

Abstract
We describe two unrelated patients aged 9 and 12 years. The first patient presented with multiple congenital contractures not associated with webbing (pterygia). Interestingly, his genetic testing showed the typical genotypic criteria of Escobar syndrome (CHRNG heterozygous mutation). The characteristics of the second child were compatible with the phenotypic and genotypic criteria for Escobar syndrome. Both patients manifested the typical facial features suggestive of Escobar syndrome. The aim of this paper is twofold: first, to illustrate that the absence of popliteal webbing is not a sufficient reason to exclude Escobar syndrome in patients with multiple contractures and second, dysmorphic facial features and the presence of certain radiological abnormalities might be considered baseline diagnostic tools in favor of this syndromic entity.
Keywords: CHRNG heterozygous mutation, Congenital multiple contractures, Escobar syndrome
Introduction
Multiple pterygium syndrome (MPS) is a rare genetic disorder, most cases of which are sporadic. The most consistent malformations seen in MPS are webbing (pterygia) involving the neck (100%), antecubital (90%), and popliteal (90%) areas; syndactyly (74%) and camptodactyly (84%) of the fingers; numerous joint flexion contractures (74%); and foot deformities (74%). Other possible anomalies include umbilical and inguinal hernias (26%), congenital hip dislocation (21%), and hypoplastic nipples (11%). Scoliosis and kyphosis may develop and the stance become crouched. The inheritance is autosomal recessive1, 2, 3. The first clinical description of this entity was published in 1969 by Norum et al.4.
Multiple contractures without webbing can occur in a great many syndromic entities, including Freeman–Sheldon syndrome, congenital muscular dystrophy, Tel Hashomer camptodactyly, and Schwartz–Jampel syndrome1. Webbing (multiple and local) can be also associated with a number of genetic syndromes (popliteal pterygium syndrome, Noonan syndrome, fetal akinesia deformation sequence) and is an important differential diagnostic sign.
Escobar syndrome, a variant of MPS, manifests as an autosomal‐recessive condition with multiple congenital anomalies and is caused by homozygous or compound heterozygous mutation of the CHRNG gene. The CHRNG gene encodes the gamma subunit of the acetylcholine receptor (AChR) and is localized on chromosome 2 (2q37.1). MPS is traditionally divided into prenatally lethal multiple pterygium syndrome (LMPS) and nonlethal, or Escobar variant multiple pterygium syndrome (EVMPS)3. Both variants are characterized by webbing (pterygia) of the elbows and/or knees and joint contractures involving the neck and are usually labeled as arthrogryposis. Vogt et al. demonstrated a correlation between clinical manifestations of MPS and genetic data. In this cohort, 27% of kindred of subjects with EVMPS had a detectable CHRNG mutation5. These authors stated that 94% of patients with Escobar syndrome with CHRNG mutation show the typical phenotype of multiple pterygia.
In this paper we present two patients who both satisfied the criteria for diagnosis of Escobar syndrome but had different phenotypic manifestations.
Case Reports
Patient 1
The first patient was referred to our department from the Department of Neurology for the first time at the age of 6 years. This child was the first live offspring of a‐29‐year‐old woman with a history of multiple spontaneous abortions who was married to a 33‐year‐old related man. Soon after birth, he had manifested progressive contractures with no pterygia: the original clinical diagnosis had been congenital myopathy. He had undergone a series of thorough investigations, all of which had proved negative. Accordingly, a diagnosis of arthrogryposis had been made. His subsequent motor development had been severely retarded.
Clinical examination at the age of 6 years showed his growth parameters were within two standard deviations of the mean. Craniofacial anomalies included a brachycephalic skull, high forehead, thin lips, low‐set ears, multiple caries and high arched palate. Orofacial anomalies include immobile face, epicanthal folds, palpebral fissures, low‐set ears, ptosis, micrognathia and down‐turned corners of the mouth. Joint movements in the upper limbs were limited. Similarly, he had marked restrictions in movement of his hips and knees that had resulted in development of valgus deformity. Severe feet deformities (left equino‐plano‐valgus deformity with forefoot abduction, right equino‐cavo‐varus deformity) were notable (Fig. 1). Examination of the oral cavity showed severe gingival fibromatosis, high vault palatine and dental caries (Fig. 2). Viewed from behind, a brachycephalic head (flat occiput), low‐set‐ears, bilateral whorls and a low‐hair line were evident (Fig. 3). Hearing, vision and intelligence were normal.
Figure 1.
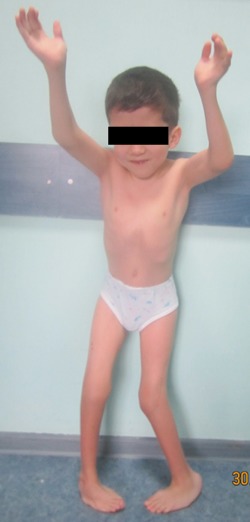
Patient 1. Photo showing craniofacial features of brachycephalic skull, high forehead, thin lips and low‐set ears, orofacial anomalies including epicanthal folds, palpebral fissures, ptosis, micrognathia and down‐turned corners of the mouth The patient is short and musculoskeletal examination showed multiple flexion contractures, vertebral anomalies (thoracolumbar S‐shaped scoliosis of the spine), limited joint movements in the upper limbs and marked restrictions in the movement of the hips and knees with valgus deformity. He has left equino‐plano‐valgus deformity with forefoot abduction and right equino‐cavo‐varus deformity.
Figure 2.
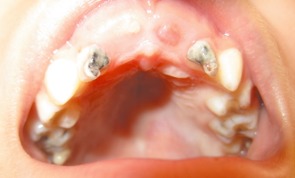
Patient 1. Open mouth photo showing severe gingival fibromatosis, high vault palatine and dental caries.
Figure 3.
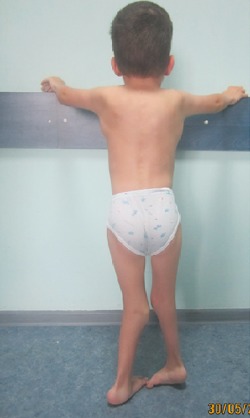
Patient 1. Posterior photo showing a brachycephalic head (flat occiput), low‐set‐ears, bilateral whorls and a low‐hair line.
Musculoskeletal examination showed multiple flexion contractures, vertebral anomalies (thoracolumbar S‐shaped scoliosis of the spine) and limited joint movements in the upper limbs. Lower limb standing radiograph showed bilateral hip dislocation associated with marked acetabulo‐femoral dysplasia and bilateral knee subluxation (Fig. 4). Foot radiograph revealed talipes equinovarus associated with defective ossification of the metatarsals and osteoporosis; varus of the hindfoot, supination of the forefoot and rocker‐bottom deformity of the midfoot were also evident (Fig. 5). Despite the absence of knee flexion deformity with popliteal webbing, the overall clinicoradiographic features were suggestive of Escobar syndrome. Accordingly, genetic testing was organized. The boy was found to have a CHRNG mutation (heterozygous mutations c.459dupA [{p.Val154Serfs*24}/c.794T>G{p.Leu265Ser}]). His father had a heterozygous mutation (N/c.459dupA [p.Val154Serfs*24]), as did his mother (N/c.794T>G[p.Leu265Ser]).
Figure 4.
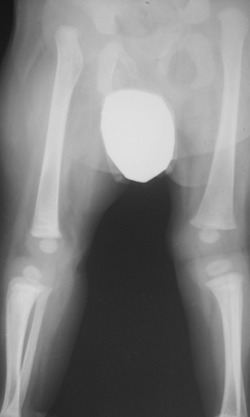
Patient 1. Lower limb radiograph at age of 4 years showing bilateral hip dislocation associated with marked acetabulo‐femoral dysplasia and bilateral knee dislocation.
Figure 5.
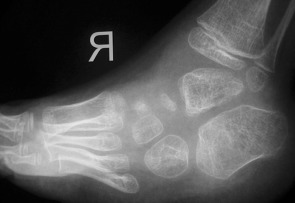
Patient 1. Foot radiograph showing talipes equinovarus associated with defective ossification of the metatarsals and osteoporosis, varus of the hindfoot, supination of the forefoot and rocker‐bottom deformity of the midfoot.
Patient 2
The second patient was first referred to our department at the age of 6 years because of extensive webbing associated with widespread massive multiple contractures. His family history was non‐contributory; however, his parents were first‐degree cousins. He had severely retarded development of gross and fine motor skills. The child had been investigated thoroughly during his first years of life by the Department of Neurology, the suspected diagnosis being congenital myopathy. However, all tests had been negative, ruling out this diagnosis. As with the first case, a diagnosis of arthrogryposis multiplex had been made.
Clinical examination at the age of 6 years showed severe growth deficiency (‐3 standard deviations). Craniofacially, he had deep‐seated eyes, depressed nasal bridge, long philtrum and large low‐set ears. His hearing and vision were normal and neurological examination showed nothing of significance.
Musculoskeletal examination showed remarkable limitations in movement of all joints because of the extreme degree of webbing. He had thoraco‐lumbar kyphoscoliosis associated with remarkable pectus excavatum and weakening of the pectoral muscles. There was webbing across multiple flexion areas, including the neck, axilla, antecubital fossae, fingers and popliteal areas (flexion contracture of 150°, Fig. 6). The boy underwent hamstring tenotomy at the level of the ischial tuberosity. Tenotomy of the flexor hallucis longus and Z‐lengthening of the Achilles tendon have also been performed bilaterally as a single‐stage surgery. The procedures were accomplished by release of the posterior fibrous band. Because of tension on the sciatic nerve and vascular bundle, full extension of the knee joints was achieved by several weeks postoperatively by means of serial casting. Gradual soft tissue lengthening with an Ilizarov external fixator considered contraindicated by the intertwining of the sciatic nerve and associated vessels along this fibrotic band. Postoperatively the child developed the ability to walk freely and his lower legs functions were much improved (Fig. 7).
Figure 6.
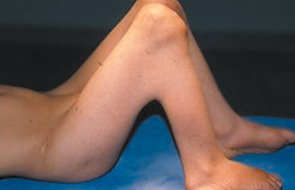
Patient 2. Photo showing webbing across the popliteal areas (flexion contracture of 150°).
Figure 7.
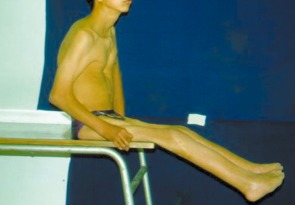
Patient 2. Postoperatively the release of popliteal webbing allowed the child to walk freely and his lower functions were much improved.
Discussion
Escobar syndrome is usually diagnosable at birth; however, the characteristic webs (pterygia) sometimes do not develop until later in infancy. At birth there may be multiple joint contractures with camptodactyly, talipes equinovarus and rocker‐bottom feet. The neck is short and may be webbed. The external genitalia can be hypoplastic in both males and females. The face is relatively immobile with downturned corners, epicanthic folds and mild ptosis4. These typical characteristics were present in our first patient, but he did not develop pterygia as he grew older.
Several reports have described the various presentations of patients with Escobar syndrome5, 6, 7, 8, 9, 10, 11, 12, 13. None of these reports describe the pattern of pathology seen in our first patient.
The condition has now been genetically mapped and mutations found in the embryonal gamma subunit of the acetylcholine receptor (CHRNG) by Morgan et al.14 and Hoffmann et al.15.
Neck pterygium can be associated with trisomy 21 syndrome, trisomy 18, Turner syndrome, Noonan syndrome and Nielson syndrome; all these disorders are characterized by short stature, cleft palate, camptodactyly and vertebral fusions. Another syndromic entity characterized by neck webbing is Golden–Lakin syndrome (dolichocephaly, small mandible, pointed facies, bifid uvula, pectus excavatum, kyphoscoliosis, flexion deformity of the knees without webbing, pes cavus and club foot). Escobar syndrome should be differentiated from pterygium syndromes with limb pterygia such as popliteal pterygium syndrome (autosomal dominant inheritance). Other syndromic entities that are associated with congenital knee flexion deformities are popliteal pterygium syndrome, Beal congenital contractual arachnodactyly and arthrogryposis multiplex congenita.
There are various methods for treating popliteal pterygia. These include serial casting or traction, the outcome of which is reportedly unsatisfactory. Among possible surgical approaches, lengthening of soft tissues such as skin, muscles and ligaments by resection of fibrous bands and Z‐plasty is the preferred technique16. The Ilizarov technique is an improvement on conservative methods. It allows progressive correction of the most complex deformities of the knee with simultaneous correction of associated foot deformities and limb lengthening17. Oppenheim et al. reported that excision of fibrous bands and Z‐plasty were possible in only two of seven cases of patellofemoral pain syndrome (four patients); the remaining cases required secondary operations such as femoral extension osteotomy, femoral shortening or amputation because of severe adhesions to the sciatic nerve18.
Mennen et al. considered arthrogryposis to be a catch‐all term used to describe various conditions that present with joint contractures19. Hall listed dozens of clinical entities that share the element of congenital contractures and have been labeled as arthrogryposis20. He considered such contractures to be the end result of decreased fetal intra‐uterine movement after a period of normal development. However, we believe that the conclusions of these studies are outdated because they encourage bypassing of the importance of phenotypic characterization as a baseline tool in the management of children with multiple deformities. Arthrogryposis multiplex is a symptom complex rather than a diagnosis; it has various causes and presents as many different syndromes21.
Disclosure: The authors declare that they have no competing interests. The patients gave the informed consent prior to being included into the study. The study was authorized by the local Ethics Committee and was performed in accordance with the ethical standards of the 1964 Declaration of Helsinki as revised in 2000.
References
- 1. Wynne‐Davies R, Gormley J. The prevalence of skeletal dysplasias. An estimate of their minimum frequency and the number of patients requiring orthopaedic care. J Bone Joint Surg Br, 1985, 67: 133–137. [DOI] [PubMed] [Google Scholar]
- 2. Bussiere JA. Developement Abnormal D'un Faisceau Musculare Acromio‐Mastoidien Rudimentaire, Malformation Congenitale Rare, Observe'e A Pondicherry (Indes Orientales). Annales d'Hygiene et de Medecine Coloniales, 1902, 5: 686–688. (In French) [Google Scholar]
- 3. Escobar V, Bixler D, Gleiser S, Weaver DD, Gibbs T. Multiple pterygium syndrome. Am J Dis Child, 1978, 132: 609–611. [DOI] [PubMed] [Google Scholar]
- 4. Norum RA, James VL, Mabry CC. Pterygium syndrome in three children in a recessive pedigree pattern. Birth Defects, 1969, 5: 233–235. [Google Scholar]
- 5. Vogt J, Morgan NV, Rehal P, et al CHRNG genotype‐phenotype correlations in the multiple pterygium syndromes. J Med Genet, 2012, 49: 21–26. [DOI] [PubMed] [Google Scholar]
- 6. Beckerman RC, Buchino JJ. Arthrogryposis multiplex congenita as part of an inherited symptom complex: two case reports and a review of the literature. Pediatrics, 1978, 61: 417–422. [DOI] [PubMed] [Google Scholar]
- 7. Kousseff BG, Nichols P. A new autosomal recessive syndrome with Noonan‐like phenotype, myopathy, with congenital contractures and malignant hyperthermia. Birth Defects Orig Artic Ser, 1985, 21: 111–117. [PubMed] [Google Scholar]
- 8. Ramer JC, Ladda RL, Demuth WW. Multiple pterygium syndrome. An overview. Am J Dis Child, 1988, 142: 794–798. [DOI] [PubMed] [Google Scholar]
- 9. Willems PJ, Colpaert C, Vaerenbergh M, et al Multiple pterygium syndrome with body asymmetry. Am J Med Genet, 1993, 47: 106–111. [DOI] [PubMed] [Google Scholar]
- 10. Scott H, Hunter A, Bedard B. Non‐lethal arthrogryposis multiplex congenita presenting with cystic hygroma at 13 weeks gestational age. Prenat Diagn, 1999, 19: 966–971. [DOI] [PubMed] [Google Scholar]
- 11. Spranger S, Spranger M, Meinck HM, Tariverdian G. Two sisters with Escobar syndrome. Am J Med Genet, 1995, 57: 425–428. [DOI] [PubMed] [Google Scholar]
- 12. Thompson EM, Donnai D, Baraitser M, Hall CM, Pembrey ME, Fixsen J. Multiple pterygium syndrome: evolution of the phenotype. J Med Genet, 1987, 24: 733–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rozin MM, Hertz M, Goodman RM. A new syndrome with camptodactyly, joint contractures, facial anomalies, and skeletal defects: a case report and review of syndromes with camptodactyly. Clin Genet, 1984, 26: 342–355. [DOI] [PubMed] [Google Scholar]
- 14. Morgan NV, Brueton LA, Cox P, et al Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am J Hum Genet, 2006, 79: 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoffmann K, Muller JS, Stricker S, et al Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor gamma subunit. Am J Hum Genet, 2006, 79: 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCall RE, Budden J. Treatment of multiple pterygium syndrome. Orthopedics, 1992, 15: 1417–1422. [DOI] [PubMed] [Google Scholar]
- 17. Damsin JP, Carlioz H. Treatment of limb deformities by the Ilizarov method. Rev Chir Orthop Reparatrice Appar Mot, 1994, 80: 324–333. [PubMed] [Google Scholar]
- 18. Oppenheim WL, Larson KR, McNabb MB, Smith CF, Setoguchi Y. Popliteal pterygium syndrome: an orthopaedic perspective. J Pediatr Orthop, 1990, 10: 58–64. [PubMed] [Google Scholar]
- 19. Mennen U, van Heest A, Ezaki MB, Tonkin M, Gericke G. Arthrogryposis multiplex congenita. J Hand Surg Br, 2005, 30: 468–474. [DOI] [PubMed] [Google Scholar]
- 20. Hall JG. Arthrogryposis. Am Fam Physician, 1989, 39: 113–119. [PubMed] [Google Scholar]
- 21. Al Kaissi A, Kalchhauser G, Grill F, Klaushofer K. Arthrogryposis multiplex congenital in a child manifesting phenotypic features resembling dysosteosclerosis/osteosclerosis malformation complex; 3DCT scan analysis of the skull base. Cases J, 2008, 1: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]