

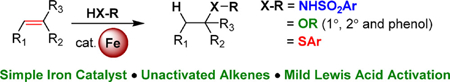
Abstract
The hydrofunctionalization of alkenes, explored for over 100 years, offers the potential for a direct, atom-economical approach to value-added products. While thermodynamically favored, the kinetic barrier to such processes necessitates the use of catalysts to control selectivity and reactivity. Modern variants typically rely on noble metals that require different ligands for each class of hydrofunctionalization, thereby limiting generality. This Letter describes a general iron-based system that catalyzes the hydroamination and hydroetherification of simple unactivated olefins.
Graphical Abstract
The hydrofunctionalization of alkenes offers a direct method to forge beneficial carbon—heteroatom bonds. Starting from abundant alkene or alkyne building block thermodynamically favorable addition1 o f a hydrogen—heteroatom bond (H – N, H – O, or H – S) across a unit of unsaturation builds molecular complexity succinctly. Within the context of hydrofunctionalization, hydroamination is the most studied,2 with less attention given to hydroetherification3 and hydrothiolation.4 Traditionally, precious metals such as palladium, rhodium, ruthenium, and gold have been used to activate the π-system (Figure 1a).5 Recently, earth-abundant, first-row transition metals have enabled unique variants of these reactions,6,7 with iron offering new vistas in hydrofunctionalization over a diverse range of X–H bonds.8
Figure 1.

(a) Hydroamination, hydroetherification, and hydrothiolation require different conditions for each reaction. (b) This work offers a single catalyst for sulfonamides, alcohols, and a thiophenol.
Of the first-row transition metal-catalyzed methods, copper and iron catalysts offer the most generality. While copper catalysts can require specific ligands9 or substrates10 for difficult hydrothiolation reactions, simple iron salts can be used under “ligandless” conditions to provide Markovnikov selectivity.8 Furthermore, iron(III) salts enable the hydrofunctionalization of styrene derivatives or strained alkenes to form carbon–nitrogen,11,12 carbon–xygen,13 and carbon–sulfur14 bonds under different sets of conditions. Similar approaches rely on Bronsted acids,15–17 such as trifluoromethanesulfonic acid18 or those generated in situ from metal triflate salts.19–24 In spite of these advances, functionalizing unactivated alkenes with iron(III) catalysts is limited to intramolecular reactions.25–28 Previously, we have disclosed a powerful, yet mild, iron system capable of catalytically activating aliphatic alcohols toward substitution reactions.29–31 In our studies of alcohol substitution with sulfonamide nucleophiles, reaction monitoring revealed that cyclohexanol can undergo an iron-promoted E1 elimination, forming cyclohexene.32 The in situ-generated alkene also proved competent in the reaction. Enticed by this promising lead, we postulated that alkene could be used directly in hydrofunctionalization reactions. Here, we report a general iron catalyst capable of the intermolecular hydrofunctionalization of unactivated alkenes with sulfonamides, alcohols, and select thiols (Figure 1b).
To begin, we evaluated the hydroamination of cyclohexene with p-toluenesulfonamide in the presence of select acid catalysts (Table 1). Strong Lewis acids, such as AlCl3, were unable to promote the desired reaction (Table 1, entry 1). Likewise, mild Lewis acid FeCl3 provided only trace yield (Table 1, entry 2). The combination of FeCl3 with noncoordinating silver salts33 greatly enhanced the Lewis acidity of the iron catalyst, providing the hydroamination product in modest-to-good yields (Table 1, entries 4–10). While the combination of FeC l3 with AgAsF6 gave marginally higher yield (Table 1, entry 5), AgSbF6 (Table 1, entry 4) was chosen due to the significantly lower cost compared to AgAsF6. Catalytic amounts of strong Bronsted acids, such as an aqueous solution of H SbF6 (Table 1, entry 10) or concentrated H Cl (Table 1, entry 11), were unable to promote hydroamination. Furthermore, the reaction does not seem to be driven by “hidden Bronsted acid catalysis”34 as evidenced by product formation in the presence of Cs2CO3 (Table 1, entry 13) and 2,6-di-tert-butyl-4-methylpyridine (Table 1, entry 14).23,35 While these bases imposed a slight decrease in yield, the retention of catalytic activity suggests that iron is the primary catalyst.
Table 1.
Evaluation of Acid Catalysts
 | ||
|---|---|---|
| entry | catalyst | yield (%)a |
| 1 | A1C13 | 0 |
| 2 | FeCl3 | <5 |
| 3 | AgSbF6 | 0 |
| 4 | FeCI3w/3 AgSbF6 | 78 |
| 5 | FeCI3 w/3 AgAsF6 | 82 |
| 6 | FeCl3 w/3 AgBF4 | 43 |
| 7 | FeCl3 w/3 AgOTf | 17 |
| 8 | FeCI3-6H2O w/3 AgSbF6 | 58 |
| 9 | FeBr3 w/3 AgSbF6 | 79 |
| 10 | FeCl2 w/2 AgSbF6 | 33 |
| 11b | HSbF6 | 0 |
| 12c | HCI | 0 |
| 13d | FeCl3 w/3 AgSbF6 | 68 |
| 14e | FeCl3 w/3 AgSbF6 | 42 |
NMR yields using 1,3,5-trimethoxybenzene standard.
65–75% aqueous solution.
12 M concentrated.
Cs2CO3(0.15equiv) added.
2,6-Di-terf-butyl-4-methylpyridine (0.15 equiv) added.
With suitable conditions for the hydroamination of cyclohexene, a variety of sulfonamide nucleophiles were evaluated (Scheme 1). Sulfonamides were a privileged amine source for our catalytic system. Other amine classes such as electrondeficient anilines, amides, and carbaates provided no hydroamination products, likely due to strong binding to the iron catalyst (see Supporting Information). The highest yields were achieved with p-toluenesulfonamide (1a) affording hydroamination product (2a) in good yield, even on 5 mmol scale. Similar sulfonamides, such as o-toluenesulfonamide (1b) and benzenesulfonamide (1c), gave reasonable yields. More easily removable 2-nitrobenzenesulfonamide (1d) was tolerated.36 Sterically bulky (1e), electron-rich (1f), as well as electron-poor (1g–h) sulfonamides gave moderate-to-good yields. Heterocycles, such as the thiophene in 1i, could be incorporated as in 1i. Additionally, secondary sulfonamides 1j and 1k produced tertiary amine products (2j–k) in modest yields.
Scheme 1. Hydroam ination with Sulfonamide Nucleophiles.

Reactions run at 0.5mmol scale.
With a wide range of viable sulfonamides, we next evaluated the scope of alkenes (Scheme 2). Smaller cyclic alkenes cyclopentene (3a) and cycloheptene (3b) worked well, while larger cyclooctene and cyclododecane surprisingly failed to produce product (data not shown). Strained norbornene (3c) reacted smoothly to afford 4c in 80% yield. Unsymmetric alkenes, such as 1-hexene (3d), gave a mixture of the 2–and 3–substituted products (4d), likely through a carbocation rearrangement.37 Ester-containing substrates (3e) appear to inhibit the reaction, producing relatively low yields. More reactive trisubstituted alkenes (3f–j) proceeded in moderate- to-good yields. Cyclic, trisubstituted alkenes (3f–g) gave superior yields with FeCl3 alone. The modest yields of this substrate class are due to competitive dimerization of the alkene. Furthermore, the addition of AgSbF6 led to increased dimerization and gave little to no hydroamination products. Derivatives of citronellol, elaborated either with a tosylate leaving group (3i) or protected with TIPS (3j), were tolerated without nucleophilic displacement of the tosylate (4i) or deprotection (4j). Additionally, 2,2-disubstituted alkenes, such as 3k, could be used to produce 4k in serviceable yield.
Scheme 2. Alkene Scope for Hydroam ination.

Reactions run at 0.5 mmol scale, [a] Mixture of 2- and 3-substituted products, [b] FeCI3 only, [c] FeCI3 only, reaction run at r.t.
We next sought to translate this methodology to form C–O through hydroetherification reactions (Scheme 3). Under our reaction conditions, the combination of FeCl3 and AgSbF6 can activate alcohols, leading to deleterious substitution reactions instead of the desired hydroetherification products. To eliminate this side reaction, primary alcohols—a challenging substrate for substitution reactions32—were chosen as the class of nucleophile for hydroetherification (Scheme 3a). Primary alcohols were less reactive than sulfonamides under our catalytic conditions. In order to achieve suitable yields, excess alkene was necessary. Primary alcohols with pendant benzene rings (5a–b) gave the desired hydroetherification products (6a–b) with the majority of the remaining mass balance being recovered starting alcohol. Placing an electron-withdrawing group on the pendant benzene ring, such as fluoride (5c), bromide (5d), or nitro (5e), gave the highest yields. Simple primary alcohols, such as n-pentanol (5f), were also competent nucleophiles.
Scheme 3. Hydroetherification and Hydrothiolation.

Reactions run at 0.5 mmol scale, [a] Yield obtained by GC using dodecane internal standard.
In a quest to expand the nucleophile scope to secondary alcohols, phenols, and thiophenols, milder conditions were employed. Since FeCl3 alone does not activate secondary alcohols, these reactions could employ this cheap catalyst system. Using strained alkene norbornene, the hydroetherification of secondary alcohols proceeded with good yields (Scheme 3b). Secondary alcohol 4-phenylcyclohexanol (5g) afforded the desired product 6g in excellent yield, while 4- phenyl-2-butanol (5h) gave only modest yield. Additionally, p- nitrophenol (5i) formed the hydroetherification product in 74% yield. The strongly withdrawing nitro group proved critical to promote hydroetherication and inhibit Friedel- Crafts side products—even p-fluorophenol led primarily to Friedel-C rafts products (data not shown). Excitingly, p- nitrothiophenol (5j) gave the desired hydrothiolation product 6i. Thiols represent a difficult substrate class since disulfide formation competes under the reaction conditions.14
To evaluate the alkene scope for hydroetherification, primary alcohol 5e was evaluated over a range of alkenes. Similar to the hydroamination, small cyclic alkenes cyclopentene (3a) and cycloheptene (3b), as well as strained norbornene (3c) performed well. 1-Hexene (3d) produced a mixture of carbocation-rearranged products. Trisubsituted olefins produced moderate yields of the hydroetherification products 7f–1. While 2-methyl-2-butene (3h) reacted efficiently at 40 °C with only FeCl3, 2-methyl-2-pentene (3l) proceeded in high yield at room temperature. Finally, tosylated citronellol 3i afforded 7i without excess olefin (Scheme 4).
Scheme 4. Alkene Scope for Hydroetherification.

PNP = 4-Nitrophenyl. Reactions run at 0.5 mmol scale, [a] Mixture of 2- and 3-substituted products. [b] FeCI3 only, [c] FeCk only, run at r.t. [d] 1 equiv alkene used.
In summary, we have developed an efficient iron-based catalytic system for the hydrofunctionalization of unactivated alkenes. Using a simple, air- and moisture-tolerant catalyst, the efficient construction of C–N, C–O, and C–S bonds can be accomplished under the same conditions. This modular approach functionalizes mono-, di-, and trisubstituted olefins with a wide range of sulfonamides along with primary and secondary alcohol nucleophiles. Proceeding with Markovnikov selectivity, this method offers a mild alternative to strong Bronsted acid catalysts.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funds from Indiana University in partial support of this work. We also gratefully acknowledge the NSF CAREER Award (CHE-1254783). Eli Lilly & Co. and Amgen supported this work through the Lilly Grantee Award and the Amgen Young Investigator Award. P.T.M. was supported by the Graduate Training Program in Quantitative and Chemical Biology (T32 GM 109825) and Indiana University. IU mass spectrometry for HRMS (NSF Grant CHE1726633).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.or-glett.9b00427.
Experimental details, compound characterization, and NMR data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Johns AM; Sakai N; Ridder A; Hartwig JF J. Am. Chem. Soc 2006, 128, 9306–9307. [DOI] [PubMed] [Google Scholar]
- (2).Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Chem. Rev 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- (3).Hintermann L Recent Developments in Metal-Catalyzed Additions of Oxygen Nucleophiles to Alkenes and Alkynes In C-X Bond Formation; Vigalok A, Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2010; pp 123–155. [Google Scholar]
- (4).Kondo T; Mitsudo T.-a. Chem. Rev 2000, 100, 3205–3220. [DOI] [PubMed] [Google Scholar]
- (5).Huang L; Arndt M; Gooßen K; Heydt H; Gooßen L J. Chem. Rev 2015, 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- (6).Lepori C; Hannedouche J Synthesis 2017, 49, 1158–1167. [Google Scholar]
- (7).Chen J; Lu Z Org. Chem. Front 2018, 5, 260–272. [Google Scholar]
- (8).Greenhalgh MD; Jones AS; Thomas SP ChemCatChem 2015, 7, 190–222. [Google Scholar]
- (9).Delp SA; Munro-Leighton C; Goj LA; Ramírez MA; Gunnoe TB; Petersen JL; Boyle PD Inorg. Chem 2007, 46, 2365–2367. [DOI] [PubMed] [Google Scholar]
- (10).Xi H; Ma E; Li Z Tetrahedron 2016, 72, 4111–4116. [Google Scholar]
- (11).Michaux J; Terrasson V; Marque S; Wehbe J; Prim D; Campagne J-M Eur. J. Org. Chem 2007, 2007, 2601–2603. [Google Scholar]
- (12).Cheng X; Xia Y; Wei H; Xu B; Zhang C; Li Y; Qian G; Zhang X; Li K; Li W Eur. J. Org. Chem 2008, 2008, 1929–1936. [Google Scholar]
- (13).Ke F; Li Z; Xiang H; Zhou X Tetrahedron Lett 2011, 52, 318–320. [Google Scholar]
- (14).Cabrero-Antonino JR; Leyva-Pérez A; Corma A Adv. Synth. Catal 2012, 354, 678–687. [Google Scholar]
- (15).Anderson LL; Arnold J; Bergman RG J. Am. Chem. Soc 2005, 127, 14542–14543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rosenfeld DC; Shekhar S; Takemiya A; Utsunomiya M; Hartwig JF Org. Lett 2006, 8, 4179–4182. [DOI] [PubMed] [Google Scholar]
- (17).Schlummer B; Hartwig JF Org. Lett 2002, 4, 1471–1474. [DOI] [PubMed] [Google Scholar]
- (18).Li Z; Zhang J; Brouwer C; Yang C-G; Reich NW; He C Org. Lett 2006, 8, 4175–4178. [DOI] [PubMed] [Google Scholar]
- (19).Taylor JG; Whittall N; Hii KK Org. Lett 2006, 8, 3561–3564. [DOI] [PubMed] [Google Scholar]
- (20).Weïwer M; Coulombel L; Duñach E Chem. Commun 2006, 332–334. [DOI] [PubMed] [Google Scholar]
- (21).Zhang J; Yang C-G; He C J. Am. Chem. Soc 2006, 128, 1798–1799. [DOI] [PubMed] [Google Scholar]
- (22).Giner X; Nájera C Org. Lett 2008, 10, 2919–2922. [DOI] [PubMed] [Google Scholar]
- (23).Michon C; Medina F; Capet F; Roussel P; Agbossou- Niedercorn F Adv. Synth. Catal 2010, 352, 3293–3305. [Google Scholar]
- (24).Chen J; Goforth SK; McKeown BA; Gunnoe TB Dalton Trans 2017, 46, 2884–2891. [DOI] [PubMed] [Google Scholar]
- (25).Komeyama K; Morimoto T; Takaki K Angew. Chem., Int Ed. 2006, 45, 2938–2941. [DOI] [PubMed] [Google Scholar]
- (26).Komeyama K; Morimoto T; Nakayama Y; Takaki K Tetrahedron Lett 2007, 48, 3259–3261. [Google Scholar]
- (27).Bernoud E; Oulié P; Guillot R; Mellah M; Hannedouche J Angew. Chem., Int Ed. 2014, 53, 4930–4934. [DOI] [PubMed] [Google Scholar]
- (28).Pérez SJ; Purino MA; Cruz DA; López-Soria JM; Carballo RM; Ramírez MA; Fernández I; Martín VS; Padrón JI Chem. - Eur. J 2016, 22, 15529–15535. [DOI] [PubMed] [Google Scholar]
- (29).Jefferies LR; Cook SP Org. Lett 2014, 16, 2026–2029. [DOI] [PubMed] [Google Scholar]
- (30).Jefferies LR; Cook SP Tetrahedron 2014, 70, 4204–4207. [Google Scholar]
- (31).Cook S; Jefferies L; Weber S Synlett 2015, 26, 331–334. [Google Scholar]
- (32).Marcyk PT; Jefferies LR; AbuSalim DI; Pink M; Baik M-H; Cook SP Angew. Chem, Int Ed. 2019, 58, 1727–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Beck W; Suenkel K Chem. Rev 1988, 88, 1405–1421. [Google Scholar]
- (34).Dang TT; Boeck F; Hintermann L J. Org. Chem 2011, 76, 9353–9361. [DOI] [PubMed] [Google Scholar]
- (35).Bowring MA; Bergman RG; Tilley TD Organometallics 2011, 30, 1295–1298. [Google Scholar]
- (36).Wuts PGM; Greene TW; Greene TW Greene’s protective groups in organic synthesis, 4th ed.; Wiley-Interscience: Hoboken, N.J., 2007; p xxviii, 1082 p. [Google Scholar]
- (37).Whitmore FC J. Am. Chem. Soc 1932, 54, 3274–3283. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.