

Abstract
Importance
Infantile nystagmus syndrome (INS) is a group of disorders presenting with genetic and clinical heterogeneities that have challenged the genetic and clinical diagnoses of INS. Precise molecular diagnosis in early infancy may result in more accurate genetic counseling and improved patient management.
Objective
To assess the accuracy of genomic data from next-generation sequencing (NGS) and phenotypic data to enhance the definitive diagnosis of INS.
Design, Setting, and Participants
A single-center retrospective case series was conducted in 48 unrelated, consecutive patients with INS, with or without associated ocular or systemic conditions, who underwent genetic testing between June 1, 2015, and January 31, 2017. Next-generation sequencing analysis was performed using a target panel that included 113 genes associated with INS (n = 47) or a TruSight One sequencing panel that included 4813 genes associated with known human phenotypes (n = 1). Variants were filtered and prioritized by in-depth clinical review, and finally classified according to the American College of Medical Genetics and Genomics guidelines. Patients underwent a detailed ophthalmic examination, including electroretinography and optical coherence tomography, if feasible.
Main Outcomes and Measures
Diagnostic yield of targeted NGS testing.
Results
Among the 48 patients (21 female and 27 male; mean [SD] age at genetic testing, 9.2 [10.3] years), 8 had a family history of nystagmus and 40 were simplex. All patients were of a single ethnicity (Korean). Genetic variants that were highly likely to be causative were identified in 28 of the 48 patients, corresponding to a molecular diagnostic yield of 58.3% (95% CI, 44.4%-72.2%). FRMD7, GPR143, and PAX6 mutations appeared to be the major genetic causes of familial INS. A total of 10 patients (21%) were reclassified to a different diagnosis based on results of NGS testing, enabling accurate clinical management.
Conclusions and Relevance
These findings suggest that NGS is an accurate diagnostic tool to differentiate causes of INS because diagnostic tests, such as electroretinography and optical coherence tomography, are not easily applicable in young infants. Accurate application of NGS using a standardized, stepwise, team-based approach in early childhood not only facilitated early molecular diagnosis but also led to improved personalized management in patients with INS.
This case series assesses the accuracy of genomic data from next-generation sequencing and phenotypic data to enhance the definitive diagnosis of infantile nystagmus syndrome.
Key Points
Question
What is the diagnostic rate of targeted next-generation sequencing for infantile nystagmus syndrome?
Findings
In this case series of 48 consecutive patients with infantile nystagmus syndrome, the diagnostic yield of targeted next-generation sequencing was 58.3%. The initial clinical diagnosis was revised in 10 of 48 patients after targeted next-generation sequencing.
Meaning
Findings from this case series suggest that early application of targeted next-generation sequencing can be a front-line diagnostic tool to differentiate causes of infantile nystagmus syndrome.
Introduction
Infantile nystagmus syndrome (INS) is a heterogeneous disorder characterized by continuous oscillation of the eyes with variable visual acuity. Infantile nystagmus syndrome is an important clinical diagnosis because nystagmus is a common presenting sign of many ocular, neurologic, and systemic diseases.1 Nystagmus can be associated with low vision in early infancy owing to several causes, such as Leber congenital amaurosis (LCA), PAX6 (OMIM 607108)–related phenotypes, or albinism, which makes the differential diagnosis of INS more complex. Furthermore, ophthalmologists can miss subtle retinal abnormalities because continually oscillating eyes hinder the performance of detailed clinical examination. Therefore, meticulous clinical workups using optical coherence tomography (OCT), visual evoked potential, and electroretinography (ERG), are important to differentiate underlying causes of nystagmus. However, results of electrophysiological testing are often inconclusive in pediatric patients. In addition, young children often have been deemed unsuitable for conventional tabletop OCT.
Determining the precise genetic cause of INS has significant clinical relevance in terms of defining a firm molecular diagnosis and clarifying inheritance patterns, thereby guiding genetic counseling and precision medicine. Conventional Sanger sequencing of FRMD7 (OMIM 300628) and GPR143 (OMIM 300808) genes has been shown to be helpful for genetic diagnosis in familial INS, but no putative mutations have been identified in sporadic cases through Sanger sequencing of FRMD7 and GPR143, to our knowledge.2 To date, more than 100 genes have been associated with INS, making it an ideal target for clinical application of next-generation sequencing (NGS). The aim of this study was to assess the clinical utility of NGS in the diagnosis of INS and to determine the clinical relevance of early molecular diagnosis in these patients.
Methods
Recruitment and Selection of Patients With INS
The cohort for this study included 48 consecutive patients of a single ethnicity (Korean) with infantile-onset (within 6 months of birth) nystagmus with or without associated ocular or systemic conditions who were willing to agree to undergo genetic testing between June 1, 2015, and January 31, 2017. All patients underwent ophthalmologic examinations, which included slitlamp examination if feasible, determination of the presence and type of nystagmus, fundus examination, and measurement of visual acuity. Electroretinography (Reti-port, Roland Consult, or RetEval, LKC Technologies) was performed according to International Society for Clinical Electrophysiology of Vision standards.3 The ERG used skin electrodes in children younger than 6 years, and sedation with oral chloral hydrate was used in patients younger than 2 years. Spectral domain OCT (Heidelberg Engineering) was performed if applicable. A previous study described the OCT acquisition and analysis protocol in patients with nystagmus.4 Eye movement recordings were performed with electro-oculography (Nicolet Compact Four/CA2000, Nicolet). Informed written consent was provided by patients and/or parents and peripheral blood samples were obtained for genetic analysis from all patients. This study was approved by Severance Hospital Institutional Review Board and adhered to the tenets of the Declaration of Helsinki.5
Targeted Gene Sequencing Using NGS Panel
For the customized NGS panel, we selected 113 candidate causative genes known to cause INS based on literature reviews, the RetNet database (available at https://sph.uth.edu/Retnet/),6 and the Online Mendelian Inheritance in Man (OMIM) database (https://www.ncbi.nlm.nih.gov/omim) (eTable 1 in the Supplement). The TruSight One sequencing panel, which included 4813 genes associated with known clinical phenotype, was used in 1 patient (P15) owing to multiple systemic features. Target enrichment was performed with molecular inversion probe–based capture method using a customized target enrichment kit (Celemics). Pooled libraries were sequenced using a MiSeq sequencer (Illumina) and the MiSeq Reagent Kit, version 2 (300 cycles).
NGS Data Analysis
Data analysis was performed primarily through our custom bioinformatic workflow. Briefly, raw sequence data were mapped to GRCh37 (hg19) using the Burrows-Wheeler Aligner algorithm,7 followed by removal of duplicate reads, realignment of insertions and deletions, base quality recalibration, and variant calling using the Genome Analysis Toolkit.8 Every variant that was suspected to be pathogenic, likely pathogenic, or a variant of unknown significance was confirmed by visual inspection of the BAM file using Integrated Genomics Viewer, version 2.3 (Broad Institute). Quality metrics were calculated for each sample using the FastQC software and Target Enrichment Quality Control package (eTable 2 in the Supplement). On average, more than 8.4 million reads were sequenced per sample, and approximately 8.3 million reads (97.7%) were mapped on references using the customized panel. The mean horizontal coverage, which was interpreted as percentage of regions with more than 30 × coverage, was 99.7%.
Split-read–based detection of large insertions and deletions was conducted using the Pindel9 and Manta algorithms,10 and both results were finally crosschecked. Read-depth–based detection of structural rearrangements was conducted using the ExomeDepth software.11 Chromosomal copy number variations detected by ExomeDepth were further crosschecked using our custom bioinformatic workflow; this crosscheck retrieved base-level depth of coverage for each BAM file using SAMtools software12 and normalized the depths against those of other samples in the same batch. We performed off-target analysis of chromosomal copy number alterations using the CopywriteR software.13
Variant Filtering and Classification
The following databases were used for variant annotation: OMIM, Human Gene Mutation Database, ClinVar, dbSNP, 1000 Genomes, Exome Aggregation Consortium, Exome Sequencing Project, and Korean Reference Genome Database. The pathogenicity of missense variants was predicted using 5 in silico prediction algorithms, including SIFT (Sorting Intolerant From Tolerant), PolyPhen2, MutationTaster, MutationAssessor, and Functional Analysis through Hidden Markov Models implemented in dbNSFP version 3.0a.14 Splice site analysis was determined using the MaxEntScan, NNSPLICE (Neural Network Splice Prediction), Human Splice Finder, GeneSplicer, and SpliceFinder-like algorithms implemented in the Alamut Visual software (Interactive Biosoftware).15 The interpretation of variants followed the 5-tier classification system recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology using a step-by-step approach (eFigure 1 in the Supplement).16 The systematic approaches for variant classification are also described in the eAppendix in the Supplement. For the purpose of this study, patients were grouped in the following 3 categories: (1) probable molecular diagnosis: patients with pathogenic or likely pathogenic disease-associated variant(s); (2) possible molecular diagnosis: patients with 2 heterozygous mutations without segregation analysis, or patients harboring a single pathogenic or likely pathogenic disease–associated variant in a gene linked with recessive traits, provided the patient phenotype matches the known spectrum of clinical features for this gene; or (3) unsolved: all other patients for whom no pathogenic or likely pathogenic disease–associated variants were detected.
Confirmation Using Other Methods
For small nucleotide variations, pathogenic and likely pathogenic variants as well as variants of uncertain significance needing data from parents were examined using Sanger sequencing on a 3730 DNA Analyzer with the BigDye Terminator version 3.1 Cycle Sequencing kit (Applied Biosystems). Large exonic deletions and duplications were confirmed using the available multiplex ligation-dependent probe amplification kit (MRC-Holland). As a result, all variants from NGS presented in this study were further confirmed by Sanger sequencing or multiplex ligation-dependent probe amplification.
Results
This study included 48 patients (27 males and 21 females) with INS. None of the probands were of consanguineous parentage, and family history of nystagmus was noted in 8 of the 48 patients (17%). Six patients were consistent with an X-linked inheritance pattern, and 2 patients (P22 and P43) appeared to have autosomal dominant inheritance. The pedigrees of all 8 patients who had a family history of nystagmus are shown in eFigure 2 in the Supplement. Thirty-two patients (67%) had undergone ERG testing and OCT was performed in 27 patients (56%). The mean (SD) age at genetic analysis was 9.2 (10.3) years (range, 0.3-39.8 years [eFigure 3 in the Supplement]). Seven patients (15%) exhibited extraocular features.
Diagnostic Rate of Targeted NGS in INS
Pathogenic or likely pathogenic alleles were identified in 36 patients; 4 of these patients had no segregation analysis in compound heterozygous mutations (eTables 3 and 4 in the Supplement) and 4 others had only 1 pathogenic or likely pathogenic variant in recessive traits that were classified as a possible diagnosis. Probable molecular diagnosis was made in 28 of the 48 patients, corresponding to a molecular diagnostic rate of 58.3% (95% CI, 44.4%-72.2%) (Figure 1), while a possible molecular diagnosis was made in 8 patients, and diagnosis was unsolved in 12 patients. Twenty-four variants could be identified as likely pathogenic and 19 variants were identified as pathogenic. The clinical phenotypes and genotype results of the 28 patients with a probable molecular diagnosis are summarized in Table 1 and Table 2.17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33 Among all 48 patients, initial clinical diagnosis of presumed idiopathic infantile nystagmus was made in 15 (31%) and 23 patients (48%) were diagnosed with LCA. Of the remainder, 4 patients were diagnosed with ocular albinism, 3 with PAX6-related phenotype, and 3 with achromatopsia. The initial clinical diagnosis was revised in 10 patients (21%) (Figure 2). Representative cases are shown in Figure 3, which depicts phenotypical similarities in 4 patients who all had different mutations. These patients were diagnosed with idiopathic infantile nystagmus at the initial visit.
Figure 1. Diagnostic Rate and Mutated Genes in Patients With Infantile Nystagmus Syndrome (INS).
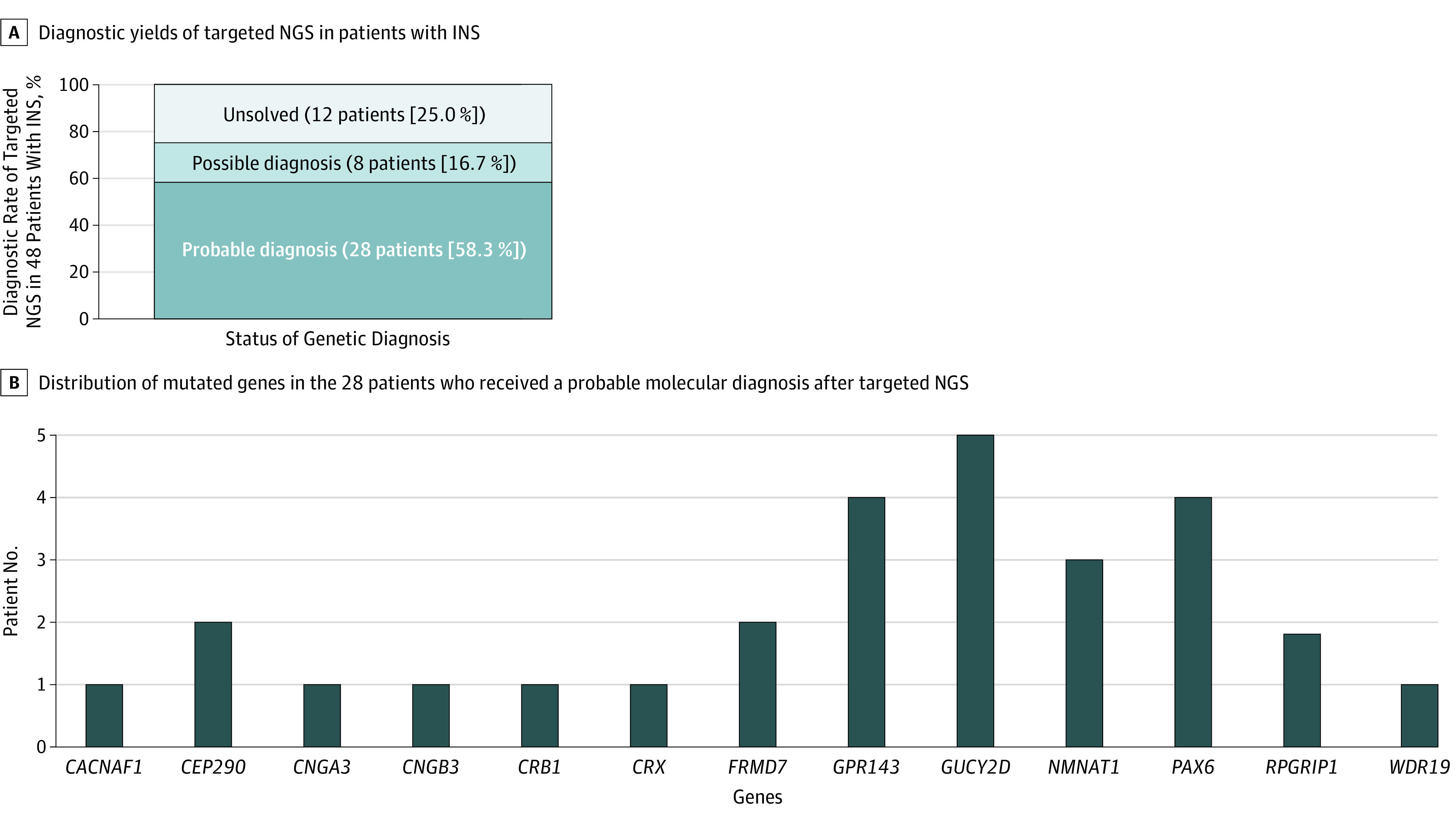
NGS indicates next-generation sequencing.
Table 1. Clinical Features of 28 Patients With Probable Molecular Diagnosis in Infantile Nystagmus Syndrome.
| Patient No./Sex/Age, ya | Initial Clinical Impression | Molecular Diagnosis | Diagnosis After Revisit | Nystagmus | Refraction | BCVA | Fundus | Oculodigital Sign | ERG | Additional Phenotype | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | ||||||||||
| 1/M/2 | LCA | GUCY2D | LCA | Roving | +0.50 | +1.50 | NA | Grossly normal | Y | Extinguished | N |
| 2/F/1.6 | LCA | GUCY2D | LCA | Roving | +5.50 | +5.50 | NA | Grossly normal | Y | Extinguished | N |
| 3/F/1 | LCA | GUCY2D | LCA | Roving | +5.00 | +5.00 | NA | Grossly normal | Y | Extinguished | N |
| 4/M/1.6 | LCA | GUCY2D | LCA | HP | +5.50 | +5.50 | NA | Grossly normal | Y | Extinguished | N |
| 5/F/21.5 | LCA | GUCY2D | LCA | HP | +2.25 | +2.25 | 0.05 | Yellowish fovea | Y | Extinguished | N |
| 6/F/1.5 | LCA | NMNAT1 | LCA | Roving | +3.50 | +3.50 | NA | Atrophic macula | Y | Extinguished | N |
| 7/M/2.3 | LCA | NMNAT1 | LCA | HJ | +5.50 | +5.50 | NA | Atrophic macula | Y | Extinguished | N |
| 8/F/0.8 | LCA | NMNAT1 | LCA | Roving | +6.00 | +6.00 | NA | Atrophic macula | Y | Extinguished | N |
| 9/M/3.8 | LCA | RPGRIP1 | LCA | HJ | +5.25 | +5.25 | CF | Grossly normal | Y | Extinguished | N |
| 10/M/1.5 | IIN | RPGRIP1 | LCA | HJ | +4.25 | +4.25 | NA | Grossly normal | Y | Extinguished | N |
| 11/F/1 | LCA | CEP290 | LCA | Roving | +5.50 | +5.50 | NA | Grossly normal | Y | Extinguished | Hypotonia |
| 12/F/1.6 | LCA | CEP290 | LCA | Roving | +6.00 | +6.00 | NA | Grossly normal | Y | Extinguished | N |
| 13/M/4 | IIN | CRB1 | LCA | HP | +5.75 | +5.75 | HM | Mottling granular | Y | Extinguished | N |
| 14/F/1.8 | LCA | CRX | LCA | HJ | +3.00 | +2.50 | NA | Pigmentary changes | N | Extinguished | N |
| 15/F/8.5 | LCA | WDR19 | Senior-Loken syndrome | HP changed to VJ | +10.00 | +10.00 | 0.05 | Pigmentary changes | Y | Extinguished | Renal failure and liver failure |
| 16/M/10.5 | OA | GPR143 | OA | HP | +1.75 | +1.25 | 0.2 | No foveal reflex | N | NA | N |
| 17/M/14.6 | IIN | GPR143 | OA | HJ | –2.00 | –1.00 | 0.2 | Grossly normal | N | NA | N |
| 18/M/19.8 | OA | GPR143 | OA | HJ | 0 | –0.25 | 0.1 | No foveal reflex | N | NA | N |
| 19/M/18.3 | OA | GPR143 | OA | HP | –0.75 | –0.25 | 0.4 | No foveal reflex | N | NA | N |
| 20/F/9.8 | IIN | PAX6 | PAX6 | VJ | +6.50 | +6.50 | 0.4 | Grossly normal | N | NA | Cataract |
| 21/M/26.6 | PAX6 | PAX6 | PAX6 | HJ | NA | NA | 0.5 | No foveal reflex | N | NA | Cataract |
| 22/F/8.8 | PAX6 | PAX6 | PAX6 | HP | +3.00 | +2.50 | 0.6 | No foveal reflex | N | NA | N |
| 23/M/18.4 | IIN | PAX6 | PAX6 | HJ | +1.625 | +1.625 | 0.5 | No foveal reflex | N | NA | N |
| 24/M/3 | ACHM | CNGA3 | ACHM | Rotary | +5.00 | +5.00 | NA | Grossly normal | N | Photopic: absent | N |
| 25/F/12.3 | ACHM | CNGB3 | ACHM | HJ | +3.00 | +3.00 | 0.05 | Grossly normal | N | Photopic: absent | N |
| 26/F/33.1 | IIN | FRMD7 | IIN | HJ | 0 | 0 | 0.8 | Grossly normal | N | NA | N |
| 27/M/2.4 | IIN | FRMD7 | IIN | HP | +0.50 | +0.50 | NA | Grossly normal | N | NA | N |
| 28/M/6 | IIN | CACNA1F | CSNB | HJ | –2.50 | –2.50 | 0.2 | Grossly normal | N | Negative ERG | N |
Abbreviations: ACHM, achromatopsia; BCVA, best-corrected visual acuity; CF, counting fingers; CSNB, congenital stationary night blindness; ERG, electroretinography; F, female; HJ, horizontal jerk; HM, hand motion; HP, horizontal pendular; IIN, idiopathic infantile nystagmus; LCA, Leber congenital amaurosis; M, male; N, no; NA, not available; OA, ocular albinism; VJ, vertical jerk; Y, yes.
Age indicates age at last visit.
Table 2. Pathogenic or Likely Pathogenic Mutations Identified in 28 Patients With Infantile Nystagmus Syndrome.
| Patient No. | Initial Diagnosis | Gene | Zygosity | Mutations | Segregation Analysis | ExAC (MAF) |
In Silico Prediction (FATHMM) | Previous Literature |
Accession ID for Transcripta |
|---|---|---|---|---|---|---|---|---|---|
| 1 | LCA | GUCY2D | Compound; heterozygous | c.1978C>T: p.Arg660Ter; c.3038G>A: p.Gly1013Glu | Paternal; maternal | 0.0002; none |
NA; D (–2.12) |
Reference17; novel |
NM_000180.3 |
| 2b | LCA | GUCY2D | Compound; heterozygous | c.1991A>C: p.His664Pro; c.2649delT: p.Phe883LeufsTer13 | Paternal; maternal | None; none |
D (–1.86); NA |
Novel; novel |
NM_000180.3 |
| 3 | LCA | GUCY2D | Homozygous | c.2649delT: p.Phe883LeufsTer13 | NA | None | NA | Novel | NM_000180.3 |
| 4b | LCA | GUCY2D | Compound; heterozygous | c.2649delT: p.Phe883LeufsTer13; c.3038G>A: p.Gly1013Glu | Paternal; maternal | None; none |
NA; D (–2.12) |
Novel; novel |
NM_000180.3 |
| 5 | LCA | GUCY2D | Compound; heterozygous | c.2818C>T: p.Arg940Trp; c.3038G>A: p.Gly1013Glu | Paternal; maternal | 0.0000083; none |
D (–1.55); D (–2.12) |
Novel; novel |
NM_000180.3 |
| 6b | LCA | NMNAT1 | Compound; heterozygous | c.196C>T: p.Arg66Trp; c.709C>T: p.Arg237Cys | Maternal; paternal | 0.00013; 0.000074 |
D (–6.60); D (–4.67) |
References18,19; references18,19,20 |
NM_022787.3 |
| 7b | LCA | NMNAT1 | Compound; heterozygous | c.196C>T: p.Arg66Trp; c.709C>T: p.Arg237Cys | Maternal; paternal | 0.00013; 0.000074 |
D (–6.60); D (–4.67) |
References18,19; references18,19,20 |
NM_022787.3 |
| 8 | LCA | NMNAT1 | Compound; heterozygous | c.709C>T: p.Arg237Cys; exon 4, 5 deletion |
Paternal; maternal | 0.000074; none |
D (–4.67); NA |
References18,19,20; reference21 |
NM_022787.3 |
| 9 | LCA | RPGRIP1 | Compound; heterozygous | c.2302C>T: p.Arg768Ter; c.3565_3571del: pArg1189GlyfsTer7 | Paternal; maternal | 0.000039; 0.000017 |
NA; NA |
Novel; reference22 |
NM_020366.3 |
| 10 | IIN | RPGRIP1 | Homozygous | c.3565_3571del: p.Arg1189GlyfsTer7 | NA | 0.000017 | NA | Reference22 | NM_020366.3 |
| 11 | LCA | CEP290 | Compound; heterozygous | c.1666delA: p.Ile556PhefsTer17; c.6012-12T>A | Maternal; paternal | 0.0142; 0.000025 |
NA; 0.25177 |
References23,24; references24,25 |
NM_025114.3 |
| 12b | LCA | CEP290 | Compound; heterozygous | c.-1G>A; c.6012-12T>A | Paternal; maternal | 0.0000089; 0.000025 |
0.90875; 0.25177 |
Novel; references24,25 |
NM_025114.3 |
| 13 | IIN | CRB1 | Compound; heterozygous | c.1208C>G: p.Ser403Ter; c.1576C>T: p.Arg526Ter | Paternal; maternal | None; none |
NA; NA | Reference26; reference23 |
NM_201253.2 |
| 14b | LCA | CRX | Heterozygous | c.442delG: p.Gly148AlafsTer39 | De novo | None | NA | Novel | NM_000554.5 |
| 15 | LCA | WDR19 | Homozygous | c.3533G>A: p.Arg1178Gln | NA | 0.00058 | Tolerated; (–0.91) |
Reference27 | NM_025132.3 |
| 16 | OA | GPR143 | Hemizygous | c.514G>C: p.Gly172Arg | NA | None | D (–6.09) | Novel | NM_000273.2 |
| 17 | IIN | GPR143 | Hemizygous | c.659-3C>G | NA | None | 0.90355 | Novel | NM_000273.2 |
| 18 | OA | GPR143 | Hemizygous | Exon 2, 3 deletion | NA | None | NA | Reference28 | NM_000273.2 |
| 19 | OA | GPR143 | Hemizygous | Exon 2, 3 deletion | NA | None | NA | Reference28 | NM_000273.2 |
| 20 | IIN | PAX6 | Heterozygous | c.113G>C: p.Arg38Pro | NA | None | D (–6.36) | Novel | NM_000280.4 |
| 21 | PAX6 | PAX6 | Heterozygous | c.622C>T: p.Arg208Trp | NA | 0.0000082 | D (–3.83) | References29,30 | NM_000280.4 |
| 22 | PAX6 | PAX6 | Heterozygous | c.782G>T: p.Arg261Leu | NA | None | D (–4.43) | Novel | NM_000280.4 |
| 23 | IIN | PAX6 | Heterozygous | c.1088C>A: p.Ser363Ter | NA | None | NA | Novel | NM_000280.4 |
| 24 | ACHM | CNGA3 | Compound; heterozygous | c.1262delA: p.Lys421SerfsTer44; c.1642G>A: p.Gly548Arg | Paternal; maternal | None; 0.000025 |
NA; D (–6.35) |
Novel; novel |
NM_001298.2 |
| 25 | ACHM | CNGB3 | Homozygous | c.1928 + 2T>C | NA | None | 0.99601 | Novel | NM_019198.4 |
| 26 | IIN | FRMD7 | Heterozygous | c.901T>C: p.Tyr301His | NA | None | D (–4.63) | Novel | NM_194277.2 |
| 27 | IIN | FRMD7 | Hemizygous | c.910C>T: p.Arg304Ter | NA | None | NA | References18,31,32 | NM_194277.2 |
| 28 | IIN | CACNA1F | Hemizygous | c.342delC: p.Phe115SerfsTer22 | Maternal | None | NA | Novel | NM_005183.2 |
Abbreviations: ACHM, achromatopsia; D, damaging; ExAC, Exome Aggregation Consortium; FATHMM, functional analysis through hidden Markov models; IIN, idiopathic infantile nystagmus; LCA, Leber congenital amaurosis; MAF, minor allele frequency; NA, not available; OA, ocular albinism.
NM numbers were the accession identification numbers for longest transcript of the gene provided by Ensembl.
Previously reported with different next-generation sequencing methods using clinical exome sequencing (TruSight One sequencing panel).33
Figure 2. Schematic Diagram of Initial Clinical Diagnosis and Molecular Diagnosis After Targeted Next-Generation Sequencing (NGS) in 48 Patients With Infantile Nystagmus Syndrome (INS).
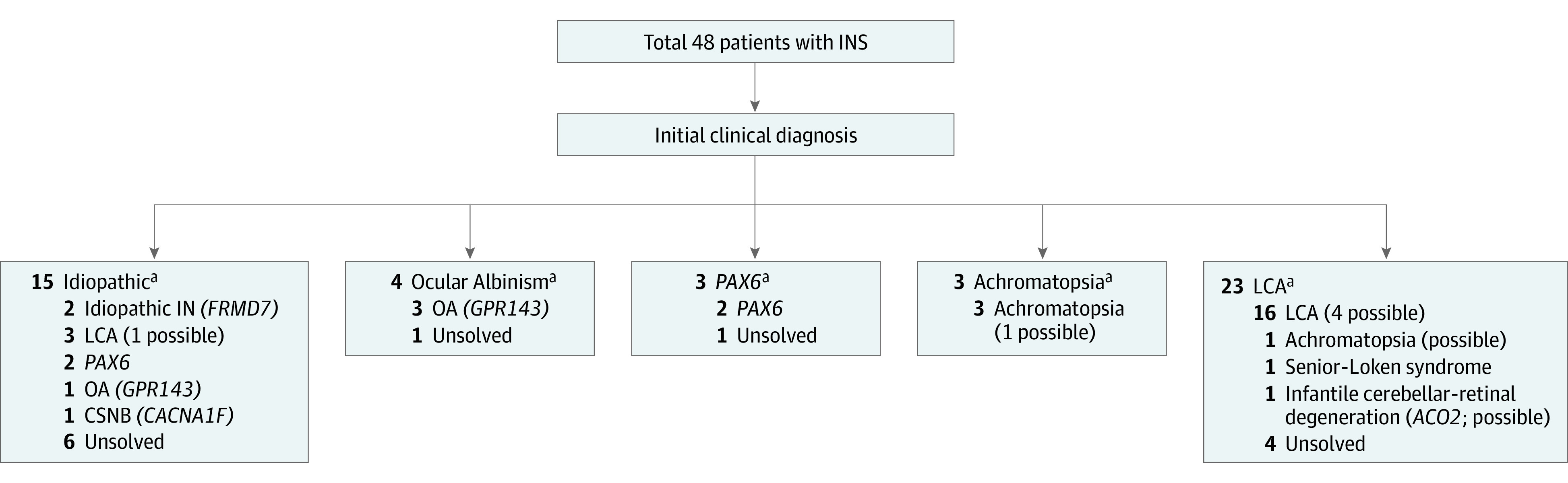
The correct molecular diagnosis was made in patients with INS through NGS. CSNB indicates congenital stationary night blindness; IN, infantile nystagmus; LCA, Leber congenital amaurosis; OA, ocular albinism; and possible, possible molecular diagnosis.
aAfter molecular diagnosis by targeted NGS, the initial clinical diagnosis was revised for some patients.
Figure 3. Fundus Photographs, Optical Coherence Tomography (OCT), and Electrooculography in Various Causes of Infantile Nystagmus Syndrome.

All 4 patients had jerky nystagmus, initially diagnosed as idiopathic infantile nystagmus. P indicates patient in P26 (A), P17 (B), P20 (C), and P28 (D).
Four patients (8%) had single causative variants in a gene associated with recessive traits. Of the remainder (12 of 48 [25%]), we were unable to designate variants responsible for the compatible phenotypes (eTable 5 in the Supplement). Among 8 patients with familial INS, pathogenic or likely pathogenic variants were identified in 7 patients (88%). In the 40 patients with simplex, probable diagnosis was made in 21 patients, for a the diagnostic yield of targeted NGS of 53% (95% CI, 37.0%-68.0%).
Phenotypes According to Affected Genes With Pathogenic or Likely Pathogenic Mutations
All patients with LCA had compatible clinical phenotypes according to causative mutations. The most frequently mutated gene was GUCY2D (OMIM 600179) (n = 5), followed by PAX6 (n = 4). Two patients (P10 and P30) with RPGRIP1 (OMIM 605446) mutations were misdiagnosed with an idiopathic form of nystagmus at the initial visit. Compound heterozygous mutations of RPGRIP1 were identified in P30 (c.2079C>G, p.Tyr693Ter and c.2209_2215 + 18del) and P9 (c.2302C>T, p.Arg768Ter and c.3565_3571del, p.A rg1189GlyfsTer7), while a homozygous mutation of RPGRIP1 was found in P10 (c.3565_3571del). All 3 patients had horizontal jerk nystagmus and normal fundus appearance at the initial visit. One patient (P9) exhibited pigmentary retinopathy at the periphery and diffuse loss of ellipsoid zone on OCT findings at a subsequent visit (eFigure 4 in the Supplement). Another patient (P13) with CRB1 (OMIM 604210) mutations (c.1208C>G, p.Ser403Ter and c.1576C>T, p.Arg526Ter) first visited our clinic owing to horizontal nystagmus at 1 year of age. This patient’s fundus was normal at the initial visit. At the subsequent visit, results of a dilated fundus examination revealed pigmentary retinopathy and thickened retina with degeneration of photoreceptor layers (eFigure 5 in the Supplement). One patient (P15) visited our clinic owing to horizontal nystagmus at 1 year of age. She had high hyperopia and a presumptive diagnosis of LCA was made. At 2 years of age, this patient developed acute renal failure and required kidney transplantation. Next-generation sequencing analysis revealed a homozygous c.3533G>A (p.Arg1178Glu) WDR19 (OMIM 608151) mutation. The clinical diagnosis was revised to Senior-Loken syndrome (eFigure 6 in the Supplement).
eFigure 7 in the Supplement shows the genotypes and clinical phenotypes in 4 patients with GPR143 mutations. All 4 patients exhibited absence of foveal reflex and grade 4 foveal hypoplasia detected by OCT. None exhibited an iris transillumination defect. In P17, pigmentation of the retina was normal, and he was initially diagnosed with idiopathic infantile nystagmus. At 14 years of age, OCT findings demonstrated grade 4 foveal hypoplasia. Next-generation sequencing revealed a noncanonical splice site c.659-3C>G GPR143 mutation. The normal 3′ splice site had broken and a new splice acceptor site was introduced at the c.659-3C>G position (eFigure 8 in the Supplement).
Four patients with PAX6 mutations had grade 1-3 foveal hypoplasia according to the type of mutation. Fundus photographs revealed retained foveal reflex in 3 patients (eFigure 9 in the Supplement). One patient (P20) exhibited presenile posterior subcapsular cataract, and another (P21) underwent cataract surgery at 24 years of age. Except for P22, 3 patients had normal iris structures. One patient (P23) had only isolated foveal hypoplasia.
A hemizygous frameshift (c.342delC, p.Phe115SerfsTer22) mutation in CACNA1F (OMIM 300110) was identified in P28. This patient visited our clinic owing to horizontal nystagmus at 1 year of age. The fundus was normal except for a large cup-disc ratio, and findings of ERG testing were ambiguous at that time. This patient was initially thought to have idiopathic infantile nystagmus. At 5 years of age, ERG findings revealed the characteristic negative waveform in dark-adapted 3.0 ERG and double peak sign in light-adapted 30-Hz flicker, consistent with incomplete congenital stationary night blindness (eFigure 10 in the Supplement).
Mutation Hotspots in Koreans With INS
A total of 6 mutations in 4 genes (GUCY2D c.2649delT, c.3038G>A; NMNAT1 (OMIM 608700) c.196C>T, c.709C>T; CNGA3 (OMIM 600053) c.1001C>T; and RPGRIP1 c.3565_3571del) were detected in more than 2 unrelated patients in this study. Two variants in GUCY2D have not been described in any previous reports or reputable database, to our knowledge, nor were they detected in a normal population database. Because the 3 patients with these variants in GUCY2D were not related, these mutations may be candidate variants for founder mutation evaluation in the Korean population, fulfilling the criteria for segregation quantification for pathogenicity according to the recently refined guideline known as Sherloc.34
Discussion
In this study, we analyzed 48 patients with INS to determine an effective strategy for making a firm molecular diagnosis of this disorder. Probable molecular diagnosis was made in 28 patients, corresponding to a diagnostic yield of 58.3%. Moreover, the results of genetic diagnoses enabled revision of initial clinical diagnoses, which eventually led to improvement of personalized care and genetic counseling. A previous study that evaluated the clinical utility of NGS in familial infantile nystagmus reported a detection rate of 80% (12 of 15 patients).35 This finding was consistent with our results in familial cases (7 of 8 patients [88% detection rate]).
We found that none of the female obligate carriers of the GPR143 mutation had nystagmus. A previous study also reported that all obligate carriers with the GPR143 mutation exhibited no sign of nystagmus.28,36 Female carriers with the GPR143 mutation exhibit only a mottled pattern of retinal pigmentation.37 A female with FRMD7 mutation (P26) was affected in our study, and it has been reported that the penetrance of the FRMD7 mutation is 53% in female obligate carriers.38 Ocular albinism can be misdiagnosed as idiopathic because hypopigmentation of the fundus and iris transillumination defects are relatively rare in Asian patients.28,36 Therefore, NGS will facilitate genetic counseling and future family planning in X-linked familial INS.
Most patients with LCA exhibit severe visual impairment in early childhood, which is accompanied by characteristic features such as roving eye movement, oculodigital sign, or amaurotic pupil. Unfortunately, pupillary response is difficult to evaluate in infants because of their small resting size.39 In addition, patients with LCA may have fundi that appear normal during infancy.40 Moreover, patients with LCA with CEP290 (OMIM 610142) mutations have a wide spectrum of visual acuity abnormalities from light perception to greater than 0.5.41 Therefore, early NGS testing will aid in differentiating LCA from other nonprogressive diseases with nystagmus such as achromatopsia.
Nystagmus waveform may provide a clue to the sensory causes of nystagmus,42 and handheld OCT will help clinicians locate retinal structural abnormalities.43 A high-frequency, low-amplitude nystagmus, which is regarded as a characteristic waveform of sensory origin, may not be evident early in these patients.39 In addition, obtaining the fovea center using OCT is difficult, especially in young children with nystagmus, and the interpretation of OCT findings can be misleading owing to poorly centered fovea imaging. Pediatric electrophysiology testing and handheld OCT are labor-intensive endeavors conducted in a limited number of specialized facilities. Electrophysiology testing under sedation carries a risk for adverse events such as respiratory depression or ventricular arrhythmia.44 Therefore, NGS has potential value in avoiding unnecessary ERG testing if NGS testing reveals FRMD7 or GPR143 mutations, and the inheritance is consistent with an X-linked recessive pattern. If causative mutations associated with retinal disorders are found, imaging or ERG testing will be needed to correlate genotypes and phenotypes.
Many studies have confirmed the clinical utility of NGS genetic testing in ophthalmic diseases.45,46,47 Should targeted NGS be considered when a clinical context supports a specific diagnosis such as aniridia syndrome or ocular albinism? In these circumstances, NGS techniques may be more cost-effective than Sanger sequencing if the responsible gene has many exons. Next-generation sequencing can also detect copy number variations or variants in cis-regulatory regions of PAX6, known to cause phenotypes. The significant potential advantage of massive parallel sequencing in INS should be emphasized, as illustrated in patients with syndromic ciliopathic findings. Next-generation sequencing can enable physicians to increase surveillance for treatable manifestations of the disease if the correct diagnosis is made in a timely fashion.48 In addition, decisions regarding brain neuroimaging can be tailored to individual patients with LCA through NGS testing.
Targeted NGS is relatively focused and inexpensive, and can reveal deep intronic regions known to cause disease if particular deep intronic regions were included in the panel. Meanwhile, whole-exome sequencing has several strengths in that it does not require any gene list update, and data from new regions can be reanalyzed at a later time to discover novel causative genes. Whole-genome sequencing has the advantages of better detection of copy number variants and screening more broadly. However, the interpretation of variants in noncoding regions are limited; therefore, the clinical use of whole-genome sequencing for clinical molecular diagnosis is not yet warranted.
Limitations
Because CACNA1F, PAX6, and SLC38A8 (OMIM 615585) mutations also cause nystagmus without overt retinal dystrophy,49,50 more comprehensive genetic analysis, including all possible genes, should be conducted. A recent study included 336 genes associated with nystagmus.35 In addition, our panel did not include the SLC38A8 gene associated with INS; known deep intronic variants such as CEP290 c.2991 + 1665A>G, FRMD7 c.285-118C>T, and GPR143 c.659-131T>G, which are thought to activate a cryptic splice donor50,51,52,53; or an upstream or downstream cis-element of PAX6 or TYR (OMIM 606933) genes.54,55 Another limitation is difficulty in detecting copy number variations through targeted NGS.35,45 Our panel did not include the GJC2 (OMIM 608803) gene, which has been suggested to cause infantile nystagmus.56 Finally, more patients who had poor vision with nystagmus were willing to undergo genetic testing in this study. Therefore, there may have been a bias toward patients with inherited retinal disorders. These factors should be considered when interpreting our results.
Conclusions
This study demonstrated that NGS has a potential role in early diagnosis of both familial and sporadic cases involving INS. It also demonstrated that NGS is a useful tool for making accurate diagnoses, which in turn have made revision of initial clinical diagnoses possible. Early molecular diagnosis of INS will enable physicians to conduct genetic counseling for further family planning and personalized care in a timely fashion. Given the high prevalence of nystagmus in various causes of ocular and systemic diseases and the phenotypical similarities of nystagmus, greater attention should be devoted to making a clinical diagnosis in these patients. With the advent of NGS technologies and analysis methods, NGS may serve as a standard initial assessment tool in patients with INS.
eTable 1. Target Genes Associated With Infantile Nystagmus Syndrome
eTable 2. Quality Control Matrices of Next-Generation Sequencing Results for all Patients in This Study
eTable 3. The Clinical Features of Patients Who Had 2 Pathogenic Variants With No Segregation Analysis
eTable 4. Pathogenic or Likely Pathogenic Variants in 4 Patients Who Had Compound Heterozygous Mutations With No Segregation Analysis
eTable 5. The Clinical Features of Patients With Infantile Nystagmus Syndrome With Only 1 Putative Pathogenic Variant Or Unsolved Cases
eAppendix. Annotation, Interpretation of Variants, Phenotype Review, and Consensus Discussion
eFigure 1. Schematic Diagram of Next-Generation Sequencing Analysis Work Flow
eFigure 2. Pedigree of 8 Patients Who Had Family History of Nystagmus
eFigure 3. The Distribution of Age at Referral for Genetic Testing
eFigure 4. Wide-Field Fundus Photograph and Optical Coherence Tomography in a Patient With RPGRIP1 Mutations
eFigure 5. Fundus Photograph and Optical Coherence Tomography in a Patient With CRB1 Mutations
eFigure 6. Fundus Photograph, Optical Coherence Tomography, Ultrasonogram of the Kidney, and Abdominal Computed Tomography in a Patient With Senior-Loken Syndrome Caused by Homozygous p.Arg1178Glu WDR19 Mutation
eFigure 7. Phenotypic Variability of 4 Patients With GPR143 Mutations
eFigure 8. In silico Prediction of Intronic Mutation Within GPR143 Intron 5
eFigure 9. Fundus Photographs and Optical Coherence Tomography in 4 Patients With PAX6 Mutations
eFigure 10. Fundus Photograph and Electroretinogram in a Patient With CACNA1F Mutation
References
- 1.Richards MD, Wong A. Infantile nystagmus syndrome: clinical characteristics, current theories of pathogenesis, diagnosis, and management. Can J Ophthalmol. 2015;50(6):400-408. [DOI] [PubMed] [Google Scholar]
- 2.Soens ZT, Li Y, Zhao L, et al. Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet Med. 2016;18(10):1044-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130(1):1-12. [DOI] [PubMed] [Google Scholar]
- 4.Han J, Lee T, Lee JB, Han SH. Retinal microstructures are altered in patients with idiopathic infantile nystagmus. Graefes Arch Clin Exp Ophthalmol. 2017;255(8):1661-1668. [DOI] [PubMed] [Google Scholar]
- 5.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. [DOI] [PubMed] [Google Scholar]
- 6.RetNet: Retinal Information Network. https://sph.uth.edu/retnet/. Accessed on January 26, 2016.
- 7.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Schulz-Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220-1222. [DOI] [PubMed] [Google Scholar]
- 11.Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Handsaker B, Wysoker A, et al. ; 1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuilman T, Velds A, Kemper K, et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing: the human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lotery AJ, Namperumalsamy P, Jacobson SG, et al. Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Arch Ophthalmol. 2000;118(4):538-543. [DOI] [PubMed] [Google Scholar]
- 18.Falk MJ, Zhang Q, Nakamaru-Ogiso E, et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44(9):1040-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki Y, Margolin Z, Borgo B, Havranek JJ, Milbrandt J. Characterization of Leber congenital amaurosis–associated NMNAT1 mutants. J Biol Chem. 2015;290(28):17228-17238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrault I, Hanein S, Zanlonghi X, et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet. 2012;44(9):975-977. [DOI] [PubMed] [Google Scholar]
- 21.Coppieters F, Todeschini AL, Fujimaki T, et al. Hidden genetic variation in LCA9-associated congenital blindness explained by 5'UTR mutations and copy-number variations of NMNAT1. Hum Mutat. 2015;36(12):1188-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seong MW, Kim SY, Yu YS, Hwang JM, Kim JY, Park SS. Molecular characterization of Leber congenital amaurosis in Koreans. Mol Vis. 2008;14:1429-1436. [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Wang X, Zou X, et al. Comprehensive molecular diagnosis of a large Chinese Leber congenital amaurosis cohort. Invest Ophthalmol Vis Sci. 2015;56(6):3642-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brancati F, Barrano G, Silhavy JL, et al. ; International JSRD Study Group . CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81(1):104-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsurusaki Y, Kobayashi Y, Hisano M, et al. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013;58(2):113-115. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Zhao K, Sheng X, et al. Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Invest Ophthalmol Vis Sci. 2013;54(3):2186-2197. [DOI] [PubMed] [Google Scholar]
- 27.Lee JM, Ahn YH, Kang HG, et al. Nephronophthisis 13: implications of its association with Caroli disease and altered intracellular localization of WDR19 in the kidney. Pediatr Nephrol. 2015;30(9):1451-1458. [DOI] [PubMed] [Google Scholar]
- 28.Fang S, Guo X, Jia X, Xiao X, Li S, Zhang Q. Novel GPR143 mutations and clinical characteristics in six Chinese families with X-linked ocular albinism. Mol Vis. 2008;14:1974-1982. [PMC free article] [PubMed] [Google Scholar]
- 29.Hanson IM, Seawright A, Hardman K, et al. PAX6 mutations in aniridia. Hum Mol Genet. 1993;2(7):915-920. [DOI] [PubMed] [Google Scholar]
- 30.Lim HT, Seo EJ, Kim GH, et al. Comparison between aniridia with and without PAX6 mutations: clinical and molecular analysis in 14 Korean patients with aniridia. Ophthalmology. 2012;119(6):1258-1264. [DOI] [PubMed] [Google Scholar]
- 31.Li N, Wang L, Cui L, et al. Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol Vis. 2008;14:733-738. [PMC free article] [PubMed] [Google Scholar]
- 32.AlMoallem B, Bauwens M, Walraedt S, et al. Novel FRMD7 mutations and genomic rearrangement expand the molecular pathogenesis of X-linked idiopathic infantile nystagmus. Invest Ophthalmol Vis Sci. 2015;56(3):1701-1710. [DOI] [PubMed] [Google Scholar]
- 33.Han J, Rim JH, Hwang IS, et al. Diagnostic application of clinical exome sequencing in Leber congenital amaurosis. Mol Vis. 2017;23:649-659. [PMC free article] [PubMed] [Google Scholar]
- 34.Nykamp K, Anderson M, Powers M, et al. ; Invitae Clinical Genomics Group . Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas MG, Maconachie G, Sheth V, McLean RJ, Gottlob I. Development and clinical utility of a novel diagnostic nystagmus gene panel using targeted next-generation sequencing. Eur J Hum Genet. 2017;25(6):725-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zou X, Li H, Yang L, et al. Molecular genetic and clinical evaluation of three Chinese families with X-linked ocular albinism. Sci Rep. 2017;7:33713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oetting WS. New insights into ocular albinism type 1 (OA1): mutations and polymorphisms of the OA1 gene. Hum Mutat. 2002;19(2):85-92. [DOI] [PubMed] [Google Scholar]
- 38.Thomas S, Proudlock FA, Sarvananthan N, et al. Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain. 2008;131(pt 5):1259-1267. [DOI] [PubMed] [Google Scholar]
- 39.Lambert SR, Taylor D, Kriss A. The infant with nystagmus, normal appearing fundi, but an abnormal ERG. Surv Ophthalmol. 1989;34(3):173-186. [DOI] [PubMed] [Google Scholar]
- 40.Winkelman JE, Horsten GPM. Congenital blindness in the presence of a normal fundus. Ophthalmologica. 1959;137(6):423-425. doi: 10.1159/000303584 [DOI] [Google Scholar]
- 41.McAnany JJ, Genead MA, Walia S, et al. Visual acuity changes in patients with Leber congenital amaurosis and mutations in CEP290. JAMA Ophthalmol. 2013;131(2):178-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pieh C, Simonsz-Toth B, Gottlob I. Nystagmus characteristics in congenital stationary night blindness (CSNB). Br J Ophthalmol. 2008;92(2):236-240. [DOI] [PubMed] [Google Scholar]
- 43.Lee H, Sheth V, Bibi M, et al. Potential of handheld optical coherence tomography to determine cause of infantile nystagmus in children by using foveal morphology. Ophthalmology. 2013;120(12):2714-2724. [DOI] [PubMed] [Google Scholar]
- 44.Nordt SP, Rangan C, Hardmaslani M, Clark RF, Wendler C, Valente M. Pediatric chloral hydrate poisonings and death following outpatient procedural sedation. J Med Toxicol. 2014;10(2):219-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor RL, Parry NRA, Barton SJ, et al. Panel-based clinical genetic testing in 85 children with inherited retinal disease. Ophthalmology. 2017;124(7):985-991. [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Xiao J, Huang H, et al. Molecular genetic testing in clinical diagnostic assessments that demonstrate correlations in patients with autosomal recessive inherited retinal dystrophy. JAMA Ophthalmol. 2015;133(4):427-436. [DOI] [PubMed] [Google Scholar]
- 47.Scott AF, Mohr DW, Kasch LM, et al. Identification of an HMGB3 frameshift mutation in a family with an X-linked colobomatous microphthalmia syndrome using whole-genome and X-exome sequencing. JAMA Ophthalmol. 2014;132(10):1215-1220. [DOI] [PubMed] [Google Scholar]
- 48.Ellingford JM, Sergouniotis PI, Lennon R, et al. Pinpointing clinical diagnosis through whole exome sequencing to direct patient care: a case of Senior-Loken syndrome. Lancet. 2015;385(9980):1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez Y, Gradstein L, Flusser H, et al. Isolated foveal hypoplasia with secondary nystagmus and low vision is associated with a homozygous SLC38A8 mutation. Eur J Hum Genet. 2014;22(5):703-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poulter JA, Al-Araimi M, Conte I, et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93(6):1143-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79(3):556-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas MG, Crosier M, Lindsay S, et al. Abnormal retinal development associated with FRMD7 mutations. Hum Mol Genet. 2014;23(15):4086-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naruto T, Okamoto N, Masuda K, et al. Deep intronic GPR143 mutation in a Japanese family with ocular albinism. Sci Rep. 2015;5:11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray K, Chaki M, Sengupta M. Tyrosinase and ocular diseases: some novel thoughts on the molecular basis of oculocutaneous albinism type 1. Prog Retin Eye Res. 2007;26(4):323-358. [DOI] [PubMed] [Google Scholar]
- 55.Bhatia S, Bengani H, Fish M, et al. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet. 2013;93(6):1126-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Owczarek-Lipska M, Juschke C, Mulahasanovic L, et al. Whole exome sequencing towards a rapid identification of genetic variants associated with congenital nystagmus. Presented at: ARVO Annual Meeting; May 9, 2017; Baltimore, MD. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Target Genes Associated With Infantile Nystagmus Syndrome
eTable 2. Quality Control Matrices of Next-Generation Sequencing Results for all Patients in This Study
eTable 3. The Clinical Features of Patients Who Had 2 Pathogenic Variants With No Segregation Analysis
eTable 4. Pathogenic or Likely Pathogenic Variants in 4 Patients Who Had Compound Heterozygous Mutations With No Segregation Analysis
eTable 5. The Clinical Features of Patients With Infantile Nystagmus Syndrome With Only 1 Putative Pathogenic Variant Or Unsolved Cases
eAppendix. Annotation, Interpretation of Variants, Phenotype Review, and Consensus Discussion
eFigure 1. Schematic Diagram of Next-Generation Sequencing Analysis Work Flow
eFigure 2. Pedigree of 8 Patients Who Had Family History of Nystagmus
eFigure 3. The Distribution of Age at Referral for Genetic Testing
eFigure 4. Wide-Field Fundus Photograph and Optical Coherence Tomography in a Patient With RPGRIP1 Mutations
eFigure 5. Fundus Photograph and Optical Coherence Tomography in a Patient With CRB1 Mutations
eFigure 6. Fundus Photograph, Optical Coherence Tomography, Ultrasonogram of the Kidney, and Abdominal Computed Tomography in a Patient With Senior-Loken Syndrome Caused by Homozygous p.Arg1178Glu WDR19 Mutation
eFigure 7. Phenotypic Variability of 4 Patients With GPR143 Mutations
eFigure 8. In silico Prediction of Intronic Mutation Within GPR143 Intron 5
eFigure 9. Fundus Photographs and Optical Coherence Tomography in 4 Patients With PAX6 Mutations
eFigure 10. Fundus Photograph and Electroretinogram in a Patient With CACNA1F Mutation