

SUMMARY
Channelrhodopsin2 (ChR2) optogenetic excitation is widely used to study neurons, astrocytes, and circuits. Using complementary approaches in situ and in vivo, we found that ChR2 stimulation leads to significant transient elevation of extracellular potassium ions by ~5 mM. Such elevations were detected in ChR2-expressing mice, following local in vivo expression of ChR2(H134R) with adeno-associated viruses (AAVs), in different brain areas and when ChR2 was expressed in neurons or astrocytes. In particular, ChR2-mediated excitation of striatal astrocytes was sufficient to increase medium spiny neuron (MSN) excitability and immediate early gene expression. The effects on MSN excitability were recapitulated in silico with a computational MSN model and detected in vivo as increased action potential firing in awake, behaving mice. We show that transient, physiologically consequential increases in extracellular potassium ions accompany ChR2 optogenetic excitation. This coincidental effect may be important to consider during astrocyte studies employing ChR2 to interrogate neural circuits and animal behavior.
In Brief
Using multiple approaches, Octeau et al. discover that optogenetic excitation of ChR2-expressing cells leads to significant transient extracellular potassium ion elevations that increase neuronal excitability and immediate early gene expression in neurons following in vivo stimulation.
Graphical Abstract
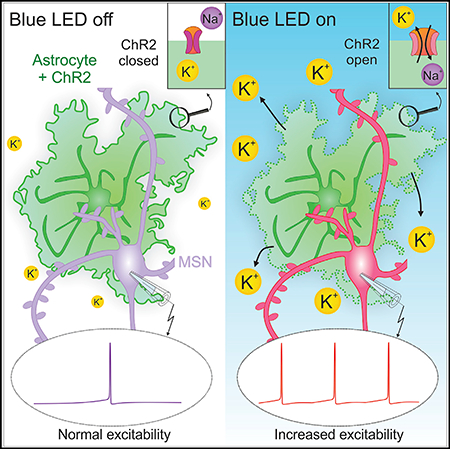
INTRODUCTION
The emergent field of optogenetics has revolutionized neuroscience since the landmark discovery that optical stimulation of channelrhodopsin2 (ChR2)-expressing cells elicits membrane potential depolarization (Nagel et al., 2003) and by the subsequent demonstration that such responses can be used to evoke action potential (AP) firing in neurons (Boyden et al., 2005). ChR2 and its variants are now widely used to explore neurons, glia, and multiple other cell types, permitting straightforward optical stimulation of genetically targeted cell populations and exploration of their contributions to neural circuit properties and animal behaviors (Bernstein and Boyden, 2011; Deisseroth, 2015).
The usual interpretation of neuronal studies that employ ChR2 is that selective, brief optical excitation of the targeted neurons causes sufficient membrane depolarization to engage endogenous voltage-gated ion channels and lead to AP firing in ChR2- expressing neurons, which then may cause ensuing changes in neural circuits controlling behavior. Thus, ChR2 provides an initial depolarizing inward current to bring the neurons to threshold. In this way, ChR2 has become a powerful tool for functional interrogation of neural circuits in vivo in a multitude of scenarios (Bernstein and Boyden, 2011; Deisseroth, 2015). Given its ease of use, ChR2-mediated excitation has also been used in a similar manner to explore the roles of astrocytes in regulating mouse behavior and circuits. For example, ChR2- driven astrocyte excitation has been used to implicate astrocytes in breathing (Gourine et al., 2010), response selectivity in the visual cortex (Perea et al., 2014), regulation of blood flow (Masamoto et al., 2015), sleep (Pelluru et al., 2016; Yamashita et al., 2014), and cerebellar motor function (Sasaki et al., 2012). It has also been used to regulate neuronal activity by engaging astrocyte mechanisms (Deemyad et al., 2018; Mederos et al., 2019; Shen et al., 2017) and to mimic the effects of neuronal activity on astrocyte glutamate transport (Gomez et al., 2019). However, astrocytes have few if any voltage-gated ion channels and display no propagated voltage signals. In the absence of a physiologically relevant endogenous voltage-dependent excitability mechanism to engage following brief ChR2 stimulation, several astrocyte studies have instead used quite long periods of ChR2 stimulation lasting from seconds to tens of seconds in order to observe subsequent effects on single neurons, circuits, and, in some cases, animal behavior. In some of these cases, ChR2-mediated excitation of astrocytes has suggested that astrocytes regulate circuits via gliotransmission, which was evoked by optical stimulation (Bang et al., 2016; Figueiredo et al., 2011).
We have been exploring astrocyte-neuron interactions in striatal microcircuits (Chai et al., 2017; Jiang et al., 2016; Octeau et al., 2018a; Srinivasan et al., 2016; Tong et al., 2014; Yu et al., 2018). As part of this, buoyed by the aforementioned studies, we used Thy1-ChR2-YFP mice (Arenkiel et al., 2007) to drive trains of APs in premotor cortical neurons and to study the consequences of this increased excitation upon striatal MSNs and astrocytes. However, during the experiments, we identified an unexpected slow inward current in both striatal medium spiny neurons (MSNs) and, surprisingly, in astrocytes. We describe the properties of the slow inward current and subsequent experiments that suggested that it was due to release of K+ into the extracellular space, which we carefully explored. The data reveal a potentially important consequence of ChR2-mediated optogenetic excitation, which is likely an important confounding feature for astrocyte studies employing prolonged ChR2 stimulation, especially for behavioral experiments where electrophysiological studies are not performed. Elevated K+ is expected to affect the physiology of not only neurons, but also diverse brain cell types (McDonough and Youn, 2017), as well as affect local blood flow and functional hyperemia (Girouard et al., 2010). These could influence the interpretation of, and possibly even cause, endpoint measures at different temporal scales, especially in vivo, in studies that employ ChR2 in astrocytes.
RESULTS
Transient Extracellular K+ Elevations during Optical Stimulation in Thy1-ChR2-YFP Mice
ChR2 and its variants are the most commonly used tools for optogenetic excitation (Zhang et al., 2011). Immunohistochemistry (IHC) revealed strong expression of ChR2-YFP in Thy1-ChR2-YFP mice (Arenkiel et al., 2007) with strong expression in layer V cortical neurons (Figure 1A; n = 3 mice). As expected with the Thy1 promoter, there was little expression in the striatum (Figure 1A), but at higher magnification, there was strong colocalization between ChR2-YFP and neurofilament (Nflm) in corticostriatal white matter tracts, but no significant colocalization between ChR2-YFP and neuronal nuclei (NeuN) or with the astrocyte marker S100β (Figures 1B and 1C; n = 12 images, n = 4 mice). Next, we recorded in whole-cell voltage-clamp mode from MSNs in sagittal brain slices and employed wide-field optical stimulation using parameters comparable to those used in vivo (Gibson et al., 2014). We used 473 nm light, ~0.02 mW/mm2 power, and 25 ms flashes at 20 Hz for 30 s and recorded optically evoked excitatory postsynaptic currents (EPSCs) in all MSNs from Thy1-ChR2-YFP mice, but not from wild-type (WT) mice (Figure 1D; n = 15 cells, n = 8 mice). The evoked EPSCs were accompanied by the development of a slow inward current (at −75.6 mV after correcting for the tip potential) during the optical stimulation train (Figure 1D; see Figure S1 for expanded traces). This inward current peaked at −90 ± 19 pA, developed with a time constant (τ) of 1.1 ± 0.1 s, was maintained during the stimulation train, and decayed with a τ of 3.7 ± 0.2 s (Figure 1D; n = 15 cells, n = 4 mice). Furthermore, recordings from striatal astrocytes revealed a similar inward current of −281 ± 48 pA and with rise and decay τ values of 1.0 ± 0.1 and 4.3 ± 0.2 s, respectively (Figure 1E; n = 15 cells, n = 8 mice). The finding that optically evoked inward currents could be measured in striatal MSNs and astrocytes, even though neither cell type expressed ChR2-YFP (Figures 1A–1C), argues against photocurrents; the kinetics of the inward currents were also nearly three orders of magnitude slower than ChR2 activation and deactivation (Zhang et al., 2011). Additionally, the astrocyte responses argue against ionotropic glutamate receptor activation during corticostriatal axon stimulation (see later section) because such ion channels are unlikely to be expressed in striatal astrocytes (Chai et al., 2017). Moreover, the finding that the peak inward currents were larger in astrocytes than in MSNs suggests they may be due to elevation of K+. The astrocyte membrane has high resting K+ conductance and is thus sensitive to extracellular K+ increases.
Figure 1. Optical Stimulation of ChR2-Expressing Cells Evokes Transient Elevation of Extracellular K+.

(A) Representative image showing brain distribution of channelrhodopsin2 (ChR2-YFP; green) in a Thy1-ChR2-YFP mouse brain section together with neurofilament (Nflm, red). The inset shows strong colocalization of ChR2 and NeuN positive neurons in cortex.
(B) The images show colocalization of ChR2 with Nflm positive axonal tracts in the striatum, but not with NeuN or S100β positive neurons and astrocytes.
(C) Pearson’s colocalization correlation coefficient (r) for images such as those shown in (B).
(D) Left: schematic depicting patch-clamp of medium spiny neurons (MSNs) in the striatum. Middle: representative current waveforms from MSNs in Thy1-ChR2-YFP or WT mice in response to a train of flashes to stimulate corticostriatal axons. Right: peak amplitude of the first EPSC upon blue light flashes.
(E) Left: schematic depicting patch-clamp of astrocytes in the striatum. Middle: representative current waveforms from striatal astrocytes in Thy1-ChR2-YFP or WT mice in response to a train of flashes. Right: quantification of the steady-state inward current (Iss) from MSNs and astrocytes in the striatum from WT or Thy1-ChR2-YFP mice.
(F) Left: schematic depicting the experimental setup for recording of K+ dynamics in brain slices with blue light activation ofChR2. Right: graph plots the response of the K+ electrode to various K+ concentrations in the extracellular fluid.
(G) Left: waveforms of changes in K+ concentration in response to a single flash of various light intensities. Right: quantification of the peak amplitude of K+ in WT and Thy1-ChR2-YFP mice in response to single flashes of various light intensities.
(H) Left: waveform depicting changes in K+ in response to different numbers of flashes in Thy1-ChR2 and WT mice. Right: quantification of the peak amplitude of the K+ elevation in WT and Thy1-ChR2-YFP mice in response to different numbers of flashes.
(I) Left: waveform depicting K+ responses in the striatum with TTX or TTX, CNQX, and APV. Right: average data for experiments in the left panel.
(J) Left: schematic depicting patch-clamp of pyramidal neurons in the cortex. Middle: representative voltage waveform in cortical pyramidal cells from WT or Thy1-ChR2 mice with light flashes. Right: quantification of the number of action potentials.
(K) Left: quantification of the peak K+ concentration in the cortex. Middle: representative waveform of K+ concentrations in the cortex. Right: average data from the experiment depicted in the left panel. Data shown as mean ± SEM. In some cases, the error bars are smaller than the symbol used to represent the mean. See also Figures S1—S3.
We made K+ selective microelectrodes to directly measure K+ in the extracellular space of striatal slices before, during, and after optical stimulation in Thy1-ChR-YFP mice. The calibrated electrodes displayed the expected ~54 mV change for a 10-fold change in K+ (Figure 1F; mean of 4 electrodes shown). Furthermore, single 25 ms flashes at a range of powers (0–0.09 mW/mm2) elicited clear, transient extracellular K+ elevations in Thy1-ChR2-YFP mice, but not in WT mice (Figure 1G; n = 14, n = 4 mice). Such transients peaked and decayed over seconds, as expected for release and subsequent washout of K+ in the extracellular space (Figure 1G). At power of 0.02 mW/mm2, 30 s trains of light flashes at 20 Hz elicited significant increases in extracellular K+ that were maintained for the duration of the stimulus, with rise and decay τ values of 2.1 ± 0.6 and 6.1 ± 0.5 s (Figure 1H; Table 2). The kinetics of the K+ elevations were thus similar to the steady-state inward currents recorded from MSNs and astrocytes (Figures 1D and 1E): the slightly slower time course measured with the electrodes may reflect sampling from the tip of the K+ electrode, whereas the MSN and astrocyte recordings sampled within the tissue. At peak, the extracellular K+ levels were elevated significantly to 7.2 ± 0.3 mM from a bath concentration of 4.5 mM (Figure 1H). Furthermore, the change in K+ evoked by a train of stimuli was not blocked by tetrodotoxin (TTX; 500 nM) or a combination of TTX, cyanquixaline (6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μM), and D 2-Amino-5-phosphonopentanoic acid (APV; 10 μM) to block APs and ionotropic glutamate receptors (Figure 1I; n = 14–23 slices, n = 4 mice). Similar, albeit as expected smaller, changes were measured when the flash duration was reduced to 2 ms (Figures S2A-S2D; n = 14–23 slices, n = 4 mice, 20 Hz). We also performed a set of experiments in the cortex to ascertain if the K+ elevations were a striatal specific effect. However, during optical stimulation, we measured slow-seconds time scale cortical pyramidal neuron depolarization (rise and decay 0.3 ± 0.1 and 6.8 ± 0.7 s, respectively) and significant-seconds time scale elevations in extracellular K+ (8.7 ± 0.5 mM, with rise and decay 2.8 ± 0.2 and 9.1 ± 1.1 s, respectively) that were not blocked by TTX (Figures 1J and 1K; n = 16 slices, n = 4 mice). Responses were also observed with 2 ms flashes (Figures S2E and S2F; n = 12 slices, n = 4 mice).
Table 2.
Model Parameters Used in MSN Computations Shown in Figure 4
| S∞ | τ(V) | ||||||
|---|---|---|---|---|---|---|---|
| Current | Gate | (nS.μm−2) | Vs (mV) | ks (mV) | τ0 (ms) | Vτ (mV) | kτ (mV) |
| INat | , hNat | 1.61 | See STAR Methods | ||||
| INap | mNap | 3.1e−3 | −47.8 | 3.1 | 1.0 | - | - |
| INas | mNas | 0.02 | −16.0 | 9.4 | 637.8 | −33.5 | 26.3 |
| IKDR | 0.11 | See STAR Methods | |||||
| IKIR | mKIR | 0.33e−3 | −100 | −10 | – | – | – |
| IKRP | mKRP | 0.155 | −13.4 | 12.1 | 206.2 | −53.9 | 26.5 |
| hKRP | −55.0 | −19.0 | See STAR Methods | ||||
| IKAf | mKAf | 0.0017 | −33.1 | 7.5 | 1.0 | – | – |
| hKAf | −70.4 | −7.6 | 25.0 | – | – | ||
| IKAS | mKAS | 6.7e−6 | −25.6 | 13.3 | 131.4 | −37.4 | 27.3 |
| hKAS | −78.8 | −10.4 | See STAR Methods | ||||
Full details are in the STAR Methods. Here, PNa, leak = 1.96e−7, PCl, leak = 1.5e−7, and PK, leak = 0.6e−7(μm/ms). The equilibrium potentials for Na+, K+ and Cl− were 56.64 mV, −87.06 mV, and −95.07 mV at 295.15 K, respectively, and were calculated using the Nernst equation for each ion with concentrations identical to those used in the experiments.
Transient Extracellular K+ Elevations after AAV-Driven Expression of ChR2
Channelrhodopsin is frequently deployed using adeno-associated viruses (AAVs) for localized expression. We explored extracellular K+ changes following local expression of the commonly used ChR2 variant, ChR2(H134R), in the striatum. Intrastriatal hSynapsin1-driven expression of ChR2(H134R)-YFP was robustly localized to NeuN positive neurons (Figures 2A and 2B), and optical stimulation with trains of increasing duration resulted in significant elevations of extracellular K+ to 8.8 ± 0.8 mM (Figure 2C; n = 16 slices, n = 4 mice). The rise and decay τ values of these elevations were 3.2 ± 0.3 and 9.5 ± 1.4 s, respectively (Figures 2A–2C; n = 16 slices, n = 4 mice). Furthermore, the K+ elevations were not significantly affected by TTX (Figure 2D; p > 0.05, n = 11–12 slices, n = 4 mice), showing they were not due to AP firing. Next, we expressed ChR2(H134R)-mCherry in striatal astrocytes using AAV2/5 and the GfaABC1D promoter (Haustein et al., 2014; Shigetomi et al., 2013): as expected, we detected no colocalization with NeuN positive neurons but detected significant colocalization with S100β positive astrocytes (Figures 2E and 2F). Optical stimulation with trains of increasing duration resulted in significant elevation of extracellular K+ to 7.4 ± 0.2 mM (Figure 2G; n = 16 slices, n = 4 mice). The rise and decay τ values of such elevations were 3.0 ± 0.2 and 11.0 ± 1.4 s, respectively (Figures 2E–2G; n = 16 slices, n = 4 mice). Responses were also observed with 2 ms flashes (Figures S2G and S2H; n = 16 slices, n = 4 mice). Astrocytes are proposed to regulate extracellular K+ dynamics via Ba2+ sensitive Kir4.1 ion channels (Nwaobi et al., 2016). Ba2+ block (100 μM) of Kir4.1 did not significantly alter the peak change in K+ evoked by ChR2(H134R) activation; however, suggesting a possible role for Kir4.1 in K+ clearance, Ba2+ significantly increased the rise and decay times of the extracellular K+ elevations (Figure 2H; n = 16 slices, n = 4 mice). To explore the possibility that the ChR2-mediated extracellular K+ elevations may be due to brain slicing, we repeated the experiments following expression of ChR2(H134R) in cortical astrocytes in vivo (Figure 2I). We measured significant, albeit smaller, slow K+ elevations in the cortex in vivo of awake head-fixed mice (rise and decay times of 0.5 ± 0.4 and 1.4 ± 0.7 s, respectively), but not in mice that received a control AAV (Figure 2J; n = 4 mice each). Finally, we compared the K+ elevations measured at room temperature and at more physiological temperatures (34°C): we found significant elevations at both temperatures and only subtle effects of temperature on the kinetics of extracellular K+ (Table 1; Figure S4).
Figure 2. Transient Extracellular K+ Increases Occur Following AAV-Driven Expression of ChR2.

(A) AAV vector for neuron-specific expression of ChR2(H134R).
(B) Left: representative image of the dorsolateral striatum showing the colocalization of hSyn-ChR2(H134R)-YFP (red) and NeuN (green). Right: representative image of hSyn-ChR2(H134R)-YFP (red) and S100β (green).
(C) Left: waveforms depicting striatal K+ elevations in mice expressing the hSyn-ChR2. Right: average data for hSyn-ChR2(H134R)-YFP K+ elevations.
(D) Left: waveforms of K+ elevations with and without TTX. Right: quantification of various properties of the responses shown in the left panel.
(E) AAV vector for astrocyte-specific expression of ChR2(H134R).
(F) Left: representative image showing the distribution of GfaABC1D-ChR2 showing overlap with S100β (green). Right: representative image of the dorsolateral striatum showing colocalization of GfaABC1D-ChR2 (red) and NeuN (green).
(G) Left: waveforms of K+ elevations in mice expressing the GfaABC1D-ChR2. Right: average data for the experiment in the left panel.
(H) Left: waveform of K+ elevations with and without Ba2+ applied to the slice. Right: quantification of various properties of the responses in the left panel.
(I) Representative images from animals expressing ChR2(H134R) in cortical astrocytes, showing overlap with S100β in green.
(J) Left: schematic depicting the experimental setup for in vivo monitoring of K+ concentrations. Middle: waveforms of K+ concentration changes in the cortex from in vivo recordings. Right: average peak K+ concentration in the in vivo recordings. The LED power was 0.33 mW/mm2. Data shown as mean ± SEM. In some cases, the error bars are smaller than the symbol used to represent the mean.
See also Figures S2 and S4.
Table 1.
Comparison of ChR2-Mediated Extracellular K+ Elevations at Room Temperature and at More Physiological Temperatures
| # Slices [# Mice] |
Peak Change in K+ (mM) |
Rise Time (τ; s) |
Decay Time (τ; s) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 20°C | 34° C | 20°C | 34°C | p | 20°C | 34° C | p | 20°C | 34° C | p | |
| Transgenic mouse | 15 [4] | 18 [4] | 2.7 ± 0.2 | 2.7 ± 0.3 | 0.55 | 2.1 ± 0.6 | 5.3 ± 0.7 | < 0.001 | 7.2 ± 0.5 | 14.4 ± 3.8 | 0.07 |
| Thy1 ChR2 | |||||||||||
| AAV hSynapsin1 ChR2 | 12 [4] | 16 [4] | 4.3 ± 0.8 | 3.5 ± 0.3 | 0.29 | 3.2 ± 0.3 | 4.7 ± 0.5 | 0.02 | 9.5 ± 1.4 | 18.9 ± 1.5 | < 0.001 |
| AAV GfaABC1D ChR2 | 16 [4] | 20 [4] | 2.9 ± 0.2 | 2.7 ± 0.1 | 0.52 | 3.0 ± 0.2 | 2.9 ± 0.3 | 0.74 | 11.0 ± 1.4 | 12.8 ± 1.0 | 0.12 |
The peak change in K+ concentrations were not significantly different at room (20°C) versus a more physiological temperature (34°C) in acute slice experiments in the striatum. However, some aspects of the kinetics (τ) were. All values are reported as mean ± SEM from the number of slices indicated (n) from 4 mice in each condition.
ChR2-Mediated Extracellular K+ Elevations Increased MSN Excitability in Brain Slices
The finding that ChR2(H134R) activation in astrocytes caused an increase in extracellular K+ (~7.4 mM) comparable to that caused by neuronal ChR2 activation (~8.8 mM; Figures 2C and 2G) provided an opportunity to evaluate if such K+ fluxes were consequential for neuronal function. To this end, we expressed ChR2(H134R)-mCherry in adult striatal astrocytes using AAV2/5 and the GfaABC1D promoter (Haustein et al., 2014; Shigetomi et al., 2013). We confirmed in these specific experiments that ChR2(H134R)-mCherry was expressed in ~80% of striatal S100β positive astrocytes, but not in striatal NeuN positive neurons (Figures 3A and 3B; n = 12 sections, n = 4 mice). In this experimental approach, we identified and recorded from MSNs but optically stimulated ChR2(H134R)-mCherry expressing astrocytes nearby in the dorsolateral striatum. A single brief train of ten 2 ms flashes significantly and slowly depolarized MSNs by ~2 mV (Figure 3D; n = 14 MSNs, n = 4 mice). A longer 30 s train of flashes depolarized MSNs by ~7 mV for the duration of the stimulation train (Figure 3D; n = 14 MSNs, n = 4 mice). The depolarization also peaked and decayed with seconds time scale kinetics similar to those observed for extracellular K+ elevations (Figures 2G and 3D). Some past studies have suggested that ChR2 stimulation of astrocytes evokes astrocyte glutamate release, which is proposed to be a gliotransmitter acting on neurons. However, we detected no change in excitatory postsynaptic potentials (EPSPs) arriving onto MSNs during optical stimulation of astrocytes, and of most relevance here, the change in MSN membrane potential was not affected by the bath application of CNQX and APV (10 μM), which completely blocked all EPSPs (Figure 3D). These data suggest that ChR2- mediated stimulation of striatal astrocytes depolarized MSNs significantly, likely by K+ elevations and not via glutamate release (Chai et al., 2017; see also Nedergaard and Verkhratsky, 2012 and Sloan and Barres, 2014). To explore neuronal consequences more carefully in the striatum, we applied slow ramps to determine the amount of current required to trigger APs in MSNs (rheobase) before, during, and after optical stimulation of ChR2-expressing astrocytes (Figure 3E). In all cases, we measured significant MSN membrane potential depolarization and a significant drop in the rheobase (Figure 3E; n = 14 MSNs, n = 4 mice). Furthermore, following in vivo stimulation of astrocytes expressing ChR2(H134R), we detected a significant increase in the expression of the immediate early gene, cFos, in 20% of DAPI-positive cells in the striatum (Figures 3F–3H; n = 7 slices, n = 3 mice), which was significantly greater than that observed in mice expressing tdTomato in astrocytes (Figures 3G and 3H). Of the cells with elevated cFos expression following in vivo astrocyte optogenetic stimulation, ~35% were S100β positive astrocytes (Figures 3F and 3H), whereas ~18% were NeuN positive neurons (Figures 3G and 3H). These data are consistent with the interpretation that optogenetic excitation of ChR2(H134R)-expressing astrocytes in vivo was sufficient to alter astrocyte and neuronal excitability, resulting in significantly elevated cFos expression in a proportion of the cells. During our experiments, we observed no overt change in open-field activity or general behavior during optical stimulation of ChR2(H134R)-expressing astrocytes relative to control mice (Figure S3).
Figure 3. ChR2-Mediated Extracellular K+ Increased Neuronal Excitability.

(A) Left: representative image of a striatal section from mice expressing GfaABC1D-ChR2(H134R) (red), showing overlap with S100β (green). Right: fraction of S100β positive cells expressing GfaABC1D-ChR2(H134R) in the dorsolateral striatum.
(B) Left: representative image from mice expressing GfaABC1D-ChR2(H134R) (red), showing no overlap with NeuN (green). Right: fraction of NeuN positive cells expressing GfaABC1D-ChR2(H134R) in the dorsolateral striatum.
(C) Left: schematic depicting patch-clamp of medium spiny neurons (MSNs) in the striatum near astrocytes expressing ChR2(H134R). Right: waveform of membrane potential changes of a MSN to a series of stepwise current injections.
(D) Left: traces of membrane potential changes from MSNs in response to a train of 10 flashes with and without glutamate antagonists in mice expressing GfaABC1D-ChR2. Traces for 30 s of flashes with and without glutamate antagonists are also shown. Right: average data from experiments shown with the traces.
(E) Left: waveforms of membrane potential responses of a MSN to a ramp current injection with and without blue light. Inset: highlighted region showing rheobase shift. The scatter graphs show average data for rheobase and membrane potential.
(F) Left: representative images showing overlap between cFos (upper left inset, white) and S100β (lower left inset, red) from animals in which ChR2 was expressed in astrocytes and activated with blue light. Right: as in left panels, but for NeuN (green) instead of S100β.
(G) As in (F), but for control experiments expressing tdTomato instead of ChR2.
(H) Summary plots of cFos expression following optical stimulation of astrocytes expressing ChR2 in relation to control experiments where astrocytes expressed tdTomato. For the experiments in (F)-(H), the stimulation conditions were 0.05 Hz (10 s on and 10 s off for 1 hr). The power was ~3.5 mW from the tip of the cannula before implantation. Data shown as mean ± SEM. In some cases, the error bars are smaller than the symbol used to represent the mean.
See also Figures S3 and S4.
Computational Modeling: MSN Excitability Increase Recapitulated by K+ Elevation Alone
We adapted a MSN model (Mahon et al., 2000) (Figure 4A; Table 2), which comprised a single compartment representing a MSN soma and proximal dendrites, and eight active Hodgkin-Huxley (HH)-based models of variable membrane conductances. The modeled currents are persistent sodium (INap); slowly inactivating sodium (INas); transient sodium (INat); delayed rectifier potassium (IKDR); inwardly rectifying potassium (IKIR); persistent potassium (IKRP); fast inactivating A-type potassium (IKAf); slow inactivating A-type potassium (IKAs); and a lumped, non-specific ion leak current. We replaced the non-specific leak current with three Goldman-Hodgkin-Katz (GHK)-based specific ion leaks (Cl−, Na+, and K+). We used the model to explore how K+ concentration changes measured in the experiments reported in Figure 3 affect MSN excitability in silico. We adjusted the model’s parameters to replicate basal MSN spiking features (Mahon et al., 2000) (see STAR Methods). We then varied conductance and permeability parameters to constrain a population of MSN models to reproduce the observed ranges of resting membrane potential values and resting levels of excitability during light off and light on conditions. We required that each model reproduce these measures at both 4.5 and 7.5 mM extracellular K+ to be included in the population. By asking if models exist sensitive to K+ changes within experimentally observed ranges from Figures 1 and 2, we aimed to test if the modeled range of extracellular K+ was sufficient to explain the data. Current injection ramp protocols were replicated for each model instance using two extracellular K+ concentration parameters related to Figure 3 (i.e., 4.5 and 7.5 mM). A model instance comprised a unique set of ionic conductances and specific leak permeability parameters. Using a modified version of the parameter optimization via evolutionary algorithms approach (Rumbell et al., 2016) (see STAR Methods), we identified a population of model instances that reproduced the range of excitability features measured before and after optical stimulation of astrocytes expressing ChR2 (Figure 3) when the extracellular K+ increased from 4.5 to 7.5 mM K+ (Figure 4B), respectively. The resulting population of model instances recapitulates a range of excitability features measured empirically, which are modulated consistently in the same direction by extracellular K+ across all model instances, thus establishing extracellular K+ as an independent parametric change in the model sufficient to explain changes in MSN excitability observed during ChR2 excitation (Figure 4). We further optimized these models, constraining with specific spiking features from one of the experimental traces. One such outcome generated the waveforms depicted in Figure 4C, which closely recalls experimental data of Figure 3. To explore the full range of extracellular K+ concentrations, we incrementally varied K+ in the model used to generate Figure 4C. Increases in extracellular K+ depolarized MSNs and decreased rheobase in the model (Figure 4D). K+ above 9 mM caused seizure-like bursting activity in the MSN model; to generate plots in Figure 4D, we thus did not exceed 7.5 mM K+. Overall, this MSN model supports the hypothesis that extracellular K+ elevations are sufficient per se to reproduce the increased excitability changes measured during experiments in Figure 3 without the need to invoke additional factors.
Figure 4. Computational Modeling of K+ Effects In Silico.

(A) Diagrammatic representation of the circuit model of a MSN with the indicated active conductances and ion-specific leak currents. Key values used in the model are listed in Table 1: kinetic parameters were unchanged from those reported in published work (Mahon et al., 2000).
(B) Scatter graphs plot the effect of K+ elevations from 4.5 to 7.5 mM on the MSN model population’s resting membrane potentials and rheobases. Each conductance parameter set (model instance) is represented as a pair of points joined by a line. Across scatterplots, colors represent the same model instance.
(C) Traces of MSN excitability from the computational model at the indicated levels of K+. The arrows emphasize the shift in rheobase with increasing K+.
(D) Summary plots of how resting membrane potential and rheobase change as a function of extracellular K+ in the MSN model depicted in (C). Full details of the MSN model are provided in the STAR Methods section.
ChR2-Mediated Extracellular K+ Elevations Increased MSN Excitability In Vivo
Experiments reported in the preceding sections show that optical stimulation of ChR2-expressing astrocytes causes extracellular K+ increases (Figure 2) and MSN depolarization (Figure 3D), as well as increased MSN excitability in brain slices (Figure 3E) and in silico (Figure 4). In order to determine whether such responses translated into increased MSN AP firing in vivo, we recorded MSN activity in awake behaving mice (Figure 5) using multisite silicon probes (Bakhurin et al., 2016). ChR2 was selectively expressed in striatal astrocytes using approaches described in the preceding sections. Silicon probes with an inbuilt light-emitting diode (LED) were used for optical excitation and electrophysiological recording (Figure 5A) in awake behaving mice on a spherical treadmill. MSN APs were analyzed offline, sorted using established criteria (Bakhurin et al., 2016) and AP frequency assessed before, during, and after optical stimulation (Figure 5A). Post hoc, we verified in each mouse that AAV-driven ChR2 expression was overlapping anatomically with the site at which the silicon probes were placed for electrophysiological assessments (Figure 5B). As a control group, we used mice in which tdTomato was identically expressed in astrocytes: MSN APs from tdTomato and ChR2 groups were identified and recorded (Figure 5C). Figure 5D shows AP firing of 60—70 MSNs before, during, and after optical stimulation for the control tdTomato and the ChR2 groups. From the raster plots, it was clear that optical stimulation did not evoke alterations in MSN AP firing in the control group, but optical stimulation evoked clear increases in AP firing in the ChR2 group (Figure 5D; n = 5 mice in each group). These data are summarized as average firing over time (Figure 5E) or as histograms before, during, and after optical stimulation (Figure 5F). Together with the studies of extracellular K+ increases, MSN excitability, and computational modeling, these in vivo recordings strongly indicate that optical stimulation of ChR2-expressing astrocytes can cause significantly elevated MSN firing. Our brain slice data and modeling suggest that astrocyte ChR2-mediated K+ elevations are the underlying cause.
Figure 5. In Vivo Activation of ChR2 in Astrocytes Enhances MSN AP Firing Measured with Silicon Probes in Awake Behaving Mice.

(A) Schematic depicting experimental workflow using multi-site silicon probes in vivo. Mice were injected with AAVs to express tdTomato or ChR2 within astrocytes. Light flashes were produced by 30 s of 25 ms flashes at 20 Hz and at 470 nm using a LED light source.
(B) IHC staining of coronal sections from mice injected with AAVs expressing tdTomato (left) or ChR2 (right) in astrocytes of the dorsolateral striatum (white dashed outline) and used for in vivo recordings.
(C) Representative waveforms from narrow spiking single units in either tdTomato (left) or ChR2 (right) expressing mice. The average trace is the thick colored line.
(D) Raster plots of single unit recordings from 5 mice expressing tdTomato (top graph) or ChR2 (bottom graph) during 30 s periods with the LED off or on.
(E) Average firing rate of all units over the recording period (in 5 s bins) in tdTomato or ChR2 groups of mice. The period optical stimulation is highlighted in blue.
(F) Bar graphs of average activity of all units in the 30 s periods prior to, during, or following optical stimulation in either condition. Average data are represented as mean ± SEM from n = 5 mice.
DISCUSSION
We show that transient extracellular K+ elevations accompany optogenetic excitation with two widely used variants of ChR2 (wild-type and H134R) in a common transgenic mouse model, following AAV-mediated delivery in brain slices and in vivo, regardless of whether the ChR2 is expressed in neurons or astrocytes. The simplest interpretation is that K+ flows down its electrochemical gradient through the ChR2 pore from the inside to the outside of cells. The ChR2 pore displays a K+ to Na+ relative permeability of ~0.4 (Lin et al., 2009; Nagel et al., 2003) and a minimal open pore diameter of ~6.2 Å (Richards and Dempski, 2012). Thus, opening of the ChR2 pore carries inward Na+ current and outward K+ current under physiological conditions as determined by the respective ion equilibrium potentials relative to the cell’s membrane potential. Our data show that this outward K+ flux is significant and elevates K+ concentrations in the extracellular space to consequential levels during seconds-long optical stimulations usually employed in astrocyte studies.
Interestingly, our observations recall work at the neuromuscular junction where a slow endplate potential due to K+ release directly by nerve terminal APs was detected (Katz and Miledi, 1982). Several other papers have similarly documented K+ elevations during neuronal activity (Heinemann et al., 1990; McCreery and Agnew, 1983; Walton and Chesler, 1988). However, the K+ elevations we report are not due to K+ release during the AP because they occurred when APs were blocked and when ChR2 was expressed in astrocytes devoid of APs. Furthermore, although K+ is expected to flow through open endogenously expressed voltage-dependent K+ channels that open during ChR2-mediated depolarization, this cannot account for our observations: astrocytes have little if any rectification indicative of voltage-gated K+ conductance, and owing to their low membrane resistance and high resting ohmic K+ conductance, they rarely deviate from near the K+ equilibrium potential. In light of these considerations, we discuss the relevance of ChR2-mediated K+ fluxes in relation to the diverse uses of optogenetic excitation strategies, but with a focus on astrocyte studies.
A logical interpretation of our astrocyte studies is that optogenetic stimulation increased neuronal excitability via extracellular K+ elevations, which was recapitulated in a computational model. Our studies should not be overinterpreted, however, as indicating that we dismiss gliotransmission (Araque et al., 2014), but our studies highlight an important consideration with the use of ChR2 in these regards. It will be important to candidly consider if cellular and behavioral responses ascribed to ChR2-evoked gliotransmission onto neurons (reviewed by Bang et al., 2016) could instead reflect elevations of extracellular K+. Even subtle changes in K+ will change neuronal excitability directly (Hille, 2001) and indirectly by reducing glutamate clearance by electrogenic transporters (Barbour et al., 1988; Szatkowski et al., 1990). This will lead to elevated ambient glutamate levels, which could conceivably be interpreted as glutamate gliotransmission. If so, the relevance of this phenomenon to circuits and behaviors may have been misinterpreted. Furthermore, strong excitation of astrocytes with ChR2 may change extracellular electric fields, which may affect neuronal ephaptic signaling (Jefferys, 1995). The use of optogenetic excitation approaches to study astrocytes, therefore, requires careful consideration not least because these cells are largely electrically silent (Kuffler, 1967). It is not immediately clear why artificially changing membrane potential is appropriate to explore astrocyte physiology, although such approaches are obviously meaningful for neurons.
Our goals in this study were not to identify a past behavioral study based on ChR2 and highlight it as being better explained by transient K+ elevations. Instead, we followed empirical observations that show K+ elevations occur, and we suggest this should be considered in the interpretation of future studies using ChR2, especially in astrocytes. We also did not perform detailed behavior analysis, but during our experiments, we observed no overt change in open-field activity or general behavior during optical stimulation of ChR2(H134R)-expressing striatal astrocytes relative to control mice. This is unsurprising because astrocytes do not have axonal projections and do not discriminate between D1 and D2 MSNs. Thus, we suggest that the elevation of K+ would not discriminate between D1 and D2 MSNs, both of which exist within single astrocyte territories (Chai et al., 2017; Octeau et al., 2018a) and may drive compensatory signals in relation to simple behaviors. Instead, we interpret the in vivo data to show that elevated cFos responses most likely reflect neuronal firing caused by transient K+ elevations, a finding confirmed by recording neuronal activity in vivo.
It is problematic to search for a specific striatal microcircuit-related behavior that may be caused by astrocyte-mediated K+ increases because unlike a neurotransmitter, K+ will affect the functions of all cells, not just neurons. Instead, we explored the effect of astrocyte ChR2-mediated K+ elevations on the behavior of single neurons using modeling and experiments. These data strongly suggest that ChR2-mediated optical excitation of astrocytes can affect the excitability of neurons via K+ elevations. Since K+ is tightly regulated, even small changes are expected to affect neurons, astrocytes, microglia, and blood vessels (McDonough and Youn, 2017). For example, K+ is potently ictogenic at around 9 mM (de Curtis et al., 2018), has vasoconstrictor and vasodilator actions on brain vasculature (Girouard et al., 2010), contributes to neurological disease phenotypes (Nwaobi et al., 2016; Tong et al., 2014), and will synchronize neuronal activity (Jefferys, 1995). Subtle changes in K+ will depolarize neurons and alter the open and closed probabilities of the activation and inactivation gates of voltage-gated K+ and Na+ channels, which of course will alter AP properties through Hodgkin-Huxley outcomes (Hodgkin and Huxley, 1952). Such effects may explain why ChR2-mediated stimulation of astrocytes silenced neurons in the subthalamic nucleus in pioneering early studies (Gradinaru et al., 2009). Hence, long-term optogenetic stimulation in vivo may cause complex effects on cells in parallel with the effects desired by expression of ChR2 in specific cell types. K+ changes may also engage local circuits that modulate behavior through ionic or ephaptic mechanisms (Jefferys, 1995) in addition to the pathways targeted by ChR2 expression. These are relevant considerations with the use of optogenetic strategies in vivo and may predict the emergence of unrelated, possibly pathogenic, effects during use. Additional consequences of the larger and more compartmentalized intracellular Na+ fluxes also merit consideration. Perhaps, infrared induced electro-mechanical effects (Plaksin et al., 2018) may provide opportunities for remote neuronal excitation (Shapiro et al., 2012) without reliance on substantial ionic currents. In the case of astrocytes, new tools are clearly needed to remotely manipulate physiological levels of activity.
It would be egregiously misleading of us to suggest that transient K+ elevations markedly contribute to most optogenetic excitation studies. This is not our intention. More parsimoniously, we suggest that the impact of extracellular K+ dynamics will depend on the membrane properties of the neurons in question (e.g., their input resistance, levels of open K+ channels, and rectification), as well as the activation and inactivation thresholds for voltage-gated ion channels. Local neuroanatomy will also be relevant, as this will determine how K+ elevations are dissipated in the extracellular milieu and will affect local electric fields. Overall, our insights echo caution by others emphasizing effects of light itself (Rungta et al., 2017) and the importance of depolarization block (Herman et al., 2014) and of circuit-related off-target effects in the case of silencing approaches (Otchy et al., 2015; Südhof, 2015). Admittedly, our work may have little relevance to the use of ChR2 in neuronal studies where optical stimuli are usually brief, but we suggest transient K+ increases are likely to be markedly consequential in astrocyte and behavioral studies when optical stimuli are typically prolonged. The impact of such changes may have been missed when independent electrophysiological evaluations were not employed to measure cellular level electrical changes and extracellular K+ levels. We suggest that cellular neurophysiology is necessary in parallel with in vivo studies in order to make adequate interpretation of neural circuit and behavioral outcomes in studies employing ChR2 and other related optogenetic strategies.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Baljit S. Khakh (bkhakh@mednet.ucla.edu). Adeno-associated viruses and plasmids are, or will, be made available on Addgene or through the University of Pennsylvania Vector Core.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Chancellor’s Animal Research Committee at the University of California Los Angeles. Mice were housed with food and water available ad libitum in a 12-hour light/dark environment. Mice were healthy with no abnormal behavioral phenotypes and were not involved in prior studies. For all experiments, mice were sacrificed during the light cycle. Both male and female mice were used for experiments (between 8–16 weeks of age).
METHODS DETAILS
Mouse lines
Most experiments were conducted on C57BL/6N mice (from Taconic) from an in-house colony. Heterozygous Thy1-ChR2-YFP mice were obtained from Jackson Laboratories (strain B6.Cg-Tg(Thy1-COP4/EYFP)9Gfng/J; stock #007615) and have been published by others (Arenkiel et al., 2007). Thy1-ChR2 mice were maintained as heterozygous × wt breeding pairs in an in-house colony. For all experiments, wt mice were C57BL/6N.
Plasmid and adeno-associated viruses (AAV)
All plasmid constructs were generated using standard molecular biology techniques and the In-Fusion HD Cloning Kit (Clontech). All constructs were sequenced before AAV generation. For all constructs, we modified plasmid pZac2.1 GfaABC1D Lck-GCaMP6f (Addgene plasmid # 52924). Briefly, we generated adeno-associated virus plasmids (AAV2/5) capable of expressing the selected cDNA using the following strategy. For the plasmid, GfaABC1D-ChR2(H134R)-mCherry, we removed Lck-GCaMP6f using Xho1 and Xba1 restriction enzymes and infused the ChR2-(H134R)-mCherry cDNA. The fully sequenced “pZac2.1” plasmids were sent to the Penn Vector Core, which used them to generate AAV serotype 2/5 for this construct yielding a concentration of ~1 × 1013 genome copies/ml (gc/ml). This construct has been deposited at Addgene in the Khakh lab repository, and the AAVs are available from the UPenn Vector Core Catalog.
In vivo microinjections of AAVs
Male and female P42-P56 C57BL/6N mice were used in all AAV experiments. Briefly, animals were deeply anesthetized using isoflurane (induction at 5%, maintenance at 1%−2.5% v/v). After induction of anesthesia, the mouse’s head was carefully placed into a stereotaxic frame and gently secured by blunt ear bars and their nose was placed into an anesthesia system (David Kopf Instruments). Before the surgery, mice were administered 0.05 mL of buprenorphine (Buprenex, 0.1 mg/ml) subcutaneously. 10% povidone iodine and 70% ethanol were used to sterilize the incision site. Short (5–8 mm) skin incisions were made, followed by craniotomies ~2 mm in diameter using a small steel burr (Fine Science Tools) powered by a highspeed drill (K.1070; Foredom) directly above the site of the injection. Sterile saline was applied onto the skull to lessen heating caused by drilling. Viral injections were performed using a stereotaxic apparatus (David Kopf Instruments) which guided the placement of angled bevel glass pipettes (1B100 – 4; World Precision Instruments) into the appropriate brain region. Our stereotaxic coordinates (Axes: Anterior-Posterior / Medial-Lateral / Dorsal-Central; relative to Bregma) for AAV microinjection were as follows: dorsolateral striatum: +0.8 / +2.0 / −2.4 and superficial motor cortex: −0.1 /+0.7 / −0.1. In this study, the following viruses and their titers were used: AAV2/5 GfaABC1D-ChR2(H134R)-mCherry (8.1 × 1012 gc/ml), AAV2/5 hSynapsin1-ChR2(H134R)-YFP (2.9 × 1012 gc/ml, Lot# AV4319J, UNC GTC Vector Core). AAVs were injected by using a syringe pump at 200 nl/min (Pump11 PicoPlus Elite; Harvard Apparatus). The glass needles were left in place for at least 6 min and slowly removed over the course of 1 minute. The incisions were closed with single external 6 – 0 nylon sutures. After surgery, animals recovered overnight in cages which were placed halfway on a low-voltage heating pad. As an analgesic, subcutaneous buprenorphine was given twice daily for 48 hours following surgery. In addition, trimethoprim/sulfadiazine diet (275 & 1,365 ppm, TD.06596, Teklad) was fed ad lib prophylactically for one week after surgery. Following AAV injection and recovery, the mice were sacrificed 22–28 days later for all experiments.
Acute brain slice preparation and single cell recordings
Acute brain slices were prepared from P42–70 wt, Thy1-ChR2 mice or AAV microinjected mice. Briefly, animals were deeply anesthetized and decapitated. The brains were placed in ice-cold modified artificial CSF (aCSF, containing in mM: 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose) and cut into 300 μm-thick coronal or parasagittal slices containing areas of the striatum and cortex. Brain slices equilibrated for ~30 min at 32–34°C in normal aCSF (containing in mM; 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose) which was continuously bubbled with a mixture of 95% O2/5% CO2, stored at room temperature in the same buffer. All slices were used for experiments within ~6 hr of animal death. Cells were visualized using IR-DIC optics on an upright microscope (BX61WI, Olympus). pCLAMP10 software and a MultiClamp 700B amplifier and Digidata 1322A was used for electrophysiology (Axon Instruments). For all recordings we used normal aCSF at 20–25°C. For patch recordings, the intracellular solution contained the following (in mM): 135 potassium gluconate, 3 KCl, 0.1 CaCl2, 10 HEPES, 1 EGTA, 8 Na2-phosphocreatine, 4 Mg-ATP, 0.3 Na2-GTP, pH 7.3 adjusted with KOH. The initial access resistances were < 20 MΩ for all cells; if this changed by > 20% the cell was discarded.
Recording K+ ion dynamics in slices and in vivo
To record slice K+ dynamics, we made K+ selective microelectrodes (Octeau et al., 2018b). Briefly, borosilicate glass capillaries were pulled to a fine tip of ~2–5 μm and then baked at ~200°C in the presence of 5% dichlorodimethylsilane for 1 hour up to overnight to enhance the hydrophobicity of the glass by silanization. The silanized capillaries were then allowed to cool to room temperature and then broken to a diameter of approximately 15 μm using a blunt instrument. The capillaries were then backfilled with a solution containing 300 mM NaCl and 10mM HEPES, pH 7.4. The microelectrode tip was then immersed in a small volume (~1 μl) of potassium ionophore cocktail (5% w/v valinomycin, 93% v/v 1,2-dimethyl-3-nitrobenzene, 2% w/v potassium tetrakis(4-chlorophenyl)borate) and this solution was drawn approximately 1 mm into the tip by capillary action. The primed microelectrode was then fixed into an electrode holder on the connected to an amplifier and digitizer which transmitted voltage responses to a PC recording the responses in pCLAMP10. The voltage responsiveness of all microelectrodes were calibrated with solutions of aCSF which had various concentrations (in mM 0.1, 1.0, 10 and 100) of KCl, for these solutions iso-osmolar quantities of NaCl were subtracted to maintain total solution osmolarity at ~310 mOsm. The K+-selective microelectrodes were then slowly inserted into prepared brain slices which were superfused with 4.5 mM K+ aCSF and the voltage responses to blue LED flashes were measured. For in vivo measurement of K+ ion dynamics mice were prepared with a cranial bar fixed to the skull to maintain brain position. Mice were anesthetized with isoflurane (3%−5% induction, 1.5% maintenance) ten minutes after injection of a systemic analgesic (carprofen, 5 mg per kg of body weight) and placed in a stereotaxic frame. During surgery, mice were always kept at 37°C using a feedback-controlled heating pad. Pressure points and incision sites were injected with lidocaine (2%), and eyes were protected from desiccation using artificial tear ointment. The skin above the skull was cut and a custom-made lightweight metal head holder was implanted on the skull using Vetbond (3M) and a recording chamber was built using dental cement (Ortho-Jet, Lang). Access to the brain was closed using a thin layer (~2 mm) fast drying silicone elastomer (Kwik-sil, WPI) and a top layer of dental cement. Mice had a recovery period from surgery of five days, during which they were administered amoxicillin (0.25 mg per ml in drinking water through the water supply). After the recovery period, mice were habituated to head fixation on the spherical treadmill. 20–25 days following viral injection mice were placed onto the spherical treadmill and maintained in a head fixed position using a metal arm to grip the head holder attached to the mouse’s skull. Following head restriction, the dental cement and silicone covering was removed and ACSF was applied to the surface of the brain to prevent tissue drying. To assess potassium ion changes a K+ selective microelectrode was prepared, calibrated and then lowered ~50 microns perpendicularly into the cortical surface. Electrode potentials were allowed to stabilize over the course of 8–10 minutes before recording. Light flashes from a LED-Blue optogenetics light source were delivered via a fiber which was controlled remotely using a TTL pulse from Pulser software version 2.3.1 (Prizmatix, Israel). Electrode responses were captured and recorded using an Axopatch 700B amplifier connected via a digitizer to a computer with pCLAMP9 software. After completion of the experiments, the mice were euthanized by pentobarbital injection and immunostaining was performed on the brains to confirm viral expression.
In vivo ChR2 activation and behavioral assessment
To deliver blue light to the striatum we constructed a fiber optic cannula using a DIY-cannula kit (Prizmatix). The fiber optic was cleaved into the desired length, which was approximately 10 mm. A small, ~1 μL droplet of epoxy resin was applied to the flat opening of a 6.5 mm long cannula. The fiber was then inserted through the epoxy bead into the cannula until it protruded approximately 1 mm from the opposite end of the cannula. Using a heat gun, we then cured the epoxy resin and secured the fiber in place. When the epoxy cooled, the fiber on the convex end of cannula was made transparent using polishing paper of increasing grits; from 4500 to 60000 final grit strength. Cannulated fibers were tested for their integrity and maximum light output (≥8 mW) before being used for the implantation. To implant the cannulated fiber optic, mice were first anesthetized with isoflurane (induction at 5%, maintenance at 1%−3%). The scalp was shaved and animals were gently fitted into a stereotaxic frame with their heads secured by ear bars. Ophthalmic ointment was applied to eyes to prevent dryness and postoperative discomfort. The surgical area was sterilized thrice with 10% povidone iodine and 70% isopropanol. A skin incision was made on top of the head and the periosteum was removed by swiping with a cotton swap wetted with 3% hydrogen peroxide. A mark was made on the skull at coordinates above the dorsolateral striatum using the stereotactic apparatus. At this mark, a small opening (~1 mm in diameter) was drilled using small steel burr powered by high speed drill; during which saline was applied to the skull to diffuse heating and remove bone debris. Afterward, 1 μL of AAV was injected at 200 nl/min (dorsolateral striatum coordinates relative to bregma: in mm +0.8/+1.8/−2.4). The needle was kept in place for 10 minutes and slowly removed over a 5 minute period. Subsequently, the cannulated fiber optic was slowly lowered into the striatum and secured in place using Vetbond tissue adhesive (3M Maplewood, MN) and dental cement. Three weeks after the surgery the mouse was connected to optical stimulation system (Prizmatix). The optical cannula was connected to the patch cord through the mating sleeve (Prizmatix). Blue light pulses were applied in a stimulation paradigm consisting of 10 s light on and 10 s light off for a period of 60 min. During this period the mouse behavior in an opaque open chamber (L × W × H in cm: 29 × 29 × 15) was recorded using a camera placed above the field. Locomotion was tracked by ANY-maze software (Stoelting Co.) and the tracking accuracy was verified by the investigator.
Immunohistochemistry (IHC)
Prior to transcardial perfusion, mice were euthanized with 200 mg/kg pentobarbitol (i.p.). Once reflexes were lost, the abdominal cavity was opened and heparin (0.1 ml, 100 USP) was injected into the heart to avert blood clotting. Mice were perfused with ~50 mL of 0.1 M phosphate buffered saline (PBS) followed by ~50 mL 10% buffered formalin (Fisher Scientific). After careful removal from the skull, the brain was post fixed in 10% buffered formalin overnight. The tissue was cryoprotected (0.1 M phosphate buffered saline with 30% sucrose) the following day for at least 2 days at 4°C until use. 40 μm sections were sectioned using a cryostat (Leica) and processed for immunohistochemistry. Sections were washed 4 times in 0.1 M PBS for 5 min each, and then incubated in a blocking solution comprising 5% NGS in 0.1 M PBS with 0.2% Triton X-100 for 1 hr at room temperature with gentle rocking. Sections were then incubated in primary antibodies diluted in ‘blocking solution overnight at 4°C. The following primary antibodies were used: chicken anti-GFP (1:1000; Abcam Cat# ab13970 RRID:AB_300798), rabbit anti-NeuN (1:1000; Cell Signaling; RRID: AB_2630395), rabbit anti S100β (1:2000, Abcam Cat# ab41548 RRID:AB_956280), guinea pig anti-cFOS (1:500, Synaptic systems #226004 RRID:AB_2619946), rabbit anti-dsRed (1:1000, Clontech Laboratories Cat# 632496 RRID:AB_10013483), mouse anti-mCherry (1:1000, St. John’s Cat# STJ97087), guinea pig anti-NeuN (1:500; Synaptic Systems Cat # 266 004; RRID:AB_2619988), mouse anti-neurofilament (1:250, BioLegend Cat# SMI-312R, RRID:AB_2314906). The next day the sections were washed 4 times in 0.1 M PBS for 5 min each before incubation at room temperature for 1–2 hr with secondary antibodies diluted in 0.1 M PBS with 5% NGS and 0.2% Triton X-100. The following Alexa conjugated secondary antibodies were used: goat anti-guinea pig 488 (1:1000, Molecular Probes Cat# A-11073 RRID:AB_142018), goat anti-rabbit 546 (1:1000, Molecular Probes Cat# A-11035 RRID:AB_143051), goat anti-chicken 488 (1:1000, Molecular Probes Cat# A-11039 RRID:AB_142924), goat anti-guinea pig 546 (1:1000, Thermo Fisher Scientific Cat# A-11074 RRID:AB_2534118) and goat anti-mouse 546 (1:1000, Molecular Probes Cat# A-11030 RRID:AB_144695). The sections were then rinsed 4 times in 0.1 M PBS for 5 min each, and briefly dipped in distilled water before being dried, mounted and coverslipped on microscope slides. Fluorescence images were taken using UPlanSApo 20X 0.85 NA and UPlanFL 40X 1.30 NA oil immersion objective lens and FV1000 Fluoview confocal laser-scanning microscope. We used the 488 nm emission wavelength of an Argon laser to excite Alexa488, with the intensity adjusted to 0.5%−10% of the maximum output, which was 10 mW. The sample emission light path consisted of a band-pass emission filter (505–525 nm) before the photomultiplier tube. Alexa 546 was excited by the 543 nm wavelength of a Helium-neon laser at 2%−30% of the maximum output (1 mW). The emitted light pathway consisted of a dichroic mirror (SDM560) and a 560–600 nm band-pass filter. For imaging Alexa 647, we used a 635 nm light emitting diode (LD635, Olympus) at 0.1%−15% of its power output which was 23 mW. The emitted light pathway consisted of a dichroic mirror (SDM640) and a 655–755 band-pass filter. For fluorescence images of brain hemisections, individual images were acquired using identical settings and stitched together to show a single composite image of the tissue section.
Computational model of MSNs
The computational model of the medium spiny neuron (MSN) was refined from a past study (Mahon et al., 2000), but is reported in full here. The model comprises a single compartment, representing a MSN soma and proximal dendrites, and eight active Hodgkin-Huxley (HH) based models of variable membrane conductances associated with MSN ion channel currents. The eight modeled currents are the persistent sodium current (INap), the slowly inactivating sodium current (INas), the transient sodium current (INat) the delayed rectifier potassium current (IKDR), the inward rectifying potassium current (IKIR), the persistent potassium current (IKRP), the fast inactivating A-current (IKAf), the slow inactivating A-current (IKAs), and a lumped, non-specific ion leak current.
The model is computationally inexpensive compared to multicompartment models (Wolf et al., 2005), and in its original form captured MSN spike timing dynamics such as characteristic latency to first spike and short-term facilitation of intrinsic excitability. A major drawback of the original model is its assumption of different reversal potentials (EK or ENa) among ion channels selective for the same ion species, rendering it biophysically unrealistic, because these channels likely share the same intracellular and extracellular ion concentrations. To resolve this discrepancy, we replaced the model’s non-specific leak current with specific leak currents for potassium, sodium and chloride, each based on the Goldman-Hodgkin-Katz (GHK) equation:
where ϕs is the current density (flux) (amperes per unit area) of ion S, Ps is the permeability of ion S (μm/ms), [S]i is the intracellular concentration of ion S (mM), [S]o is the extracellular concentration of ion S (mM), and Vm is the membrane potential (mV). Reversal potentials for each ion species are then calculated once for all currents using the Nernst equation:
where R is the universal gas constant 8.314 J⋅K−1⋅mol−1, T is the temperature 295.15 K, z is the valence of the ionic species, and F is the Faraday’s constant 96,485 C⋅mol-1. We set external and internal ion concentrations to those used for the experiments reported in preceding sections. The calculation of MSN membrane potential in our revised model then follows:
where V is the membrane potential. Ion channel currents followed , where each g is the conductance of an ion channel current (noted by subscript). IInj is an injected current. IX is either the specific leak current for each ion species or the ion channel current. Specific leak current is derived by multiplying current density (Φs) from Equation 1 by the compartment surface area (assumed to be 1 μm2). Ion channel currents are derived by multiplying channel conductance (gX) by membrane potential minus the ion specific reversal potential (V − EX) by compartment surface area. The activation m and optional inactivation h gating variables were as reported in Mahon et al. (2000) followed:
where p represents (m, h, n). For channel Nat and KDR, the gating parameters were as follows:
All other currents obeyed the Boltzmann equation for steady state activation
and the voltage dependent time constants followed
except for the time constant of IAs and IKRP. The inactivation for these currents, followed
and
For numerical integration, we chose a time step of 0.01 ms. The datasets used for the model were archival at the time they were shared with researchers from IBM, and no new experiments were suggested, designed, or performed based on these models and analyses. All simulations were performed using the IBM Neural Tissue Simulator (NTS) (Kozloski and Wagner, 2011) on IBM Cloud. NTS executes simulations based on model descriptions (written in the Model Description Language) and resource allocation scripts (written in the Graph Specification Language), both of which are available in Supplemental Information. The software is experimental, and readers are therefore encouraged to contact the IBM authors if interested in using the tool.
We employed an optimization technique to search parameter space of this model. Each optimization targeted features extracted from measures taken during experiments under differing conditions as reported in Figure 3. Specifically, we optimized the single neuron model to reproduce both the Light off and Light on experimental conditions. By first setting the extracellular K+ concentrations to 4.5 mM and 7.5 mM, respectively for each condition, we guided automatic changes to model conductance parameters toward models that replicated both experimental measurements and conditions simultaneously. The optimizations accessed and varied 11 parameters, the maximal conductances , , , , , , , and GHK based permeability coefficients PCl, leak, PNa, leak and PK, leak.
The optimization employed the non-dominated sorting (NS) differential evolution (DE) algorithm (NSDE) (Deb et al., 2002; Price et al., 2005), previously used to search parameter space of compartmental neuron models in Rumbell et al. (2016). To run the algorithm, we used a modified version of the BluePyOpt (Van Geit et al., 2016) python framework for single neuron optimization. Each parameter set was simulated for 5 s. Trial-and-error was used to assess optimization metaparameters such as population size and number of generations. A single optimization of ~500 generations of a population of ~100 models took approximately ~12 hours of computing time on an X86_64 Intel architecture (2 Ghz, 64 bit, 56 cores, 128 Gb of RAM). The neuron model error score for each target feature was calculated by extracting feature measures and subtracting them from the exact target values based on empirical measures and dividing the absolute value of this quantity by a deviation variable based on variability of the experimental measures. Dominance ranking according to the NS algorithm was used as the first criterion for model selection, and total error was used to sort models within dominance ranks. We ran three optimizations, first to validate the modified version of the model with specific ion leaks to reproduce MSN model spiking features as in the original implementation (Mahon et al., 2000). All of the relevant features targeted and parameters thus obtained are reported in Tables S1-S5. A second optimization to find a population of parameter sets that reproduced the range of empirically observed excitability measures with Light off and on conditions by the two extracellular K+ conditions, and third to target several spiking features from one neuron recording to produce a more detailed characterization of MSN firing, sufficiently accurate to allow interpolation of excitability measures when tested with various extracellular K+ values. We targeted 4 features for the second optimization: empirical measurements of resting membrane potential (RMP) and rheobase (Rh) in both light on and light off conditions, modeled here as 4.5 mM K+ and 7.5 mM K+ respectively. Target feature values were chosen as (target value ± deviation) 70.0 ± 4.0 (RMP, 4.5 mM K+), 60.4 ± 4.0 (RMP, 7.5 mM K+), 310.23 ± 10.0 pA (Rh, 4.5 mM K+), and 200.89 ± 8.10 pA, to approximate the variability around the empirical median of these features. These models were also constrained by change in initial RMP voltage variability ~0. For this optimization, the NSDE algorithm (Deb et al., 2002; Price et al., 2005) used in Rumbell et al. (2016) was modified by subtracting 2.0 from the calculated error score for each feature, rounding up to 0.0. This ensured that any feature values calculated from models that lie within two standard deviations of the mean would be considered equally acceptable as a potential model of an empirical observation. As this meant that any models with all 4 feature values within 2 standard deviations of the target mean would have a total error score of 0.0, we used a crowdedness function to sort models with equal total error score, calculated as Euclidean distance to the nearest neighbor in feature space. This additional selection criterion encouraged the algorithm to find models filling the range of acceptable features within 2 standard deviations of the target mean. We ran this optimization for 1000 generations with population size 110, simulating 110,000 unique parameter sets, among which 1119 acceptable models with total error score of 0.0 were identified. From among these, 20 were selected randomly and their resulting feature values displayed in Figure 4B. For the third optimization, we chose 14 features to optimize in the models, which characterized measurements of neuron properties (Figure 4) such as Rheobase, resting membrane potential, inter-spike interval distributions, action potential shape. Here the target feature values were precise and extracted from one empirical recording, and the ‘standard deviations’ used here was selected as 5% of the target feature value for each feature, except RMP where smaller ‘standard deviation’ values ~0.1 were used, to prevent the optimization from entering local minima (Tables S1-S5). As this optimization targeted one particular voltage trace, the above method for reducing errors to 0.0 within 2 standard deviations was not used. We ran this optimization for 5000 generations with a population size of 220, testing 1,100,000 unique parameter sets, at which point we terminated the optimization and selected the best fitting member of the population to demonstrate the voltage traces shown in Figure 4C. This model was then simulated in conditions with extracellular K+ between 4.5 and 7.5 mM in increments of 0.5mM, demonstrating reliability of the change in excitability produced by this modulation (Figure 4D).
In vivo multi-unit recordings of neuronal activity
Three weeks following AAV microinjection in the dorsolateral striatum, adult 9 week old male C57Bl6N mice were anesthetized with isoflurane (3%−5% induction, 1.5% maintenance). Ten minutes after injection of a systemic analgesic (carprofen, 5 mg per kg of body weight) mice were placed in a stereotaxic frame. Mice were maintained at 37°C at all times using a feedback-controlled heating pad. Pressure points and incision sites were injected with lidocaine (2%), and eyes were protected from desiccation using artificial tear ointment. The skin above the skull was incised, a custom-made lightweight metal head holder was implanted on the skull using Vetbond (3M) and a recording chamber was built using dental cement (Ortho-Jet, Lang). Mice had a recovery period from surgery of five days, during which they were administered amoxicillin (0.25 mg per ml in drinking water through the water supply). After the recovery period, mice were habituated to head fixation on the spherical treadmill. On the day of the recording, mice were anesthetized with isoflurane. To fix the ground wire, a small craniotomy (0.5 mm diameter) was made above the right cerebellum and a silver wire was implanted at the surface of the craniotomy and fixed with dental cement. A circular craniotomy 1 mm in diameter was made at a location above the dorsolateral striatum and the mice was allowed to recover from anesthesia. The recording chamber was filled with cortical buffer containing (in mM) 135 NaCl, 5 KCl, 1.8 CaCl2 and 1 MgCl2. The head-bar was fixed to a post and the mouse was placed on the spherical treadmill consisting of a ball floating on small cushion of air allowing for 2D movement.
All recordings were performed at least two hours after the end of anesthesia, when the mouse was alert and could actively behave. A 128-channel silicon nanoprobe was lowered into the brain (Shobe et al., 2015). Each nanoprobe had 2 shanks with 64 electrodes on each shank. Each shank was 1.05 mm long and 86 μm at its widest point and tapered to a tip. Each prong was separated by 400 μm. A multiplexed analog output signal was sent from the head stage (Intan Technologies, RHA-2164B) of each probe via thin flexible cables to a 16-bit analog to digital conversion cards (USB-6356, National Instruments), which sampled each signal at a rate of 25 kHz per channel. Signals were then filtered offline and a background signal subtraction was performed. Unit clustering was performed using JRClust (Jun et al., 2017), a fully-automated method for clustering units based on unit spike properties across multiple channels. We only included high-quality units (Butterworth filter 300–6000 Hz, spikes count > 1000, SNR > 7.0). We performed firing rate analysis in MATLAB; we averaged each unit firing rates across 5 trials with 5 s activity bins. For each experimental condition a raster graph was plotted by combining all spike times from 5 trials for each unit and down sampling in 5:1 ratio.
General reagents
All general chemicals used were from Cayman Chemical, Tocris, Thermo-Fisher or Sigma. Other specific reagents and kits are listed in the methods.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data from every experiment represents a minimum of n animals with a balanced number of male and female mice. In all the experiments, n was ≥ 4 mice; sample sizes were not calculated a priori. For AAV injections, mice were randomly assigned to each experimental group. No experimental data points were excluded. Statistical tests were run in OriginPro 2015. Summary data are presented as mean ± SEM. Note that in some of the graphs, the bars representing the s.e.m. are smaller than the symbols used to represent the mean. For every dataset, we determined within Origin whether the data were normally distributed or not using the Shapiro-Wilk test. For tests of statistical significance, we used parametric tests for normally distributed data and non-parametric tests for data that was not normally distributed. Paired and unpaired Student’s two-tailed t tests (as appropriate) and two tailed Mann-Whitney or Wilcoxon paired sign tests were used for most statistical analyses with significance declared at P < 0.05. When a statistical test was employed that was not a Student’s t test or a Mann-Whitney test for a specific case, then it is stated as such in the text. Specific P values are stated in the figures, however, when the P value was less than 0.001, it is stated as p < 10−3 to save space on the figure panels and text. However, where appropriate key statistics are also reported in the text. If the P value was greater than 0.05, then it is stated as p > 0.05. The sample size, n, is defined as the number of cells, numbers of slices, fields-of-view or numbers of mice depending on the experiment on a case-by-case basis.
DATA AND SOFTWARE AVAILABILITY
The AAV expression vector for ChR2(H134R)-mCherry using the GfaABC1D promoter will be available from Addgene (#112496). Data analyses and software guidance is available upon request. The MSN model is available from James R. Kozlozki at IBM.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| Rabbit anti-NeuN | Cell Signaling | Cat# 12943; RRID: AB_2630395 |
| Mouse anti-mCherry | Saint John’s | Cat#STJ97087; RRID:AB_2687534 |
| Rabbit anti-DsRed | Clontech Laboratories | Cat# 632496; RRID:AB_10013483 |
| Rabbit anti-S100b | Abcam | Cat# ab41548; RRID: AB_956280 |
| Guinea pig anti-Neun | Synaptic Systems | Cat# 266 004; RRID:AB_2619988 |
| Guinea pig anti-cFOS | Synaptic Systems | Cat# 226 004; RRID:AB_2619946 |
| Rabbit anti-cFOS | Abcam | Cat# ab190289; RRID:AB_2737414 |
| Mouse anti-neurofilament | Biolegend | Cat# SMI-312R; RRID:AB_2314906 |
| Alexa goat anti-rabbit 488 | Molecular Probes | Cat# A11008; RRID: AB_143165 |
| Alexa goat anti-chicken 488 | Molecular Probes | Cat# A11039; RRID: AB_142924 |
| Alexa goat anti-rabbit 546 | Molecular Probes | Cat#A11010; RRID: AB_143156 |
| Alexa goat anti-mouse 546 | Molecular Probes | Cat#A11003; RRID: AB_141370 |
| Alexa goat anti-guinea pig 488 | Molecular Probes | Cat# A-11073; RRID:AB_142018 |
| Alexa goat anti-guinea pig 546 | Molecular Probes | Cat# A-11074 RRID:AB_2534118 |
| Bacterial and Virus Strains | ||
| AAV2/5 GfaABC1D ChR2(H134R)-mCherry SV40 | This manuscript, UPenn Vector Core | Available upon request from UPenn |
| AAV2/1 hSynapsinl ChR2(H134R)-eYFP SV40 | UNC GTC vector core | Lot# AV4319J |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TTX | Cayman Chemical Company | Cat# 14964 |
| CNQX | Tocris | Cat# 0190 |
| D-AP5 | Tocris | Cat# 0106 |
| Barium chloride | Sigma | Cat# 202738 |
| Normal goat serum | Vector | Cat# S-1000 |
| Critical Commercial Assays | ||
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28104 |
| QIAGEN Mini kit | QIAGEN | Cat# 27104 |
| QIAGEN Endofree MAXI kit | QIAGEN | Cat# 12362 |
| QIAquick Gel extraction Kit | QIAGEN | Cat# 28704 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6N inbred mice | Taconic | JAX Stock # 005304 |
| Mouse: Thy1-ChR2-YFP | Arenkiel et al., 2007 | JAX Stock # 007615 |
| Recombinant DNA | ||
| pZac2.1 GfaABC1D ChR2(H134R) SV40 | This manuscript; Addgene | Plasmid # 112496 |
| Software and Algorithms | ||
| OriginPro 8.5/9/2015 | Origin Lab | N/A |
| GraphPad Instat 3 | GraphPad Software | N/A |
| pCLAMP10 | Molecular Devices | N/A |
| ClampFit 10.5 | Molecular Devices | N/A |
| Fluoview FV3000 | Olympus | N/A |
| ImageJ v1.30 | ImageJ | N/A |
| ANY-maze behavioral tracking software | Stoelting Co. | N/A |
| Prizmatix Pulser | Prizmatix Ltd. | N/A |
| MSN model parameter file | GSL file submitted here | N/A |
Highlights.
Multiple approaches reveal transient K+ elevations during ChR2 excitation
ChR2-mediated K+ elevations increase neuronal excitability and cFos expression
Neuronal effects of K+ are recapitulated with a model and in vivo
Increased K+ may contribute to astrocyte experiments employing ChR2 in vivo
ACKNOWLEDGMENTS
This work was supported by NIH grants MH099559, NS060677, and MH104069 (to B.S.K.). B.S.K. is also supported by a Paul G. Allen Distinguished Investigator Award. S.H. was supported by the UCLA Cross-Disciplinary Scholars in Science and Technology Program. M.G. was supported partly by a CHDI Award (to B.S.K.). S.L.A., T.H.R., T.M.H.-T., and J.R.K. were supported by IBM. The GfaABC1D ChR2(H134R)-mCherry AAV was first tested by A.D. Johnston in the Khakh lab. Thanks to Dr. M.V. Sofroniew for sharing equipment. Thanks to Dr. S. Masmanidis for help with manufacturing silicon multielectrode probes and for the recording hardware. The design and manufacture of the silicon probes was supported by UCLA NSF NeuroNex Hub (1707408). B.S.K. thanks Ege Kavalali for insightful discussions during the early stages on the shores of Lake Tahoe.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.078.
REFERENCES
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, and Volterra A (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ, Ehlers MD, and Feng G (2007). In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron 54, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhurin KI, Mac V, Golshani P, and Masmanidis SC (2016). Temporal correlations among functionally specialized striatal neural ensembles in reward-conditioned mice. J. Neurophysiol 115, 1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang J, Kim HY, and Lee H (2016). Optogenetic and Chemogenetic Approaches for Studying Astrocytes and Gliotransmitters. Exp. Neurobiol 25, 205–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Brew H, and Attwell D (1988). Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature 335, 433–435. [DOI] [PubMed] [Google Scholar]
- Bernstein JG, and Boyden ES (2011). Optogenetic tools for analyzing the neural circuits of behavior. Trends Cogn. Sci 15, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, and Deisseroth K (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whitelegge JP, et al. (2017). Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531–549.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis M, Uva L, Gnatkovsky V, and Librizzi L (2018). Potassium dynamics and seizures: Why is potassium ictogenic? Epilepsy Res. 143, 50–59. [DOI] [PubMed] [Google Scholar]
- Deb K, Pratap A, Agarwal S, and Meyarivan T (2002). A Fast and Elitist Multiobjective Genetic Algorithm: NSGA-II. IEEE Trans. Evol. Comput 6, 182–197. [Google Scholar]
- Deemyad T, Lüthi J, and Spruston N (2018). Astrocytes integrate and drive action potential firing in inhibitory subnetworks. Nat. Commun 9, 4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K (2015). Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci 18, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo M, Lane S, Tang F, Liu BH, Hewinson J, Marina N, Kasymov V, Souslova EA, Chudakov DM, Gourine AV, et al. (2011). Optogenetic experimentation on astrocytes. Exp. Physiol 96, 40–50. [DOI] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, and Nelson MT (2010). Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc. Natl. Acad. Sci. USA 107, 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez JA, Perkins JM, Beaudoin GM, Cook NB, Quraishi SA, Szoeke EA, Thangamani K, Tschumi CW, Wanat MJ, Maroof AM, et al. (2019). Ventral tegmental area astrocytes orchestrate avoidance and approach behavior. Nat. Commun 10, 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, and Kasparov S (2010). Astrocytes control breathing through pH-dependent release of ATP. Science 329, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, and Deisseroth K (2009). Optical deconstruction of parkinsonian neural circuitry. Science 324, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haustein MD, Kracun S, Lu XH, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O’Dell TJ, Marvin JS, et al. (2014). Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Stabel J, and Rausche G (1990). Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog. Brain Res 83, 197–214. [DOI] [PubMed] [Google Scholar]
- Herman AM, Huang L, Murphey DK, Garcia I, and Arenkiel BR (2014). Cell type-specific and time-dependent light exposure contribute to silencing in neurons expressing Channelrhodopsin-2. eLife 3, e01481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). Ion Channels of Excitable Membranes, Third Edition (Sinauer Associates, Inc.). [Google Scholar]
- Hodgkin AL, and Huxley AF (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferys JG (1995). Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiol. Rev 75, 689–723. [DOI] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Looger LL, and Khakh BS (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JJ, Mitelut C, Lai C, Gratiy S, Anastassiou C, and Harris TD (2017). Real-time spike sorting platform for high-density extracellular probes with ground-truth validation and drift correction. bioRxiv. 10.1101/101030. [DOI]
- Katz B, and Miledi R (1982). An endplate potential due to potassium released by the motor nerve impulse. Proc. R. Soc. London 216B, 497–507. [DOI] [PubMed] [Google Scholar]
- Kozloski J, and Wagner J (2011). An Ultrascalable Solution to Large-scale Neural Tissue Simulation. Front. Neuroinform 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffler SW (1967). Neuroglial cells: physiological properties and a potassium mediated effect of neuronal activity on the glial membrane potential. Proc. R. Soc. London 168B, 1–21. [DOI] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, and Tsien RY (2009). Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys. J 96, 1803–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S, Deniau JM, Charpier S, and Delord B (2000). Role of a striatal slowly inactivating potassium current in short-term facilitation of corticostriatal inputs: a computer simulation study. Learn. Mem 7, 357–362. [DOI] [PubMed] [Google Scholar]
- Masamoto K, Unekawa M, Watanabe T, Toriumi H, Takuwa H, Kawaguchi H, Kanno I, Matsui K, Tanaka KF, Tomita Y, et al. (2015). Unveiling astrocytic control of cerebral blood flow with optogenetics. Sci. Rep 5, 11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreery DB, and Agnew WF (1983). Changes in extracellular potassium and calcium concentration and neural activity during prolonged electrical stimulation of the cat cerebral cortex at defined charge densities. Exp. Neurol 79, 371–396. [DOI] [PubMed] [Google Scholar]
- McDonough AA, and Youn JH (2017). Potassium Homeostasis: The Knowns, the Unknowns, and the Health Benefits. Physiology (Bethesda) 32, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos S, Hernández-Vivanco A, Ramírez-Franco J, Martín-Fernández M, Navarrete M, Yang A, Boyden ES, and Perea G (2019). Melanopsin for precise optogenetic activation of astrocyte-neuron networks. Glia 67, 915–934. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, and Bamberg E (2003). Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 100, 13940–13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, and Verkhratsky A (2012). Artifact versus reality-how astrocytes contribute to synaptic events. Glia 60, 1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, and Olsen ML (2016). The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 132, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, and Khakh BS (2018a). An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron 98, 49–66.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Faas G, Mody I, and Khakh BS (2018b). Making, Testing, and Using Potassium Ion Selective Microelectrodes in Tissue Slices of Adult Brain. J. Vis. Exp 135, e57511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otchy TM, Wolff SB, Rhee JY, Pehlevan C, Kawai R, Kempf A, Gobes SM, and Ölveczky BP (2015). Acute off-target effects of neural circuit manipulations. Nature 528, 358–363. [DOI] [PubMed] [Google Scholar]
- Pelluru D, Konadhode RR, Bhat NR, and Shiromani PJ (2016). Optogenetic stimulation of astrocytes in the posterior hypothalamus increases sleep at night in C57BL/6J mice. Eur. J. Neurosci 43, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Yang A, Boyden ES, and Sur M (2014). Optogenetic astrocyte activation modulates response selectivity of visual cortex neurons in vivo. Nat. Commun 5, 3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaksin M, Kimmel E, and Shoham S (2018). Thermal transients excite neurons through universal intramembrane mechano-electrical effects. Phys. Rev. X 8, 011043. [Google Scholar]
- Price KV, Storn RM, and Lampinen JA (2005). Differential Evolution: A Practical Approach to Global Optimization (Springer; ). [Google Scholar]
- Richards R, and Dempski RE (2012). Re-introduction of transmembrane serine residues reduce the minimum pore diameter of channelrhodopsin-2. PLoS ONE 7, e50018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbell TH, Draguljić D, Yadav A, Hof PR, Luebke JI, and Weaver CM (2016). Automated evolutionary optimization of ion channel conductances and kinetics in models of young and aged rhesus monkey pyramidal neurons. J. Comput. Neurosci 41, 65–90. [DOI] [PubMed] [Google Scholar]
- Rungta RL, Osmanski BF, Boido D, Tanter M, and Charpak S (2017). Light controls cerebral blood flow in naive animals. Nat. Commun 8, 14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Beppu K, Tanaka KF, Fukazawa Y, Shigemoto R, and Matsui K (2012). Application of an optogenetic byway for perturbing neuronal activity via glial photostimulation. Proc. Natl. Acad. Sci. USA 109, 20720–20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MG, Homma K, Villarreal S, Richter CP, and Bezanilla F (2012). Infrared light excites cells by changing their electrical capacitance. Nat. Commun 3, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Nikolic L, Meunier C, Pfrieger F, and Audinat E (2017). An autocrine purinergic signaling controls astrocyte-induced neuronal excitation. Sci. Rep 7, 11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S,Xu J, Sofroniew MV, Ellisman MH, and Khakh BS (2013). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol 141, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shobe JL, Claar LD, Parhami S, Bakhurin KI, and Masmanidis SC (2015). Brain activity mapping at multiple scales with silicon microprobes containing 1,024 electrodes. J. Neurophysiol 114, 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan SA, and Barres BA (2014). Looks can be deceiving: reconsidering the evidence for gliotransmission. Neuron 84, 1112–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu T-Y, Chai H, Xu J, Huang BS, Golshani P, Coppola G, and Khakh BS (2016). New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron 92, 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2015). Reproducibility: Experimental mismatch in neural circuits. Nature 528, 338–339. [DOI] [PubMed] [Google Scholar]
- Szatkowski M, Barbour B, and Attwell D (1990). Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348, 443–446. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, and Khakh BS (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Geit W, Gevaert M, Chindemi G, Rössert C, Courcol JD, Muller EB, Schürmann F, Segev I, and Markram H (2016). BluePyOpt: Leveraging Open Source Software and Cloud Infrastructure to Optimise Model Parameters in Neuroscience. Front. Neuroinform 10, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KD, and Chesler M (1988). Activity-related extracellular potassium transients in the neonatal rat spinal cord: an in vitro study. Neuroscience 25, 983–995. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Moyer JT, Lazarewicz MT, Contreras D, Benoit-Marand M, O’Donnell P, and Finkel LH (2005). NMDA/AMPA ratio impacts state transitions and entrainment to oscillations in a computational model of the nucleus accumbens medium spiny projection neuron. J. Neurosci 25, 9080–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Hamada A, Suhara Y, Kawabe R, Yanase M, Kuzumaki N, Narita M, Matsui R, Okano H, and Narita M (2014). Astrocytic activation in the anterior cingulate cortex is critical for sleep disorder under neuropathic pain. Synapse 68, 235–247. [DOI] [PubMed] [Google Scholar]
- Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, and Khakh BS (2018). Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Vierock J, Yizhar O, Fenno LE, Tsunoda S, Kianianmomeni A, Prigge M, Berndt A, Cushman J, Polle J, et al. (2011). The microbial opsin family of optogenetic tools. Cell 147, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The AAV expression vector for ChR2(H134R)-mCherry using the GfaABC1D promoter will be available from Addgene (#112496). Data analyses and software guidance is available upon request. The MSN model is available from James R. Kozlozki at IBM.