

Abstract
As a unique rocaglate (flavagline) natural product, aglaroxin C displays intriguing biological activity by inhibiting HCV viral entry. To further elucidate structure-activity relationships and diversify the pyrimidinone scaffold, we report a concise synthesis of aglaroxin C utilizing a highly regioselective pyrimidinone condensation. We have prepared more than forty aglaroxin C analogues utilizing various amidine condensation partners. Through biological evaluation of analogues, we have discovered two lead compounds, CMLD012043 and CMLD012044, which show preferential bias for the inhibition of HCV viral entry vs. translation inhibition. Overall, the study demonstrates the power of chemical synthesis to produce natural product variants with both target inhibition bias and improved therapeutic indexes.
Keywords: aglaroxin, natural product synthesis, late-stage modification, viral entry, HCV, rocaglate, natural product analogues, inhibition bias
Graphical Abstract
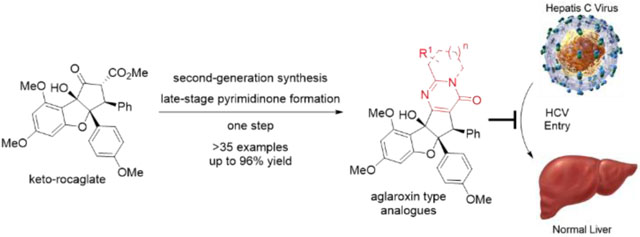
INTRODUCTION
Rocaglates (flavagline) natural products were first isolated from the dried roots and stems of Aglaia elliptifolia Merr. (family Meliaceae) in 1982.1 Since then, over thirty rocaglate natural products have been identified with unique structures and intriguing biological activities.2, 3, 4 For instance, silvestrol (1, Figure 1)5 was identified as an excellent translation inhibitor for cancer chemotherapy,6 whereas other related natural products and analogues (2-4) displayed similar activities.7 In our previous studies, translation inhibition was found to be associated with inhibition of the DEAD box RNA helicase eIF4A.8 Owing to the interesting structures and biological activities of rocaglates, a number of total syntheses9 and medicinal chemistry studies10, 11 have been disclosed. Our group has a longstanding interest in the synthesis of rocaglates and derivatives as well as investigations of their biology.6, 7, 8, 9g, i, k o–q, 10a, c, e–f In 2015, we reported the enantioselective synthesis of aglaiastatin (5) and aglaroxin C (6) through biomimetic kinetic resolution.9pSubsequent studies revealed that 5 was a promising translation inhibitor. In contrast, 6, containing a fully substituted pyrimidi-none core, showed only moderate translation inhibition.9p In separate studies, we discovered that 6 inhibited hepatitis C viral (HCV) entry into host cells at a low μM concentration, potentially through inhibition of the prohibitins (PHBs) as viral entry factors.12, 13 Notably, prohibitins 1 and 2 have been reported as general viral entry factors for other viruses including dengue virus type 2 (DENV-2)14 and Chikungunya.15 We were further encouraged by the observation that 6 did not induce cytotoxicity by translation inhibition at a concentration that is effective for inhibition of viral entry, providing a promising therapeutic in dex (TI) for potential treatment of HCV infection.12
Figure 1.

Representative Biologically Active Rocaglate Natural Products and Synthetic Analogues
HCV is a widespread viral pathogen. The recent estimated number of viral carriers is 143 million worldwide,16 and over 2% of the population in North America has been infected. In addition, chronic HCV infection causes severe liver diseases in carriers, including liver cancer and cirrhosis.17 In 2015, over a half million people died from diseases due to HCV infection.18 Recently, direct-acting antiviral agents (DAAs) have become available to cure HCV infection19 by inhibiting the function of non-structural (NS) viral proteins. However, treatment effects of DAAs vary across different genotypes of HCV, and HCV potentially may develop resistance to these agents.20 For example, the NS3 protease inhibitor simeprevir only treats genotypes 1 and 4 of HCV, and several signature resistance mutations have been identified against this treatment.21 Sofosbuvir, a top NS5B inhibitor, failed in HCV treatment which was associated with resistance mutations (Figure SI1).22 Accordingly, discovery of alternative treatments for HCV is sorely needed. As PHB-mediated cellular signaling pathways12 are required by all HCV genotypes to infect host cells, it is conceivable that a small molecule such as aglaroxin C (6) may block infection from multiple HCV genotypes.23 Additionally, targeting the host component for viral entry may create higher genetic barriers against resistance.24 Lastly, aglaroxin C and analogues may also be valuable for dissecting the mechanisms of HCV entry and may inhibit other viruses sharing the same entry mechanism.
Our goal was to reengineer the structure of aglaroxin C (6) to increase activity against HCV entry while minimizing translation inhibition, which may lead to undesired cytotoxicity.5, 6, 7, 8 As the oxidation state change between 6 and aglaiastatin (5) led to different biological profiles, we speculated that the pyrimidinone subunit of 6 may be important for inhibition of HCV viral entry vs. translation inhibition. Our first-generation synthesis of aglaroxin C (6) utilized a key intermediate, keto-rocaglate 9,25 which was synthesized through the excited state intramolecular proton transfer (ESIPT) [3+2] cycloaddition between 3-hy-droxyflavone 7 and methyl cinnamate followed by α-ketol shift of the aglain intermediate 8 (Scheme 1).9i However, the late stage pyrimidinone synthesis in our first-generation synthetic route is not ideal for rapid analog synthesis, affording 6 in low yield on a multi-milligram scale (Scheme 1).26 We also envisioned that stepwise pyrimidinone synthesis may suffer from poor functional group tolerance in analogues due to the use of both acidic and oxidative conditions. Moreover, use of tethered aminoacetals such as 10 in the ester-amide exchange to produce intermediate 11 considerably narrows the diversity of accessible analogues due to limited availability of such building blocks. We therefore considered a streamlined synthesis of aglaroxin analogues through late stage, one-step pyrimidinone formation (Scheme 2).
Scheme 1.

First-Generation Synthesis of Aglaroxin C Stepwise Pyrimidinone Formation
Scheme 2.

Retrosynthetic Analysis for Aglaroxin C Analogues
RESULTS AND DISCUSSION
Development of a Direct Pyrimidinone Formation.
Our second-generation synthetic route (Scheme 2), which relies on condensation of keto-rocaglate (9) with commercially available amidines 13, was expected to flexibly provide analogues (12) for biological experiments. Pyrimidinones have served as biologically important substructures in both drugs and natural products.27, 28 There are many well-established and practical methods for the synthesis of pyrimidinones and pyrimidines, including the Traube synthesis of purines,29 which inspired us to consider late stage construction of the pyrimidinone core of aglaroxin analogues.30 However, we anticipated that condensation between the structurally complex substrate 9 and amidines 13 would be challenging. In particular, the highly ionizable tertiary, benzylic alcohol adjacent to the ketoester moiety may provide unexpected reactivity under the reaction conditions.
Our initial model study for pyrimidinone formation employed benzamidine as a simplified condensation partner. Microwave conditions (Table 1, entry 1) produced minimal amounts of pyrimidinone 12a in an unsatisfactory yield of 28% (determined by 1H NMR analysis) due to low conversion.9p Thermal conditions (entry 2) resulted in full consumption of 9, while cyclopentapyrimidinedione 16 and 3,5-dimethox-yphloroglucinol were found as the major identified side products. The formation of fragmentation product 16 was found to correlate with the amount of 4-dimethylaminopyridine (DMAP) employed; reduction to 0.3 equivalents of DMAP minimized production of 16 (entry 3) but also led to formation of an unexpected cyclization product, oxazoline 15. The structure of 15 was confirmed by single crystal X-ray crystal structure analysis.26 Reduced equivalents of the benzamidine reaction partner resulted in an incomplete reaction, with increased production of decarboxylation product 14 and retro-Nazarov products 17 (entries 4 & 5).26 We next found that diluting the reaction eightfold improved the yield of 12a (entry 6). Increasing both temperature and reaction time under dilute conditions eliminated the production of 15, but also led to a lower yield of 12a in favor of significant amounts of 16 (entry 7). Gratifyingly, by reducing the reaction time to 45 minutes, we obtained a 90% NMR yield of 12a with minimal fragmentation to 16 (entry 8); ultimately, an 81% isolated yield of 12a was obtained on a 100-mg scale. Interestingly, we found that the model reaction gave the almost identical yield in absence of DMAP under the optimized conditions (entry 9), but we acheived better reproducibility when amidine hydrochloride salt was used with DMAP (vide infra).
Table 1.
Optimization of the Direct Pyrimidinone Formation
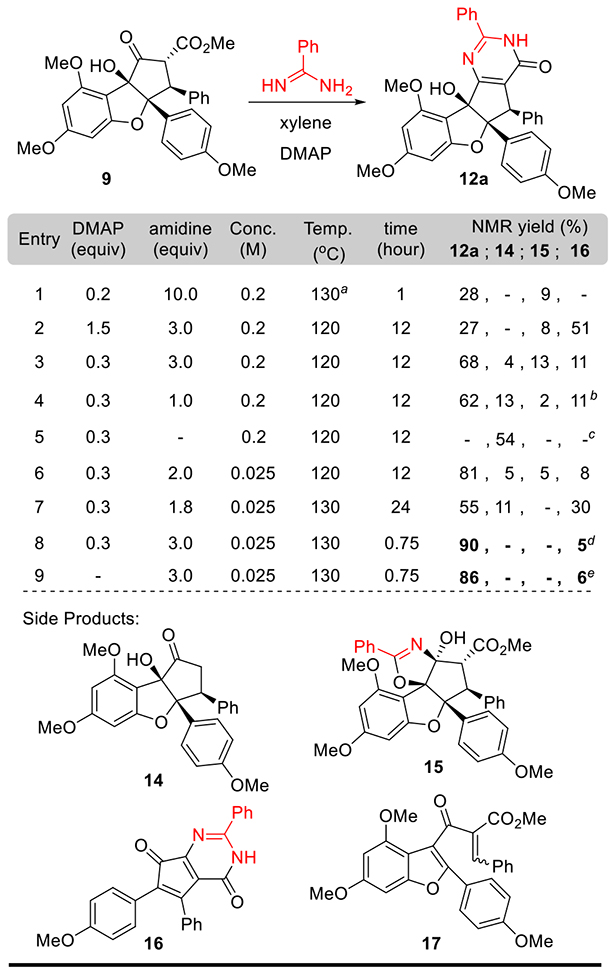
|
Microwave, toluene as solvent;
10% of 9 recovered;
20% of 9 recovered, 24% yield of 17;
81% isolated yield of 12a;
78% isolated yield of 12a.
Proposed Mechanistic Pathway.
According to trends observed during reaction optimization, we propose the mechanistic pathway depicted in Scheme 3. We postulate that water generated from the pyrimidinone condensation may hydrolyze 9 to the β-keto-acid intermediate 21, which undergoes decarboxylation to ketone 14. When no nucleophile was used, 14 and retro-Nazarov product 17 were obtained. Presumably DMAP may promote intramolecular proton transfer to 19 followed by the extrusion of water to afford the retro-Nazarov precursor 20, whereas the formed water triggered decarboxylation of 9 (an alternative mechanism is shown in the SI). In the presence of the amidine, the desired formation of hemiaminals 22 and epi- 22appears to compete with decarboxylation and retro-Nazarov reaction, which can be enhanced through use of increased equivalents of the amidine. Based on the Katrizky mechanism determined for the Traube reaction of β-keto esters and amidines,31 two possible pathways may be envisioned for the formation of pyrimidinone 12a. In one mechanism, hemiaminal epi-22, generated from disfavored concave-addition of the amidine to 9,9i may cyclize to dihydropyrimidinone 25 followed by extrusion of water. In contrast, hemiaminal 22, obtained from amidine addition to the convex face of 9, has an anti-relationship between the aminal and ester thereby preventing direct cyclization. Instead, loss of water would produce the observed enamine 23, which we propose then cyclizes to 12a through extrusion of methanol generating imidoyl ketene 24 followed by a 6π-electrocyclization to pyrimidinone 12a.32 To support the formation of hemiaminal 22, the oxazoline byproduct 15 was formed through cyclization of the tertiary alcohol of 22 to the amidine carbon followed by extrusion of ammonia (blue arrow). The formation of 22 is also supported by our isolation of enamine 23 from a 250 mg scale reaction, where a 28% yield of 23 was found to precipitate after 10 minutes; we subsequently found that 23 can also be synthesized in 60% yield using 9 and benzamidine at 60 °C in toluene.26 Interestingly, isolated 23 was found to solely produce 12a with no observed formation of 15 when 23 was resubjected to the reaction conditions. We rationalize that the sp3-hybridized hemiaminal carbon of 22 allows for a conformation necessary for intramolecular cyclization, whereas the sp2-hybridized enamine carbon of 23 prevents a similar cyclization. This conforms to the observation that elevated temperatures facilitate extrusion of water generating enamine 23 and minimize formation of byproduct 15. In contrast, 15 did not generate 12a under the reaction conditions. In a control experiment, DMAP was found to accelerate the fragmentation of 12a into 16 and 3,5-dimethoxyphloroglucinol.26
Scheme 3.

Possible Mechanistic Pathways and products of Direct Pyrimidinone Formation.
Second-Generation Synthesis of Aglaroxin C.
After determining optimal conditions for direct pyrimidinone formation, we next sought to apply these conditions to synthesize aglaroxin C (6). In this case, the requisite pyrrolidin-2-imine was commercially available as an HCl salt (Table 2). Initial attempts using stepwise free-basing protocols failed due to the high-water solubility of the amidine; accordingly, we focused on in-situ free-basing conditions for optimization studies. Using excess base (entries 1 and 2), no conversion of substrate 9 was observed after refluxing for 12 h. As 9 exists as mixture of enol/keto isomers, presumably excess base favors formation of an unreactive enolate. In contrast, use of excess pyrrolidin-2-imine HCl salt (entry 3) did not induce pyrimidinone formation. We assume that DMAP is protonated by the amidine salt which negated its function. When a slight excess of amidine vs. Na-OMe (entry 4) was used, we obtained 6 in 19% isolated yield. Consistent with our earlier optimization efforts, use of increased concentration and 12 h reaction time resulted in a complex reaction mixture containing fragmentation products (entry 5) Finally, use of 3.0 equiv. of amidine salt and 2.95 equiv. of NaOMe along with a catalytic amount of DMAP (40 mol%, 130 °C) afforded 6 in 76% isolated yield on a 100-mg scale (entry 6).
Table 2.
Optimization of Second-Generation Synthesis of Aglaroxin C
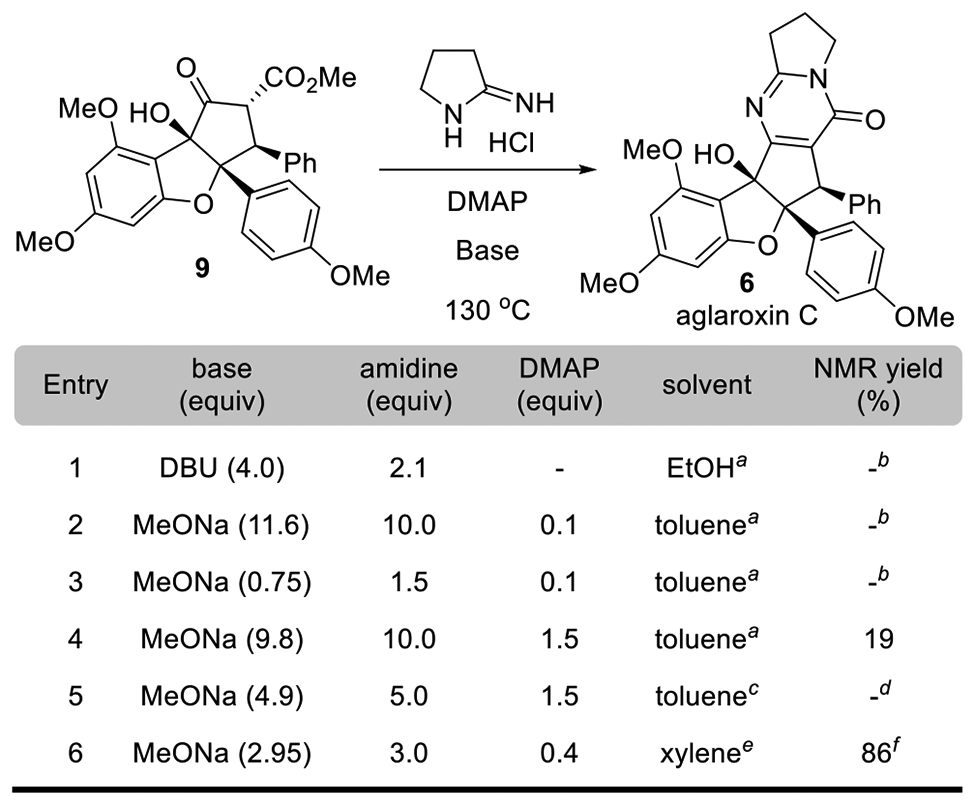 |
0.025 M concentration, reflux 12 h;
no conversion;
0.2 M concentration, reflux 12 h;
complex reaction; only isolated the fragmentation products;
0.025 M concentration, 130°C 1 h;
76% isolated yield for aglaroxin C; 9% decarboxylated product also obtained.
Synthesis of Aglaroxin Analogues.
With reliable annulation protocols available using both amidines and amidine salts, we next synthesized a library of aglaroxin analogues (Table 3). In general, direct pyrimidinone formation was found to tolerate both aromatic and aliphatic amidines. We observed consistent yields for formation of C-substituted pyrimidinones (12b-12f). For instance, simple aliphatic amidines reacted with 9 to afford C-Me (12b) and C-Bn (12c) pyrimidinones. Hindered amidines such as iso-butanamidine and cyclohexylcarboxamidine afforded the desired products 12d and 12e in excellent yields. Interestingly, condensation of carbomethoxyformamidine with 9 was followed by an unexpected decarboxylation to produce the non-substituted product 12f in 53% yield. Moreover, we discovered that the pyrimidinone condensation could tolerate various functional groups, including ester 12g (41%), cyclopropane 12h (91%), and methoxyl methylene 12i (52%). Ester exchange was observed when ethyl amidinoacetate was employed, producing compound 12g. Additionally, guanidine-type reaction partners successfully underwent condensation, yielding products such as 12j (90% yield).
Table 3.
Synthese of Aglaroxin C Analogues Using Direct Pyrimidinone Formationa
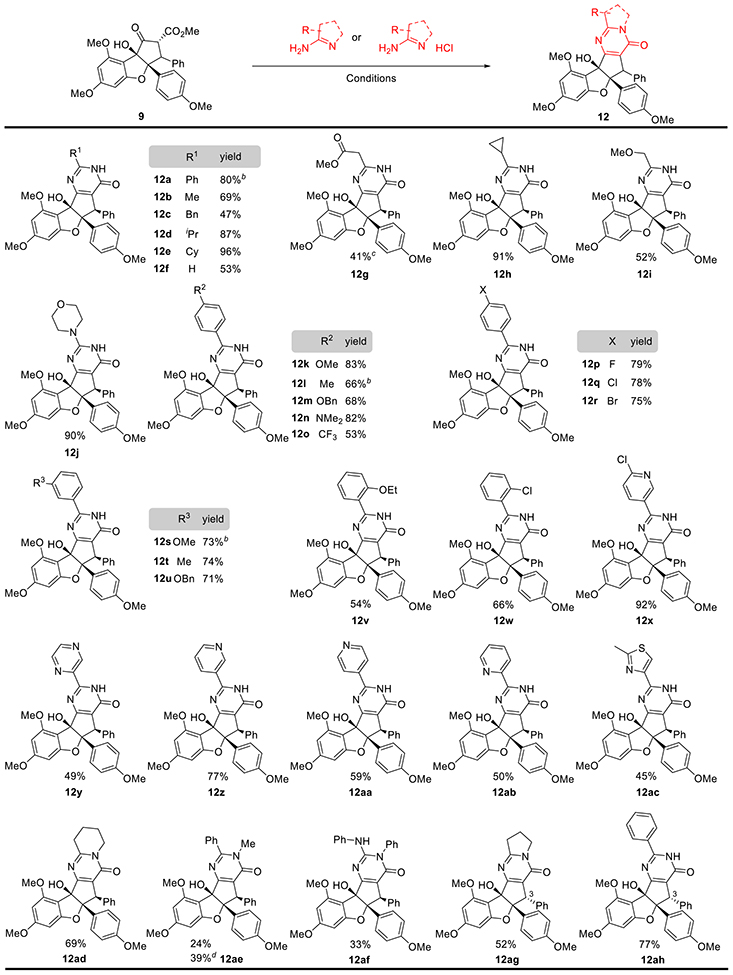 |
Standard conditions: 25 mg of 9, 3 equiv amidine, 0.3 equiv DMAP, m-xlyene (0.025 M), 130°C, 45 min; 2.95 equiv NaOme (1.0 M in MeOH) was used for amidine;
250 mg of 9 was used;
ethyl 2-amidinoacetate HCI Salt was used, and 8% of the ethyl ester products was isolated;
m-xlyene (0.2 M)
Preliminary biological studies of the analog set indicated that C-aryl pyrimidinones consistently possessed good inhibition of HCV infection with low cytotoxicity (vide infra).26 Accordingly, we synthesized additional C-aryl pyrimidinone analogues with a variety of substitution patterns, with an initial focus on para-substituted aryl benzamidines (Table 3, 12k-r). Generally, condensations tolerated both electron-rich and deficient benzamidines; however, we found that greater amounts of fragmentation products were generated using electron-deficient amidines (cf. compounds 12k-n 66–83% yield) with compound 12o, which was generated in 53% yield along with the corresponding fragmentation products. Compound 12m was further subjected to hydrogenolysis to afford the free phenol which may serve as a potential handle for further modifications.26 As expected, analogues including para-halides (12p-12r) were synthesized in 70–80% yields. Such compounds may also allow for late stage functionalization via aminations and SNAr reactions. We found that meta-substituted benzamidines were also workable, affording products (12s-12u) using the standard protocol in reasonable yields (71%−73%). Using the sterically hindered ortho-substituted benzamidines, 12v and 12w were synthesized in moderate yields (54% and 66%). Six C-heteroaryl substituted analogues (12x−12ac) were also synthesized; in these cases, we observed substantial fragmentation of the desired products. Only 12x and 12z were obtained in reasonable yields (92% and 77%, respectively), while compounds 12y and 12aa-12ac were obtained in yields averaging 50%.
As we found that aglaroxin C (6, Table 2) was synthesized regioselectively using an unsymmetrical amidine, we also tested additional unsymmetrical amidines in the pyrimidinone formation. Among all products formed, we found that the N- substituent was situated exclusively on the nitrogen adjacent to the pyrimidinone carbonyl (12ad-af). During the synthesis of 12ae, a small amount of oxazoline 15 was obtained as the only observable side product. This result supports our mechanistic proposal (cf. Scheme 3) wherein the less sterically hindered, unsubstituted nitrogen likely engages in initial hemiaminal formation with 9. Along these lines, we also synthesized 12ac, a ring-expanded analog of 6, in 69% yield. Unlike 12ad, adducts 12ae and 12af were synthesized in 24 and 33% yields, respectively; these reactions generated significant amounts of retro-Nazarov and decarboxylated products, suggestive of lower overall reactivities among the N-substituted amidines.26 We also optimized the yield of 12ae to 39% by increasing the reaction concentration to 0.2 M. Finally, using exo-keto-rocaglate as the starting material, we synthesized compounds 12ah and 12ag, the C-3 epimers of 12a and 6, in 77 and 52% yields, respectively.
As a comparison to the direct condensation with unsymmet-rical amidines, we also attempted alkylations of 12a to produce compounds 12ai-aj (Table 4) 12al-an (see Supporting Information).26 Unfortunately, all attempted alkylations displayed poor chemo-and regioselectivity, favoring the undesired O-al-kylation products such as O-methoxypyrimidine 12ai.
Table 4.
Structure-Activity Relationship of Racemic Aglaroxin C Analogues in the HCV infection inhibition and cytotoxicity Assaysa
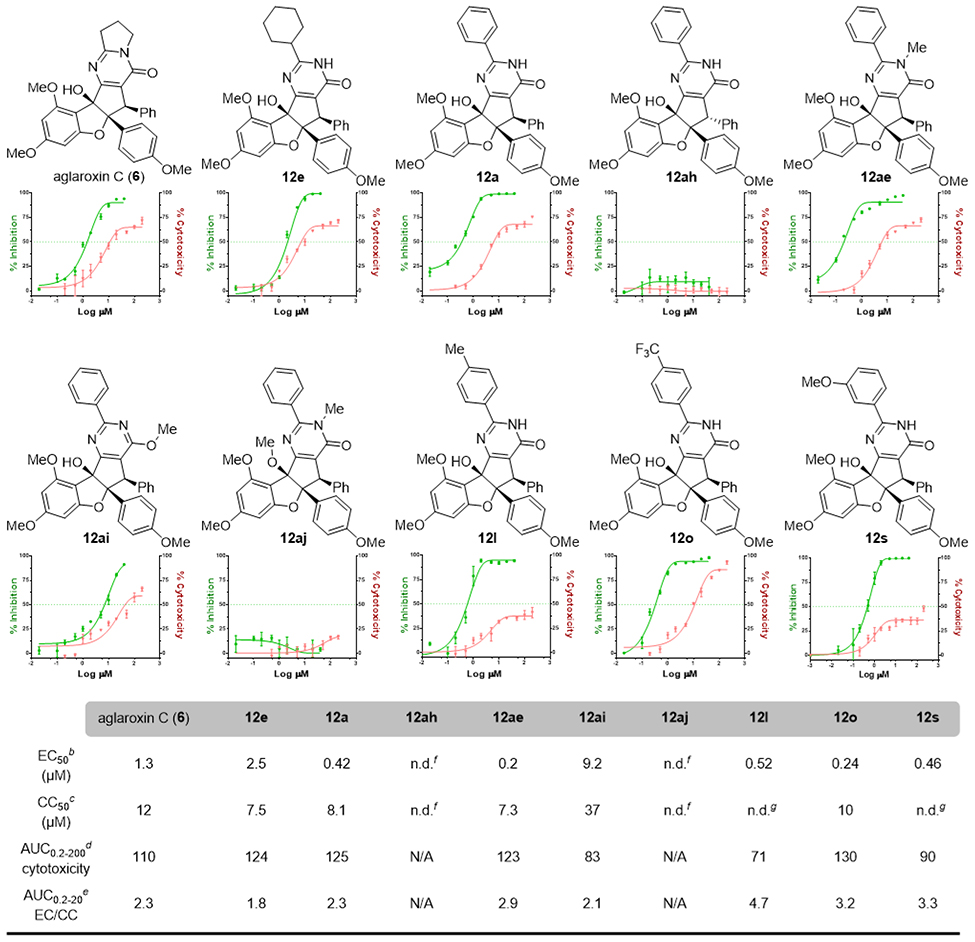 |
Absolute EC50 and CC50 were determined from fitted sigmoid curves;
Absolute EC50 indicates the concentration of compound that inhibits HCV infection by 50%. For its determination, HCVcc-Luc was added to Huh7.5.1 cells at 37 °C together with serially diluted compounds for 3 h prior to removal. Infected cells were incubated at 37 °C for an additional 48 h prior to luciferase assay (mean of n = 3; error bars, s.d.);
Absolute CC50 indicates the concentration of compound required to effect 50% cell death. The numbers of viable cells in culture were determined using the CellTiter-Glo Cell Viability Luminescent Assay kit according to the manufacturer’s (Promega) instruction (mean of n = 3; error bars, s.d.);
AUC0.2–200 (cytotoxicity) is the integration of area under the cytotoxicity curve from 0.2 to 200 μM concentration;
AUC0.2–20 (EC/CC) is the ratio (fold-difference) of the integraded area under curve (AUC) values from 0.2 to 20 μM concentration for the effective curve vs. the cytotoxicity curve;
n d = not determined; no detectable inhibition or cytotoxicity was observed;
n.d. = CC50 was not determined; some cytotoxicity was observed, but cytotoxicity curve did not reach 50% inhibition.
BIOLOGICAL STUDIES
Structure-Activity Relationships.
We next evaluated the library of (±)−aglaroxin analogues and side products against HCV infection, and also tested their corresponding cytotoxicity in human hepatic (Huh) 7.5.1 cells.26 In comparison to aglaroxin C (6), C-alkyl substituted pyrimidinones 12b-d and 12h (Table 3) exhibited increased inhibition against HCV infection, whereas 12e-g had decreased activities. Nevertheless, the C-aryl substituted products (12a, k-w) showed a promising increase of inhibition of HCV infection with similar or reduced cytotoxicity in comparison to 6. Excitingly, 12l (CMLD012043) and 12s (CMLD012044) showed a three-fold greater inhibition of HCV infection in comparison to 6, while 12l and 12s exhibited relatively low cytotoxicity to Huh-7.5.1 cells (less than 50% cell death at 200 μM). Thus, the two lead compounds (12l and 12s) provided excellent selectivity indexes (SI) in inhibiting HCV infection (vide infra). Notably, among all the C-heteroaryl substituted aglaroxin analogues (12x−12ac), only 12y and 12ab displayed slightly increased inhibition of HCV infection relative to 6. The six-membered ring analog 12ad and guanidine-type adducts 12j and 12af were found to have moderate antiviral activity.
To further understand structure-activity relationships (SAR) among aglaroxin analogues, we highlight ten compounds in Table 4 along with dose-response curves depicting their effectiveness in both HCV infection and cytotoxicity assays. As a benchmark compound, 6 was found to inhibit HCV infection with an EC50 of 1.3 μM and showed a CC50 of 12 μM. Of note, EC50 and CC50 values were calculated according to the fitted sigmoid curves, which are reported as absolute values (indicating the concentration of compounds providing 50% inhibition and 50% cell death, respectively). Next, we compared the EC50 of cyclo-hexyl- (12e) and phenyl-substituted (12a) pyrimidinones and found that C-aryl substitutions led to improved potency against viral infection (420 nM vs. 2.5 μM). The C-3 epimer (12ah) of 12a was found to be inactive against HCV infection and no cytotoxicity. It is apparent that a syn-relationship of the two aryl rings on the cyclopenta[b]benzofuran core is crucial for HCV inhibition.
Based on the observation that N-methylated isomer 12ae was one-fold more active than 12a (EC50 0.2 μM vs. 0.42 μM) with similar cytotoxicities (CC50 7.3 μM vs. 8.1 μM),33 we next utilized methylation to evaluate other sites for introduction of target identification tags. In contrast to N-methylation, we observed a decrease in inhibition of HCV infection (EC50 = 9.2 μM) with the O-methylated pyrimidine 12ai. The doubly methylated product 12aj was also found inactive and non-toxic. Finally, we studied the impact of substituting the conjugated C-aryl group of the pyrimidinone (cf. 12l, 12o, and 12s). Compound 12o, with an electron-withdrawing CF3-group, had higher cytotoxicity (CC50 = 10 μM), and only a one-fold better EC50 (0.24 μM) for HCV inhibition than 12l and 12s (EC50 ~ 0.5 μM). The two lead compounds 12l and 12s displayed improved EC50s in the viral infection assay but displayed low cytotoxicity; neither compound reached 50% cell death up to 200 M concentration (maximum cytotoxicity plateau: 40% and 35% cell death for 12l and 12s, respectively). Of note, we observed a similar plateauing of cytotoxicity using aglaroxin C (6) and its analogues (12e, 12a, and 12ae). In contrast, the 4’-trifluoro-methylphenyl analog 12o achieved close to 100% cell death. For a more reliable comparison of cytotoxicities among these compounds, we also calculated the area under the cytotoxicity curve (AUC) by integration,34 an alternative method for accurately quantifying low cytotoxicity.35 In this manner, AUC0.2–200 analysis indicates that 12e, 12a, and 12ai share similar cytotoxicities to 6, whereas 12l and 12s exhibit relatively lower cytotoxicities. Extending this analysis to compare the SI among the analogues, we next calculated the AUC0.2–20(EC/CC) ratios, determining that 12ae, 12l, 12o, and 12s had wider therapeutic windows (0.6 to greater than 2-fold increase in the SI) than 6. Based on the performance of 12l and 12s using these metrics, we utilized these two lead compounds for further biological investigations including mechanism studies.
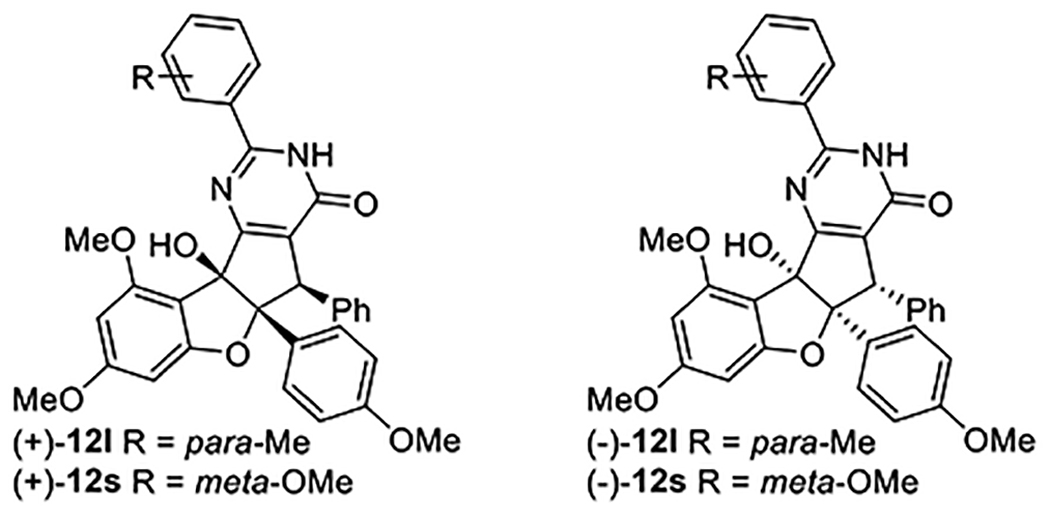
To further characterize the mode of action of the two lead compounds 12l and 12s, we independently synthesized each of their respective enantiomers and investigated their biological activities in both the HCV viral infection and cytotoxicity assays (Table 5). As expected, only (+)−12l and (+)−12s displayed HCV inhibition, whereas the other enantiomers (−)−12l, and (−)−12s were found to be inactive and non-toxic.26 Interestingly, we noticed that maximum cytotoxicity plateau of both active enantiomer (+)−12l and (+)−12s increased substantially compared to their racemic counterparts (±)−12l and (±)−12s. To verify this result, a secondary MTS cell viability assay was also performed on racemic compounds (±)−12k, (±)−12l, and (±)−12o, and showed nearly identical toxicity curves as were observed in the CellTiter-Glo assay.26 While these results warrant further investigation, they suggest a potential rationale for the observed SI enhancement in which the “inactive” enantiomers may reduce or otherwise mitigate the cytotoxic effects of the active species through an as-yet undefined mechanism.
Table 5.
Chirality-Based Biological Profiles of Lead Compoundsa
 | ||||||
|---|---|---|---|---|---|---|
| compound | (±)−12l | (+)−12l | (−)−12l | (±)−12s | (+)−12s | (−)−12s |
| EC50b (μM) | 0.5 | 0.3 | n.d.e | 0.5 | 0.5 | n.d.e |
| CC50c (μM) | n.d.d | 10 | n.d.e | n.d.d | 7.0 | n.d.e |
EC50 and CC50 were collected using the same assay in Table 4 and were calculated based on the fitted sigmoid curves;
EC50 indicates the concentration of a compound that inhibits 50% HCV infection;
CC50 indicates the concentration of a compound leads 50% cell death;
n.d. = CC50 was not determined; some cytotoxicity was observed, but cytotoxicity curve did not reach 50% inhibition;
n.d. = not determined; no detectable inhibition or cytotoxicity was observed.
Aglaroxin Analogues Do Not Affect HCV RNA Replication and Translation.
We subsequently evaluated the ability of the aglaroxin C analogues to inhibit HCV RNA replication and mRNA translation using an HCV replicon system which harbors a full-length HCV Genotype 1b RNA genome.36 In replicon cells, HCV RNA replicates and is translated into viral proteins without forming infectious viruses. Hence, this system permits accurate assessment of any effects on viral RNA replication and translation. As shown in Figure 2, compounds (+)−12l and (+)−12s when used at 2 μM for 3 h did not inhibit HCV RNA replication or protein synthesis. This data implies that the observed inhibitory effects of (+)−12l and (+)−12s were unlikely due to inhibition of viral RNA replication or mRNA translation. To corroborate these findings, we assembled an in vitro translation inhibition assay where a bicistronic reporter mRNA was programmed for translation in Krebs-2 extracts (Figure 3).37 In this system, translation of the Firefly luciferase (FLuc) translation is cap-dependent whereas Renilla luciferase (RLuc) translation is dependent upon the HCV internal ribosome entry site (IRES) (Fig. 3A). It was observed that translation inhibition of firefly luciferase by 6 and analogues 121 and 12s were minimal in comparison with CR-1–31-B (Figure 1, 4), a highly potent rocaglate translation inhibitor showing strong inhibition of cap-dependent protein synthesis (Figure 3B).10c This data suggests that HCV mRNA translation is not inhibited by 6 and its analogues 12l and 12s.
Figure 2.

Aglaroxin C (6) and analogues (121 and 12s) neither inhibit HCV RNA replication nor protein translation. HCV replicon cell line 2-3+ was treated with indicated compounds for 3 h. Cells were incubated for an additional 48 (A & C) or 72 h (B & D). Cellular RNA or protein was isolated for quantifications of HCV viral RNA by real-time PCR (A & B) or non-structural protein NS5 by western blotting (C & D).
Figure 3.

Aglaroxin-type analogues displayed minimal translation inhibition in the comparison to (−)−4 (CR-1–31-B), (A) Schematic representation of the bicistronic reporter FF/HCV/Ren mRNA used to monitor translation. In this system, Firefly luciferase (FLuc) translation is cap-dependent whereas Renilla luciferase (RLuc) expression is HCV-IRES dependent. (B) Assessment of cap or HCV dependent translation of FF/HCV/Ren mRNA in the presence of 1 μM of the indicated compounds in Krebs-2 extracts. Results are presented relative to values obtained in the presence of DMSO and expressed as mean ± error of 2 biological replicates.
Aglaroxin Analogues Inhibit HCV Entry.
As aglaroxin C was previously reported to inhibit HCV entry,12 we carried out two sets of experiments to test whether compounds 12l and 12s also inhibit viral entry. Firstly, we generated infectious lentiviral pseudotypes bearing glycoproteins of HCV (HCVpp), Chikungunya virus (CHIKVpp), Ebola virus (Ebolapp), and vesicular stomatitis virus (VSVpp). Compositionally, these pseudotyped viruses differ only in viral envelopes because they are packaged using the same lentiviral reporter construct with a separate construct expressing specified viral envelope protein. Hence, the only difference among these pseudoviral particles is the mode of entry, which is dictated by the particular viral glycoprotein found on the viral envelopes. Compounds 12l and 12s, as both the racemates and single (+)−enantiomers, specifically inhibited HCVpp and CHIKVpp but not Ebolapp and VSVpp (Figure 4, A and B). Interestingly, like rocaglamide, compounds (+)−12l and (+)−12s as well as their racemic versions also inhibited Dengue virus infection (Figure 4C). It is worth mentioning that HCV, Dengue virus, and CHIKV all utilize PHBs to enter cells; further studies are warranted to determine whether compounds 12l and 12s directly target PHBs. Secondly, to confirm inhibition of HCV entry, we performed time-of-addition experiments using the lead compounds 12l and 12s wherein compounds were added at different times relative to when the virus was added to cells. Similar to 6, 12l and 12s displayed maximal anti-HCV activity when added together with the virus, but partially lost their activity when added 3 h after infection was initiated (Figure 4D). This finding confirmed that 12l and 12s are preferentially inhibiting viral entry.
Figure 4.

Aglaroxin C (6) and its analogues (121 & 12s) inhibit viral entry. (A) Huh7.5.1 cells were infected by indicated pseudovirus in the presence of compounds (1 μM) for 3 h. After an additional 48 h infection, cells were lysed for luciferase assay. Relative infectivity was calculated by normalizing against the values obtained from cells treated with DMSO (arbitrarily set to 1.0) (mean ± SD, *p < 0.05). (B) Same as in a except (±)−121 and (±)−12s were added at 2 μM. (C) Huh7.5.1 cells were infected by Dengue virus (serotype 2, Thailand 16681 strain, MOI 0.1) treated with 2 μM compounds for 3 h. 48 h post-infection, RNA was isolated for quantitative RT-PCR analysis. Viral RNA levels were normalized against GAPDH levels. Data presented are representatives of two independent experiments. (D) (±)−6, (±)−12l, and (±)−12s inhibited HCV infection when added together with the virus. Details on this time-of-addition experiment can be found in Supporting Information.
CONCLUSION
In summary, we have developed a second-generation synthesis of aglaroxin C using late-stage, direct pyrimidinone formation of a keto-rocaglate scaffold. Using this method, we have used commercially available amidines as reaction partners to synthesize a library of over forty aglaroxin C analogues. Among newly synthesized analogues, we successfully demonstrated SAR for inhibition of HCV infection and identified two aryl pyrimidinone lead compounds, 12l and 12s, which have low cytotoxicities to Huh 7.5.1 cells. Additional biological studies with 12l and 12s indicate that the mechanism of inhibition of HCV infection is through inhibition of HCV viral entry, rather than by blocking viral replication and translation. Finally, 12l and 12s are also effective against infection of other viruses including Dengue and Chikungunya, both of which have been found to use prohibitins (PHBs) as an entry factor. These studies illustrate the power of chemical synthesis to bias inhibition of HCV viral entry vs. translation inhibition and improve properties including therapeutic index. Further studies toward target identification of 12l, 12s, and related compounds is currently in progress and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (R01 DK088787 and R35 GM-118173) and the Canadian Institutes of Health Research (FDN-148366) for research support, Dr. Jeffrey Bacon (Boston University) for X-ray crystal structure analyses, Dr. Norman Lee (Boston University) for high-resolution mass spectrometry data, and Nicholas Grigoriadis (Boston University) for preliminary studies NMR (CHE-0619339) and MS (CHE0443618) facilities at Boston University are supported by the NSF. Research at the BU-CMD was supported by NIH grant GM-067041. We thank Dr. Kyle Reichl (Boston University) for proofreading the manuscript.
Footnotes
Supporting Information
Experimental procedures and characterization data for all new compounds described herein, including a CIF file for compound 15. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Lu King M; Chiang C-C; Ling H-C; Fujita E; Ochiai M; McPhail AT X-Ray Crystal Structure of Rocaglamide, a Novel Antileulemic 1-Cyclopenta[b]benzofuran from Aglaia elliptifolia J. Chem. Soc., Chem. Commun 1982, 1150. [Google Scholar]
- (2).For isolation of aglaroxin C, see: Kokpol U; Venaskul-chai B; Simpson J; Weavers RT Isolation and X-Ray Structure Determination of a Novel Pyrimidinone from Aglaia odo-rata. J. Chem. Soc., Chem. Commun 1994, 773; Ohse T; Ohba S; Yamamoto T; Koyano T; Umezawa K Cyclopen-tabenzofuran Lignan Protein Synthesis Inhibitors from Aglaia odorata. J. Nat. Prod 1996, 59, 650; Nugroho BW; Edrada RA; Gussregen B; Wray V; Witte L; Proksch P Insecticidal Rocaglamide Derivatives from Aglaia duppereana. Phytochemistry 1997, 44, 1455.
- (3).For general reviews of rocaglate natural products, see: Ebada SS; Lajkiewicz N; Porco JA Jr.; Li-Weber M;Proksch P Chemistry and Biology of Rocaglamides (= Flavaglines) and Related Derivatives from Aglaia Species (Me-liaceae) In Progress in the Chemistry of Organic Natural Products Vol. 94; Kinghorn AD, Falk H, Kobayashi J, Eds.; Springer Vienna: Vienna, 2011, p 1; Ribeiro N; Thuaud F; Nebigil C; Desaubry L Recent Advances in the Biology and Chemistry of the Flavaglines. Bioorg. Med. Chem 2012, 20, 1857. Pan L; Woodard JL; Lucas DM; Fuchs JR; Kinghorn AD Rocaglamide, Silvestrol and Structurally Related Bioactive Compounds from Aglaia Species Nat. Prod. Rep 2014, 31, 924.
- (4).For reviews of the synthesis of rocaglates, see: Peter P; RuAngelie E; Rainer E; Frank IB; Bambang WN Chemistry and Biological Activity of Rocaglamide Derivatives and Related Compounds in Aglaia Species (Meliaceae). Curr. Org. Chem 2001, 5, 923; Cai X.-h.; Xie B; Guo H Progress in the Total Synthesis of Rocaglamide. ISRN Org. Chem 2011, 2011, 7; Zhao Q; Abou-Hamdan H; Desaubry L Recent Advances in the Synthesis of Flavaglines, a Family of Potent Bioactive Natural Compounds Originating from Traditional Chinese Medicine. Eur. J. Org. Chem 2016, 2016, 5908.
- (5).Hwang BY; Su B-N; Chai H; Mi Q; Kardono LBS; Afriastini JJ; Riswan S; Santarsiero BD; Mesecar AD; Wild R; Fairchild CR; Vite GD; Rose WC; Farnsworth NR; Cordell GA; Pezzuto JM; Swanson SM; Kinghorn AD Silvestrol and Episilvestrol, Potential Anticancer Rocaglate Derivatives from Aglaia silvestris. J. Org. Chem 2004, 69, 3350. [DOI] [PubMed] [Google Scholar]
- (6).(a) Cencic R; Carrier M; Trnkus A; Porco JA Jr.; Minden M; Pelletier J Synergistic Effect of Inhibiting Translation Initiation in Combination with Cytotoxic Agents in Acute Myelogenous Leukemia Cells. Leukemia Res 2010, 34, 535; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lin C-J; Nasr Z; Premsrirut Prem K.; Porco JA Jr.; Hippo Y; Lowe Scott W.; Pelletier J Targeting Synthetic Lethal Interactions between Myc and the eIF4F Complex Impedes Tu-morigenesis. Cell Rep 2012, 1, 325; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sadlish H; Galicia-Vazquez G; Paris CG; Aust T; Bhullar B; Chang L; Hel-liwell SB; Hoepfner D; Knapp B; Riedl R; Roggo S; Schuierer S; Studer C; Porco JA Jr.; Pelletier J; Movva NR Evidence for a Functionally Relevant Rocaglamide Binding Site on the eIF4A-RNA Complex. ACS Chem. Bio 2013, 8, 1519; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Robert F; Roman W; Bramoulle A; Fellmann C; Roulston A; Shustik C; Porco JA Jr.; Shore GC; Sebag M; Pelletier J Translation Initiation Factor eIF4F Modifies the Dexamethasone Response in Multiple Myeloma. PNAS 2014, 111, 13421; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pelletier J; Graff J; Ruggero D; Sonenberg N Targeting the eIF4F Translation Initiation Complex: A Critical Nexus for Cancer Development. Cancer Res 2015, 75, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Cencic R; Carrier M; Galicia-Vazquez G; Bordeleau M-E; Sukarieh R; Bourdeau A; Brem B; Teodoro JG; Greger H; Tremblay ML; Porco JA Jr.; Pelletier J Antitumor Activity and Mechanism of Action of the Cyclo-penta[6]benzofuran, Silvestrol. PLOS ONE 2009, 4, e5223; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wolfe AL; Singh K; Zhong Y; Drewe P; Rajasekhar VK; Sanghvi VR; Mavrakis KJ; Jiang M; Roderick JE; Van der Meulen J; Schatz JH; Rodrigo CM; Zhao C; Rondou P; de Stanchina E; Teruya-Feldstein J; Kelliher MA; Speleman F; Porco JA Jr.; Pelletier J; Ratsch G; Wen-del H-G RNA G-quadruplexes Cause eIF4A-dependent Oncogene Translation in Cancer. Nature 2014, 513, 65; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chu J; Cencic R; Wang W; Porco JA Jr.; Pelletier J Translation Inhibition by Rocaglates Is Independent of eIF4E Phosphorylation Status. Mol. Cancer Ther 2016, 15, 136; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Langlais D; Cencic R; Moradin N; Kennedy JM; Ayi K; Brown LE; Crandall I; Tarry MJ; Schmeing M; Kain KC; Porco JA Jr.; Pelletier J; Gros P Rocaglates as Dual-targeting Agents for Experimental Cerebral Malaria. PNAS 2018, 115, e2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Iwasaki S; Floor SN; Ingolia NT Rocaglates Convert DEAD-box Protein eIF4A into a Sequence-selective Translational Repressor. Nature 2016, 534, 558; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chu J; Galicia-Vazquez G; Cencic R; Mills, John R.; Katigbak, A.; Porco, J. A., Jr., Jr.; Pelletier, J. CRISPR-Mediated Drug-Target Validation Reveals Selective Pharmacological Inhibition of the RNA Helicase, eIF4A. Cell Rep 2016, 15, 2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For chemical synthesis of rocaglate natural products, see: Kraus GA; Sy JO A Synthetic Approach to Rocaglamide via Reductive Cyclization of S-Keto Nitriles. J. Org. Chem 1989, 54, 77; Trost BM; Greenspan PD; Yang BV; Saulnier MG An Unusual Oxidative Cyclization. A Synthesis and Absolute Stereochemical Assignment of (−)−Rocaglamide. J. Am. Chem. Soc 1990, 112, 9022; Davey AE; Schaeffer MJ; Taylor RJK Enantioselective Synthesis of Cyclo-penta[6]benzofurans via An Organocatalytic Intramolecular Double Cyclization. J. Chem. Soc., Chem. Commun 1991, 1137; Davey AE; Schaeffer MJ; Taylor RJK Synthesis of the Novel Anti-leukaemic Tetrahydrocyclopenta[b]benzofuran, Rocaglamide and Related Synthetic Studies J. Chem. Soc., Perkin Trans 1 1992, 2657; Dobler MR; Bruce I; Cederbaum F; Cooke NG; Diorazio LJ; Hall RG; Irving E Total Synthesis of (±)−Rocaglamide and Some Aryl Analogues. Tetrahedron Lett 2001, 42, 8281; Thede K; Die-drichs N; Ragot JP Stereoselective Synthesis of (±)−Rocaglaol Analogues. Org. Lett 2004, 6, 4595; Gerard B; Jones G; Porco JA Jr. A Biomimetic Approach to the Rocaglamides Employing Photogeneration of Oxidopyryliums Derived from 3-Hydroxyflavones. J. Am. Chem. Soc 2004, 126, 13620; Diedrichs N; Ragot JP; Thede K A Highly Efficient Synthesis of Rocaglaols by a Novel a-Arylation of Ketones. Eur. J. Org. Chem 2005, 2005, 1731; Gerard B; Sangji S; O’Leary DJ; Porco JA Jr. Enantioselective Photocycloaddition Mediated by Chiral Brensted Acids: Asymmetric Synthesis of the Rocaglamides. J. Am. Chem. Soc 2006, 128, 7754; Sous ME; Khoo ML; Holloway G; Owen D; Scammells PJ; Rizzacasa MA Total Synthesis of (−)−Episilvestrol and (−)−Silvestrol. Angew. Chem. Int. Ed 2007, 46, 7835; Gerard B; Cencic R; Pelletier J; Porco JA Jr. Enantioselective Synthesis of the Complex Rocaglate (−)−Sil-vestrol. Angew. Chem. Int. Ed 2007, 46, 7831; Malona JA; Cariou K; Frontier AJ J. Am. Chem. Soc 2009, 131, 7560; Magnus P; Freund WA; Moorhead EJ; Rainey T Nazarov Cyclization Initiated by Peracid Oxidation: The Total Synthesis of (±)−Rocaglamide. J. Am. Chem. Soc 2012, 134, 6140;22444715 Magnus P; Freund WA; Moorhead EJ; Rainey T Formal Synthesis of (±)−Methyl Rocaglate Using an Unprecedented Acetyl Bromide Mediated Nazarov Reaction. J. Am. Chem. Soc 2012, 134, 6140; Lajkiewicz NJ; Roche SP; Gerard B; Porco JA Jr. Enantioselective Photocycloaddi-tion of 3-Hydroxyflavones: Total Syntheses and Absolute Configuration Assignments of (+)−Ponapensin and (+)−Elliptifoline. J. Am. Chem. Soc 2012, 134, 13108; Stone SD; Lajkie-wicz NJ; Whitesell L; Hilmy A; Porco JA Jr. Biomimetic Kinetic Resolution: Highly Enantio- and Diastereoselec-tive Transfer Hydrogenation of Aglain Ketones to Access Flavagline Natural Products. J. Am. Chem. Soc 2015, 137, 525; Wang W; Clay A; Krishnan R; Lajkiewicz NJ; Brown LE; Sivaguru J; Porco JA Jr. Total Syntheses of the Isomeric Aglain Natural Products Foveoglin A and Perviridisin B: Selective Excited-State Intramolecular Proton-Transfer Photo-cycloaddition. Angew. Chem. Int. Ed 2017, 56, 14479.
- (10).For representative studies of rocaglate analogues from academia, see: Roche SP; Cencic R; Pelletier J; Porco JA Jr. Biomimetic Photocycloaddition of 3-Hydroxyflavones: Synthesis and Evaluation of Rocaglate Derivatives as Inhibitors of Eukaryotic Translation. Angew. Chem. Int. Ed 2010, 49, 6533; Thuaud F; Ribeiro N; Gaiddon C; Cresteil T; De-saubry L Novel Flavaglines Displaying Improved Cytotoxicity. J. Med. Chem 2011, 54, 411; Rodrigo CM; Cencic R; Roche SP; Pelletier J; Porco JA Jr., Synthesis of Rocaglamide Hydroxamates and Related Compounds as Eukaryotic Translation Inhibitors: Synthetic and Biological Studies. J. Med. Chem 2012, 55, 558; Hawkins BC; Lindqvist LM; Nhu D; Sharp PP; Segal D; Powell AK; Campbell M; Ryan E; Chambers JM; White JM; Rizzacasa MA; Lessene G; Huang DCS; Burns CJ Simplified Silvestrol Analogues with Potent Cytotoxic Activity. ChemMedChem 2014, 9, 1556; Lajkiewicz NJ; Cognetta AB; Niphakis MJ; Cravatt BF; Porco JA Jr. Remodeling Natural Products: Chemistry and Serine Hydrolase Activity of a Rocaglate-Derived β-Lactone. J. Am. Chem. Soc 2014, 136, 2659; Wang W; Cencic R; Whitesell L; Pelletier J; Porco JA Jr. Synthesis of Aza-Rocaglates via ESIPT-Mediated (3+2) Photocycloaddition. Chem. Eur. J 2016, 22, 12006; Zhao Q; Tijeras-Raballand A; de Gramont A; Raymond E; De-saubry L Bioisosteric Modification of Flavaglines. Tetrahedron Lett 2016, 57, 2943.
- (11).For representative medicinal remodeling of rocaglates from industry, see: Bruce I; Cooke NG; Diorazio LJ; Hall RG; Irving E Synthesis of the Carbocyclic Analogue of (±)−Rocaglamide. Tetrahedron Lett 1999, 40, 4279; Liu T; Nair SJ; Lescarbeau A; Belani J; Peluso S; Conley J; Tillotson B; O’Hearn P; Smith S; Slocum K; West K; Helble J; Douglas M; Bahadoor A; Ali J; McGovern K; Fritz C; Palombella VJ; Wylie A; Castro AC; Tremblay MR Synthetic Silvestrol Analogues as Potent and Selective Protein Synthesis Inhibitors. J. Med. Chem 2012, 55, 8859.
- (12).(a) Liu S; Wang W; Brown LE; Qiu C; Lajkiewicz N; Zhao T; Zhou J; Porco JA Jr.; Wang TT A Novel Class of Small Molecule Compounds that Inhibit Hepatitis C Virus Infection by Targeting the Prohibitin-CRaf Pathway. EBi-oMedicine 2015, 2, 1600; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang TT; Liu S; Wang W; Lajkiewicz N; Porco JA Jr Aglaroxin C and Derivatives as HCV Entry Inhibitors US patent 2018 US 10,085,988 B1. [Google Scholar]
- (13).For other natural product inhibitors of HCV viral entry, see: Calland N; Sahuc M-E; Belouzard S; Pene V; Bonnafous P; Mesalam AA; Deloison G; Descamps V; Sahpaz S; Wychowski C; Lambert O; Brodin P; Duverlie G; Meuleman P; Rosenberg AR; Dubuisson J; Rouille Y; Seron K Polyphenols Inhibit Hepatitis C Virus Entry by a New Mechanism of Action. J. Virol 2015, 89, 1005; Lin L-T; Chung C-Y; Hsu W-C; Chang S-P; Hung T-C; Shields J; Russell RS; Lin C-C; Li C-F; Yen M-H; Tyrrell DLJ; Lin C-C; Richardson CD Saikosaponin b2 is a Naturally Occurring Terpenoid That Efficiently Inhibits Hepatitis C Virus Entry. J. Hepatol 2015, 62, 541; Qian X-J; Zhang X-L; Zhao P; Jin Y-S; Chen H-S; Xu Q-Q; Ren H; Zhu S-Y; Tang H-L; Zhu Y-Z; Qi Z-T A Schisandra-Derived Compound Schizandronic Acid Inhibits Entry of Pan-HCV Genotypes into Human Hepatocytes. Sci. Rep 2016, 6, 27268; Bose M; Kamra M; Mullick R; Bhattacharya S; Das S; Karande AA A Plant-derived Dehydrorotenoid: a New Inhibitor of Hepatitis C Virus Entry. FEBSLett 2017, 591, 1305.
- (14).Kuadkitkan A; Wikan N; Fongsaran C; Smith DR Identification and Characterization of Prohibitin as a Receptor Protein Mediating DENV-2 Entry into Insect Cells. Virology 2010, 406, 149. [DOI] [PubMed] [Google Scholar]
- (15).Wintachai P; Wikan N; Kuadkitkan A; Jaimipuk T; Ubol S; Pulmanausahakul R; Auewarakul P; Kasinrerk W; Weng W; Panyasrivanit M; Paemanee A; Kittisenachai S; Roytrakul S; Smith DR Identification of Prohibitin as a Chikungunya Virus Receptor Protein. J. Med. Virol 2012, 84, 1757. [DOI] [PubMed] [Google Scholar]
- (16).Vos T; Allen C; Arora M; Barber RM; Bhutta ZA; Brown A; Carter A; Casey DC; Charlson FJ; Chen AZ; Coggeshall M; Cornaby L; Dandona L; Dicker DJ; Dilegge T; Erskine HE; Ferrari AJ; Fitzmaurice C; Fleming T; Forouzanfar MH; Fullman N; Gething PW; Goldberg EM; Graetz N; Haagsma JA; Hay SI; Johnson CO; Kassebaum NJ; Kawashima T; Kemmer L; Khalil IA; Kinfu Y; Kyu HH; Leung J; Liang X; Lim SS; Lopez AD; Lozano R; Marczak L; Mensah GA; Mokdad AH; Naghavi M; Nguyen G; Nsoesie E; Olsen H; Pigott DM; Pinho C; Rankin Z; Reinig N; Salomon JA; Sandar L; Smith A; Stanaway J; Steiner C; Teeple S; Thomas BA; Troeger C; Wagner JA; Wang H; Wanga V; Whiteford HA; Zoeckler L; Abajobir AA; Abate KH; Abbafati C; Abbas KM; Abd-Allah F; Abraham B; Abubakar I; Abu-Raddad LJ; Abu-Rmeileh NM; Ackerman IN; Adebiyi AO; Ademi Z; Adou AK; Afanvi KA; Agardh EE; Agarwal A; Kiadaliri AA; Ahmadieh H; Ajala ON; Akinyemi RO; Akseer N; Al-Aly Z; Alam K; Alam NKM; Aldhahri SF; Alegretti MA; Alemu ZA; Alexander LT; Alhabib S; Ali R; Alkerwi A. a.; Alla F; Allebeck P; Al-Raddadi R; Alsharif U; Altirkawi KA; Alvis-Guzman N; Amare AT Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 310 Diseases and Injuries, 1990–2015: a Systematic Analysis for the Global Burden of Disease Study 2015. The Lancet 2016, 388, 1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Denniston MM; Jiles RB; Drobeniuc J; Klevens RM; Ward JW; McQuillan GM; Holmberg SD Chronic Hepatitis C Virus Infection in the United States, National Health and Nutrition Examination Survey 2003 to 2010. Ann. Intern. Med 2014, 160, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang H; Naghavi M; Allen C; Barber RM; Bhutta ZA; Carter A; Casey DC; Charlson FJ; Chen AZ; Coates MM; Coggeshall M; Dandona L; Dicker DJ; Erskine HE; Ferrari AJ; Fitzmaurice C; Foreman K; Forouzanfar MH; Fraser MS; Fullman N; Gething PW; Goldberg EM; Graetz N; Haagsma JA; Hay SI; Huynh C; Johnson CO; Kassebaum NJ; Kinfu Y; Kulikoff XR; Kutz M; Kyu HH; Larson HJ; Leung J; Liang X; Lim SS; Lind M; Lozano R; Marquez N; Mensah GA;Mikesell J; Mokdad AH; Mooney MD; Nguyen G; Nsoesie E; Pigott DM; Pinho C; Roth GA; Salomon JA; Sandar L; Silpakit N; Sligar A; Sorensen RJD; Stanaway J; Steiner C; Teeple S; Thomas BA; Troeger C; VanderZanden A; Vollset SE; Wanga V; Whiteford HA; Wolock T; Zoeckler L; Abate KH; Abbafati C; Abbas KM; Abd-Allah F; Abera SF; Abreu DMX; Abu-Rad-dad LJ; Abyu GY; Achoki T; Adelekan AL; Ademi Z; Adou AK; Adsuar JC; Afanvi KA; Afshin A; Agardh E; Agarwal A; Agrawal A; Kiadaliri AA; Ajala ON; Akanda AS; Akinyemi RO; Akinyemiju TF; Akseer N; Lami FHA; Alabed S; Al-Aly Z; Alam K; Alam NKM; Alasfoor D; Aldhahri SF; Aldridge RW; Alegretti MA; Aleman AV; Alemu ZA; Alexander LT Global, Regional, and National Life Expectancy, All-cause Mortality, and Cause-specific Mortality for 249 Causes of Death, 1980–2015: a Systematic Analysis for the Global Burden of Disease Study 2015. The Lancet 2016, 388, 1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).For a comprehensive review of DAAs for HCV, see: Gotte M; Feld JJ Direct-acting Antiviral Agents for Hepatitis C: Structural and Mechanistic Insights. Nat. Rev. Gastroenterol. Hepatol 2016, 13, 338.
- (20).Xiao F; Fofana I; Thumann C; Mailly L; Alles R; Robinet E; Meyer N; Schaeffer M; Habersetzer F; Doffoel M; Leyssen P; Neyts J; Zeisel MB; Baumert TF Synergy of Entry Inhibitors with Direct-acting Antivirals Uncovers Novel Combinations for Prevention and Treatment of Hepatitis C. Gut 2015, 64, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Romano KP; Ali A; Aydin C; Soumana D; Ozen A; Deveau LM; Silver C; Cao H; Newton A; Petropoulos CJ; Huang W; Schiffer CA The Molecular Basis of Drug Resistance against Hepatitis C Virus NS3/4A Protease Inhibitors. PLOSPathog 2012, 8, e1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Tong X; Le Pogam S; Li L; Haines K; Piso K; Baronas V; Yan J-M; So S-S; Klumpp K; Najera I In Vivo Emergence of a Novel Mutant L159F/L320F in the NS5B Polymerase Confers Low-Level Resistance to the HCV Polymerase Inhibitors Mericitabine and Sofosbuvir. J. Infect. Dis 2014, 209, 668; [DOI] [PubMed] [Google Scholar]; (b) Walker A; Filke S; Lubke N; Obermeier M; Kaiser R; Haussinger D; Timm J; Bock HH Detection of a Genetic Footprint of the Sofosbuvir Resistance-associated Substitution S282T after HCV Treatment Failure. Virol. J 2017, 14, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).For a recent review of current HCV entry inhibitor development, see: Qian X-J; Zhu Y-Z; Zhao P; Qi Z-T Entry Inhibitors: New Advances in HCV Treatment. Emerg. Microbes Infect 2016, 5, e3.
- (24).He S; Li K; Lin B; Hu Z; Xiao J; Hu X; Wang AQ; Xu X; Ferrer M; Southall N; Zheng W; Aube J; Schoenen FJ; Marugan JJ; Liang TJ; Frankowski KJ Development of an Aryloxazole Class of Hepatitis C Virus Inhibitors Targeting the Entry Stage of the Viral Replication Cycle. J. Med. Chem 2017, 60, 6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yueh H; Gao Q; Porco JA; Beeler AB A Photochemical Flow Reactor for Large Scale Syntheses of Aglain and Rocaglate Natural Product Analogues. Bioorg. Med. Chem 2017, 25, 6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).See the Supporting Information for complete details.
- (27).For approved drugs containing the pyrimidinone substructure and their synthesis, see: risperidone: Kim D.-m.; Kang M-S; Kim JS; Jeong J-H An Efficient Synthesis of Risper-idonevia Stille Reaction: Antipsychotic, 5-HT2, and Dopamine-D2-antagonist. Arch. Pharm. Res 2005, 28, 1019; paliperidone: Solanki PV; Uppelli SB; Pandit BS; Mathad VT An Improved and Efficient Process for the Production of Highly Pure Paliperidone, a Psychotropic Agent, via DBU Catalyzed N-Alkylation. ACS Sustainable Chem. Eng 2013, 1, 243; Fimasartan: Kim TW; Yoo BW; Lee JK; Kim JH; Lee K-T; Chi YH; Lee JY Synthesis and Antihypertensive Activity of Pyrimidin-4(3H)-one Derivatives as Losartan Analogue for New Angiotensin II Receptor Type 1 (AT1) Antagonists. Bioorg. Med. Chem. Lett 2012, 22, 1649.
- (28).For selected synthesis of natural products containing pyrimidinones, see: Doveston RG; Steendam R; Jones S; Taylor RJK Total Synthesis of an Oxepine Natural Product, (±)−Janoxepin. Org. Lett 2012, 14, 1122; Santos MFC; Harper PM; Williams DE; Mesquita JT; Pinto EG; da Costa-Silva TA; Hajdu E; Ferreira AG; Santos RA; Murphy PJ; Andersen RJ; Tempone AG; Berlinck RGS Anti-parasitic Guanidine and Pyrimidine Alkaloids from the Marine Sponge Monanchora arbuscular. J. Nat. Prod 2015, 78, 1101.
- (29).For the Traube purine synthesis, see: Traube W Ueber Guanidinderivate Zweibasischer Sauren. Ber. Dtsch. Chem. Ges 1893, 26, 2551; Traube W; Schottlander F; Goslich C; Peter R; Meyer FA; Schluter H; Steinbach W; Bredow K Uber Ortho-diamino-pyrimidine und ihre Uberfuhrung in Purine. Ann. Chem 1923, 432, 266.
- (30).For selected pyrimidinone syntheses from 1,3-dicarbonyl substrates, see: Lal B; D’Sa AS; Kulkarni BK; de Souza NJ A Convenient One-pot Entry into Novel 2-Substi-tuted-6,7-dihydro-4H-Pyrimido(2,1-a) Isoquinolin-4-ones. Tetrahedron 1990, 46, 1323; Venkatesan AM; Levin JI; Baker JS; Chan PS; Bailey T; Coupet J Substituted 4H-Pyrido[1,2-a]pyrimidin-4-one Angiotensin II Receptor Antagonists. Bioorg. Med. Chem. Lett 1994, 4, 183; Taylor EC; Zhou P; Tice CM 6-Trifluoromethanesulfonyloxy-4(3H)-pyrimidinones as Versatile Intermediates for the Synthesis of 6-Functionalized 4(3H)-Pyrimidinones . Tetrahedron Lett 1997, 38, 4343; Puig-de-la-Bellacasa R; Gimenez L; Pettersson S; Pascual R; Gonzalo E; Este JA; Clotet B; Borrell JI; Teixido J Diverse Combinatorial Design, Synthesis and In Vitro Evaluation of New HEPT Analogues as Potential Nonnucleoside HIV-1 Reverse Transcription Inhibitors. Eur. J. Med. Chem 2012, 54, 159; Guirado A; Alarcon E; Vicente Y; Andreu R; Bautista D; Galvez J A New Convenient Synthetic Approach to Diarylpyrimidines. Tetrahedron 2016, 72, 3922
- (31).(a) Katritzky AR; Yousaf TI A C-13 Nuclear Magnetic Resonance Study of the Pyrimidine Synthesis by the Reactions of 1,3-Dicarbonyl Compounds with Amidines and Ureas Can. J. Chem 1986, 64, 2087. [Google Scholar]; (b) Katritzky AR; Os-tercamp DL; Yousaf TI The Mechanisms of Heterocyclic Ring Closures. Tetrahedron 1987, 43, 5171. [Google Scholar]
- (32).For imidoyl ketene generation and subsequent cyclization, see: Ham S; Birney DM Imidoylketene: An ab Initio Study of Its Conformations and Reactions. J. Org. Chem 1996, 61, 3962; Alajarin M; Ortin M-M; Sanchez-Andrada P; Vidal A; Bautista D From Ketenimines to Ketenes to Quinolones: Two Consecutive Pseudopericyclic Events. Org. Lett 2005, 7, 5281; Abe T; Kida K; Yamada K A Copper-Catalyzed Ritter-type Cascade via Iminoketene for the Synthesis of Quinazolin-4(3H)-ones and Diazocines. Chem. Commun 2017, 53, 4362.
- (33).Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions. Angew. Chem. Int. Ed 2013, 52, 12256. [DOI] [PubMed] [Google Scholar]
- (34).Dye JF; Somers SS; Guillou PJ Simplified Quantitation of Cytotoxicity by Integration of Specific Lysis against Effector Cell Concentration at a Constant Target Cell Concentration and Measuring the Area Under the Curve. J. Immunol. Methods 1991, 138, 1. [DOI] [PubMed] [Google Scholar]
- (35).(a) Sheeran TP; Jackson FR; Dawes PT; Collins M; Shadforth MF Measurement of Natural Killer Cell Cytotoxicity by Area Under a Cytotoxic Curve: A Method Suitable for Rheumatoid Arthritis. J. Immunol. Methods 1988, 115, 95; [DOI] [PubMed] [Google Scholar]; (b) Brown C; Havener T; Everitt L; McLeod H; Motsinger-Reif A A Comparison of Association Methods for Cytotoxicity Mapping in Pharmacogenomics. Front. Genet 2010, 2; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang S; Pang L Comparing Statistical Methods for Quantifying Drug Sensitivity Based on In Vitro Dose-Re-sponse Assays. Assay Drug Dev. Technol 2012, 10, 88. [DOI] [PubMed] [Google Scholar]
- (36).Scholle F; Li K; Bodola F; Ikeda M; Luxon BA; Lemon SM Virus-Host Cell Interactions during Hepatitis C Virus RNA Replication: Impact of Polyprotein Expression on the Cellular Transcriptome and Cell Cycle Association with Viral RNA Synthesis. J. Virol 2004, 78, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Novac O; Guenier AS; Pelletier J Inhibitors of Protein Synthesis Identified by a High Throughput Multiplexed Translation Screen. Nucleic Acids Res 2004, 32, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.