

INTRODUCTION
Glioblastoma is the most common malignant primary brain tumor in adults. Despite aggressive initial treatment, recurrence is inevitable and almost always fatal. Treatment options for recurrent disease are limited and do not substantially improve survival. In recent years, molecular characterization of gliomas has revealed recurrent somatic mutations, structural rearrangements, and epigenetic alterations. These genomic alterations have implications for prognosis and response to treatment. IDH1/2 mutations are associated with a more favorable prognosis among glioblastomas, whereas tumors with MGMT promoter hypermethylation are more likely to respond to treatment with alkylating agents.1,2 The pattern of genomic alterations can also provide insight into the origin and evolution of the tumor. For example, the presence of coexistent mutations in IDH1 and ATRX suggest a secondary glioma that has progressed from a lower-grade glioma.3-5 In addition to informing prognosis and diagnostic classification, some genomic alterations in glioma represent potential therapeutic targets. Specifically, recurrent alterations in one of several receptor tyrosine kinases (RTKs), including EGFR, PDGFRA, MET, and FGFR1/2/3, have been observed in the majority of glioblastomas.6 Unfortunately, efforts to develop genomically targeted therapy for the treatment of glioblastoma have been unsuccessful to date.7 It remains unknown the extent to which the lack of activity of targeted therapy may be attributed to so-called key alterations that do not represent true clonal drivers that are critical to tumor growth and progression compared with the simple inability of existing drugs to achieve therapeutic exposures within the CNS.
In addition to these more common genomic alterations, less common fusions that involve the kinase domain of tropomyosin receptor kinases (TRKs; A/B/C) encoded by the genes, NTRK1, NTRK2, and NTRK3 have also been identified in gliomas.8-11 In other cancer types, selective inhibition of these TRK fusions has resulted in remarkable efficacy12; however, it remains unknown whether this therapeutic strategy is effective in primary brain tumors. Here, we report the case of a young woman with recurrent glioblastoma treated with larotrectinib, a first-in-class, potent, highly selective TRK inhibitor. To our knowledge, this is the first report of the use of a TRK inhibitor in a patient with a TRK fusion–positive glioblastoma.
CASE REPORT
A 30-year-old woman was initially diagnosed in 2011 with WHO grade III anaplastic astrocytoma of the right temporal lobe. She underwent gross total resection, followed by adjuvant radiation as part of RTOG 0834.13 Figure 1 includes a detailed clinical history. In 2015, routine surveillance magnetic resonance imaging (MRI) showed increased enhancement in the right temporal lobe that was consistent with disease recurrence. She received seven cycles of temozolomide, which was subsequently terminated as a result of both clinical and radiographic progression. Resection of the recurrent tumor demonstrated malignant progression to WHO grade IV glioblastoma that harbored IDH1 R132 and was ATRX null by immunohistochemistry. The patient was treated on a clinical trial of temozolomide and an indoleamine-pyrrole 2,3-dioxygenase inhibitor but experienced progression within 6 weeks. MRI demonstrated a large mass that was centered at the medial margin of the right frontal to parietal resection cavity with extension into the basal ganglia, thalamus, and right superior cerebral peduncle with associated cerebral edema. This precipitated an urgent piecemeal debulking as a result of rapid clinical deterioration. The tumor was resected in three specimens: temporal pole, hippocampal area, and central tumor. Pathologic review confirmed glioblastoma in the central and hippocampal components as well as infiltrating astrocytoma in the temporal pole (Data Supplement). She was then treated with combined lomustine and bevacizumab.
Fig 1.
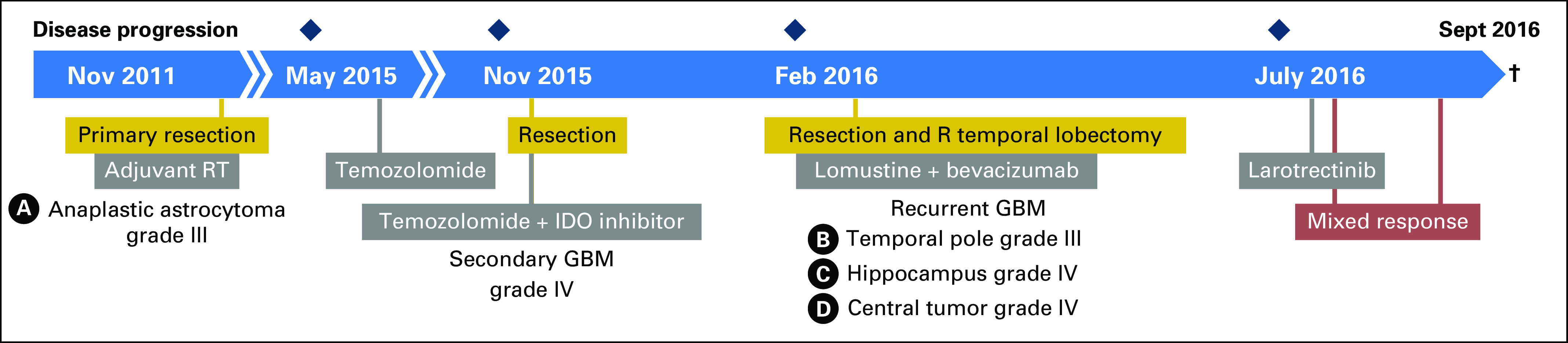
Timeline of clinical course. Chronology of major clinical events and lines of therapy along the glioblastoma (GBM) disease course. Gold boxes indicate surgeries, gray boxes show radiation therapy (RT) and systemic treatment, and the red box indicates response to targeted therapy. Resected specimens are labeled A to D. IDO, indoleamine 2,3-dioxygenase; R, right. (†) Patient death.
Given the rapid clinical deterioration, to identify novel therapeutic options, we performed prospective molecular characterization of the central tumor using targeted anchored multiplex RNA sequencing, which revealed an in-frame EML4-NTRK3 fusion that involved the kinase domain of NTRK3 in the dominant clone of the central tumor (85.4% read support). The EML4-NTRK3 fusion was orthogonally confirmed by Sanger sequencing of the central tumor. The patient continued on lomustine and bevacizumab until disease progression several months later. At this time, she consented to, and was started on, larotrectinib through an institutional review board–approved emergency expanded access application. Three weeks into the treatment, a brain MRI indicated a response to treatment, the degree of which was radiologically discrepant between disease sites (Fig 2). Whereas there was significant periventricular response to larotrectinib—tumor shrinkage of 67 × 52 mm to 8 × 4 mm, which corresponded to a partial response by Response Assessment in Neuro-Oncology criteria, and of 162 cm3 to 57 cm3, which represented a 65% volumetric decrease—there was no shrinkage in the right supraorbital area and occipital lobe. A repeat MRI 1 month later demonstrated disease progression with increasing tumor in the latter sites. Ultimately, the patient experienced clinical deterioration as a result of these two areas of progression and discontinued larotrectinib. She died of her disease shortly thereafter.
Fig 2.

Response to larotrectinib. Serial contrast T1-weighted images at pretreatment (left column) show enhancing disease in the infundibulum (green arrow #1), basal ganglia (green arrow #2), frontal lobe (green arrow #3), subependyma, and corpus callosum (green arrow #4), which is improved by day 18 of treatment with larotrectinib (middle column). At day 40 (right column), there is worsening enhancing disease in the inferior frontal lobe (red arrow #1), occipital lobe (red arrow #2), leptomeninges (red arrow #3), and parietal lobe (red arrow #4).
We hypothesized that intratumoral heterogeneity of the fusion oncoprotein was the basis for the marked response to therapy observed in some areas of her tumor but not others. To explore this, we performed RNA sequencing and fluorescence in situ hybridization on the hippocampal and temporal pole specimens. Of interest, the NTRK fusion was not detected in either lesion, a finding that is consistent with the radiographic effects of larotrectinib and, overall, a subclonal sensitizing fusion. To better characterize the full extent of intratumoral heterogeneity, we subsequently performed whole-exome sequencing of her original tumor from the right temporal lobe and the three resected areas of her recurrence. Although all four tumors shared truncal and astrocytoma-defining mutations in IDH1, ATRX, and TP53, the regions evolved divergently thereafter (Fig 3A). Only the hippocampal and central tumors contained a focal CDKN2A/B deletion and a mutational repertoire indicative of temozolomide-induced hypermutation (Figs 3B and 3C and Data Supplement). Moreover, the hippocampal and central tumors had distinct RTK alterations, including a PDGFRA amplification in the hippocampal specimen and the EML4-NTRK3 fusion in the central tumor. Furthermore, although both lesions harbored numerous C>T and G>A transitions at CpN sites, which was consistent with a signature of temozolomide-induced hypermutation,14 the majority of these mutations were private to each tumor, which suggests that multiple clonally related tumors underwent independent waves of therapy-associated mutagenesis. The relatively few shared alterations did not seem to be temozolomide induced and likely preceded this therapy. Thus, our genomic characterization of the patient’s tumor confirmed profound intratumoral heterogeneity, including subclonality of the NTRK fusion.
Fig 3.

Molecular characterization of initial and recurrent glioma. (A) Evolutionary relationships among sequenced tumor specimens inferred from somatic mutational data. Key functional alterations are shown for the branches in which they arose (bold). Among hypermutated specimens (c and d), branches are abbreviated and the number of temozolomide (TMX) -induced mutations in each lesion is specified. (B) Pattern of hallmark truncal mutations in IDH1, TP53, and ATRX across the initial tumor (a) and recurrent specimens (b-d) as well as private driver events in postprogression samples (c and d, as indicated). (C) Overall somatic mutational burden (top) of all tumor samples is shown, which indicates hypermutation in lesions c and d, but not in the initial pretreatment tumor or post-TMZ temporal pole tumor (b). At bottom is the fraction of mutations per sample attributable to the mutagenic effect of TMZ. SNV, single nucleotide variant.
DISCUSSION
Gene fusions that involve the TRK family of kinases have been identified in a wide array of adult and pediatric cancers in which they result in constitutive kinase activation and drive tumor growth.8,15-20 Larotrectinib is a novel small-molecule inhibitor that specifically targets TRK and has demonstrated unprecedented efficacy in TRK fusion–positive cancers irrespective of tumor histology, with the majority of patients achieving rapid and durable clinical responses12,21; however, the efficacy of TRK inhibition in gliomas is currently unknown. Although this patient ultimately succumbed to her disease, the observed mixed response to therapy suggests that larotrectinib penetrated the blood-brain barrier and had activity against the cell population in her heterogeneous tumor that harbored the EML4-NTRK3 fusion.
Intratumoral heterogeneity in glioblastoma has become increasingly appreciated as a contributor to therapeutic resistance and disease recurrence.22,23 This case highlights that molecular profiling of a single area may not capture the full genomic landscape of a tumor. Kinase fusions, including TRK fusions, are typically clonal when present in other diseases and thus represent some of the most vulnerable targets of novel therapeutics.8 The presence of the TRK fusion in only one subclone suggests that distinct cell populations in this tumor relied on different driver alterations; therefore, genomic heterogeneity limits, but does not preclude, clinical benefit from a targeted agent. The extent to which this is true for other fusion-positive glioblastomas remains unclear. Another aspect of the profound intratumoral heterogeneity observed here was somatic hypermutation, a well-recognized consequence of temozolomide therapy that occurs in a subset of treated patients.24 When present, this may increase the risk of malignant progression via the introduction of novel driver mutations that activate pathways that are quiescent at diagnosis, but as evident here, does not preclude sensitivity to molecularly targeted therapy.25 The observation that hypermutation does not occur in all patients whose tumors are exposed to this therapy suggests that certain cancers are more vulnerable to the mutagenic effect of alkylating agents. Our report demonstrates that susceptibility can differ even within a single tumor, resulting in hypermutation of some, but not all, subclones; however, precisely what molecular context underlies these differences is not yet known. An improved understanding of what permits this phenotypic change and whether a biomarker of susceptibility to therapy-induced hypermutation exists may ultimately lead to more informed treatment decisions for patients with glioma.
In conclusion, these data suggest that TRK fusions may represent potential therapeutic targets in glioblastoma, a disease with few effective therapies, and that larotrectinib has the potential for antitumor activity in CNS tumors that harbor these fusions. Whereas patients with TRK fusion–positive primary brain tumors are eligible for the ongoing NAVIGATE phase II larotrectinib trial (ClinicalTrials.gov identifier: NCT0257643), our data indicate that identifying tumors with truncal TRK fusions is necessary for realizing the true potential of TRK-directed therapy in this disease.
ACKNOWLEDGMENT
We thank the members of the Marie-Josée and Henry R. Kravis Center for Molecular Oncology for discussions and support.
Footnotes
Funded by the Sontag Foundation, American Cancer Society Grant No. RSG-15-067-01-TBG, and National Institutes of Health Grants No. R01-CA204749 and T32-CA009207.
Presented at the 2017 American Association for Cancer Research Annual Meeting, Washington, DC, April 1-5, 2017.
AUTHOR CONTRIBUTIONS
Conception and design: Alison M. Schram, Alexander Drilon, Nora Ku, David M. Hyman, Barry S. Taylor
Financial support: David M. Hyman
Administrative support: Bethany Hanusch
Provision of study materials or patients: Christian Grommes, Nora Ku, David M. Hyman
Collection and assembly of data: Alison M. Schram, Philip Jonsson, Alexander Drilon, Ryma Benayed, Bethany Hanusch, Robert J. Young, Christian Grommes, David M. Hyman, Barry S. Taylor
Data analysis and interpretation: Alison M. Schram, Philip Jonsson, Alexander Drilon, Tejus A. Bale, Jaclyn F. Hechtman, Robert J. Young, Thomas J. Kaley, David M. Hyman, Barry S. Taylor
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Alison M. Schram
No relationship to disclose
Philip Jonsson
No relationship to disclose
Alexander Drilon
Consulting or Advisory Role: Ignyta, Loxo Oncology
Consulting or Advisory Role: TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Genentech, Takeda Pharmaceuticals, Helsinn Therapeutics, BeiGene
Tejus A. Bale
No relationship to disclose
Jaclyn F. Hechtman
Honoraria: Medscape
Consulting or Advisory Role: Navigant Consulting
Ryma Benayed
No relationship to disclose
Bethany Hanusch
No relationship to disclose
Robert J. Young
Stock and Other Ownership Interests: Agios, Alexion Pharmaceuticals, Biogen, Celgene, Gilead Sciences, Karyopharm Therapeutics, Spark Therapeutics, Regeneron, Stemline Therapeutics, Vertex
Consulting or Advisory Role: Agios, Puma Biotechnology
Research Funding: Agios (Inst)
Christian Grommes
Consulting or Advisory Role: BTG
Nora Ku
Employment: Loxo Oncology
Stock and Other Ownership Interests: Loxo Oncology, Novartis (I), BioTheranostics (I), Pfizer (I), Amgen (I), Genentech (I), AstraZeneca (I), Macrogenics (I), Peregrine Pharmaceuticals (I), Pierian Bioscience (I), Cascadian Therapeutics (I)
Travel, Accommodations, Expenses: Novartis (I), BioTheranostics (I), Pfizer (I), Amgen (I), Genentech (I), AstraZeneca (I), Macrogenics (I), Peregrine Pharmaceuticals (I), Pierian Bioscience (I)
Thomas J. Kaley
No relationship to disclose
David M. Hyman
Consulting or Advisory Role: Atara Biotherapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiopharm Group, ArQule, Genentech
Research Funding: AstraZeneca, Puma Biotechnology, Loxo Oncology
Barry S. Taylor
No relationship to disclose
REFERENCES
- 1.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network. Brat DJ, Verhaak RG, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kannan K, Inagaki A, Silber J, et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012;3:1194–1203. doi: 10.18632/oncotarget.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan CW, Verhaak RGW, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. Erratum: Cell 157:753, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Touat M, Idbaih A, Sanson M, et al. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann Oncol. 2017;28:1457–1472. doi: 10.1093/annonc/mdx106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schram AM, Chang MT, Jonsson P, et al. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735–748. doi: 10.1038/nrclinonc.2017.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Lee Y, Cho HJ, et al. NTRK1 fusion in glioblastoma multiforme. PLoS One. 2014;9:e91940. doi: 10.1371/journal.pone.0091940. Erratum: PLoS One 9:e101140, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731–739. doi: 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Bent MJ, Baumert B, Erridge SC, et al. Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: A phase 3, randomised, open-label intergroup study. Lancet. 2017;390:1645–1653. doi: 10.1016/S0140-6736(17)31442-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter C, Smith R, Cahill DP, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006;66:3987–3991. doi: 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell JP, Powell DJ, Cunnane M, et al. The TRK-T1 fusion protein induces neoplastic transformation of thyroid epithelium. Oncogene. 2000;19:5729–5735. doi: 10.1038/sj.onc.1203922. [DOI] [PubMed] [Google Scholar]
- 16.Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 17.Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469–1472. doi: 10.1038/nm.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116. doi: 10.1038/ncomms4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. doi: 10.1158/2159-8290.CD-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shukla N, Roberts SS, Baki MO, et al. Successful targeted therapy of refractory pediatric ETV6-NTRK3 fusion-positive secretory breast carcinoma. JCO Precis Oncol. doi: 10.1200/PO.17.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sottoriva A, Spiteri I, Piccirillo SGM, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171:1042.e10–1056.e10. doi: 10.1016/j.cell.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]