced SOCE was approximately 80% blocked in fura-2 loaded mouse pancreatic acini by CM4620. (C) Fura-2-loaded mouse pancreatic acini were pretreated with or without CM4620 and then treated with cholecystokinin (CCK) as indicated in A. (D) Fura-2-loaded mouse pancreatic acini were pretreated with CM4620 and then treated with carbachol (CCh) as indicated in A. Levels of store-operated Ca2+ entry were quantitatively analyzed by comparing the areas under traces recorded in the presence or absence of CM4620 and with 1.8 mM Ca2+ in the extracellular media. Graphs show mean ±SD; n=3 independent cell preparations.
We next tested CM4620 efficacy in response to the classic acinar cell secretagogues CCK and the acetylcholine receptor agonist carbachol (CCh). These agents elicit both physiological and pathological responses in acinar cells in a concentration-dependent manner. Fura-2 loaded mouse acini were stimulated with supramaximal concentrations of either CCK (10 nM) or CCh (250 μM) in the presence or absence of CM4620; [Ca2+ ]c was measured as described above. As shown in Figures 1C–1D, CM4620 at concentrations as low as 3 μM was highly effective to block the SOCE triggered by these stimuli. We recently reported similar results in human acini stimulated with CCh and the bile acid TLCS (Lugea, et al., 2017b).
Orai1 is a transmembrane protein originally identified as forming the channel portion of the molecular complex that mediates SOCE in various cell types. Additional members of this protein family, Orai2 and Orai3, have been identified in mammals with variable cell expression patterns (Trebak and Putney, 2017). All Orai homologs produce CRAC currents; the sizes of their respective current amplitudes follow the rank order Orai1 > Orai2 > Orai3 (Mercer, et al., 2006, Trebak and Putney, 2017). Orai1 and STIM1 expression in mouse and human pancreatic acini was previously reported (Lur, et al., 2009, Hong, et al., 2011). Here, we show that mouse and rat pancreatic acini express Orai1, Orai2 and STIM1, and their expression is not affected by CM4620 treatment (Figure 2). The relative potency of CM4620 to block Orai1/STIM1- versus Orai2/STIM1-mediated CRAC currents was previously tested in HEK-293 cells stably overexpressing the corresponding human proteins. Thus, CM4620 was reported previously to be approximately 7-fold more potent at inhibiting Orai1/STIM1-mediated Ca2+ currents compared to Orai2/STIM1-mediated currents, as judged by IC50 values near 120 and 900 nM, respectively (Wen, et al., 2015). These data imply that CM4620 will inhibit SOCE in pancreatic acini that express Orai1 and Orai2, but with a higher potency on Orai1/STIM1 channels based on the channel selectivity exhibited by this compound. Further, CM4620 blocked SOCE in acinar cells triggered by a Ca2+ pump inhibitor with 70–80% efficacy, consistent with our data demonstrating the presence of both Orai1 and Orai2 channels in these cells.
Figure 2.
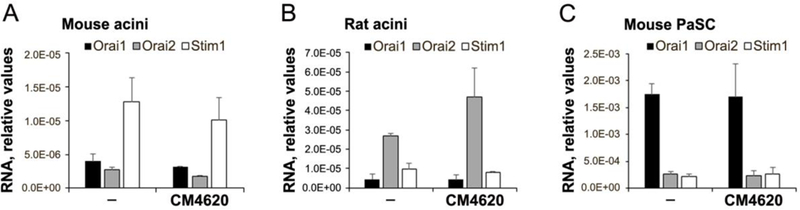
Expression levels of Orai1, Orai2 and Stim1 in primary mouse (A) and rat (B) pancreatic acini and immortalized mouse pancreatic stellate cells (PaSC; C) were measured by qPCR. PCR products were electrophoresed in 2% agarose gels and detected as single bands at the expected molecular weights (not shown). Primer sequences are indicated in Table 1.
CM4620 prevents cell death and trypsinogen activation in primary rodent and human acini
We next tested whether CM4620 could prevent cell death responses evoked by toxic stimuli in pancreatic acini prepared from mouse, rat and human pancreata. Shortly after isolation, acini were preincubated for 30 min with and without CM4620, and then stimulated for up to 3 h with toxic concentrations of TLCS (0.5 mM) or CCK (100 nM). These stimuli have previously shown to induce acinar cell necrosis at least in part through Ca2+ store depletion and associated triggering of SOCE (Voronina, et al., 2002, Perides, et al., 2010). We found that CM4620 significantly prevented or attenuated TLCS- and CCK-induced cell death, as determined by PI uptake, in acini preparations from the three species (Figure 3A).
Figure 3. CM4620 decreases cell death and trypsin activity in pancreatic acini.
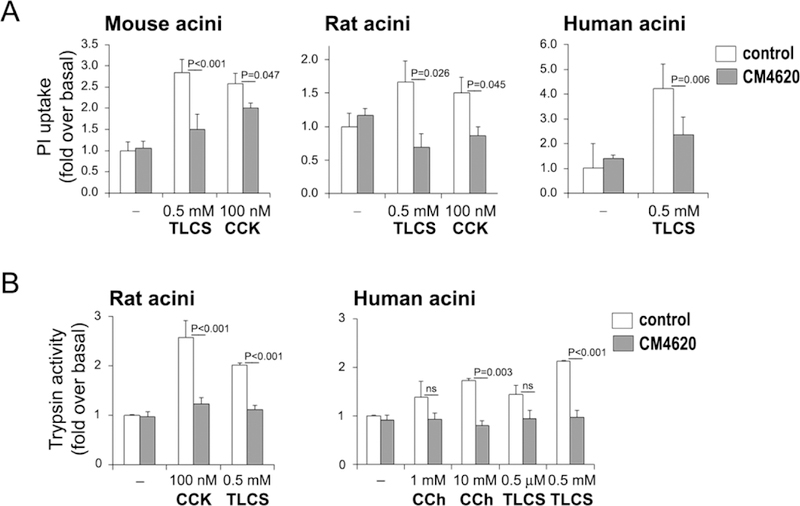
(A) Freshly isolated rodent and human acini were pretreated with 3 μM CM4620 for 30 min and then treated with toxic doses of CCK or TLCS for 3 h. Cell death was estimated by measuring propidium iodide (PI) intake into the cells. Graphs show mean ± SEM, n=3 independent cell preparations. (B) Pancreatic acini were pretreated with 3 μM CM4620 for 30 min and then treated with CCK, TLCS or carbachol (CCh) for 1 h. Trypsin activity was measured by enzymatic chromogenic assay. Graph shows mean ± SEM; n=3–4 independent cell preparations.
We further explored the effects of distinct pathophysiologic stimuli to trigger rapid intracellular trypsin activation in pancreatic acini and tested the dependence of these responses on SOCE. Human acini were preincubated with and without 3 μM CM4620 for 30 min, then stimulated with either CCh (1 mM and 10 mM) or TLCS (0.5 μM and 0.5 mM) for 1 h. As shown in Figure 3B, these agents dose-dependently enhanced the levels of intracellular trypsin activity, and this effect was completely blocked by CM4620 treatment, indicating dependence of intracellular trypsin activation by these stimuli on SOCE. Similar CM4620 inhibitory effects on trypsin activity were found in rat acini stimulated with toxic concentrations of CCK or TLCS. We did not attempt to test the effects of CCK in human acini, due to our previous experience with our preparations obtained from cadaveric pancreata which suggested they respond poorly to CCK and express only very low levels of the CCK-A receptor (Lugea, et al., 2017b). This is a controversial issue, with some evidence indicating poor CCK responses supported by molecular analysis of low CCK-A expression levels (Ji, et al., 2001, Lugea, et al., 2017b) and other studies, which rely on extremely fresh or intact human pancreatic acinar tissue obtained from surgical resections (Murphy, et al., 2008) reporting excellent responses to physiological concentrations of CCK.
CM4620 administration reduces pancreatitis responses in mouse cerulein acute pancreatitis
Previous studies have shown the potential of transient SOCE blockade as a therapeutic strategy in acute pancreatitis (Gerasimenko, et al., 2013, Wen, et al., 2015). The concept that a Ca2+ influx inhibitor could be beneficial in this disease is based on the well-established role of elevated [Ca2+]c as a pathophysiologic trigger mechanism in pancreatitis (Petersen, et al., 2009, Gerasimenko, et al., 2014a). The CRAC channel blockers GSK-7975A (developed by GlaxoSmithKline), administered by subcutaneous infusion as the pro-drug GSK-6288B, and CM4620, administered by intraperitoneal injection, were found to be effective in reducing pancreas damage in three mouse experimental models of AP (Wen, et al., 2015). We used the cerulein acute pancreatitis model as an initial test platform to confirm the efficacy of CM4620 to treat acute pancreatitis. Mice receiving 7-hourly intraperitoneal (IP) cerulein injections (50 μg/kg) were treated with CM4620 (20 mg/kg; 2 IP injections, 30 min before the 1st and 4th cerulein administration), and then tissue samples collected at 1 h after the last cerulein injection. As anticipated, measures of AP were attenuated by CM4620 (Figure 4). Cerulein-induced increases in blood amylase and lipase levels were attenuated by ~20% and ~45%, respectively, and intrapancreatic trypsin activation was reduced by at least 65%. These data suggest a level of AP inhibition comparable to that achieved in mice in previous studies with GSK-7975A and CM4620 (Wen, et al., 2015).
Figure 4. CM4620 administration significantly reduces pancreatitis responses in mouse cerulein acute pancreatitis.
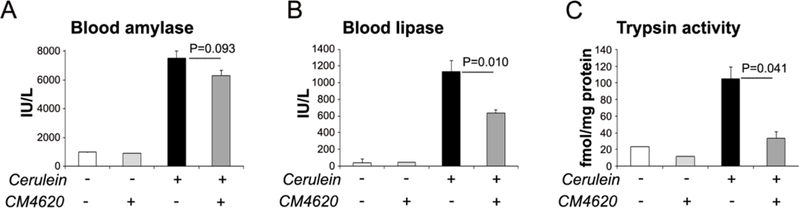
Pancreatitis was induced in mice by supramaximal cerulein administration (50 μg/kg, 7 hourly IP injections); saline-treated mice were used as controls (−). CM4620 (20 mg/kg) or vehicle control were administered intraperitoneally 30 min before the 1st and 4th cerulein injection; blood and tissues were collected 1 h after the last cerulein injection. Graphs show levels of amylase (A) and lipase (B) in blood, and trypsin activity in pancreatic tissue homogenates. Data in graphs are mean ± SD of the values from of 3 mice per group.
Therapeutic intravenous (IV) administration of CM4620 to rats ameliorates cerulein-induced acute pancreatitis
We next tested the ability of CM4620 to limit the pancreas damage in two rat models of AP, i.e., one in which CM4620 was provided therapeutically after the insult (as scheduled in Figure 5A), and one in which the agent was administered prophylactically before the insult (see Figure 5B). To better mimic the therapeutic regime in AP patients during hospitalization, the Orail inhibitor was administered in both models as a continuous IV infusion into the jugular vein. Efficacy of CM4620 was first investigated in the therapeutic AP model with rats receiving 4 hourly IP injections of cerulein (50 μg/kg). CM4620 IV infusion (at three different dose levels, 5, 10, or 20 mg/kg) was begun 30 min after the first cerulein injection and continued for 4 h; animals were then sacrificed 30 min after the end of the infusion period (Figure 5A). In this model, we documented the efficacy of CM4620. istological examination of the pancreas showed a dramatic reduction of pathophysiologic features in cerulein/CM4620-treated compared to cerulein-treated rats (Figures 6A–D). CM4620 significantly reduced pancreatic edema, acinar cell vacuolization, necrosis and the extent of inflammatory cell infiltration found in cerulein-treated animals. Major effects were seen in histologic scoring of rats infused with CM4620, with higher doses showing similar effects to the lowest CM4620 dose in these experiments. Collective scoring of these features measured overall effects near 50% reduction at the lowest CM4620 dose employed and 60% reduction at higher doses (Figure 6D). Cerulein-induced elevated blood amylase and lipase levels were only marginally inhibited at increasing CM4620 dose levels, a feature that may reflect a unique aspect of the rat model or the timing of treatment (Figure 6E). However, intrapancreatic trypsin activation, a major manifestation of acute pancreatitis, was diminished with CM4620 by as much as ~70%, although there was no apparent dose dependence of CM4620 (Figure 6F), indicating that even the lowest doses were at or near maximum efficacy.
Figure 5. Schematic showing the experimental protocol for cerulein-induced acute pancreatitis in rats.
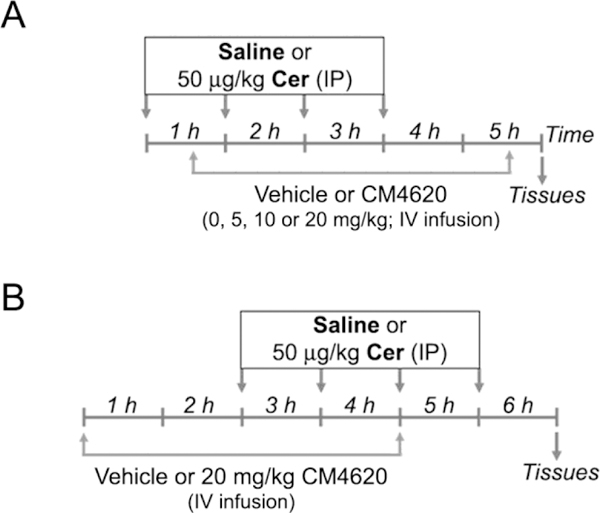
Acute pancreatitis was induced in male Sprague-Dawley rats by intraperitoneal (IP) administration of cerulein (Cer; 50 μg/kg; 4 hourly injections); control mice received saline as vehicle control. In a subset of rats, the Orai/Stiml inhibitor CM4620 was administered at the indicated doses as a 4-h continuous intravenous (IV) infusion into the jugular vein. The IV infusion was initiated 30 min after the first cerulein injection (A, therapeutic administration of CM4620) or 2 h before the first cerulein injection (B, prophylactic administration of CM4620). Animals remained conscious during the whole procedure. Blood and tissue samples were collected post-mortem at the indicated times.
Fig. 6. Therapeutic administration of CM4620 reduces pathologic responses in cerulein-induced acute pancreatitis.
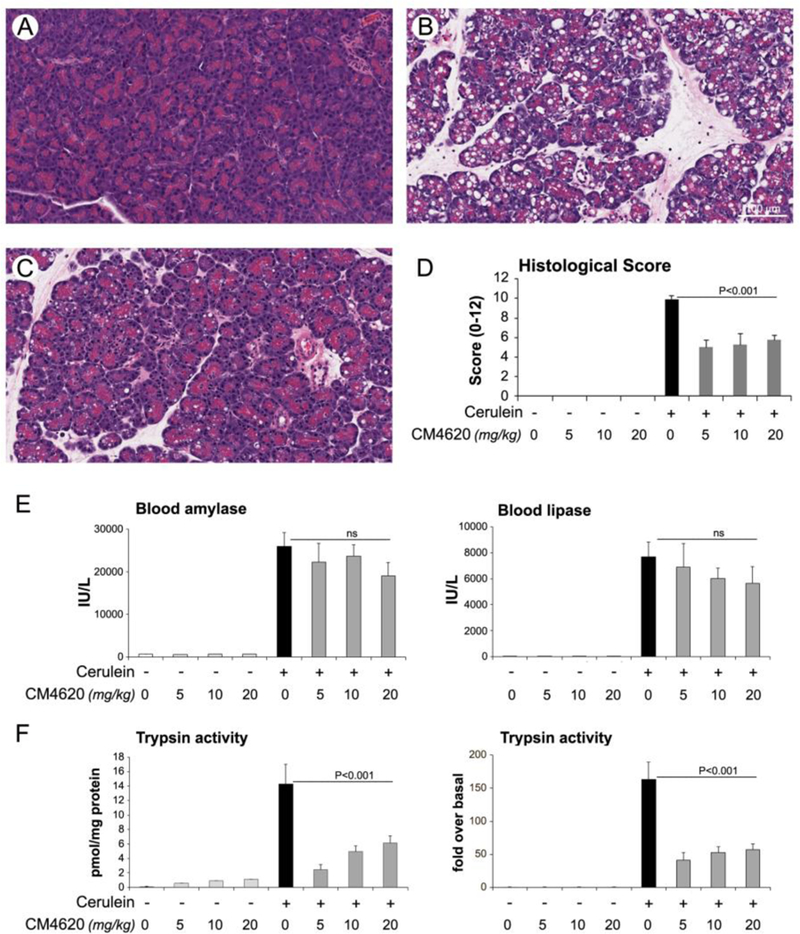
Cerulein acute pancreatitis was induced in male Sprague-Dawley rats as indicated in Figure 3A. Cerulein or saline control (−) were administered as 4 hourly IP injections, and CM4620 (at the indicated doses) or vehicle control as a 4-h continuous intravenous (IV) infusion into the jugular vein. The IV infusion was initiated 30 min after the first cerulein injection, and blood and tissue samples were collected 30 min after the end of the infusion period. (A-C) Representative photomicrographs from H&E stained tissues depicting pancreatic injury in the following groups: saline/vehicle (A), cerulein/vehicle (B) and cerulein/20mg/kg CM4620 (C). Graph in (D) shows quantitative analyses (mean ± SD; n=4–12 rats per group) for pancreatic injury scored in H&E stained tissue sections following the score system indicated in Experimental Procedures. (E) Graphs show levels of amylase and lipase activities in blood from rats subjected to cerulein acute pancreatitis. (F) Graphs illustrate trypsinogen activation in pancreatic tissue homogenates from rats subjected to acute pancreatitis. Trypsinogen activation is expressed as pmol of trypsin/mg of total protein (left graph) or fold over saline-treated control groups (right graph). Data in graphs are mean ± SD, n=4–12 rats/group.
Acinar cell death, both apoptosis and necrosis, accompanied by elevated levels of pancreas tissue inflammation, is a prominent feature in cerulein AP in rats (Mareninova, et al., 2006). We therefore examined pancreas tissues for cell death pathway activation and effects of CM4620 in the rat AP model. We analyzed for markers of cell death by Western blot: CHOP expression, as a marker of ER stress-associated cell death, loss of full-length PARP and caspase 3 cleavage as markers of DNA damage and apoptosis (Figure 7A). Levels of the MAPK, ERK1/2 were also analyzed as a loading control. We found that pancreatic CHOP expression, largely absent under control conditions, was potently induced by cerulein (Figures 7A–B), supporting activation of ER stress pathways during AP. CHOP mRNA and protein levels were much reduced in the Cerulein + CM4620 groups, suggesting that SOCE blockade ameliorates cerulein-induced ER stress. Full- length Poly(ADP-ribose) polymerase (PARP) was abundant in controls and diminished almost to zero in the pancreas of cerulein injected rats (Figure 7A). CM4620 caused a partial restoration of the control PARP level, with greater restoration at the higher CM4620 dose level. Caspase-3 is cleaved during triggering of apoptosis, which could be observed upon AP induced by cerulein injections, and CM4620 administration at different doses inhibited this response (Figure 7A). Taken together, the data support that blockade of Orai channels inhibited multiple cell death signaling pathways in the rat model of AP.
Figure 7. Therapeutic administration of CM4620 diminishes cell death signaling in cerulein-induced acute pancreatitis.
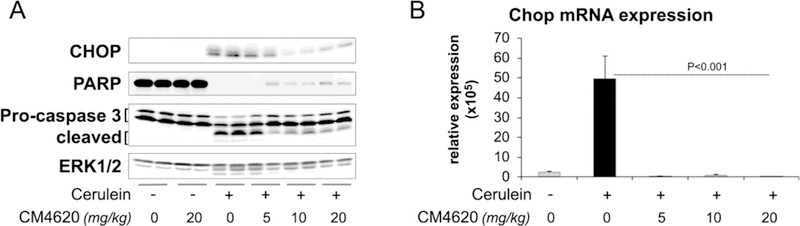
Cerulein acute pancreatitis was induced in rats as indicated in Figure 3A. (A) Pancreatic levels of CHOP, a transcription factor that regulates ER stress associated cell death, and the apoptotic markers PARP and caspase 3 were assessed by Western blotting. Levels of ERK1/2 were measured as loading control. Each lane shows data from an individual rat, 2 rats per group. (B) Graphs shows Chop mRNA levels in pancreas tissues as determined by qPCR. Data is representative of 3–4 rats/group. Data in graphs are mean ± SD.
We next analyzed CM4620 effects on local and systemic inflammatory responses. Myeloperoxidase (MPO) activity, a marker of the extent of neutrophil infiltration, in pancreas and lung tissues was virtually abolished at all levels of CM4620 infusion (Figure 8A–B). To further substantiate these data, we also measured mRNA levels of MPO as well as two cytokines, TNFα and IL-6 commonly found elevated in pancreatitis patients and experimental pancreatitis (Szatmary and Gukovsky, 2016, Lugea, et al., 2017b). As expected, cerulein robustly induced expression of MPO, IL-6, and TNFα mRNA in both pancreas and lung, and these elevated levels were dramatically reduced in the animals infused with CM4620 (Figure 8C–D).
Figure 8. Therapeutic administration of CM4620 reduces local and systemic inflammation in cerulein-induced acute pancreatitis.
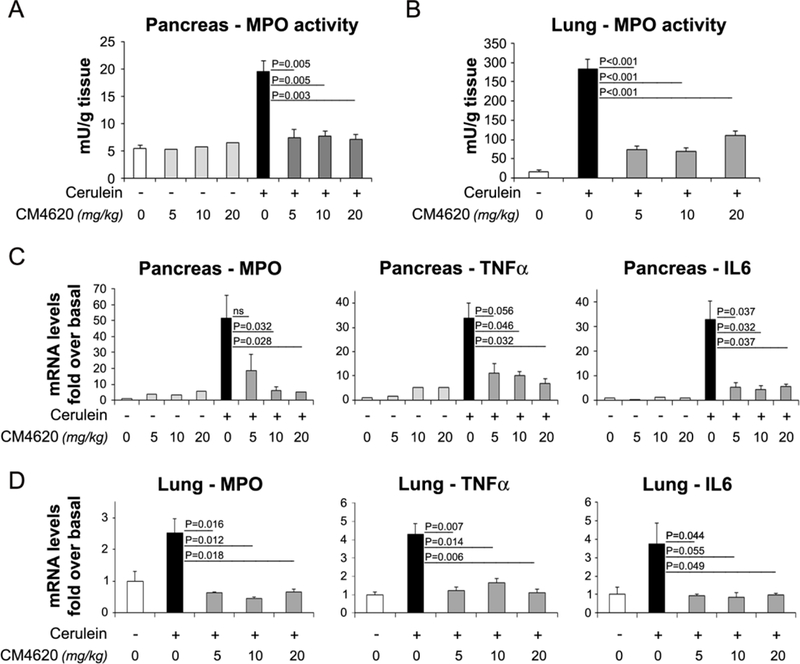
Cerulein acute pancreatitis was induced in rats as indicated in Figure 3A. Neutrophil infiltration within pancreas (A) or lungs (B) was assessed by measuring myeloperoxidase (MPO) activity in tissue homogenates and expressed as units of MPO activity per tissue weight. Expression levels of MPO, TNFa and IL6 were measured in pancreas (C) and lung (D) tissues by qPCR. Data in graphs represent mean ± SD of the values from 4–8 rats per group.
Results shown in Figure 9 illustrate inhibitory effects of prophylactic administration of CM4620 on AP responses (see Figure 5B for experimental design). In these studies, cerulein was administered as 4 hourly IP injections (50 μg/kg each injection), and CM4620 IV infusion (at 20 mg/kg) was begun 2 h before the first cerulein injection and continued for 4 h; animals were then sacrificed 2 h after the end of the infusion period. Compared to cerulein alone, CM4620 administration markedly reduced the total histological scoring in H&E-stained pancreatic tissues, and the pancreatic trypsin and MPO activities elicited by cerulein administration (Figure 9). Taken together, these data show that both prophylactic and therapeutic administration of CM4620 is effective in reducing pancreatic damage and inflammation in the rat cerulein AP model, an effect likely consistent with overall prevention of acinar cell damage due to massive SOCE (Figures 1 and 3) and beneficial effects of Orai channel inhibition by CM4620 in other cell types participating in the initiation and development of AP, mainly immune cells infiltrating the pancreas and PaSC.
Figure 9. Prophylactic administration of CM4620 to rats ameliorates severity of acute pancreatitis.
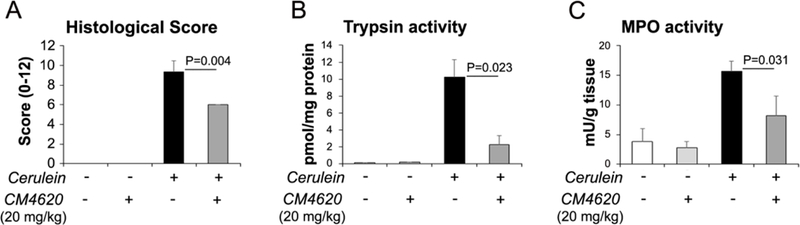
Cerulein acute pancreatitis was induced in rats as indicated in Figure 3B. Cerulein or saline control (−) were administered as 4 hourly IP injections, and CM4620 (20 mg/kg) or vehicle control as a 4-h continuous intravenous (IV) infusion into the jugular vein. The IV infusion was initiated 2 h before the first cerulein injection, and blood and tissue samples were collected 2 h after the end of the infusion period. (A). Graph shows quantitative analyses (mean ± SD; n=3 rats per group) for pancreatic injury scored in H&E stained tissue sections following the score system indicated in Material and Methods. Graphs in (B) and (C) show levels of pancreatic trypsin and myeloperoxidase activities, respectively. Data in graphs are mean ± SD, n=3 rats/group.
Orai inhibition reduces NFAT and NF-κB activities induced by pancreatitis stimuli in pancreatic acinar cells
Activation of nuclear factor of activated T-cells (NFAT) and nuclear factor-kappa B (NF-κB) are early events during acute pancreatitis development, and regulate pathogenic responses including trypsinogen activation, acinar cell damage, inflammatory cell recruitment and the progression of inflammation (Gurda, et al., 2008, Awla, et al., 2012, Muili, et al., 2013a, Muili, et al., 2013b, Jakkampudi, et al., 2016). NFAT activation is dependent on the Ca2+-activated phosphatase calcineurin (Pan, et al., 2013). In many cell types including acinar cells and immune cells, SOCE leads to calmodulin-dependent activation of calcineurin, that in turn dephosphorylates NFATs resulting in their nuclear translocation and transcription of inflammatory-related genes (Muili, et al., 2013a, Muili, et al., 2013b, Wen, et al., 2018, Zhu, et al., 2018). To examine whether SOCE inhibition with CM4620 reduces NFATs activation in experimental acute pancreatitis, we carried out luciferase gene reporter assays in mouse acini and the AR42J rat acinar cell line in the presence and absence of CM4620. Mouse acini were infected with virus encoding an Ad-NFAT-luc reporter construct (Muili, et al., 2013b), and 30 min later treated with or without CM4620. After 30 min, cells were left unstimulated or stimulated for 4.5 h with high concentrations of CCK (100 nM), and then the levels of luciferase activity were assessed to measure NFAT-dependent transcription. As shown in Figure 10A, NFAT activity was markedly induced by CCK, and CM4620 completely prevented this activity. Similar results were found in AR42J cells treated with 100 nM CCK (Figure 10B). These data strongly support a key role for Orai/STIM1-dependent SOCE on NFAT activation in acinar cells.
Figure 10. CM4620 inhibits CCK-induced NFAT and NF-κB activities in pancreatic acinar cells.
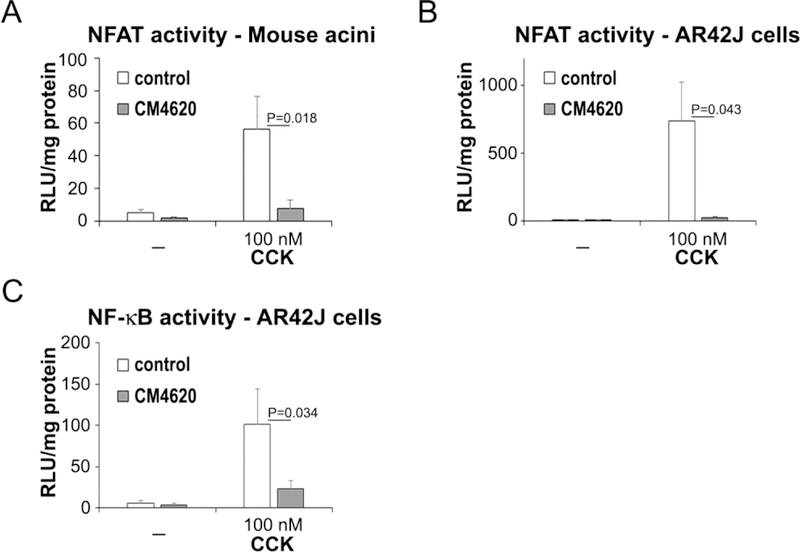
Primary mouse pancreatic acini and AR42J cells were infected with adenoviral vectors to assess NFAT and NF-κB associated luciferase activities, and then treated with 100 nM CCK in the presence or absence of CM4620. Luciferase activity was measured 4.5 h after CCK treatment and expressed as relative luciferase units (RLU) per mg of total protein. As expected, CCK induced marked NFAT-luciferase activity in mouse acini (A) and AR42J cells (B), and this effect was completely blocked by CM4620. (C) CCK also induced NF-κB activation in AR42J cells, and this effect was significantly inhibited by CM4620. Graphs show mean ± SD; n=3–4 independent experiments.
We next examined whether CM4620 inhibited CCK-induced NF-κB activation in acinar cells. Although the participation of Orai-mediated SOCE in NF-κB activation during acute pancreatitis has not been established, calcineurin has been shown to regulate bile acid-induced NF-κB activation in isolated acinar cells (Muili, et al., 2013a). Moreover, other studies indicate that Orai1/STIM1-mediated SOCE modulates NF-κB signaling and transcriptional activity in T- cells (Liu, et al., 2016). We used here AR42J cells treated with dexamethasone to render them sensitive to CCK and transduced overnight with an Ad-NF-κB-luciferase reporter, and then treated with 100 nM CCK in the presence or absence of CM4620. We found that NF-κB activation induced by CCK in these cells was partially dependent on SOCE as demonstrated by a 75% reduction in luciferase activity cells treated with CM4620+CCK compared to cells treated with CCK alone (Figure 10C).
CM4620 inhibits cytokine production in peripheral blood mononuclear cells and reduces oxidative burst in neutrophils
Acute pancreatitis is thought to originate in the pancreatic acinar cell, where damage and accompanying cell death responses give rise to proinflammatory signals that attract immune cells into the pancreas and propagate a vicious cycle involving further tissue damage and acinar cell death, sustained activation of inflammatory cells and pathways, which ultimately spread to involve systemic events and risk to multiple organ systems in the patient. As shown above, Orai/STIM1-mediated SOCE in the acinar cell participates in this sequence of events by promoting key pathological transcriptional activities and cellular processes. Orai-mediated SOCE also regulates many cellular functions in immune cells. Extensive literature indicates that Ca2+ signaling regulates functional processes in neutrophils including chemotaxis, degranulation, oxidative burst and ROS production and phagocytosis, and several of these processes are modulated by Orai/STIM1 interactions (Clemens, et al., 2017). In T-cells and mast cells, Orai channels are the major Ca2+ entry pathway and regulate cytokine production. Accordingly, we found that CM4620 markedly inhibited in human peripheral blood mononuclear cells (PBMC) the release of various cytokines commonly found elevated in acute pancreatitis, with IC50 values ranging from 47 to 952 nM (Table 2).
Table 2.
IC50 values for CM4620 on cytokine release by human peripheral blood mononuclear cells (PBMC)*
| Cytokines | CM4620 Replicate IC50Values in nM (mean) |
|---|---|
| IFNγ | 175, 102 (138) |
| IL-1β | 264, 217 (240) |
| IL-2 | 71, 47 (59) |
| IL-4 | 952, 807 (879) |
| IL-6 | 145, 126 (135) |
| IL-10 | 367, 239 (303) |
| IL-17 | 140, 100 (120) |
| TNFα | 278, 173 (225) |
Cells were stimulated with plate-bound anti-CD3/anti-CD28 in 10% FBS for 48 h.
In a second study using freshly isolated neutrophils obtained from human volunteers’ blood, oxidative burst was stimulated with either 0.5 μM N-Formyl Met-Leu-Phe (fMLF) that activates a select GPCR (in a Ca2+-dependent manner), or with 0.5 μM phorbol myristate acetate (PMA) that triggers downstream signal transduction and NADPH activity (in a Ca2+-independent manner). As shown by data in Figure 11, oxidative burst was induced by both fMLF and PMA, but only the fMLF-induced ROS production was selectively attenuated by preincubation with CM4620. These data are in agreement with previous reports indicating that ROS production induced by fMLF is reduced in neutrophils lacking STIM1, suggesting that SOCE is required for classical short-term neutrophil responses (Clemens, et al., 2017). Taken together, our results support the concept that Orai inhibitors attenuate acute pancreatitis responses by direct inhibition of cytokine and ROS production by infiltrated immune cells.
Figure 11. CM4620 diminishes the oxidative burst induced by fMLF peptide in human neutrophils.
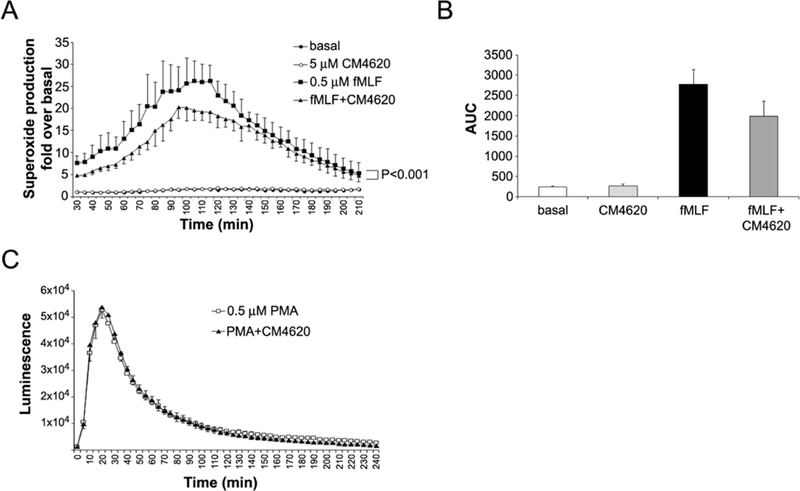
Neutrophils isolated from human blood were pretreated for 30 min with DMSO (as control) or CM4620 and then superoxide anion formation was stimulated with fMLF (A-B) or PMA (C) and measured for 4 h using the chemiluminescence LumiMax (R) superoxide anion measurement kit (Agilent Technologies, La Jolla, CA) following manufacturer’s instructions. CM4620 significantly inhibits fMLF-induced superoxide production (P<0.001; Two-way ANOVA). Data in graph are mean ± SD; n=2 independent experiments. (B) Graph shows area under the curve (AUC) for traces depicted in (A). (C) CM4620 does not affect the oxidative burst induced by PMA in human neutrophils. Data in graph are mean ± SD; n=3 independent experiments.
CM4620 inhibits fibro-inflammatory responses in activated pancreatic stellate cells
Pancreatic stellate cells (PaSC) are resident cells located in the periacinar space in close proximity with acinar cells (Apte, et al., 2013). In the healthy pancreas, PaSC display a quiescent phenotype characterized by low proliferation rates but upon tissue damage PaSC de-differentiate into a myofibroblast-like phenotype, a process termed “activation” (Omary, et al., 2007). Activated PaSC produce high amounts of collagens and inflammatory mediators that contribute to tissue repair, fibrogenesis and the inflammatory microenvironment during acute, recurrent acute and chronic pancreatitis development (Omary, et al., 2007). PaSC express a variety of membrane receptors including Toll-like receptors (TLRs) (Masamune, et al., 2008) and during pancreatitis can be activated both by cytokines and damage-associated molecular patterns (DAMPs; TLR ligands) released by damaged acinar cells or immune cells (Apte, et al., 2013). Recent studies have identified bradykinin-mediated CRAC channels in mouse PaSC within acini (Gryshchenko, et al., 2016) and we found marked expression of Orai1 and STIM1 in cultured PaSC from mouse (Figure 2). We explored here whether fibro-inflammatory responses elicited by lipopolysaccharide (LPS), a TLR4 ligand that elicits Orai1-mediated SOCE and cytokine production in mesenchymal cells (Park, et al., 2016), were altered by CM4620 in cultured mouse PaSC. As shown in Figure 12, LPS induced significant increases in the expression levels of IL6, CCL2 (MCP1), IL4, TGFβ1 and SERPINE1 (PAI-1) as well as the PaSC activation markers COL1A1 and ACTA2 (αSMA), and these effects were blocked by the Orai/STIMl inhibitor CM4620. The data suggest that Orai inhibitors can attenuate pancreatitis responses by acting on TLR-mediated fibro-inflammatory signals in PaSC.
Figure 12. CM4620 prevents expression of fibro-inflammatory genes in mouse pancreatic stellate cells.
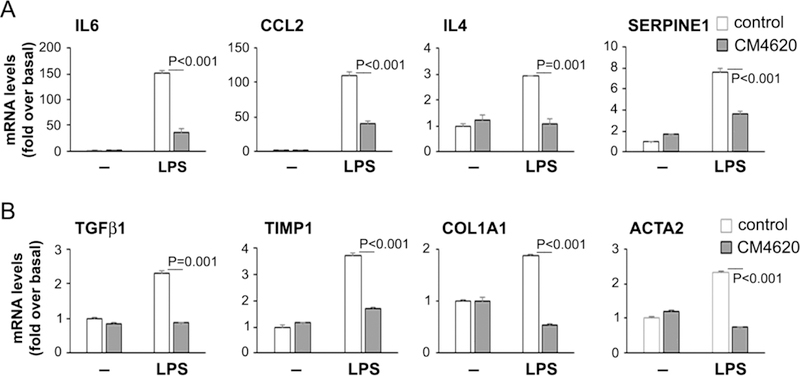
Ligands of Toll-like receptors (TLRs) are known to induce cell activation and inflammatory responses in pancreatic stellate cells (PaSC). Mouse PaSC were treated for 3 h with the TLR4 ligand LPS (1 μg/ml) in the presence or absence of 3 μM CM4620. mRNA levels of the indicated cytokines and PaSC markers were measured by qPCR. Data in graph are mean ± SD; n=3 independent experiments.
Discussion
Experiments in the present study provided new insights into the pathobiologic mechanisms of acute pancreatitis (AP). Normal free Ca2+ levels within the ER range between 100–800 μM; and a minimum level of [Ca2+]ER (estimated at 50 μM) is required to sustain optimal ER protein folding and chaperone functions (Gorlach, et al., 2006) Significant reduction in [Ca2+]ER impairs normal folding, chaperone and post-translational modification reactions and promotes ER stress (Sutton, et al., 2008). Alterations in [Ca2+]c, either linked to or independent of ER stress, cell death and proinflammatory responses may also play roles in the mechanism of AP. It is now clear that pancreatitis generating agents cause ER Ca2+ depletion and Ca2+ entry via store operated Ca2+ entry (SOCE), as reflected here and in previous studies (Gerasimenko, et al., 2013, Wen, et al., 2015). That the Orai inhibitor, CM4620, attenuates SOCE in acinar cells from mice was shown previously (Wen, et al., 2015) and is confirmed here. Whereas the inhibition achieved at the highest soluble concentration of CM4620 (i.e., 10 μM) was extensive (70–80%), the residual channel activity may be attributable to alternative SOCE mechanisms such as either Orai2/STIM1 channels, or the TrpC3 channel (Kim, et al., 2009). Here, we present novel data on Orai expression implying that CM4620 preferentially acts on Orai1 rather than Orai2 in these cells.
Effects of SOCE inhibition by CM4620 on pathobiologic responses of pancreatitis previously documented in mice (Gerasimenko, et al., 2013, Wen, et al., 2015). include histologically scored damage such as pancreatic edema and acinar cell vacuolization, and trypsin activation in pancreatic acini. Our data confirm these results and delve deeper into the mechanisms of damage induced by SOCE. In particular, we investigate ER stress/cell death responses, which are now added to the effects attenuated by SOCE inhibition by CM4620, and further extend the models in which CM4620-mediated SOCE blockade has been observed to diminish AP severity to the rat and human. We further exploit the rat model to establish the efficacy in experimental AP of CM4620 administration by the intravenous route that is closer to that to be used in humans than the IP route used in mice.
Whereas previous studies (Gerasimenko, et al., 2013, Wen, et al., 2015) first showed that SOCE plays an important role in the pancreatic acinar cell, we report here that SOCE is also required for individual contributions of an expanded range of cell types to AP pathophysiology.
In particular, we present novel data exploring the role of Orai-mediated Ca2+ entry in local pancreatic stellate cells and circulating immune cells, finding that SOCE in these distinct cell types also contributes to AP disease mechanisms. Our novel findings also include inhibition by CM4620 of myeloperoxidase (MPO) activity and inflammatory cytokine mRNA expression in pancreas and lung of a rat AP model. Our novel data probing the effects of Orai inhibition in distinct cell types that constitute individual parts of the local and systemic aspects of the pathophysiology provide a template to focus further assessments of additional components of disease mechanisms beyond Ca2+ entry.
At the level of the pancreatic acinar cell, widely hypothesized to represent the locus where disease events initiate, the proximal effects of Ca2+ depletion of the ER and sustained increases in [Ca2+]C on cellular death responses include increased CHOP expression, PARP destruction and caspase 3 cleavage. Ca2+ entry and rising [Ca2+]C have been shown to mediate mitochondrial failure and cell death responses in several models of experimental AP. The sequence of a pathologic unfolded protein response (UPR) CHOP response and mitochondrial failure appear to be universal features of AP models. Our data suggest that SOCE and sustained increases in [Ca2+]C activate a pathologic UPR branch centered on increased CHOP expression that is associated with cell death and inflammatory signaling. The increased CHOP expression can account for some, but not all the pathobiologic responses observed in AP models. Inhibition of Orai1-mediated SOCE by CM4620 prevented the increase in CHOP we observed with high-dose cerulein. Further study will be necessary to establish whether the ER-generated signal CHOP or other signals are necessary for the mitochondrial failure or other damage caused by excessively elevated [Ca2+]c in acinar cells in AP.
As CM4620 is currently in early phase clinical trials, it is important to consider the benefits and risks of the strategy of Orai blockade, especially with respect to any impact on the gut microbiome. Indeed, specific genetic deletion of Orai1 in acinar cells in mice was reported to induce intestinal bacterial outgrowth and dysbiosis (Ahuja, et al., 2017). These effects were attributed to detrimental effects of Orai1 deletion on physiologically highly relevant Ca2+ regulatory events and antibacterial peptide secretion by acinar cells. However, we emphasize that there is a major difference between nearly complete, permanent abrogation of Orai1 function from these cells via genetic deletion, and incomplete as well as temporary blockade via a small molecule inhibitor. Indeed, our data reinforce the concept that temporary SOCE inhibition and blockade of excessive Ca2+ entry in acinar cells and other cell types participating in the AP response confer substantial therapeutic benefit during AP. We note that data derived from Orai1 inducible deletion model used by Ahuja et al. (Ahuja, et al., 2017), which involves injection/gavage of tamoxifen for five days in succession, fail to explain why germline deletion of Orai1 produce mice which do not exhibit the same extent of intestinal inflammation or mortality described in the report by Ahuja et al. Thus, the reported effects of Orai1 ablation appear to be model-specific; further research should be undertaken to evaluate the effects of long-term treatments with Orai inhibitors.
Here, we also demonstrate that SOCE caused by pancreatitis-generating agents has significant and dramatic effects on signaling in immune cells and PaSC, i.e., on other cell types participating in the development of local and systemic inflammation during AP and recurrent AP. Our results revealed that the Orai inhibitor reduced cytokine release in PBMC and PaSC and partly inhibited the fMLF-induced oxidative burst of human neutrophils. This latter effect was modest but nevertheless could be of clinical significance as such agents are being developed for pancreatitis treatment in humans. In view of a recent study by Ferdek et al. (Ferdek, et al., 2016), gene expression responses requiring SOCE in immortalized PaSC will in future be independently validated in primary cells. In support of the concept that SOCE inhibition in PaSC may attenuate its negative actions on acini within the pancreatic microenvironment, a recent study (Gryshchenko, et al., 2018) reported a pathophysiologic mechanism whereby dying acinar cells promote Ca2+ signals in PaSC, which then further damage acinar cells in a vicious cycle. In summary, it is clear that SOCE responses in multiple cell types including inflammatory cells, PaSC and acinar cells contribute both to individual and integrated pathobiologic pathways during acute pancreatitis.
Key Points Summary.
This work confirms previous reports that CM4620, a small molecule inhibitor of Ca2+ entry via store operated Ca2+ entry (SOCE) channels formed by STIM1/Orai complexes, attenuates acinar cell pathology and acute pancreatitis in mouse experimental models.
Here we report that intravenous administration of CM4620 reduces the severity of acute pancreatitis in the rat, a hitherto untested species.
Using CM4620, we probe further the mechanisms whereby SOCE via STIM1/Orai complexes contributes to the disease in pancreatic acinar cells, supporting a role for ER stress/cell death pathways in these cells.
Using CM4620, we show that SOCE via STIM1/Orai complexes promotes neutrophil oxidative burst and inflammatory gene expression during acute pancreatitis including in immune cells which may be either circulating or invading the pancreas.
Using CM4620, we show that SOCE via STIM1/Orai complexes promotes activation and fibroinflammatory gene expression within pancreatic stellate cells.
Acknowledgements:
The authors thank Drs. Kevin Ferrari, Mohamed Ei-Sayed and Fouad R. Kandeel from the Southern California Islet Center at City of Hope (Duarte, CA, USA) and the Integrated Islet Distribution Program for providing human pancreatic acini. We also thank Ellen B. Klapper (Rita and Taft Schreiber Division of Transfusion Medicine; CSMC) for providing human blood cells.
Funding was provided by the National Institutes of Health (P01DK098108 to SJP; R01 AA019954 to AL; P50-A11999 to SJP); and CalciMedica Inc.
Abbreviations:
- CCh
carbachol
- CCK
cholecystokinin octapeptide
- CHOP
C/EBP homologous protein
- CPA
cyclopiazonic acid
- CRAC
calcium release-activated calcium channels
- ER
endoplasmic reticulum
- fMLF
N-Formyl-Met-Leu-Phe
- IP
intraperitoneal
- LPS
lipopolysaccharide
- PaSC
pancreatic stellate cells
- PI
propidium iodide
- SOC
store-operated Ca2+ channels
- SOCE
store-operated Ca(2+) entry
- TLCS
taurolithocholic acid 3-sulfate
Footnotes
Conflict of interest: MD, JR, SR and KS were or are affiliated with CalciMedica, which partially funded this study. The rest of the authors declare that they have no conflicts of interest with the contents of this article.
.References
- Ahuja M; Schwartz DM; Tandon M; Son A; Zeng M; Swaim W; Eckhaus M; Hoffman V; Cui Y; Xiao B; Worley PF; Muallem S (2017). Orai1-Mediated Antimicrobial Secretion from Pancreatic Acini Shapes the Gut Microbiome and Regulates Gut Innate Immunity. CellMetab 25, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte MV; Wilson JS; Lugea A; Pandol SJ (2013). A Starring Role for Stellate Cells in the Pancreatic Cancer Microenvironment. Gastroenterology 144, 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awla D; Zetterqvist AV; Abdulla A; Camello C; Berglund LM; Spegel P; Pozo MJ; Camello PJ; Regner S; Gomez MF; Thorlacius H (2012). NFATc3 regulates trypsinogen activation, neutrophil recruitment, and tissue damage in acute pancreatitis in mice. Gastroenterology 143, 1352–-1360 e7.. [DOI] [PubMed] [Google Scholar]
- Berridge MJ; Lipp P; Bootman MD (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Cahalan MD; Zhang SL; Yeromin AV; Ohlsen K; Roos J; Stauderman KA (2007). Molecular basis of the CRAC channel. Cell Calcium 42, 133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens RA; Chong J; Grimes D; Hu Y; Lowell CA (2017). STIM1 and STIM2 cooperatively regulate mouse neutrophil store-operated calcium entry and cytokine production. Blood 130, 1565–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criddle DN; Gerasimenko JV; Baumgartner HK; Jaffar M; Voronina S; Sutton R; Petersen OH; Gerasimenko OV (2007). Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death.Differ. 14, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Criddle DN; Raraty MG; Neoptolemos JP; Tepikin AV; Petersen OH; Sutton R (2004). Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc.Natl.Acad.Sci.U.S.A 101, 10738–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingsdale H; Voronina S; Haynes L; Tepikin A; Lur G (2012). Cellular geography of IP3 receptors, STIM and Orai: a lesson from secretory epithelial cells. Biochem Soc Trans 40, 108–11. [DOI] [PubMed] [Google Scholar]
- Feig C; Gopinathan A; Neesse A; Chan DS; Cook N; Tuveson DA (2012). The pancreas cancer microenvironment. Clin Cancer Res 18, 4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdek PE; Jakubowska MA; Gerasimenko JV; Gerasimenko OV; Petersen OH (2016). Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium-driven bile uptake. J Physiol 594, 6147–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV; Gerasimenko OV; Petersen OH (2014a). The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 592, 269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV; Gerasimenko OV; Petersen OH (2014b). The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 592, 269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV; Gryshchenko O; Ferdek PE; Stapleton E; Hébert TO; Bychkova S; Peng S; Begg M; Gerasimenko OV; Petersen OH (2013). Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci U S A 110, 13186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlach A; Klappa P; Kietzmann T (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. AntioxidRedox Signal 8, 1391–418. [DOI] [PubMed] [Google Scholar]
- Gryshchenko O; Gerasimenko JV; Gerasimenko OV; Petersen OH (2016). Ca(2+) signals mediated by bradykinin type 2 receptors in normal pancreatic stellate cells can be inhibited by specific Ca(2+) channel blockade. J Physiol 594, 281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryshchenko O; Gerasimenko JV; Peng S; Gerasimenko OV; Petersen OH (2018). Calcium signalling in the acinar environment of the exocrine pancreas: physiology and pathophysiology. J Physiol 596, 2663–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukovsky I; Pandol SJ; Gukovskaya AS (2011). Organellar dysfunction in the pathogenesis of pancreatitis. Antioxid Redox Signal 15, 2699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurda GT; Guo L; Lee SH; Molkentin JD; Williams JA (2008). Cholecystokinin activates pancreatic calcineurin-NFAT signaling in vitro and in vivo. MolBiol Cell 19, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG; Rao A (2015). Store-operated calcium entry: Mechanisms and modulation. Biochem Biophys Res Commun 460, 40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH; Li Q; Kim MS; Shin DM; Feske S; Birnbaumer L; Cheng KT; Ambudkar IS; Muallem S (2011). Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12, 232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper R; Soboloff J (2015). STIMATE reveals a STIM1 transitional state. Nat Cell Biol 17, 1232–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkampudi A; Jangala R; Reddy BR; Mitnala S; Nageshwar Reddy D; Talukdar R (2016). NF-kappaB in acute pancreatitis: Mechanisms and therapeutic potential. Pancreatology 16, 477–88. [DOI] [PubMed] [Google Scholar]
- Ji B; Bi Y; Simeone D; Mortensen RM; Logsdon CD (2001). Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology 121, 1380–90. [DOI] [PubMed] [Google Scholar]
- Kim MS; Hong JH; Li Q; Shin DM; Abramowitz J; Birnbaumer L; Muallem S (2009). Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137, 1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J; Kim ML; Heo WD; Jones JT; Myers JW; Ferrell JE Jr.; Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15, 1235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X; Berry CT; Ruthel G; Madara JJ; MacGillivray K; Gray CM; Madge LA; McCorkell KA; Beiting DP; Hershberg U; May MJ; Freedman BD (2016). T Cell Receptor-induced Nuclear Factor kappaB (NF-kappaB) Signaling and Transcriptional Activation Are Regulated by STIM1- and Orai1-mediated Calcium Entry. J Biol Chem 291, 8440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A; Gerloff A; Su HY; Xu Z; Go A; Hu C; French SW; Wilson JS; Apte MV; Waldron RT; Pandol SJ (2017a). The Combination of Alcohol and Cigarette Smoke Induces Endoplasmic Reticulum Stress and Cell Death in Pancreatic Acinar Cells. Gastroenterology 153, 1674–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A; Nan L; French SW; Bezerra JA; Gukovskaya AS; Pandol SJ (2006). Pancreas recovery following cerulein-induced pancreatitis is impaired in plasminogen-deficient mice. Gastroenterology 131, 885–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A; Waldron RT; Mareninova OA; Shalbueva N; Deng N; Su HY; Thomas DD; Jones EK; Messenger SW; Yang J; Hu C; Gukovsky I; Liu Z; Groblewski GE; Gukovskaya AS; Gorelick FS; Pandol SJ (2017b). Human Pancreatic Acinar Cells: Proteomic Characterization, Physiologic Responses, and Organellar Disorders in ex Vivo Pancreatitis. Am J Pathol 187, 2726–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lur G; Haynes LP; Prior IA; Gerasimenko OV; Feske S; Petersen OH; Burgoyne RD; Tepikin AV (2009). Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol 19, 1648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareninova OA; Sung KF; Hong P; Lugea A; Pandol SJ; Gukovsky I; Gukovskaya AS (2006). Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem 281, 3370–81. [DOI] [PubMed] [Google Scholar]
- Masamune A; Kikuta K; Watanabe T; Satoh K; Satoh A; Shimosegawa T (2008). Pancreatic stellate cells express Toll-like receptors. J Gastroenterol 43, 352–62. [DOI] [PubMed] [Google Scholar]
- Mercer JC; Dehaven WI; Smyth JT; Wedel B; Boyles RR; Bird GS; Putney JW Jr. (2006). Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem 281, 24979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S; Schoeffield MS; Fimmel CJ; Pandol SJ (1988a). Agonist-sensitive calcium pool in the pancreatic acinar cell. I. Permeability properties. Am J Physiol 255, G221–8. [DOI] [PubMed] [Google Scholar]
- Muallem S; Schoeffield MS; Fimmel CJ; Pandol SJ (1988b). Agonist-sensitive calcium pool in the pancreatic acinar cell. II. Characterization of reloading. Am J Physiol 255, G229–35. [DOI] [PubMed] [Google Scholar]
- Muili KA; Jin S; Orabi AI; Eisses JF; Javed TA; Le T; Bottino R; Jayaraman T; Husain SZ (2013a). Pancreatic acinar cell nuclear factor kappaB activation because of bile acid exposure is dependent on calcineurin. J Biol Chem 288, 21065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muili KA; Wang D; Orabi AI; Sarwar S; Luo Y; Javed TA; Eisses JF; Mahmood SM; Jin S.; Singh VP.; Ananthanaravanan M; Perides G; Williams JA.; Molkentin JD.; Husain SZ (2013b). Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J Biol Chem 288, 570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R; Criddle DN; Gukvoskaya A; Pandol S; Petersen OH; Sutton R (2008). Mitochondrial injury in pancreatitis. Cell Calcium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R; Mareninova OA; Odinokova IV; Huang W; Murphy J; Chvanov M; Javed MA; Wen L; Booth DM; Cane MC; Awais M; Gavillet B; Pruss RM; Schaller S; Molkentin JD; Tepikin AV; Petersen OH; Pandol SJ; Gukovsky I; Criddle DN; Gukovskaya AS; Sutton R; and NPBRU (2015). Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JA; Criddle DN; Sherwood M; Chvanov M; Mukherjee R; McLaughlin E; Booth D; Gerasimenko JV; Raraty MG; Ghaneh P; Neoptolemos JP; Gerasimenko OV; Tepikin AV; Green GM; Reeve JR Jr.; Petersen OH; Sutton R (2008). Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135, 632–41. [DOI] [PubMed] [Google Scholar]
- Omary MB; Lugea A; Lowe AW; Pandol SJ (2007). The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest 117, 50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MG; Xiong Y; Chen F (2013). NFAT gene family in inflammation and cancer. Curr Mol Med 13, 543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandol SJ; Gukovskaya A; Bahnson TD; Dionne VE (1994). Cellular mechanisms mediating agonist-stimulated calcium influx in the pancreatic acinar cell. Ann N YAcadSci 713, 41–8. [DOI] [PubMed] [Google Scholar]
- Pandol SJ; Schoeffield MS; Fimmel CJ; Muallem S (1987). The agonist-sensitive calcium pool in the pancreatic acinar cell. Activation of plasma membrane Ca2+ influx mechanism. J Biol Chem 262, 16963–8. [PubMed] [Google Scholar]
- Park KS; Kim SH; Das A; Yang SN; Jung KH; Kim MK; Berggren PO; Lee Y; Chai JC; Kim HJ; Chai YG (2016). TLR3-/4-Priming Differentially Promotes Ca(2+) Signaling and Cytokine Expression and Ca(2+)-Dependently Augments Cytokine Release in hMSCs. Sci Rep 6, 23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perides G; Laukkarinen JM; Vassileva G; Steer ML (2010). Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138, 715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH; Tepikin AV; Gerasimenko JV; Gerasimenko OV; Sutton R; Criddle DN (2009). Fatty acids, alcohol and fatty acid ethyl esters: toxic Ca2+ signal generation and pancreatitis. Cell Calcium 45, 634–42. [DOI] [PubMed] [Google Scholar]
- Prakriya M; Feske S; Gwack Y; Srikanth S; Rao A; Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–3. [DOI] [PubMed] [Google Scholar]
- Qi M; McFadden B; Valiente L; Omori K; Bilbao S; Juan J; Rawson J; Oancea AR; Scott S; Nair I; Ferreri K; Mullen Y; Dafoe D; Ei-Shahawy M; Kandeel F; Al-Abdullah H (2015). Human Pancreatic Islets Isolated From Donors With Elevated HbA1c Levels: Islet Yield and Graft Efficacy. Cell Transplant 24, 1879–86. [DOI] [PubMed] [Google Scholar]
- Roos J; DiGregorio PJ; Yeromin AV; Ohlsen K; Lioudyno M; Zhang S; Safrina O; Kozak JA; Wagner SL; Cahalan MD; Velicelebi G; Stauderman KA (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169, 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalbueva N; Mareninova OA; Gerloff A; Yuan J; Waldron RT; Pandol SJ; Gukovskaya AS (2013). Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 144, 437–446 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauderman KA (2018). CRAC channels as targets for drug discovery and development. Cell Calcium 74, 147–159. [DOI] [PubMed] [Google Scholar]
- Su HY; Waldron RT; Gong R; Ramanujan VK; Pandol SJ; Lugea A (2016). The Unfolded Protein Response Plays a Predominant Homeostatic Role in Response to Mitochondrial Stress in Pancreatic Stellate Cells. PLoS One 11, e0148999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton R; Petersen OH; Pandol SJ (2008). Pancreatitis and calcium signaling: Report of an international workshop, vol. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmary P; Gukovsky I (2016). The role of cytokines and inflammation in the genesis of experimental pancreatitis In Pancreatitis, ed. Williams JA, pp. 42–52. Pancreapedia: Exocrine Pancreas Knowledge Base, Mountain View, CA. [Google Scholar]
- Trebak M; Putney JW Jr. (2017). ORAI Calcium Channels. Physiology (Bethesda) 32, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M; Peinelt C; Beck A; Koomoa DL; Rabah D; Koblan-Huberson M; Kraft S; Turner H; Fleig A; Penner R; Kinet JP (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina S; Collier D; Chvanov M; Middlehurst B; Beckett AJ; Prior IA; Criddle DN; Begg M; Mikoshiba K; Sutton R; Tepikin AV (2015). The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem J 465, 405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina S; Longbottom R; Sutton R; Petersen OH; Tepikin A (2002). Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. JPhysiol 540, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina SG; Barrow SL; Gerasimenko OV; Petersen OH; Tepikin AV (2004). Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem 279, 27327–38. [DOI] [PubMed] [Google Scholar]
- Wen L; Javed TA; Yimlamai D; Mukherjee A; Xiao X; Husain SZ (2018). Transient High Pressure in Pancreatic Ducts Promotes Inflammation and Alters Tight Junctions via Calcineurin Signaling in Mice. Gastroenterology 155, 1250–1263 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L; Voronina S; Javed MA; Awais M; Szatmary P; Latawiec D; Chvanov M; Collier D; Huang W; Barrett J; Begg M; Stauderman K; Roos J; Grigoryev S; Ramos S; Rogers E; Whitten J; Velicelebi G; Dunn M; Tepikin AV; Criddle DN; Sutton R (2015). Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149, 481–92.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL; Yeromin AV; Zhang XH; Yu Y; Safrina O; Penna A; Roos J;Stauderman KA; Cahalan MD (2006). Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci US A 103, 9357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu ZD; Yu T; Liu HJ; Jin J; He J (2018). SOCE induced calcium overload regulates autophagy in acute pancreatitis via calcineurin activation. Cell Death Dis 9, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]