

Abstract
Rhodomyrtusials A–C, the first examples of triketone-sesquiterpene meroterpenoids featuring a unique 6/5/5/9/4 fused pentacyclic ring system were isolated from Rhodomyrtus tomentosa, along with several biogenetically-related dihydropyran isomers. Two bis-furans and one dihydropyran isomer showed acetylcholinesterase (AChE) inhibitory activity. Structures of the isolates were unambiguously established by a combination of spectroscopic data, ECD analysis, and total synthesis. Bioinspired total syntheses of six isolates were achieved in six steps utilizing a reactive enetrione intermediate generated in situ from a readily available hydroxy-endoperoxide precursor.
Keywords: biomimetic synthesis, Diels-Alder, enetrione, terpenoids, total synthesis
Graphical Abstract
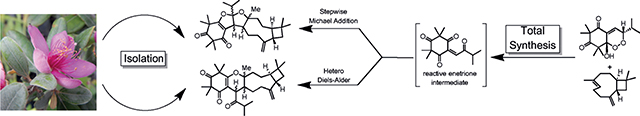
Novel meroterpenoids were isolated from R. tomentosa and characterized, and led to a hypothesis for their biosynthesis, in situ generation of a reactive enetrione in the presence of the sesquiterpene caryophyllene led to efficient chemical syntheses of four of the five meroterpenoids, as well as two biogenetically related cycloadducts, in a biomimetic, six-step sequence, Further evaluation of alkene reaction partners identified additional modes of reactivity for the enetrione.
Natural products are an important source for the discovery of new drugs.[1] Species of the Myrtaceae family are rich in structurally intriguing meroterpenoids which possess various bioactivities.[2] Interestingly, many of the natural products derived from these species are derived from a common precursor, syncarpic acid (Figure 1 a). Isolates such as callistrilones A–E, have recently attracted interest because of their promising antibacterial activity.[3] Other examples of natural products possessing the β-triketone moiety derived from syncarpic acid include rhodomyrtosone A (2),[4] tomentodione M (3),[5] and myrtucommulone K (4).[6] Compound 3 has been shown to reverse multidrug resistance in MCF-7 and K562 cells,[5] while 4 and analogues possess anticancer properties against HEP-G2 and MDA-MB-231 cell lines.[6] The structural diversity found in these natural products, coupled with their diverse biological activities, warrants continued development of new chemistry to access such scaffolds.
Figure 1.

a) Natural products derived from syncarpic acid. b) Structures of rhodomyrtusials A–C (5–7), rhotomentodiones A (8) and B (9), and tomentodiones Q (10) and R (11).
A preliminary bioactivity assay of the leaves and stems of Rhodomyrtus tomentosa revealed that the petroleum ether (PE) extract showed a significant acetylcholinesterase (AChE) inhibition rate (81%, 500 μg-mL−1))[7] Subsequent screening of the title species using a bioassay-guided method led to the isolation of three unprecedented meroterpenoids with a 6/5/5/9/4 pentacyclic ring system, rhodomyrtusials A–C (5–7), two new biogenetically related isomers, rhotomentodiones A (8) and B (9), and two known analogues, tomentodiones Q (10) and R (11) (Figure 1b),[8] all of which possess the syncarpic acid-derived moiety found in 1–4. Herein, we report the structural elucidation, AChE inhibitory activities, and biomimetic total syntheses of these metabolites using a reactive enetrione generated in situ from a hydroxyendoperoxide precursor.
Rhodomyrtusial A (5) possessed a molecular formula of C30H44O4 with nine indices of hydrogen deficiency (IHDs) based on HRESIMS data. Analysis of the :H and 13C NMR spectra (see Table S1 in the Supporting Information)[7] revealed the presence of seven tertiary and two secondary methyl groups, five methylenes, five methines, five quaternary carbon centers, two double bonds, and two carbonyl carbon centers. These functionalities accounted for four out of nine IHDs. The remaining IHDs thus required 5 to have a pentacyclic framework. Structural assignment of 5 was accomplished by analysis of two-dimensional NMR data (see Figure S1). HMBC correlations from Me-12/Me-13 to C1/C2/C3 and from Me-14/Me-15 to C3/C4/C5 revealed the presence of a β-triketone moiety. The location of an isopentyl group and the construction of one furan ring by a 1,8-oxide bridge were determined by HMBC correlations from Me-10/Me-11 to C8 and H7 to C1/C6/C9.1H-1H COSY crosspeaks of H1′/H9′/H2-10′ along with HMBC correlations from Me-12′/Me-13′ to C1′/C10′/C11′ delineated a characteristic cyclobutane ring. HMBC correlations from Me-14′ to C3′/C4′/C5′ and H2-15′ to C7′/C9′, as well as 1H-1H COSY correlations of H1′/H2-2′/H2-3′ and H5′/H2-6′/H2-7′ verified the presence of a caryophyllene unit. The key 1H-1H COSY crosspeaks of H7/H5′ verified that the triketone moiety and caryophyllene were connected by a C7–C5′ bond. The downfield chemical shifts of C8 (δC = 127.3 ppm) and C4′ (δC = 89.7 ppm) indicated that an oxygen atom was bridged at C8 and C4′ to form an additional furan ring, required to fulfil the last IHD and MS information. ROESY correlations (see Figure S1) of H9′β/H6′a; H6′a/Me-14′; and Me-14′/H7′ and H-9′ suggested that these protons were β-oriented, whereas H5′ was assigned as α-oriented based on the observed ROESY correlations of H1′α/H3′a and H3′a/H5′. Compared to the calculated ECD curve (see Figure S2), the experimental ECD spectrum of 5 with two negative Cotton effects [235 nm (Δε −0.86) and 306 nm (Δε −2.72)] and a positive Cotton effect [274 nm (Δε +8.81)] established its absolute configuration as 7R,8S,1′R,4′R,5′S,9′S.
Rhodomyrtusials B (6) and C (7) possessed the same molecular formula as 5 by HRESIMS data. Comprehensive analysis and comparison of NMR data to 5 (see Table S1) revealed that 6 and 7 shared the same planar structure with alternative stereocenters at C7, C8, C4′, and C5′ (see Figure S1). ROESY correlations of H1′α/H3′a, H3′a/Me-14′, Me-14′/H7, and H7/Me-10 supported H7, Me-14, and the isopropyl at C8 were α-oriented, whereas H5′ was β-oriented by correlations of H9′β/H5′ for 6 (see Figure S1). For 7, the observed ROESY correlations of H1′α/H3′b, H1′α/H5′, H5′/H7, and H7/Me-10 indicated these groups were α-oriented, and the β-orientation of Me-14 was established by the ROESY correlations of H3′a/Me-14′.[7] Absolute configurations of 6 and 7 were defined as 7S,8R,1′R,4′S,5′R,9′S and 7S,8R,1′R,4′R,5′S,9′S, respectively, by comparing experimental and calculated ECD spectra.[7]
In addition to 5–7, two new biogenetically related isomers (8 and 9) and two known analogues (10 and 11) were identified from R. tomentosa. LC-MS analysis indicated that all isolates were present in the cold extract of the title species (see Figure S3). The structures and absolute configurations of 8 and 11 were elucidated by spectroscopic data, ECD calculations, and X-ray crystallography)[7] The previously proposed structure and absolute configuration of 11[8] was confirmed by X-ray crystallography (Figure 2).[9] Furthermore, X-ray analysis of 8 unambiguously determined its absolute configuration as 7R,1′R,4′R,5′S,9′S (Figure 2).[9] The unambiguous assignment of 8 and analysis of ECD data[7] established the absolute configuration of 10 as 7S,1′R,4′S,5′R,9′S, which is revised from a previously reported structure.[8] Bioactivity assays of the isolates revealed that 5, 6, and 10 displayed significant AChE inhibitory activities with IC50 values of 8.8, 6.0, and 6.6 μM, respectively. These findings are in good agreement with the AChE inhibitory activity observed from the PE extract.
Figure 2.

X-ray crystal structures of 8 and 11. Thermal ellipsoids shown at 30% probability.
Proposed biosyntheses of 5–11 are shown in Scheme 1. Enetrione (14) may serve as a common precursor, allowing access to 5–11. In line with the G-factor series of endoperoxides,[10] the alkylidene 12 may undergo autoxidation to afford hydroxy-endoperoxide[4b] (13), which may subsequently undergo Kornblum-DeLaMare rearrangement[11] to 14 (Scheme 1a). A DFT model (B3LYP/6–31G**) of 14 (Figure 3) shows a twisted isopropyl ketone (C11-C12-C13-O17 = −57.1°) which appears to minimize O-O lone pair interactions.[12] (−)-trans-Caryophyllene, a common sesquiterpene, may then react with 14 at the more reactive endocyclic double bond[13] in several plausible pathways (Scheme 1 c–e, Figure 4) to furnish 5–11. The flexible, nine-membered ring is known to exist in two relevant conformations (βα/ββ = 3:1) at room temperature (Scheme 1b).[14] In the βα conformer, the olefinic methyl group and exocyclic methylene are trans with respect to one another. Conversely, the ββ conformer is oriented in a cis conformation. The assigned configurations of the dihydropyrans 8–11 indicate both conformers may react with 14 by a hetero-Diels Alder (HDA) cycloaddition (Scheme 1e).[2d,e,15a–i] In contrast stepwise Michael addition[16] of the βα conformer of caryophyllene (re-face approach Scheme 1 c) to 14 may also afford the zwitterion 15, which, after bond-rotation to 16, may undergo consecutive cyclization to 5. Bis-furan 6 may also be obtained through a similar stepwise Michael addition of the ββ conformer (si-face approach, Scheme 1 d) to 14 may afford 17, which can undergo cyclization to 6. Dihydropyrans 8–11 may also be obtained from a stepwise pathway involving zwitterionic intermediates and the bis-furans 5–7 may arise from concerted cycloadditions.
Scheme 1.

Proposed biosyntheses of 5–11. a) Synthesis of 14. b) Conformations of trans-caryophyllene. c) Stepwise Michael addition leading to 5. d) Stepwise Michael addition leading to 6. e) Hetero-Diels-Alder (HDA) cycloadditions leading to 8–11.
Figure 3.

DFT-minimized structure of 14.
Figure 4.

a) DFT-minimized structure of TS5c. b) ChemDraw rendition of TS5c.
Our synthetic studies began with the synthesis of 14. Despite literature examples reporting syntheses of enetriones,[17] there remains an absence in methodology for the synthesis of cyclic enetriones, presumably because of the reactivity of the twisted carbonyl (cf Figure 3). Alkylidene 12 may be synthesized by proline-catalyzed condensation of isovaleraldehyde with syncarpic acid.[18] Metal-mediated allylic oxidation attempts[19] on 12 were not fruitful and typically led to spontaneous oxidation to 13. After unsuccessful attempts to obtain 14 by a Kornblum-DeLaMare rearrangement, we rationalized that the elusive enetrione may be generated in situ from 13. This hydroxy-endoperoxide was previously reported by our group and was obtained as a mixture of diastereomers, albeit in 51 % yield.[4b] After experimentation, an improved set of reaction conditions for the synthesis of 13 was identified (Scheme 2 a). Subjection of 12 to photoenolization conditions[20] (390–720 nm white LEDs) in benzene as solvent and [4+2] cycloaddition with O2 afforded 13 in 72 % combined yield. Exposure of pure 13 a to O-methylation conditions[21] afforded endoperoxide 18 in 25 % yield (Scheme 2 b). Endoperoxide methyl ether (18) was characterized by NMR to assign stereochemistry.[7] In particular, a key nOe was observed between the methoxy and isopropyl methyl groups to ascertain syn stereochemistry for 13 a. By default, the OH and isopropyl group in the endoperoxide 13 b have an anti relationship.
Scheme 2.

a) Synthesis of hydroxy-endoperoxides (13). b) Méthylation and key nOe for the assignment of 13a and 13b.
With 13 a and 13 b in hand, we initiated studies towards syntheses of 5–11. Initial reactions of either endoperoxide 13 a or 13 b and trans-caryophyllene in polar solvents such as dichloromethane, dichloroethane, nitromethane, trifluoroethanol, and HFIP[15i] were unfruitful. Following reaction conditions reported by Takao and co-workers,[15c] thermolysis of 13 a in the presence of caryophyllene in toluene at 100 °C afforded 9,10, and 11, in 10, 10, and 6% yields, respectively. Thermolysis of pure 13 b (90 °C, 43 h) without caryophyllene present resulted in a mixture of 13 a and 13 b in a 1.5 to 1 ratio.[7] Additionally, thermolysis of 13 a or 13 b in the presence of trans-caryophyllene without desiccant at 60 °C gave no conversion into the natural products. These findings led us to evaluate thermolytic conditions with molecular sieves (M.S.) as a desiccant.[21] Thermolysis of 13 b in the presence of trans-caryophyllene and 4 Å M.S. in toluene at 60 °C afforded 5, 7, and 8–11, in 4, 4,19, 5, 8, and 15 % yields, respectively (55 % combined) after HPLC purification (Scheme 3).[7]
Scheme 3.

Synthesis of 5, 7, and 8–11.
Interestingly, thermolysis of 13 a with trans-caryophyllene and 4 Å M.S. resulted in a conversion that was approximately 20% as efficient when the same reaction was run with 13b.[7] The reaction conditions shown in Scheme 3 produced trace amounts of 6, but the compound was never successfully isolated. We also considered the use of Lewis and Brønsted acids as catalysts. Utilization of Ni(ClO4)26H2O, Cu-(ClO4)26H2O, Cu(BF3)26H2O, and various rare-earth metal triflates did not result in product formation. Use of trifluoroacetic acid (TFA) did not afford adducts, likely because of the strength of the acid and the propensity of caryophyllene to rearrange under strongly acidic conditions.[13] Reactions with acetic acid as additive in toluene at 60 °C afforded 5 and 8–11, however, the results were inconsistent.
Based on our experiments, we propose plausible dehydrative mechanisms for either 13 a or 13 b to produce the reactive enetrione (14) in situ (Scheme 4). The less reactive endoperoxide (13a) may form a cyclic peroxycarbenium[4b,22] intermediate (19) after protonation of the peroxy-ketal and expulsion of water. Deprotonation of the α-peroxy methine proton and resulting Kornblum-DeLaMare rearrangement[11] may afford 14 (Scheme 4 a). We believe that endoperoxide 13 b displays heightened reactivity because of the syn-stereochemical relationship of the α-peroxy hydroxy group and the α-peroxy methine hydrogen (Scheme 4b).[23] The more reactive endoperoxide (13b) may undergo formal, directed Kornblum-DeLaMare rearrangement to afford 14.[24] This intermediate can then react with caryophyllene to afford 5–11.
Scheme 4.

Proposed mechanism for enetrione formation.
We conducted initial DFT calculations (B3LYP/STO-3G) of the zwitterionic intermediates 15 and 17 (Scheme 1 c,d) to probe the proposed, stepwise Michael addition pathway. Interestingly, more than half of the conformers converged to product during the optimization process, and led us to investigate concerted pathways in silico. Indeed, we successfully located asynchronous, concerted transition states connecting 5 (Figure 4, PBEh-3C, TS5c) and 7 to 14-βα with activation barriers competitive with production of 8–11.[7] A pseudo-concerted transition state connecting 6 and 14-ββ was also located, but the calculated activation barrier was inconsistent with the observed yields. Preliminary quasi-classical trajectory analysis suggests that this inconsistency is due to a bifurcating potential energy surface (PES).[25] A more extensive computational exploration will be reported in due course.
To better understand the modes of reactivity for 14, we investigated additional alkene reaction partners (Scheme 5). Reactions involving the 1,1-disubstituted alkene of caryophyllene were not observed (Scheme 3). To study reactivity with 1,1-disubstituted alkenes, methylene cyclohexane was chosen as a simplified reaction partner (Scheme 5, entry 1). In this case, three structurally unique products were obtained in 45 % overall yield. The spirocycles 19 and 21 may arise from the concerted cycloaddition pathways previously described, and production of 20 demonstrates a new mode of reactivity for 14 involving an Alder-ene reaction.[26] To further explore the reactivity of 14, 2,3-dimethylbutadiene was evaluated as a reaction partner (Scheme 5, entry 2). The spirocycle 22 was isolated as the major product in 53 % yield, revealing that 14 also participates in normal-demand [4+2] cycloadditions. The dihydropyran diastereomers 23a/23b were also isolated as minor products in 16 % yield. This mixture of diastereomers was thermolyzed in toluene (100 °C, 24 h) and resulted in partial conversion into 22, presumably by Claisen rearrangement.[7] Use of the electron-rich reaction partner dihydrofuran (Scheme 5, entry 3) afforded the tris-furan 24 and dihydropyran 25 in 73 % combined yield. These initial studies illustrate four distinct modes of reactivity for 14 and they allow the synthesis of structurally intriguing small molecules from a common reagent.
Scheme 5.

Investigation of alkene reaction partners with 14.
In summary, we have isolated the bis-furans rhodomyrtusials A–C, three novel triketone-sesquiterpene meroterpenoids featuring a 6/5/5/9/4 pentacyclic ring system as well as four biogenetically related dihydropyran isomers from R. tomentosa. Two bis-furans and one dihydropyran were found to exhibit AChE inhibitory activities. Six of the seven isolates were synthesized in six steps utilizing a reactive enetrione generated in situ from a readily available hydroxy-endoperoxide. Further evaluation of alkene reaction partners identified additional modes of reactivity for the enetrione. Computational studies have identified a valid asynchronous, concerted pathway to 5 and 7. Further pharmacological studies, synthesis, and biological studies of triketone-sesquiterpene meroterpenoids, and exploration of the reactivity of 14 and related enetriones are currently in progress and will be reported in due course.
Supplementary Material
Acknowledgements
These studies were funded by the National Natural Science Foundation of China (U1802287 and 31600283), the National Institutes of Health (R35 GM-118173), Major Bio-Medical Project of Yunnan Province (2018ZF005), Youth Innovation Promotion Association CAS, Key Research and Development Plan of Yunnan Province-Special Project of Science and Technology in Yunnan Province (2017IB007), and Innovation Team of the Ministry of Education (IRT-17R49) and DARPA (W911NF-18–1-0025). We thank Professor Jonathan George (University of Adelaide) for helpful discussions and Thomas Purgett, Dr. Kyle Reichl, and Dr. Wenhan Zhang (BU) for assistance with the manuscript.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Xu-Jie Qin, State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences Kunming 650201 (P. R. China).
Tyler J. Rauwolf, Department of Chemistry, Center for Molecular Discovery (BUCMD), Boston University 590 Commonwealth Avenue, Boston, MA 02215 (USA).
Pan-Pan Li, College of Forestry, Southwest Forestry University Kunming 650224 (P. R. China).
Hui Liu, State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences Kunming 650201 (P. R. China).
James McNeely, Department of Chemistry, Center for Molecular Discovery (BUCMD), Boston University 590 Commonwealth Avenue, Boston, MA 02215 (USA).
Yan Hua, College of Forestry, Southwest Forestry University Kunming 650224 (P. R. China).
Hai-Yang Liu, State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences Kunming 650201 (P. R. China).
John A. Porco, Jr., Department of Chemistry, Center for Molecular Discovery (BUCMD), Boston University 590 Commonwealth Avenue, Boston, MA 02215 (USA).
References
- [1].a) Phillipson JD, Trends Pharmacol. Sci 1979, 1, 36–38 [Google Scholar]; b) Cragg GM, Grothaus PG, Newman DJ, Chem. Rev 2009,109, 3012–3043 [DOI] [PubMed] [Google Scholar]; c) Mishra BB, Tiwari VK, Eur. J. Med. Chem 2011,46,4769–4807 [DOI] [PubMed] [Google Scholar]; d) Gogineni V, Schinazi RF, Hamann MT, Chem. Rev 2015, 115, 9655–9706 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Newman DJ, Cragg GM, J. Nat. Prod 2016, 79, 629–661 [DOI] [PubMed] [Google Scholar]; f) Moloney MG, Trends Pharmacol. Sci 2016, 37, 689–701 [DOI] [PubMed] [Google Scholar]; g) Palanisamy SK, Rajendran NM, Marino A, Nat. Prod. Bioprospect 2017, 7,1–111 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Kearney SE, et al. , ACS Cent. Sci 2018, 4, 1727–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Yang SP, Zhang XW, Ai J, Gan LS, Xu JB, Wang Y, Su ZS, Wang L, Din J, Geng MY, Yue JM, J. Med. Chem 2012,55,8183–8187 [DOI] [PubMed] [Google Scholar]; b) Cao JQ, Huang XJ, Li YT, Wang Y, Wang L, Jiang RW, Ye WC, Org. Lett 2016, 18, 120–123 [DOI] [PubMed] [Google Scholar]; c) Li CJ, Ma J, Sun H, Zhang D, Zhang DM, Org. Lett 2016, 18,168–171 [DOI] [PubMed] [Google Scholar]; d) Zhang YL, Chen C, Wang XB, Wu L, Yang MH, Luo J, Zhang C, Sun HB, Luo JG, Kong LY, Org. Lett 2016, 18, 4068–4071 [DOI] [PubMed] [Google Scholar]; e) Hou LQ, Guo C, Zhao JJ, He QW, Zhang BB, Wang H, J. Org. Chem 2017, 82, 1448–1457 [DOI] [PubMed] [Google Scholar]; f) Qin XJ, Feng MY, Liu H, Ni W, Rauwolf T, Porco JA Jr., Yan H, He L, Liu HY, Org. Lett 2018, 20, 50665070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Cheng MJ, Cao JQ, Yang XY, Zhong LP, Hu LJ, Lu X, Hou BL, Hu YJ, Wang Y, You XF, Wang L, Ye WC, Li CC, Chem. Sci 2018, 9, 1488–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dethe DH, Dherange BD, Das S, Org. Lett 2018, 20, 680–683 [DOI] [PubMed] [Google Scholar]; c) Guo YH, Zhang YH, Xiao MX, Xie ZX, Org. Lett 2018, 20, 2509–2512. [DOI] [PubMed] [Google Scholar]
- [4].a) Hiranrat A, Mahabusarakam W, Tetrahedron 2008, 64, 11193–11197 [Google Scholar]; b) Gervais A, Lazarski KE, Porco JA Jr., J. Org. Chem 2015, 80, 9584–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhou X, Xia Y, Zhang Y, Luo J, Han C, Zhang H, Zhang C, Yang L, Kong L, Oncotarget 2017, 8, 101965–101983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu C, Ang S, Huang X, Tian H, Deng Y, Zhang D, Wang Y, Ye W, Wang L, Org. Lett 2016,18, 4004–4007. [DOI] [PubMed] [Google Scholar]
- [7].See the Supporting Information for complete experimental details.
- [8].Zhang YB, Li W, Jiang L, Yang L, Chen NH, Wu ZN, Li YL, Wang GC, Phytochemistry 2018,153, 111–119. [DOI] [PubMed] [Google Scholar]
- [9].CCDC 1853641 and 1855960 (11 and 8) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- [10].a) Givelet C, Bernat V, Danel M, André-Barrès C, Vial H, Eur. J. Org. Chem 2007, 3095–3101 [Google Scholar]; b) Bernat V, Coste M, André-Barrès C, New J. Chem 2009,33, 2380 [Google Scholar]; c) Ruiz J, Tuccio B, Lauricella R, Maynadier M, Vial H, André-Barrès C, Tetrahedron 2013, 69, 6709–6720 [Google Scholar]; d) André-Barrès C, Carissan Y, Tuccio B, ACS Omega 2017, 2, 5357–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yaremenko IA, Vil VA′, Demchuk DV, Terent′ev AO, Beilstein J. Org. Chem 2016,12, 1647–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ito H, MacDonald SA, Willson CG, Moore JW, Gharapetian HM, Guillet JE, Macromolecules 1986, 19, 1839–1844. [Google Scholar]
- [13].Collado IG, Hanson JR, Macias-Sanchez AJ, Nat. Prod. Rep 1998,15, 187, [DOI] [PubMed] [Google Scholar]
- [14].Shirahama H, Osawa E, Chhabra BR, Shimokawa T, Yokono T, Kanaiwa T, Amiya T, Matsumoto T, Tetrahedron Lett. 1981, 22, 1527. [Google Scholar]
- [15].a) For cycloadditions involving |3-caryophyllene, see :Lawrence AL, Adlington RM, Baldwin JE, Lee V, Kershaw JA, Thompson AL, Org. Lett 2010, 12, 1676–1679 [DOI] [PubMed] [Google Scholar]; b) Spence JTJ, George JH, Org. Lett 2011, 13, 5318–5321 [DOI] [PubMed] [Google Scholar]; c) Takao KI, Noguchi S, Sakamoto S, Kimura M, Yoshida K, Tadano KI, J. Am. Chem. Soc 2015, 137, 15971–15977 [DOI] [PubMed] [Google Scholar]; d) Lam HC, Spence JTJ, George JH, Angew. Chem. Int. Ed 2016, 55, 10368–10371; [DOI] [PubMed] [Google Scholar]; e) Liu HX, Chen K, Tang GH, Yuan YF, Tan HB, Qiu SX, RSC Adv 2016, 6, 48231–48236 [Google Scholar]; f) Lv L, Li Y, Zhang Y, Xie Z, Tetrahedron 2017, 73, 3691–3695 [Google Scholar]; g) Newton CG, Tran DN, Wodrich MD, Cramer N, Angew. Chem. Int. Ed 2017, 56, 13776–13780; [DOI] [PubMed] [Google Scholar]; h) Zhou WL, Tan HB, Qiu SX, Chen GY, Liu HX, Zheng C, Tetrahedron Lett. 2017, 58, 1817–1821 [Google Scholar]; i) Ma SJ, Yu J, Yan DW, Wang DC, Gao JM, Zhang Q, Org. Biomol. Chem 2018,16, 9454–9460. [DOI] [PubMed] [Google Scholar]
- [16].a) Onitsuka S, Nishino H, Tetrahedron 2003, 59, 755–765 [Google Scholar]; b) Gao M, Yang Y, Wu YD, Deng C, Cao LP, Meng XG, Wu AX, Org. Lett 2010,12,1856–1859 [DOI] [PubMed] [Google Scholar]; c) Fan L, Chen W, Tang K, Wu D, Chem. Lett 2012, 41, 940–942 [Google Scholar]; d) Zhu YP, Cai Q, Gao QH, Jia FC, Liu MC, Gao M, Wu AX, Tetrahedron 2013, 69, 6392–6398. [Google Scholar]
- [17].Tan H, Liu H, Zhao L, Yuan Y, Li B, Jiang Y, Gong L, Qiu S, Eur. J. Med. Chem 2017,125, 492–499. [DOI] [PubMed] [Google Scholar]
- [18].a) Mclaughlin EC, Choi H, Wang K, Chiou G, Doyle MP, J. Org. Chem 2009, 74, 730–738 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Muzart J, Tetrahedron 1987, 28, 4665–4668 [Google Scholar]; c) Patel RM, Puranik VG, Argade NP, Org. Biomol. Chem 2011, 9, 6312–6322 [DOI] [PubMed] [Google Scholar]; d) Shing TKM, Yeung YY, Su PL, Org. Lett 2006, 8, 3149–3151. [DOI] [PubMed] [Google Scholar]
- [19].Snider BB, Shi Z, J. Am. Chem. Soc 1992, 114, 1790–1800. [Google Scholar]
- [20].Bernat V, André-Barrès C, Baltas M, Saffon N, Vial H, Tetrahedron 2008, 64, 9216–9224. [Google Scholar]
- [21].Markgraf JH, Burns DB, Greeno EW, Leonard KJ, Miller MD, Synth. Commun 1984,14, 647–653. [Google Scholar]
- [22].For cyclic peroxycarbenium ions, see : Dussault P, Liu X, Org. Lett 1999,1, 1391–1393. [DOI] [PubMed] [Google Scholar]
- [23].For 1,4-syn elimination, see: Moss RJ, Rickborn B, J. Org. Chem 1986, 51, 1992–1996. [Google Scholar]
- [24].For an alkoxide-directed Kornblum - DeLaMare rearrangement, see : Palframan MJ, Kociok-Kohn G, Lewis SE, Chem. Eur. J 2012,18, 4766–4774. [DOI] [PubMed] [Google Scholar]
- [25].a) Yang Z, Dong X, Yu Y, Yu P, Li Y, Jamieson C, Houk KN, J. Am. Chem. Soc 2018,140, 3061–3067 [DOI] [PubMed] [Google Scholar]; b) Kelly KK, Hirschi JS, Singleton DA, J. Am. Chem. Soc 2009,131,8382–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu H, Zhang W, Xu Z, Chen Y, Tan H, Qiu S, RSC Adv. 2016, 6, 25882–25886. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.