

SUMMARY
The basis for region-specific neuronal toxicity in Huntington disease is unknown. Here, we show that region-specific neuronal vulnerability is a substrate-driven response in astrocytes. Glucose is low in HdhQ(150/150) animals, and astrocytes in each brain region adapt by metabolically reprogramming their mitochondria to use endogenous, non-glycolytic metabolites as an alternative fuel. Each region is characterized by distinct metabolic pools, and astrocytes adapt accordingly. The vulnerable striatum is enriched in fatty acids, and mitochondria reprogram by oxidizing them as an energy source but at the cost of escalating reactive oxygen species (ROS)-induced damage. The cerebellum is replete with amino acids, which are precursors for glucose regeneration through the pentose phosphate shunt or gluconeogenesis pathways. ROS is not elevated, and this region sustains little damage. While mhtt expression imposes disease stress throughout the brain, sensitivity or resistance arises from an adaptive stress response, which is inherently region specific. Metabolic reprogramming may have relevance to other diseases.
In Brief
Region-specific cell death is a hallmark of neurodegenerative disorders like Huntington disease, but the underlying basis is not fully understood. Here, Polyzos et al. discover that differences in neuronal susceptibility in a Huntington model arise from region-specific metabolic reprogramming of astrocytes. In the striatum, astrocytes shift to endogenous fatty acids, but in the long term, the energy benefit is outweighed by elevated oxygen damage to neurons.
Graphical Abstract
INTRODUCTION
Region-specific sensitivity characterizes Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington disease (HD), but its basis is one of the most puzzling aspects of these diseases (Ross et al., 2017). These diseases share the property that mitochondria (MT) do not keep up with the energy demands of the cell (Arun et al., 2016). However, AD, PD, and HD display region-specific cell death (Carvalho et al., 2015), and even within those regions, only select cell types are targeted (Reiner et al., 1988). In HD, for example, the toxic disease-causing protein is ubiquitously expressed, but initially, only the medium spiny neurons in the striatum (STR) are targeted for death (Arun et al., 2016; Carvalho et al., 2015; Reiner et al., 1988; Ross et al., 2017). Despite significant effort, however, researchers have yet to discover whether mitochondrial dysfunction is a cause of toxicity or a mechanism (reviewed in Brustovetsky, 2016; Liot et al., 2017; Polyzos and McMurray, 2017).
A cause-effect relationship between MT dysfunction and neuronal death has been exceptionally difficult to establish (Brustovetsky, 2016; Liot et al., 2017; Polyzos and McMurray, 2017). In postmortem brain, reduction in activity of the complexes II, III, and IV (CII, CIII, and CIV) of the electron transport chain (ETC) machinery occurs in the caudate or putamen in human HD. However, in grade 0 brains, clinically diagnosed but pre-symptomatic HD patients have no discernible micro-structural abnormalities or deficiencies in striatal ETC activity, suggesting that obvious dysfunction, if it occurs, is not measurable before the onset of clinical abnormalities (Vonsattel et al., 1985).
At what stage MT “go bad” is unknown, and whether this transition is the primary cause of the region-specific pathophysiology remains enigmatic. Postmortem samples from HD patients have the obvious and unavoidable problem that it is difficult to quantify the impact of death and postmortem delay on mitochondrial integrity. However, the results from animal models have not clarified a mechanism (Brustovetsky, 2016; Liot et al., 2017; Polyzos and McMurray, 2017). MT in mutant huntingtin (mhtt)-expressing cells often display abnormalities characterized by changes in membrane potential, mitochondrial permeability transition (Brustovetsky et al., 2003; Quintanilla et al., 2013), abnormalities in calcium dynamics (Brustovetsky et al., 2005; Panov et al., 2002), and low ATP production (Gines et al., 2003), among other bioenergetic anomalies. However, those measures have not led us closer to understanding the mechanism of region-specific neuronal toxicity. The results not only vary among animal models but also do not necessarily align with human disease (Brustovetsky, 2016; Liot et al., 2017; Polyzos and McMurray, 2017). For example, CII, CIII, and CV are documented as deficient or reduced in some human HD brains, but there are no substantial differences in the bioenergetic properties of primary striatal neurons from YAC128 (Hamilton et al., 2015), HdhQ(150/150) (Oliveira et al., 2007), and R6/2 animals (Hamilton et al., 2016).
In vivo brain imaging promises to be important in assessing whether mitochondrial defects occur at early stages of HD, but to date, it has not directly connected metabolic changes to mitochondrial dysfunction (Bonvento et al., 2017; Liot et al., 2017). Positron emission tomography (PET) provides evidence for reduced uptake of [18F] fluoro-deoxyglucose in adult HD brain (Ciarmiello et al., 2006; López-Mora et al., 2016) but does not distinguish whether the suppressed uptake is due to altered glucose transport (Covarrubias-Pinto et al., 2015), glucose-processing abnormalities (Carroll et al., 2015), or suppressed glycolysis (Gouarné et al., 2013). Based on 31phosphorous-nuclear magnetic resonance (P-NMR) and magnetic resonance imaging (MRI) (Ciarmiello et al., 2006; Liot et al., 2017), creatine, glutamate, and N-acetyl-aspartate (NAA) are altered in HD brain, but none of these small molecules are direct measures of mitochondrial metabolism.
The barriers to resolving the mechanism of region-specific toxicity and the role of MT have been substantial: (1) we lack non-invasive and direct measures of mitochondrial bioenergetics in living tissues; (2) MT, in vivo, operate within the context of a complex microenvironment, but much of what we know is based on isolated cells, which separate these organelles from the signals to which they must respond; (3) 3-nitropropionate (3-NP) toxin treatment, an irreversible CII inhibitor, transfection, and infection are useful experimental tools to probe mechanisms, but it is unknown whether MT dysfunction can be meaningfully measured when the endogenous metabolism has been altered; (4) MT synthesize ATP in proportion to the available metabolic fuel, yet a time-dependent interplay between metabolic flux, enzymology, electron transport integrity, brain region, age, and cell type are rarely measured together. Overall, we lack consensus as to whether dysfunctional MT are the cause or consequence of disease pathology, whether mitochondrial dysfunction is a loss of bioenergetics capacity (Mochel and Haller, 2011), and why, if it occurs, would the defect be region specific.
To overcome these limitations, we have applied fluorescence lifetime imaging microscopy (FLIM) (Datta et al., 2015; Lakowicz et al., 1992). FLIM is a non-invasive, high-sensitivity imaging technique that measures mitochondrial metabolism directly in living brain tissues of HdhQ(150/150) animals (Lin et al., 2001). By integrating FLIM with imaging, bioenergetics, and biochemical analysis, we uncovered unexpected drivers of region-specific toxicity in the brains of HdhQ(150/150) animals that were not possible to identify by evaluating a subset of features independently.
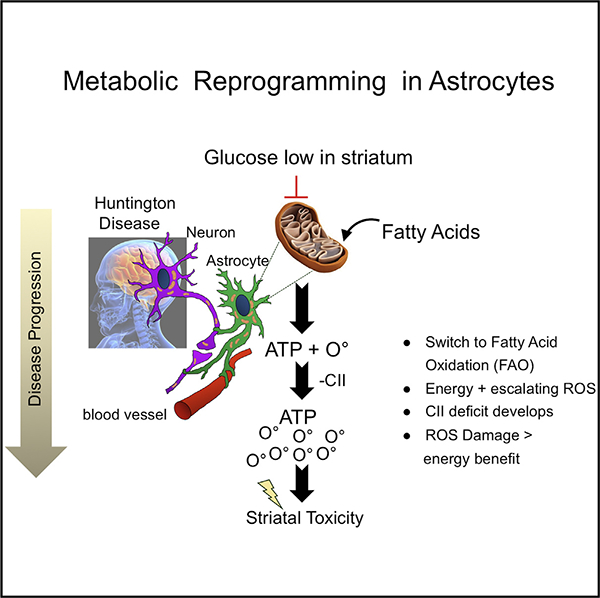
Here, we provide evidence that early region-specific neuronal susceptibility in HdhQ(150/150) animals arises from metabolic reprogramming in their astrocytes. Glucose is low in the HD brain. Astrocytes energetically adapt by reprogramming their MT to use endogenous, non-glycolytic metabolites as alternative fuel. Each region has distinct fuel, however, and astrocytes in sensitive and resistant regions use different adaptation pathways. The sensitive STR is enriched in fatty acids (FAs) and astrocyte MT switch to fatty acid oxidation (FAO) at the cost of damage from elevating reactive oxygen species (ROS), while the resistant cerebellum (CBL) is enriched in amino acid precursors for glucose regeneration without ROS-induced damage. The STR is more sensitive to toxicity. Metabolic reprogramming may be a common strategy by which use of different fuel sustains energy production during crisis but leads to damage in one region more than in another.
RESULTS
Mitochondrial Dysfunction Occurs in Living Brain Tissue of HdhQ(150/150) Animals
We used FLIM phasor analysis to create a metabolic fingerprint in susceptible (the STR) or resistant (the CBL) brain regions to HD toxicity. FLIM phasor is a non-invasive approach (Datta et al., 2015; Lakowicz et al., 1992) that directly measures reduced NADH in living brain slices, without toxin treatment, infection, or transfection, and without disruption of the in situ architecture. NADH has intrinsic autofluorescence and its lifetime is short if it is generated as a freely diffusing molecule during glycolysis in the cytoplasm, and is long when bound by CI during oxidative phosphorylation in MT (Lakowicz et al., 1992). If the free or bound fraction of reduced NADH differs between normal and diseased tissues, then the change provides a direct and sensitive measure of metabolic alterations in MT and the energetic pathways that are affected.
The FLIM lifetimes are measured at every pixel by scanning across a tissue section. Therefore, STR and CBL from HdhQ(150/150) animals were measured together in the same brain slice, with the advantage that the results are internally controlled. However, these two regions differ significantly in their cell-type composition and arrangement (von Bartheld et al., 2016), and a segmented analysis was used to compare them. The CBL comprises 80%–90% neurons and 10%–20% astrocytes, arranged in two visually distinct regions (Figure 1A): a neuron-enriched region (N), arranged in high-density layers (dark purple) adjacent to a more diffuse astrocyte-enriched region (A) (light purple) (Figure 1B, magnified image of box in A). Indeed, the average lifetime distribution of A and N over hundreds of scans was distinct (Figure 1C). In the CBL, the phasor distribution of A was centered to the right of the g = 0.5 line (Figure 1C), indicating a higher free/bound ratio (or shorter lifetimes), while the phasor distribution of N was centered to the left of the g = 0.5 line, indicating longer lifetimes (Figure 1C). The STR was primarily astrocytes (80%–90%), with 10%–20% neurons randomly dispersed throughout the region (Figure 1D) (von Bartheld et al., 2016). Scanning of the STR (Figure 1D, white box) provided only a single average NADH lifetime distribution (Figure 1E), which was similar to that of the astrocyte-rich (A) layers of the CBL (Figures S1A–S1C). Thus, we limited laser scanning to the astrocyte-enriched segments of each brain region, which were identified by diffuse purple Nissl staining (Figures 1A and 1D). The average lifetime in each pixel was calculated over the entire scanned segments, and the two FLIM averages were compared to assess the region-specific metabolic dynamics.
Figure 1. Abnormalities in Mitochondrial Metabolism Occur in Striatal MT in Living Brain Sections.

(A) Coronal section of a Nissl stained CBL illustrating dense neuronal-enriched layers in dark purple and astrocyte-enriched areas stained a diffuse purple. The segment shown in (B) is boxed in white.
(B) Magnified image of the boxed Nissl stained tissue section from (A). Scale bar is 100 μm.
(C) The two FLIM phasor distributions of NADH lifetimes (τϕ) for the neuron-rich (N) and astrocyterich (A) regions of the scanned CBL from HdhQ(150/150) (HD) mice have distinct average lifetimes. n = 3 tissue sections with two hundred scans per section, p values for all comparisons are indicated in (F). The phasors are calculated by transforming the lifetime decay data of each pixel in the imaged region to the phasor plot with co-ordinates (g = mcosϕ, s = msinϕ) where m is the modulation and ϕ is the phase shift of the fluorescence emission signal with respect to the excitation. The position of the phasor distribution indicates the free to bound NADH ratio with phasors on the right of g = 0.5 line having a higher free/bound ratio (or shorter lifetimes) than the ones in the left (longer lifetimes).
(D) Representative coronal section with prominent STR in a 15-week-old HdhQ(150/150) (HD) mouse brain. The left side is stained with NeuN antibody (neurons, green) and DAPI (all nuclei, blue). The right image is a Nissl stain. The white box indicates the scanned striatal region in the FLIM analysis in (E).
(E) Stacked FLIM phasor plots (left) from young (12- to 16-week-old) and old (>80-week-old) comparison of WT and HD mice, as in (C) for the CBL. Representative FLIM images for the STR, Scale bar, 100 mm. A pseudo-color scale for the free (Fr) and bound (Bd) fraction of NADH is shown.
(F) Box plot of the distribution of bound NADH fraction of CBL and STR of WT and HD animals at young (Y) (12–16 weeks) and old (O) ages (>80 weeks), as indicated. n = 3 tissue sections with two hundred scans per section. The box shows the quartiles of the bound NADH fraction dataset. The bars or whiskers extend to show the rest of the distribution, except outliers (shown as diamonds). The outliers are defined as points, which are more than 1.5 times the interquartile range above the third quartile or below the first quartile. p values, calculated by two-tailed Student’s t test. The p values for all comparisons in STR: HD-Y versus WT-Y (p = 7.7.× 3 10 −7); HD-O versus WT-O (p = 0.84); HD-O versus HD-Y (p = 0.11); HD-O versus WT-Y (p = 0.0001); HD-Y versus WT-O (p = 0.005); and WT-O versus WT-Y (p = 4.1 × 3 10 −6). The p values for all comparisons in the CBL: HD-Y versus WT-Y (p = 0.47); HD-O versus WT-O (p = 0.90); HD-O versus HD-Y (p = 0.007); HD-O versus WT-Y (p = 0.003); HD-Y versus WT-O (p = 0.03); and WT-O versus WT-Y (p = 0.02). See also Figure S1.
The single average NADH lifetime distribution in the STR (Figure 1E) underwent an age- and genotype-dependent increase in the free fraction of NADH in the STR of young HdhQ(150/150) animals relative to controls. The metabolic alterations were reflected in the pseudo-color change from reddish pink (wild type [WT], 20 weeks) to blue (HD, 20 weeks) (Figure 1E). No further changes were observed in the STR at older ages (Figure 1E, >80 weeks), indicating that the metabolic shift occurred early. The CBL expressed similar levels of mhtt as did the STR in HdhQ(150/150) animals (Figures S1D and S1E). However, in the same living brain sections, the fraction of available NADH in the CBL increased with age but not with genotype (Figure 1F). Thus, the metabolic shift relevant to the disease state was region specific and corresponded to a higher fraction of available NADH in the STR (Figure 1). Indeed, direct biochemical analysis confirmed that the NADH/NAD+ ratio increased in the STR of HdhQ(150/150) mice, relative to the CBL or to WT astrocytes from either region (Figure S1F). Thus, the striatal MT were fully functional.
Although an increase in the free NADH fraction has been ascribed to an elevation of glycolysis in control cells, the pathways used in most disease states are unknown. Reduced NADH is generated by glycolysis in the cytoplasm, or in the tricarboxylic acid (TCA) cycle in MT and therefore could arise from an elevation in either pathway. As measured by the extracellular acidification rate (ECAR), the activity of glycolytic enzymes was unaffected by genotype (Figures S1G–S1I). Thus, FLIM analysis provided the first direct evidence for altered mitochondrial function in the STR of HdhQ(150/150) animals. However, striatal MT did not lose activity. Instead, the striatal MT of HdhQ(150/150) animals in living tissue were characterized by a region-specific enhancement of oxidative phosphorylation and by an increase in the available fraction of NADH in the region preferentially targeted for neuronal death in HD.
Region-Specific Mitochondrial Abnormalities Occur Exclusively in Astrocytes of HdhQ(150/150) Animals and Early in Development
Astrocytes and neurons are not resolved in whole-tissue scanning. If the functional alterations in striatal MT manifest as increased oxidative phosphorylation, then we expected that the oxygen consumption rate (OCR) would increase in affected brain cells. Thus, we purified neurons and astrocytes from STR and CBL and measured their bioenergetics properties independently. Since adult primary neurons do not survive in culture, synaptosomal nerve terminals (Choi et al., 2009), which can be evaluated at any age, were used instead.
There was no mitochondrial dysfunction in MT of synaptosomes at any age, in any genotype, or in any brain region tested (Figure S1G [glycolysis], Figure S2 [ETC activity], and Figure S3 [structural integrity]). Rather, the region-specific and genotype-specific bioenergetic changes occurred exclusively in MT of astrocytes at early ages (Figures 2A and 2B). In contrast to the FLIM prediction, however, there was a reduction in CII-dependent OCR in striatal astrocytes of HdhQ(150/150) animals. When maximally activated by carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP), the addition of succinate (to permeabilized cells) selectively failed to stimulate CII in HdhQ(150/150) animals relative to controls (Figures 2B and 2C). The levels of succinate were unchanged in the STR in both genotypes (Table S2) and therefore did not limit the CII activity in the disease animals. There was no obvious morphological damage or tissue abnormalities (Figures S3K–S3N), which might account for the differential respiration between astrocytes of each genotype.
Figure 2. There Are Region- and Genotype-Specific Defects in CII Activity in Striatal Astrocytes from HdhQ(150/150) Animals.

(A) Schematic representation of inhibitors and substrates for ETC complexes shown in a hypothetical inner mitochondrial membrane. The ETC complexes and their inhibitors are indicated. Protons pumped into the intermembrane space power ATP production in CV. The number of protons produced by each step during electron transport is indicated. FCCP is an ETC uncoupler. (B and C) Bioenergetic analysis for STR (B) and CBL (C) of HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) littermates. ROT, rotenone; SUC, succinate; and ANT, antimycin-A. Blue, WT and red, HD. Plotted is oxygen consumption versus time. Error bars represent SE.
(D) Enzymatic rates for CI, CII, and CIV in astrocyte extracts of STR and CBL of HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) littermates, as indicated. The rates were calculated from the absorbance versus time (min) for background and astrocyte extracts from striatum (STR) and cerebellum (CBL) of WT and HD astrocytes of 12–16 weeks. (n = 2) Error bars represent SE. HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) are littermates.
(E and F) SDS-PAGE resolution of ETC complexes CI–V in the STR (E) and CBL (F) detected by specific antibodies for each subunit (STAR Methods), measured for four pooled animals. HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) littermates.
(G) 3D STORM images of MT in astrocytes cultures from STR and CBL from HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) of 12–16 weeks. A representative conventional image of TOM20-labeled MT in cells. (left column); STORM of CII, which was detected using SDH antibody staining in the same area as leftmost column (middle column); and an overlay of respective left and middle images (right column). Localization of CII in astrocyte MT taken from CBL of WT mice (first row), CBL of HD mice (second row), STR of WT mice (third row), and STR of HD mice (fourth row). Scale bar is 2 μm.
(H) Box-and-whisker plot of the density of mitochondrial CII STORM localizations per unit area of striatal MT in mouse astrocytes in (Figure 2G). A MATLAB program was used to count the number of single molecule events localized in each mitochondrion. Six images were taken of each sample, and the data were pooled to gather statistics on the average number of single molecule events per unit area of mitochondrion. Thus, the number of single molecule localizations was used as a proxy for the amount of mitochondrial CII. The area of MT was calculated from the TOM20 epifluorescence signal (leftmost column of Figure 2G). The samples, from left to right, were isolated from the CBL of WT mice, red; the CBL of HD mice, green; the STR of WT mice, blue; and the STR of HD mice, orange). The CII per unit area was lowest in the STR of HdhQ(150/150) (HD) animals. p values calculated by two-tailed Student’s t test are WT versus HD (STR) p = 0.046 and WT versus HD (CBL) p = 0.33. WT and HD are HdhQ(wt/wt) and HdhQ(150/150) mice, respectively.
(I) Image of mouse striatal astrocytes from the STR and CBL of HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) littermates stained with MitoTracker Deep Red (upper) and the quantification of staining intensity (lower). n = 30–50 cells, p values, calculated by two-tailed Student’s t test: STR (p = 0.44); and CBL (p = 0.43).
See also Figures S2 and S3.
A reduction was also observed in the enzymatic activity (Figure 2D) and the expressed level of CII in striatal astrocytes of HdhQ(150/150) animals (Figures 2E and 2F). This was important since there is considerable variability in the degree to which the enzymatic activity of distinct ETC complexes influences their respiration (Rossignol et al., 1999), with allowable inhibition thresholds of 10%–25%, 30%, 35%, and 50%–65% for CI–IV, respectively. Indeed, as measured by spectrophotometric assays (Figures 2D and S3P–S3Q), succinate dehydrogenase (SDH) activity was suppressed sufficiently to decrease its CII respiration (Figure 2D). SDS-PAGE analysis confirmed that the 50% reduction in striatal CII activity arose from a physical reduction of the protein in HdhQ(150/150) animals (Figure 2E), with no significant loss in the CBL or in HdhQ(wt/wt) littermates (Figure 2F). Thus, the CII deficiency was genotype, cell type, and region specific, but the bioenergetic features were inconsistent with an increase in the energetic capacity of astrocyte MT, as was predicted by FLIM.
CII Loss Occurs Specifically in Astrocytes but Does Not Entirely Account for the Deficiency of CII Activity
We considered whether there was a plausible mechanism by which a reduction in CII might lead to an increase in oxidative phosphorylation. It was possible that half of the MT in striatal astrocytes of HdhQ(150/150) animals were fully functional, with a full complement of CII, while the other half lacked CII entirely. The population of 50% functional MT might increase their activity to compensate for the 50% that were dysfunctional. To test this possibility, we used stochastic optical reconstruction microscopy (STORM) (Rust et al., 2006; Xu et al., 2013) (Figures 2G and 2H) to visualize the association of CII directly on the surface of the MT in intact cells. Astrocytes were fixed; MT were detected by antibody staining with the outer mitochondrial membrane protein-II (TOM20), while CII was detected using a specific SDH antibody (Figure 2G). Aligning pixels and image overlap were used to detect the density of CII per area MT in the STR versus the CBL of HdhQ(wt/wt) and HdhQ(150/150) animals (Figure 2G). The density of CII per MT area was reduced roughly 50% in the STR of HdhQ(150/150) animals relative to HdhQ(wt/wt) animals or to the density of CII on MT in the CBL of either genotype (Figure 2H). Genotype also did not distinguish MitoTracker Deep Red staining intensity (Figure 2I), indicating that the number of MT was unchanged in striatal astrocytes of both genotypes. Thus, most MT had lost at least some of their CII.
Overall, the results were paradoxical. CII has long been implicated as a causative factor in HD, primarily based on the phenocopy of striatal pathology induced by the 3-NP (Brouillet, 2014) or by CII subunit loss after infection of mhtt in control animals (Damiano et al., 2013). However, in unperturbed native brains from HdhQ(150/150) animals, the deficiency of CII did not entirely explain the energetic properties of astrocytes. Despite the CII deficiency, the maximum respiratory capacity (MRC) of MT in striatal astrocytes from HdhQ(150/150) (29.8 ± 0.1 pmole O2/min) and HdhQ(wt/wt) (29.2 +/−1.7 pmole O2/min) animals were indistinguishable. Thus, the energy capacity of astrocyte MT from HdhQ(150/150) animals was not compromised by the reduction of CII. Taken together with the increase in NADH, integration of the FLIM, bioenergetic, and biochemical measures suggested that a pathway operated in striatal astrocytes, which enhanced oxidative phosphorylation to compensate for the CII deficiency.
FA Metabolites Are Elevated in the STR in HdhQ(150/150) Animals
We used gas chromatography (GC)-time-of-flight (TOF) mass spectrometry (MS) to determine what kind of compensatory pathway might enhance oxidative phosphorylation in striatal astrocytes, and why such a pathway would be region specific. Flash frozen STR, hippocampus (HIP), and CBL from HdhQ(150/150) and HdhQ(wt/wt) littermates were dissected to identify the metabolites in each region with age. Multivariate statistics confirmed brain region is a dominant factor for the metabolic discrimination (Figure 3A) and that the metabolic profiles of each region differed with respect to genotype and age (Figure 3B). Indeed, the array of metabolites in each region was identified and visually displayed using MetaMapp (Figure 4A) (Fiehn, 2016; Lee et al., 2013), a published graphing approach that groups metabolites based on chemical similarity and enzymatic transformation (Figure 4) (Barupal et al., 2012; Fiehn et al., 2011; Lee et al., 2013). The resulting metabolic network consisted of six distinctive clusters (FAs, steroids, carbohydrates, organic acids, amino acids, and nucleic acids) (Figure 4A). No one m/z is a robust biomarker. However, MetaMapp allowed effective identification of a group of related metabolites in a visual fingerprint, which implicates dysregulated pathways that are altered in the disease state. Red and blue indicated metabolites in the HdhQ(150/150) brains that are higher or lower than controls, respectively (Figure 4). The disease fingerprints were compared in regions with differing sensitivity to death.
Figure 3. The Metabolic Features of HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) Littermates Differ with Genotype, Brain Region, and Age.

Score scatter plots of three brain regions in young and old animals of HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) animals (n = 6) based on orthogonal projections to latent structures discriminant analysis (OPLS-DA). The position of each data point coordinated with x- and y-axis indicates relative distances (metabolomic similarity) determined within each data set. Young is 12- to 16-week-old and old is >80-week-old (n = 6 animals for each group).
(A) The score scatter plot combining 12 different datasets: STR, HIP, and CBL of WT and HD at young and old ages. The brain region is a primary factor for the discrimination. x axis (t[1]) distinguishes the cluster of CBL from STR and HIP (53.8% total explained variance). y axis (t[2]) primarily separates the metabolite profiles between STR and HIP (6.8% total explained variance).
(B) The score scatter plots of each brain region (from left, STR, HIP, and CBL). The plot shows the clear separation of the metabolome as a function of age and genotype of STR (R2Y = 0.954, Q2 = 0.628), HIP (R2Y = 0.971, Q2 = 0.569), and CBL (R2Y = 0.663, Q2 = 0.487) as indicated.
Figure 4. A Disease Fingerprint Identifies the Metabolites that Are Altered in the Disease State in Each Brain Region.

(A) Schematic representation of chemical class grouping of metabolites from MetaMapp analysis. The reconstructed metabolic network (MetaMapp) comprises biochemical reaction pairs (Kegg substrate-product pairs) and structural similarity (Tanimoto score > 0.7).
(B) MetaMapp of STR in 12- to 16-week-old animals (young) (n = 6 animals for each region). Node colors indicate significant changes of metabolite levels (p < 0.05 by a two-tailed Student’s t test) in MetaMapp of STR in 12- to 16-week-old animals (young). Node colors indicate significant changes of metabolite levels (p < 0.05 by a two-tailed Student’s t test) in HdhQ(150/150) brain regions compared to those in WT (red, elevated metabolites and blue, decreased metabolites in HdhQ(150/150) (brain). Node size represents the fold-change of differentially expressed metabolite in HdhQ(150/150) brain compared to that of control. No one metabolite is a reliable biomarker for disease. MetaMapp is designed to identify groups of related metabolites, which is a strong indicator of a perturbed pathway. The individual metabolites are listed for inspection in (Table S1).
(C) MetaMapp of CBL and HIP in young mice, same as (B).
(D) FA and AA fingerprints among brain regions are indicated. The arrow indicates the direction of increasing sensitivity (prominent metabolites are listed in Table S1). (n = 6 animals for each region). False discovery rate and Q value for each metabolite is based on significance analysis of microarray (SAM, delta value is set to 1) (Table S1), and pathway over-representation analysis is based on global test and pathway impact score (Figures S4A and S4B).
See also Figure S4.
Three observations were significant. First, the disease fingerprint in each brain region harbored a remarkable “signature” pool of metabolites, which provided a distinct set of non-glycolytic intermediates as prominent substrates (Figures 4B and 4C). The sensitive STR (Figure 4B) was characterized by a reduction in glucose and products of glycolysis (G6P, glucose), a diminished content of essential and non-essential amino acids, and an elevated FA pool relative to the basal state (the individual metabolites are listed in Table S1). The most significant increases in the STR were identified in the lipid cluster (Table S1). Among them, the most common FAs, palmitic acid and stearic acid, were present at significantly higher levels. Thus, STR, which is inherently enriched in FAs in the basal state (Lee et al., 2013), further elevated them in the disease state. High FAs and low amino acids were region-specific features of the disease fingerprint in the STR (Figures 4B and 4D). Indeed, pathway over-representation analysis supported the significance of these changes (Figures S4A and S4B).
Second, the metabolic changes in the sensitive STR were nearly opposite to that of the resistant CBL (Figure 4C), which had little change in its metabolic profile at young ages. The metabolic content of the CBL in the disease state did not differ significantly from the basal state (Figures 4C and 4D), which was replete with glucogenic amino acid precursors for glucose renewal (Figure S4C). The HIP, which has intermediate sensitivity, shared with the STR the property that glycolytic precursors (glucose-1-phosphate [G1P], 3-phosphoglyceric acid [3-PG], and pyruvate [Pyr]) were reduced in the disease state (Figure 4C). However, the HIP lacked the increase in FAs and the decrease in AAs that were characteristic of the striatal fingerprint (Figure 4D).
Third, although there were some changes at older ages, there were, in general, no specific patterns or clustering of altered metabolites in old animals (around >80 weeks) (Figures S4D and S4E). Thus, sensitive and resistant regions have different fingerprints at young ages (Figure 4D, arrow). The resistant CBL maintained AA precursors for glucose renewal (Figure S4C), while the sensitive STR lacked AA and FAs were abundant (Figure 4D).
Striatal Astrocytes Reprogrammed Their Metabolism and Switched from Glycolysis to Fatty Acid Oxidation in the Disease State
MS indicated that FA metabolites were elevated in the disease state in a region-specific manner. However, brain cells both metabolize and synthesize FAs (Hofmann et al., 2017). Thus, the increased levels of FAs in HD STR may occur by accumulation if FA import into MT were blocked. An increased level of FAs could also occur if the demand for FAO was high. Measurements of FA transport inhibitors (Figures S5A and S5B) and FA catabolism (Figures 5B and 5C) were applied to distinguish between these two mechanisms.
Figure 5. Striatal Astrocytes Switch to Fatty Acid Oxidation as an Energy Source.

(A) Schematic representation of the electron transport chain, emphasizing FA oxidation, NADH production and NADH binding to CI. Dotted circles with β-oxidation represent steps of FA shortening (modified from Osellame et al., 2012).
(B) Generation of CO2 (top) and acid-soluble intermediates (bottom) (mostly TCA intermediates) in striatal or cerebellar astrocytes from HdhQ(wt/wt) (WT) or HdhQ(150/150) (HD) animals. Gray, CBL and black, STR. Shown are the mean and SDs for n = 3 biological replicates.
(C) Post-nuclear supernatants from brain regions were assayed for peroxisomal b-oxidation using [1-14C] lignoceric acid and for mitochondrial β-oxidation using [1-14C] palmitic acid. Shown is the residual palmitic acid β-oxidation activity after inhibition with etomoxir, antimycin A, and oligomycin in STR, CBL, hippocampus (HIP), and cortex (CTX). Error bars represent SEs for n = 6 biological replicates, each measured in triplicate.
(D) MitoSOX staining intensity for striatal astrocytes from WT and HdhQ(150/150) animals. Shown are the mean and SDs for n = 3 biological replicates, each measured in triplicate.
See also Figure S5.
Addition of 14C-oleate to MT of striatal astrocytes, when followed to completion, elevated the production of CO2 and the acid-soluble metabolites selectively in striatal astrocytes of HdhQ(150/150) relative to controls (Figure 5B). The elevated CO2 and acid-soluble metabolites (Figure 5B) were genotype and region specific, as they were not observed in astrocytes of HdhQ(wt/wt) littermates or in the resistant CBL in either genotype (Figure 5B). Astrocytes synthesize FAs in both the white and the gray matter of the brain (Hofmann et al., 2017) but normally respire on glucose and produce lactate as fuel for neurons (Bélanger et al., 2011; Supplie et al., 2017). Thus, glycolytic striatal astrocytes had reprogrammed their fuel use and switched to FA oxidation as an alternative energy source in the disease state.
As a control, we tested the level of FAO in synaptosomes, which were unaffected in the disease state by other measures (Figure 5C). Branched-chain FAs of C > 22 are shortened in the peroxisomes before entry into MT, while chain lengths of approximately C16 (palmitate) enter and are oxidized by MT directly. When provided with long- or short-chain FA substrates, 14C-labeled palmitic (C = 16) (in MT) and 14C-labeled lignoceric acid (C = 24) (in peroxisomes), b-oxidation in synaptosomes of either genotype occurred equally well in all brain regions and at any age, and activity was appropriately suppressed in the presence of inhibitors (Figure 5C). MitoSOX staining of ROS was more intense in the striatal astrocytes from HdhQ(150/150) relative to that of HdhQ(wt/wt) littermates (Figure 6D), suggesting that oxidative stress exceeded the steady-state level associated with normal homeostatic function. Thus, reprogramming to FA oxidation in striatal astrocytes of HdhQ(150/150) animals was both region and cell type specific. The striatal astrocytes used their endogenous metabolites to drive the region-specific switch (Figure 4B; Table S1).
Figure 6. FA Oxidation Restores the Succinate Response in MT of Striatal Astrocytes.

OCR for HdhQ(wt/wt) (WT) and HdhQ(150/150) (HD) astrocytes cultured in the indicated media. All data were analyzed using a two-tailed Student’s t test. (n = 3–6 per sample). GL media contains 25 mM glucose, FA media contains 1 mM glucose supplemented with FAs, and LO GL is 1 mM glucose with no added FAs. (A) Comparison of WT and HD astrocytes grown in GL media. Error bars are SE. (p < 0.004 by two-tailed Student’s t test).
(B) Comparison of WT in (GL) with HD astrocytes grown in FA media (1 mM [low] glucose supplemented with FAs); Error bars are SE.
(C) Comparison of HD astrocytes in GL and FA media. Error bars are SE. (p < 0.005 by two-tailed Student’s t test).
(D–F) MitoSOX staining intensity of striatal WT or HD astrocytes in each media condition corresponding to (A–C). Error bars are SE.
(G) Comparison of OCR of WT astrocytes in media containing 25 mM (high) GL and 1 mM (low) LO glucose media. Error bars are SE.
(H) Comparison of OCR of HD astrocytes in media containing 25 mM (high) GL and 1 mM (low) LO glucose media. Error bars are SE.
(I) Comparison of OCR of HD astrocytes cultured in GL 25 mM (high) glucose with (GL + FA) and without (GL) supplemented FAs. ROT, rotenone; SUCC, succinate; and ANT-A, anti-mycin. Error bars are SE.
See also Figures S6A–S6E.
We were unable to find other mechanisms for FA elevation. The increased levels of FAs did not arise from blocked import (Figures S5A and S5B). The carnitine palmitoyltransferase I (CPTI) inhibitor, etomoxir, inhibited FA import to the same extent in astrocytes from either HdhQ(150/150) relative to HdhQ(wt/wt) littermates (Figures S5A and S5B). The level of carnitine (Figure S5C) or carnitine palmitoyltransferase II (CPTII) (Figure S5D) was unchanged in the STR and did not affect the block of CPTI. Furthermore, if CPTII or CPTI were inhibited, accumulation of either palmitoyl-CoA or palmitoylcarnitine, respectively, is diagnostic of the deficit (Schaefer et al., 1997). However, at 12–16 weeks, palmitoyl-CoA in HdhQ(150/150) and HdhQ(wt/wt) littermates were similar, while the level of palmitoylcarnitine was lower in HdhQ(150/150) littermates (Table S1). The level of circulating free FAs in the blood plasma was also similar in HdhQ(150/150) relative to HdhQ(wt/wt) litter-mates (Figure S5E). Using real-time PCR, there were no statistical differences in the expression of 80 genes comprising enzymes of FA anabolism, transport, or catabolism (partial list is shown in Figure S5F). Thus, at the earliest stages of disease, FA accumulation in the STR of HdhQ(150/150) littermates was not due to altered expression patterns of processing enzymes, better access to FAs released from the liver, or an inability to import the available FA into MT. Since glucose was low in the STR, the metabolic fingerprint of the STR in HdhQ(150/150) animals appeared to reflect a region-specific demand for FA catabolism and was encoded in the endogenous metabolic pools.
FAs Compensate for the CII Deficiency and Restore the Succinate Response in HdhQ(150/150) Astrocytes When Glucose Is Low
Since FAO produces NADH for ETC CI binding and increases oxidative phosphorylation, we tested whether elevated FAO provided a pathway to energetically compensate for CII deficiency in the MT of striatal astrocytes (Figure 6). Astrocytes from HdhQ(wt/wt) and HdhQ(150/150) animals were allowed to respire on high glucose (25 mM, GL) or on FA media comprising low glucose (1 mM) supplemented with high FAs (Figure 6). FA media reflected the metabolic environment in the STR of HdhQ(150/150) animals, as reported by the MS-based disease signature (Figures 4B and 4D; Table S1). Similar results were obtained using physiological glucose (5 mM), which were included for comparison (Figures S6A–S6D). Since elevated FAO was accompanied by elevated ROS generation (Figure 5D), we measured OCR (Figures 6A–6C) side-by-side with the level of ROS using MitoSOX (Figures 6D–6F and S6E).
Indeed, FAs had an impact on the bioenergetics of HdhQ(wt/wt) and HdhQ (150/150) striatal astrocytes (Figures 6A–6C). As before (Figure 2B), striatal astrocytes from HdhQ(wt/wt) and HdhQ(150/150) differed in their succinate response when respiring in GL (Figure 6A). Remarkably, however, in FA media, striatal astrocytes from HdhQ(150/150) animals compensated for the CII respiratory deficiency and restored the succinatedependent OCR to that of control astrocytes (Figure 6B). OCR in striatal astrocytes of HdhQ(150/150) animals was substantially higher in the FA media relative to GL media, indicating that use of FAs was responsible for correcting the CII bioenergetic deficiency (Figure 6C). Addition of FAs also elevated CII respiration in WT astrocytes, confirming that the compensation pathway was metabolic and did not depend on the level of CII (Figure S6D). ROS was higher in striatal astrocytes of HdhQ(150/150) animals relative to controls (Figures 6D–6F). This was consistent with the idea that glycolysis was the preferred energy-producing pathway in astrocytes of WT animals, while elevated oxidative phosphorylation was used in astrocytes of HdhQ(150/150) animals.
Astrocyte MT in the CBL depended less on FAs for energy production. Although FA import into MT did not differ with genotype in animals, their response to blocking palmitate import was region specific. Etomoxir treatment led to an immediate 50%–60% reduction of OCR in striatal astrocytes (Figure S5A), while cerebellar cells retained 80%–90% of their OCR (Figure S5B).
The results indicated that the CBL had a diminished need for FAs relative to STR.
Since FA media comprised both reduced glucose and high FAs relative to GL media, we tested the relative importance of both substrates. Glucose concentration alone had little effect on OCR in astrocytes from either genotype (Figures 6G–6I), but surprisingly, if the glucose was high (25 mM or 5 mM), FAs were not needed to restore the succinate response in the astrocytes of HdhQ(150/150) animals (Figure 6, compare 6C and 6I). The results suggested that FAs were not the preferred substrate for astrocytes if glucose was available. In vitro, low-glucose conditions, as observed in the HD brain, drove the alternative use of FAs in striatal astrocytes from HdhQ(150/150) and elevated ROS. Thus, FAO energetically compensated for the CII deficiency in striatal astrocytes of HdhQ(150/150) animals and use of FAs restored the succinate-dependent OCR response to that of control cells.
Selective Accumulation of Oxidized Lipid Bodies Occurs in the STR
If an FAO pathway was relevant to neuronal toxicity, then we expected that the rise in oxidized FA products would accompany loss of neurons. Indeed, oxidized lipid species, which have been observed in B6/HdhQ111/+ mice (Carroll et al., 2015) and in Hdh(140Q) (Zheng et al., 2010) mice, were found in HdhQ(150/150) animals as they aged (Figures 7A and 7B). Oxidized proteins and lipids (referred to as lipofuscin) buildup in perinuclear bodies in the striatal tissue of HdhQ(150/150) animals. Lipofuscin is autofluorescent and readily quenched by Sudan Black B (Figure S6F). The autofluorescent signal of the lipid bodies was lost in striatal brain sections of HdhQ(150/150) animals upon Sudan BlackTrueBlack staining (Figures 7A, 7B, and S6F). Formation of the lipid bodies was region specific (Figures 7C and 7D). Their formation occurred between 60 and 100 weeks (Figures 7A and 7B), which corresponded to the onset of neuronal toxicity in the STR of HdhQ(150/150) animals (Polyzos et al., 2016). Perinuclear lipid bodies were prominent in the STR (Figure 7D, left) and were not observed in the CBL (images in Figures 7C and 7D, right), with the exception of a discrete layer at the interface of the neuronal and astrocyte regions (Figure 7D, right). Lipofuscin bodies were also observed in WT tissues but were not as large as those in HdhQ(150/150) animals and did not accumulate with age (Figures 7A and 7B). If lipid bodies were related to neuronal toxicity and ROS, both should be reversible by ROS scavengers. We have reported previously that treatment with a mitochondrial-targeted electron scavenger, XJB-5–131, suppresses ROS, attenuates neuronal decline, and reduces DNA oxidation in nuclear and mitochondrial DNA in HdhQ(150/150) animal (Polyzos et al., 2016). To test whether mitochondrial ROS promoted these events, brains from the same XJB-5–131-treated animals were evaluated for the presence of lipid bodies. Indeed, these bodies failed to accumulate in the brains of XJB-5–131-treated animals during disease progression (Figures 7A and 7B). Thus, ROS caused the accumulation of FAO products, which accompanied neuronal toxicity, and both were reversible by XJB-5–131.
Figure 7. Lipofuscin-Rich Granules Increase with Age in the Striatum of HD Animals and Are Inhibited with XJB-5–131 Treatment.

(A) Age-dependent perinuclear lipid granules (red), are quenched by TrueBlack (TB). The granules increase with age only in the STR of HD mice. Age-dependent perinuclear lipid granules are suppressed by treatment with mitochondrial targeted antioxidant XJB-5–131 (HD + XJB-5–131). Lipofuscin granule size was 2.09 ± 0.08 μm2 (WT) and 2.93 ± 0.08 μm2 (HD) at 100 weeks. WT and HD are HdhQ(wt/wt) and HdhQ(150/150) mice, respectively. Scale bar is 10 μm.
(B) Quantification of lipofuscin signal intensity in the STR from (A). Mice were treated with XJB-5–131 antioxidant (treatment for 50 weeks) (p < 0.05, two-tailed Student’s t test). (n = 3 tissue sections imaged per sample).
(C) Representative Nissl stained images of the STR or CBL as described in (Figure 1).
(D) (Top) Images from STR (left) and CBL (right) (scale bar is 50 μm) and (bottom) magnified images (scale bar is 10 μm) of red insets illustrating the large lipid-rich granules in the STR or CBL of HD animals at 100 weeks; WT and HD are HdhQ(wt/wt) and HdhQ(150/150) mice, respectively. Staining for DAPI, blue, nuclear; lipofuscin, lipid, red; and NeuN, green, neurons are visible in the image. Error bars are SE.
(E) Model for metabolic reprogramming in HdhQ(150/150) animals, as described in the text. Neuron-glia cartoon modified from (Pierre J. Magistretti, Journal of Experimental Biology. 2006. 209: 2304–2311).
See also Figure S6F.
DISCUSSION
Ubiquitous mhtt expression imparts global stress throughout the HD brain and suppresses glucose uptake. Here, we provide evidence in HdhQ(150/150) animals that the region-specific susceptibility for neuronal death is an astrocyte-driven adaptive response to compensate for diminishing brain glucose. Each brain region harbors a signature pool of non-glycolytic metabolites, which equip each region with distinct alternative fuel and a different response pathway. The vulnerable STR is enriched with FAs, and MT reprogram to FAO at a cost of elevating ROS-induced damage. Indeed, we demonstrate that low glucose drives the use of FAs in striatal astrocytes (Figure 6). The expression of mhtt imparts the same stress in the resistant CBL, but that region harbors metabolites to regenerate glucose. Vulnerability or resistance is imposed by the differential damage associated with the chosen adaptation pathway, i.e., ROS damage is more extensive in the STR.
We propose a two-stage model (Figure 7E). In early phase I, MT are fully functional in the STR of HD patients, but the metabolic precursors for glucose regeneration are limited in this region (Figure 4), and astrocytes slowly shift to enriched FA substrates as glucose declines (Figure 7E). Brain cells do not favor FAs as a fuel source because of ROS generation (Schönfeld and Reiser, 2013) and to the potential oxidative damage to ETC proteins. In phase II, MT in striatal astrocytes lose CII and increase the energy gap (Figure 7E). However, MT in striatal astrocytes adapt by hypermetabolizing FAs to maximize NADH for fueling CI. Escalation of FAO compensates in the short term and maintains bioenergetic capacity of striatal MT. In the long term, however, we envision that escalating b-oxidation creates a futile FAO-ROS cycle in which the energy-producing benefit of the FAO pathway becomes the primary source of ROS-dependent damage. Because they are the major glucose metabolizing cell type, metabolic preprogramming in astrocytes occurs early and precedes neuronal abnormalities, which develop down-stream as disease progresses (Figure 7E).
Since ETC complexes are reduced in astrocytes at the time of their isolation at P2, we are unable to precisely determine whether CII deficiency causes ROS or whether ROS elevation causes CII deficiency in striatal astrocytes. However, we favor the latter (Figure 7E). All ETC complexes harbor ironsulfur (Fe-S) clusters that are readily oxidized and inactivate enzymes in other systems (Ghezzi and Zeviani, 2012; Zorov et al., 2014). Indeed, aconitase and SDH are highly susceptible to inactivation by superoxide anion radical. Aconitate, malate, and fumarate, the products of these enzymes, are reduced in the STR of HdhQ(150/150) animals (Table S2). We find that ROS causes the oxidized lipid accumulation in HdhQ(150/150) animals (Figures 7A–7D). Treatment of brain tissues with XJB-5–131, an electron scavenger and antioxidant, attenuates the rise in oxidized lipids and neuronal toxicity (Polyzos et al., 2016), reminiscent of antioxidant reversible effects of neurodegeneration in flies (Liu et al., 2015). There are no ETC mutations in HD, and mhtt is ubiquitously expressed. Therefore, there is no obvious reason why CII should be inhibited in only one brain region and not in others, implying that the regional deficiency is metabolically driven. Although it did not restore CII expression, addition of FAs elevated the CII-dependent succinate response even in WT astrocytes (Figures 6 and S6D), indicating that rescue is metabolic. While detailed analysis will be required to sort out the rescue mechanism, reversal of Fe-S cluster oxidation, suppression of malate or other natural CII inhibitors, or pH changes provide examples of potential metabolic pathways to enhance CII activity under conditions where FAs are elevated.
These results bring to light features that are likely to have confounded previous bioenergetics analyses by us and others. Mitochondrial dysfunction is cell-type specific, yet tissues and primary neurons, the most common experimental systems, either do not resolve or do not measure both cell types. What we and others have interpreted as variable outcomes in analysis of mitochondrial function is likely, at least in part, to reflect unresolved but energetically distinct MT in astrocytes and neurons. Moreover, region-specific mitochondrial dysfunction is difficult to discern from classic measures of the glycolysis or ETC machinery. We find that the enzymes of glycolysis are active in the STR (Figures S1G–S1I), but it lacks glucose as fuel (Figure 4B; Table S1). CII of the ETC is deficient in striatal astrocytes, but MT do not lose activity. Rather, they adapt by substrate switching to their enriched FA substrates and elevate oxidative phosphorylation to energetically compensate (Figures 1 and 5).
The cell type- and region-specific nature of mitochondrial function in astrocytes argues against recent suggestions of enhanced glycolysis in dysfunctional brain cells (Oláh et al., 2008; Sameni et al., 2016). A higher free NADH level detected by FLIM was ascribed as a shift toward higher glycolysis in mhtt-transfected human embryonic kidney (HEK293) cells. The assignment was based on the NADH lifetime alone when mhtt was overexpressed in a non-neuronal cell line and without validation that glycolysis was altered under toxic conditions (Sameni et al., 2016). In primary striatal cells and in tissues from HdhQ(150/150) animals, we find that glycolysis is not increased (Figures S1G–S1I), and glucose is low (Figure 4; Table S1). FAO is elevated in the striatal astrocytes (Figure 5B), and β oxidation increases the NADH/NAD+ ratio (Figure S1F).
In other studies, the elevated activities of some glycolytic enzymes in the brains of N171–82Q transgenic mouse model of HD were accompanied by lower activity of glyceraldehyde-3-phosphate dehydrogenase (GADPH) (Oláh et al., 2008). These seemingly conflicting activities were measured in homogenates of whole brain tissues, which do not resolve affected and unaffected regions of the HD brain or the two cell-type-dependent classes of MT. Furthermore, expression of expanded polyglutamine tracts or increases in oxidative stress promotes GAPDH aggregation and catalytically inactivates the enzyme (Hwang et al., 2015; Nakajima et al., 2017). Under these conditions, cells would be expected to shift toward oxidative phosphorylation as an energy source. We show that FAs are used to restore OCR only when glucose is low in the sensitive STR (Figure 6). Indeed, overexpression of glucose-6-phosphate dehydrogenase, the major enzyme for the pentose phosphate pathway (PPP) (Legan et al., 2008), or overexpression of a glucose transporter (Besson et al., 2015) extends lifetime in flies expressing 93 glutamine repeats. The CBL in HdhQ(150/150) animals is replete with amino acid precursors for these pathways and this region is more resistant to toxicity.
Why striatal neurons rather than astrocytes die is unknown. Astrocytes are emerging as contributors to neuronal dysfunction in a growing number of brain disorders (Pekny et al., 2016). In HD, both neurons and astrocytes display inclusion markers for disease progression (Jansen et al., 2017), neurons are more prone to glutamate neuronal excitotoxicity (Jiang et al., 2016; Tong et al., 2014; Valenza et al., 2015), and there are abnormalities in neuron-astrocyte communication (Boussicault et al., 2014; Herrero-Mendez et al., 2009). Similar to most human HD MRI studies (Bender et al., 2005; Gårseth et al., 2000; Handley et al., 2016; Nambron et al., 2016; Reynolds et al., 2005), we find that lactate levels are unchanged at very early stages of disease. This result suggests that metabolic reprogramming in astrocyte MT precedes disruption of neuronal-astrocyte communication through the lactate shuttle (Bélanger et al., 2011). However, neurons may survive better in low glucose since they degrade much of their 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-3 (Pfkfb3) enzymes (Herrero-Mendez et al., 2009) and depend on oxidative phosphorylation rather than glycolysis to generate energy. FA oxidation demands more oxygen than glucose, which would provide a greater risk of neurons becoming hypoxic and may eventually inhibit neuronal MT from carrying out oxidative phosphorylation. We propose that a futile FAO-ROS cycle promotes early toxicity in the STR when the region-specific adaptation strategy in their astrocytes becomes the source of damage (Figure 7E). Loss of metabolic support from astrocytes likely sensitizes neurons over time to the many detrimental protein-protein interactions that unquestionably contribute to neuronal death at later stages (Choudhury et al., 2016).
Many diseases have region-specific sensitivities. The ability of MT to use endogenous non-glycolytic metabolites to compensate and adapt to energy imbalances provides a plausible and general adaptive strategy to sustain survival. Each distinct disease protein or susceptibility factor places its own degree of stress in its own relevant tissue and demands a region-specific response. While in HD, neurons in the STR are vulnerable to death, a region-specific metabolic “signature” fingerprint may provide a general context in which a particular disease protein is able to induce damage and susceptibility in one region better than in another.
Limitations of Study
Metabolic reprogramming occurs early in astrocytes and in brain regions that are susceptible to neuronal death. However, it remains to be determined whether the astrocyte-derived metabolic abnormalities are direct causes of neuronal toxicity. ROS is short lived and is a good source for local damage in astrocytes, yet these cells are not susceptible to death in HdhQ(150/150) animals. ROS conversion to peroxide would allow cell-cell diffusion and possibly affect neurons downstream, yet these cells are susceptible to death late in disease progression. Astrocytes may process ROS damage more efficiently than do neurons, which could be protected at early stages, develop vulnerability more slowly, or undergo damage by an independent pathway. Whatever the mechanism, the early abnormalities in MT are metabolic. In this regard, low glucose is at least one major driving force for damage and the need for alternative fuel. Solving the basis for glucose reduction in striatal tissue remains a critical future goal.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cynthia McMurray (ctmcmurray@lbl.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animals were housed in the LBNL Biosciences Animal Care Facility (ACF). All procedures involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocols were approved by the Lawrence Berkeley National Laboratories (LBNL) Animal Welfare and Research Committee. Breeding and use of HdhQ(150/150) (HD) (Lin et al., 2001) mice and HdhQ(wt/wt) (C57BL/6) (WT) mice was performed as reported previously (Polyzos et al., 2016). The specific C57BL/6 animals in this study were genetically matched with the HdhQ(150/150) mice. All HdhQ(150/150) mice were previously backcrossed over ten generations to the C57BL/6 background. Young animals were 12–16 weeks of age, while old animals were around 80 weeks of age. Since HD is paternally derived, this analysis included only male mice. Animals were used for tissue collection (n = 3–5), cell isolation (n = 20) or mass spectrometry (n = 6), as indicated.
The control HdhQ(wt/wt) (C57BL/6) was chosen as the genetic background match for HdhQ(150/150) mouse models for HD. These mice are available to us from the Jackson Laboratory (Bar Harbor, ME) and are thoroughly screened for infection and other disease prior to distribution. The HdhQ(150/150) mouse strain is a model for HD. HD is a fatal neurodegenerative disease that arises from a hereditary expansion of simple CAG trinucleotides in exon 1 of the coding sequence of the htt gene. In homozygous HdhQ(150/150) animals each allele harbors a CAG tract length of 150 CAG repeats “knocked into” the endogenous mouse huntingtin locus. The HdhQ(150/150) mice are good models for disease since the mutant gene is full length and is regulated by the native mouse promoter. They experience ‘late onset’ disease, exhibiting first phenotypic indicators at about 4–5 months of age, with progressive phenotypic decay, but living to similar lifespans as their wildtype controls. These mice are part of our breeding colony, which have been maintained for about 15 years. They are also available for purchase from the Jackson Laboratory (Bar Harbor, ME).
Animals were cared for at the Animal Care Facility (ACF) at LBNL. The diet is 5053 irradiated Pico Lab Rodent Diet 20. Bedding is changes daily. The animals are kept under a cycle of 12 hrs light/12 hours dark and housed at an average temperature of 72 (with a miminum of 69°F and a maximum of 75°F). The ACF is a state-of-the-art facility with: 1) a soft-wall design for ease of modification as the research needs change and 2) filtration systems for isolated, individual clean rooms. This configuration allows for a controlled environment and permits continuous visibility of the individual environmental units through freestanding vinyl wall Bio-Bubble transparent enclosures. The LBNL ACF is accredited by the American Association for the Accreditation of Laboratory Animal Care International (AAALAC), and the animal laboratory staff at the ACF is devoted to full-time care of housed animals.
Primary Brain Synaptosomes
Primary brain synaptosomes were isolated from male pups and pooled for analysis. Brain regional synaptosomes were isolated as described previously (Polyzos et al., 2016; Xun et al., 2012) with slight modifications. Briefly, after rapid decapitation using a guillotine, the mouse brain was quickly extracted on ice and rinsed with fresh ice-cold HBSS buffer. The cerebellum, hippocampus, striatum and cortex were obtained after immediate dissection in a buffer-filled petri dish suspended in an ice bath. The dissected brain regions were quickly transferred to a 15 mL pre-chilled Dounce glass homogenizer (size B) containing 1.5 mL of homogenization buffer (100mM Mannitol, 320mM sucrose, 0.25mM DTT, 1mM EGTA, 5mM HEPES, 2mM Glutamax (ThermoFisher Sci 35050061), 1mM sodium pyruvate and pH 7.2) and gently homogenized with 8–10 up-and-down strokes. The homogenate was centrifuged for 10 minutes at 700 rcf and 4°C. The supernatant was carefully layered on a freshly prepared discontinuous Percoll gradient of 23%, 15%, 10%, and 3% (from top to bottom) and centrifuged for 8 minutes at 32,500 rcf and 4°C (JA-17 fixed angle rotor, Beckman). Synaptosomes were obtained as the band between the 10% and 15% Percoll interface. The synaptosomal fraction was then diluted 1:10 in the homogenization buffer and centrifuged for 15 min at 15,000 rcf and 4°C to remove the Percoll solution and obtain the final synaptosomal pellet. Protein concentration of the synaptosomal pellet was determined using the Pierce 660 Protein Assay (Life Technologies 22660). Synaptosomes are small, (around 0.5–1μm) and lack nuclei and are present only in neurons. 3-D STORM super-resolution microscopy, which was also to quantify their number and average size of the (Figure S3). Each preparation is checked for the absence DAPI staining (confirming that there were no nuclei). The synaptosomal preparations were evaluated for structural cytoskeleton spacing and integrity by 3-D STORM (Figure S3). To further evaluate the viability and consistency, we checked the isolated synaptosomes from WT cells with MitoTracker Deep Red staining to establish the number of MT (Figure 2I). Bioenergetics confirmed their activity (Figure 2B).
Primary Brain Astroglia
Intact brains were collected from day 1–2 newborn pups (P1–3) of both genotypes HdhQ(150/150) mice and HdhQ(wt/wt) (C57BL/6) mice. Brain regions (hippocampus, cerebellum, cortex and striatum) were isolated in a solution of HBSS on ice. 5–8 pups for each genotype were pooled together for each region. These suspensions were digested in 5mL 0.25% Trypsin-EDTA (Gibco 25300056) for 45min at 37°C. Tissue pieces were pelleted (5min, 300 rcf, room temperature (RT)) and then gently triturated 20–30 times in pre-warmed potent media (DMEM (Gibco 10569044), 20% FBS (JRS 43635), 25mM glucose, 2mM sodium pyruvate, 2mM glutamax, 1× non-essential amino acids (Quality Biologicals 116–078–721EA), 1× antibiotic/antimycotic (Gibco #15240062)) using a 5mL pipet, to dissociate into single cells. Each cell suspension was plated into T75 culture flasks and cultured for 7–10 days (at 37°C, 5% CO2) with media exchanges every 2–3 days. Astrocyte cell purity and homogeneity was established by Immunofluorescent analysis using anti-GFAP antibody-Cy3 conjugate (Abcam ab49874). There was no detectable difference in MitoTracker™ Deep Red signal intensity among astrocyte MT (Figure 2I). There was also no detectable Iba1 (Microglial and macrophage-specific calciumbinding protein) antibody staining of isolated striatal astrocytes from WT or HdhQ(150/150) animals between 12–16 weeks (early stages), nor was there evidence of inclusions as detected by MW8 antibody raised against N-terminal aggregates of mhtt (Developmental Studies Hybridoma Bank).
METHOD DETAILS
FLIM: Instrumentation and Imaging
Frequency domain fluorescence lifetime imaging microscopy (FLIM) (Datta et al., 2015; Digman et al., 2008; Lakowicz et al., 1992; Stringari et al., 2012) was performed on a Zeiss LSM 710 microscope (Carl Zeiss, Jena, Germany) coupled to Titanium:Sapphire MaiTai laser (Spectra-Physics, Mountain View, CA) multiphoton excitation laser source with 80MHz repetition rate. NAD(P)H was excited at 740nm using 20x air objective, N/A=0.5 (EC Plan-Neofluar, Carl Zeiss, Oberkochen, Germany). The image scan speed was 25.21 μs/pixel with an image size of 256 3 256 pixels. Excitation from emission signal were separated at 690nm followed by bandpass emission filter 460/80nm (Semrock, Rochester, NY). A photomultiplier tube (H7422P-40, Hamamatsu, Japan) was used as the microscope external detector port photo-sensor unit. A320 FastFLIM FLIMbox (ISS, Champaign, IL) was employed to acquire frequency domain FLIM data. SimFCS software (LFD, Irvine) was used for FLIM data acquisition. For calibrating the FLIM system, Rhodamine 110 with known lifetime of 4 ns was measured. The (g, s) coordinate values are calculated for the first harmonic phasor plots at 80MHz (repetition rate of the laser). Imaging of each tissue sample was performed in less than an hour after the flash frozen slices were removed from −80°C freezer.
FLIM: Sample Preparations
Since tissue slices can suffer injury and hypoxic conditions during preparation, the brains were flash frozen upon dissection, sectioned on a cryostat at −15°C, brushed onto coverslips and stored at −80°C until use. In preliminary control samples, we determined how long it would take for a section to thaw (1 minute) and tested how long it took after thawing to observed loss of the starting NADH intensity (around 10 min). All test samples were scanned within this time frame. We also alternated scans according to brain region to establish that the order of regional scanning did not alter the measurements. The cellular similarity in each segment is illustrated in Figure S1. Bars are SE; n = 3 tissue sections with two hundred scans per section, p values for all comparisons are indicated.
FLIM: Data Analysis
SimFCS software, developed at the Laboratory for Fluorescence Dynamics (LFD, UC Irvine), was used to analyze the FLIM data. The phasor approach to FLIM was applied as has been described previously to quantify changes in NAD(P)H (Digman et al., 2008; Stringari et al., 2012). In the phasor method, the analysis makes no assumption on the relative concentration of NADPH versus NADH. Changes in NADH is considered together as a single pool with NAD(P)H, and the ratio of the free and bound NADH is determined in the context of the entire pool (which includes NADPH). The free/bound fraction is determined by the ratio of the average NADH lifetime between normal and diseased tissues for each brain region. Physically, the phasor of a mixture of two molecular species, each with single average exponential lifetime, will lie inside the universal circle on the line joining the phasor position of the two pure species, and its position changes according to the fractional contribution of each component. Hence, the position of the FLIM-phasor distributions (to the left or to the right in the Phasor plot) is directly related to the ratio of free to enzymebound NAD(P)H. In brief, lifetime decay information from each pixel of the image is transformed into a point on the phasor plot with co-ordinates g, which extend between 0 and 1, and s extending between 0 and 0.5. The 2-D histogram of the phasors is plotted on the phasor plot to create the phasor distribution of the image. This allows analysis of different lifetime phasor clusters, which can be mapped back onto the image to create the FLIM map. All single exponential lifetime decays fall on the universal circle defined as a semi-circle of radius 0.5 between points (0,0) and (1,0) on the phasor plot. Phasors corresponding to a fluorescence lifetime value of 0 will fall on point (1,0) while longer lifetime signals will shift towards point (0,0) with increasing lifetime. The phasor of a mixture of two molecular species, each with single exponential lifetimes, will lie inside the universal circle on the line joining the phasor position of the two pure species, depending on the fractional contribution of each component. In control cells, NAD(P)H in a free state has a shorter lifetime compared to NAD(P)H bound to enzymes (Stringari et al., 2012). Hence, the position of the FLIM-phasor is directly related to the ratio of free to enzyme-bound NAD(P)H (Lakowicz et al., 1992). This allows calculation of fraction of bound NAD(P)H (or free NAD(P)H) at each pixel or the region of interest in an image. p values were calculated by two-tailed Student’s t-test.
NADH and NAD+
NADPH is used for anabolic reactions (e.g., in lipid synthesis) and anti-oxidant defense, while NADH is needed as a reducing equivalent in energy-producing catabolic reactions (lipid breakdown and ROS) (Alberts et al., 2008). In our case, lipid catabolism and ROS are elevated, implying that astrocyte features depend most on NADH. The NADH/NAD+ ratio was measured in astrocytes. The cells were cultured in T75 flasks until they reached 80–90% confluency and harvested by trypsinization. The measurements of NAD+ and NADH was performed using a NADH/NAD+ Colorimetric Assay Kit (Abcam ab65348), according to the manufacturers protocol. Colorimetric readings were taken at 450nm over the course of 4hrs (Figure S2F).
Histology
Brain sections were cryoembedded in OCT from which they were sliced (10mm thick using a Leica Cryostat set at: −14°C for the sample and −12°C for the blade) and placed onto Histobond microscope slides (VWR 16004–416). They were immediately immersed in 100% methanol (30 sec) to help peel away the OCT. Samples were rehydrated in 75%, 50%, 25% and 0% ethanol in PBS (2 min each). Tissue were fixed in freshly prepared 4% paraformaldehyde (PFA) (15 min at RT in the dark), then incubated with 3 washes of 100mM Glycine in PBS pH 7.2 (3 washes, 5 min each). The sections were treated with Image-iT FX signal enhancer (ThermoFisher #I36933) (30min) to reduce autofluorescence and blocked (1–2 hrs) in blocking solution (PBS, 3% BSA, 5% goat serum, 0.7% donkey serum, 0.03% triton X-100). Primary antibody staining was performed overnight, followed by 3 washes with PBS (5 min each). Secondary antibody was applied later as required along with 0.5mM DAPI (1–2 hrs) followed by 3 washes in PBS. Slides were coated with Vectashield+DAPI, covered by a #1.5 coverslip, sealed with clear nail polish and stored (−20°C). Slides were imaged using a Zeiss 710 confocal microscope at 1 A.U., using either 20x air, N/A=0.8, 40x water N/A=1.2 or 100x oil N/A=1.4 lenses. Image analysis was performed using ImageJ:Fiji (Schneider et al., 2012). Primary antibodies used were mouse anti-NeuN Alexa488 conjugate (Millipore #MAB377X) (used at 1:400), mouse anti-GFAP Cy3 conjugate (Abcam #ab49874) (used at 1:400), rabbit anti-Iba1 (Wako #019–19741) (used at 1:400). Secondary antibodies used were goat anti-rabbit Alexa-555 conjugated (Invitrogen #A31630) (used at 1:400) and goat anti-mouse Alexa-488 conjugated (Invitrogen #A31620) (used at 1:400).
Sudan Black B/True Black Assay for Lipofuscin
Brain sections were cryoembedded in OCT from which they were sliced (10mm thick using a Leica Cryostat set at: −14°C for the sample and −12°C for the blade) and placed onto Histobond microscope slides (VWR 16004–416). They were immediately immersed in 100% methanol (30 sec) to help peel away the OCT. Samples were rehydrated in 75%, 50%, 25% and 0% ethanol in PBS (2 min each). Tissue were fixed in freshly prepared 4% PFA (15 min at RT in the dark), then incubated with 3 washes of 100mM Glycine in PBS pH 7.2 (3 washes, 5 min each). The brain sections were subsequently left untreated or stained with 0.1% Sudan Black B in 70% ethanol or 1X TrueBlack® for 10min. Slides were coated with Vectashield+DAPI, covered by a #1.5 coverslip, sealed with clear nail polish and stored (−20°C). Slides were imaged using a Zeiss 710 confocal microscope at 1 A.U., using either 20x air, N/A=0.8, 40x water N/A=1.2 or 100x oil N/A=1.4 lenses and imaged in the FITC (green), Cy3 (red), and Cy5 (far-red) channels. Autofluorescent perinuclear foci in either of these channels that are quenched by either Sudan Black B or True Black are indicative of lipofuscin granules.
Measurement of Extracellular Acidification Rate (ECAR) and Oxygen Consumption Rate (OCR) in Synaptosomes
ECAR and OCR were obtained by using a Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA) as previously described (Xun et al., 2012). Freshly isolated synaptosomal pellets were immediately and gently re-suspended in homogenization buffer (100mM Mannitol, 320mM sucrose, 0.25mM DTT, 1mM EGTA, 5mM HEPES, 2mM Glutamax (ThermoFisher Sci 35050061), 1mM sodium pyruvate and pH 7.2) on ice and plated (1.5mg protein/150mL/well) into a pre-chilled Seahorse XFe 96-well microplate (Agilent 102601–100). The plate was centrifuged for 50 min at 3,220 rcf and 4°C, incubated at 37°C (without CO2) for 15 min. The buffer was exchanged for a pre-warmed buffer appropriate for each assay, and the plate was transferred to the XF flux analyzer for respiration measurement. The measurement cycle consisted of 30 seconds (sec) mixing time followed by 3 measurements of 2 min each. The experimental value taken was the average of these 3 measured rates. Measurements were performed under basal conditions and/or after immediate addition of the uncoupling agent, FCCP. Inhibitors or substrates for each ETC complex were subsequently added. Three (n = 3) independent samples (ie. 3 biological replicates) were evaluated minimally in triplicate experiments.
OCR and ECAR in astrocytes from mouse striatum or cerebellum (up to passage 6), plated at 20,000 cells/well in a XFe 96-well microplate (Agilent 102601–100), were measured after growth overnight at 37°C and 5% CO2 in media (DMEM (Gibco 10569044), 20% FBS (JRS 43635), 25mM glucose, 2mM sodium pyruvate, 2mM glutamax, antibiotic/antimycotic (Gibco 15240062), and non-essential amino acids (Quality Biologicals 116–078–721EA). The media was removed, and the cells were washed prior to each assay, and replaced with the appropriate pre-warmed assay buffer. Initially identical number of astrocytes of either genotypes (HdhQ(150/150) mice or WT) were delivered into wells of a 96-well measurement plate, and grown overnight. However, differential cell growth between WT and HdhQ(150/150) mice varied the number of cells per well at the time of the assay, confounding quantification of metabolism. Thus, every well was normalized for cell number after the assay. Immediately following the Seahorse assay of astrocytes, all buffer and compounds were gently aspirated, and the cells were fixed for 10 minutes in the dark at RT using freshly prepared 4% PFA in PBS pH 7.2. The wells were incubated with 100mM Glycine, 0.05% Triton X-100 in PBS pH 7.0 for 10 minutes and stained with 0.5μg/mL DAPI in PBS pH 7.0. Cells were kept in DAPI until imaging. The images were acquired using tiling of 10x images to encompass the entire well area on a Zeiss LSM 710 confocal microscope (10x air, numerical aperture (N/A) = 0.4 objective), and DAPI stained cells were counted using ImageJ/Fiji (Schneider et al., 2012) software. The number of astrocytes in each well was used to normalize the measurements from the Seahorse assays.
Seahorse Analysis for Glycolysis
Astrocytes grown overnight in a Seahorse XFe96 well plate (Agilent 102601–100) were gently layered with pre-warmed Glycolysis buffer (XF Assay media (Agilent 102365–100), 2mM L-glutamine and pH 7.2). The plate was transferred to the XF flux analyzer for measurement of the extracellular acidification rate change (△pH/min). The order of additions for the assay was 1) 2-DG (15mM),2) Oligomycin (inhibitor of ATP synthase/complex V) (30μM), and 3) 2-deoxyglucose (2-DG) (inhibitor of hexokinase) (Xu et al., 2003).
Activity of Electron Transport Chain Proteins by Inhibition Curves
Pelleted synaptosomes or astrocyte cells in a Seahorse XFe96 well plate (Agilent 102601–100) were gently layered with pre-warmed Electron Flow buffer (XF Base media (Agilent 102353–100), 70mM sucrose, 200mM D-Mannitol, 2mM HEPES, 1mM EGTA, 0.20% BSA (Fatty acid free, Sigma A6003), 5mM L-Malate, 4mM ADP, 20mM sodium pyruvate, 1.2nM PMP (plasma membrane permeabilizer (Agilent 102504–100) at pH 7.2). The plate was transferred to the XF flux analyzer for respiration measurements. The order of additions for the assay protocol was 1) FCCP (8μM), 2) Rotenone (CI inhibitor) (4μM), 3) succinate (CII substrate) (50mM), and 4) Antimycin A (CIII inhibitor) (2μg/mL). Maximum respiratory capacity (MRC) was taken as the OCR after FCCP addition to each sample in the absence of other inhibitors.
Enzymatic Rates of ETC Complex Activity
The activities of Electron Transport Chain complexes in cells were measured using colorimetric microplate assays from Abcam (following manufacturers’ protocols). Tested were the activities of CI: NADH Dehydrogenase, CII: Succinate Dehydrogenase, and CIV: Cytochrome c Oxidase. The assays were performed on (trypsinized and pelleted) primary astrocyte cell cultures or Percoll purified synaptosomes, after lysis using the extraction/detergent supplied by the manufacturer (Abcam). These extracts were kept on ice and assayed immediately. Samples were normalized by total protein concentration (Pierce 660 Protein Assay (Life Technologies 22660)). Synaptosomes or astrocyte samples were assayed in 3 biological replicates, and each replicate was analyzed in triplicate (technical replicate) using Abcam kits. For the Complex I Assay (Abcam ab109721) 25, 70 or 200mg of protein was incubated in ‘Incubation buffer’ (300mL per well) for 3 hrs at RT and washed twice with ‘1x buffer’ (300μL). ‘Assay buffer’ (200μL) was added and wells were analyzed on a Tecan Infinite M1000 Microplate Monochromator (Abs:450nm, at 20 sec intervals for 30 min, shaking sample between each reading). For the Complex II Assay (Abcam ab109908) 60 or 120 mg of protein was incubated in ‘Incubation buffer’ (50μL per well) for 2 hrs in dark at RT washed twice with ‘1x buffer’ (300μL). ‘Lipid solution’ (40μL) was added and incubated for 30 min in dark at RT, ‘Activity solution’ (200μL) was added and wells were analyzed on a Tecan Infinite M1000 Microplate Monochromator (Abs:600nm, at 20 sec intervals for 60 min). For the Complex IV Assay (Abcam ab109911) 50mg or 150mg of protein was incubated in ‘1x buffer’ (200μL per well) for 3 hrs in dark at room temperature (RT, ~22°C) and washed 3 times with ‘1x buffer’ (300μL).’Assay solution’ (200μL) was added and wells were immediately analyzed on a Tecan Infinite M1000 Microplate Monochromator (Abs:550nm, at 1 min intervals for 120 min at 30°C).
SDS-PAGE, Western Blotting, and Quantification of Htt and Electron Transport Complexes (ETC)
Astrocyte cell culture isolated in RIPA buffer (supplemented with 1mg/mL DNaseI and HALT protease inhibitor ThermoFisher Sci #7830) (5mg total protein) or synaptosome samples in 10% homogenization buffer (in synaptosome section)(5mg total protein) were mixed with an equal volume of SDS loading dye (containing 0.35M Tris pH 6.8, 30% glycerol, 10% SDS, 0.6M Dithiothreitol, and 0.12% Bromophenol blue), heated at 95°C for 5 min, and centrifuged at 14,000 rcf for 10 min. Samples were resolved on 4–12% Tris-Glycine WedgeWell gels (Invitrogen), were transferred to nitrocellulose membranes, and stained with Ponceau S solution to visualize and quantify the total protein transferred (not shown in Figures). Membranes were blocked in PBS with 5% non-fat dry milk and 0.1% Tween-20 and probed with the corresponding antibodies. Antibodies used were mouse anti-htt (Millipore MAB-2168), anti-Complex I (subunit DNUFB8) (Invitrogen 459210), anti-Complex II (subunit 30kDa Ip) (Invitrogen 459230), anti-Complex III (Core 2) (Invitrogen 459220), anti-complex IV (OxPhos cIV subunit 1) (Invitrogen 459600), anti-complex V (ATP synthase, subunit alpha, (Invitrogen 459240), anti-histone H3 (Millipore 04–817), anti-tubulin (Sigma T5326), anti β-actin (Abcam AB8327), and sheep anti-mouse secondary antibody (NA931V, GE Healthcare), conjugated to horseradish peroxidase (HRP). Blots were imaged and quantified using a Versadoc 4000MP and Quantity One software (Biorad) (Figures 2 and S3). Experimental samples were normalized for loading to the signal from Ponceau S staining, and then normalized to the average signal for control mice. Error bars represent the mean and SE from n = 3 biological replicates.
STORM Imaging: Fixation and Immunofluorescence Labeling
Cells were fixed in 4% PFA in PBS for ~20 min. For immunofluorescence labeling, cells were first blocked with a solution of 3% bovine serum albumin and 0.1% saponin in PBS, and then stained with corresponding primary and secondary antibodies in the same blocking solution. Primary antibodies used were mouse anti-βII spectrin (BD, 612562; 1:50), rabbit anti-TOM20 (Santa Cruz, sc-11415; 1:200), and mouse anti-mitochondrial Complex II (Invitrogen, 459230; 1:400). For STORM imaging of synaptosomes, an Alexa Fluor 647-conjugated secondary antibody was used to label βII spectrin. For STORM imaging of mitochondria, Alexa Fluor 647-conjugated and Alexa Fluor 555-conjugated secondary antibodies (at 5μg/mL) were used to label mitochondria Complex II and TOM20, respectively.
STORM Imaging: Instrumentation
3D-STORM imaging (Huang et al., 2008; Rust et al., 2004) and epifluorescence imaging were performed on a homebuilt setup based on a Nikon Eclipse Ti-E inverted optical microscope using an oil immersion objective (Nikon CFI Plan Apochromat λ 100x, N/A = 1.45). Lasers at 647nm (MPB Communications) and 560 nm (MPB Communications) were coupled into an optical fiber after an acousto-optic tunable filter and then introduced into the sample through the back focal plane of the microscope. The 560nm laser (~0.2Wcm−2) was used for epifluorescence imaging of Alexa Fluor 555. For STORM of Alexa Fluor 647, the laser beams were shifted towards the edge of the objective so that emerging light reached the sample at incidence angles slightly smaller than the critical angle of the glass–water interface. Continuous illumination of 647-nm laser (~2kWcm−2) was used to excite fluorescence from labeled dye molecules and switch them into the dark state. For 3D-STORM imaging, a cylindrical lens was inserted into the imaging path so that images of single molecules were elongated in x and y for molecules on the proximal and distal sides of the focal plane (relative to the objective), respectively (Xu et al., 2013). Imaging buffer was Tris-HCl (pH 7.5) containing 100mM cysteamine, 5% glucose, 0.8mg/mL glucose oxidase, and 40μg/mL catalase.
STORM: Data Analysis
3D-STORM raw data were processed according to the previously described method (Huang et al., 2008; Xu et al., 2013). Single molecule localization analysis was performed using identical parameters between samples. Separately, a binary black-and-white image of the epifluorescence image of TOM20-Alexa Fluor 555 was generated based on a preset threshold of fluorescence intensity. Using a nearest-neighbor analysis, pixels above the threshold that were directly adjacent were considered as part of the same individual mitochondrion, and these mitochondria were indexed by the program. As each pixel is 160 × 160 nm, any structure containing fewer than 11 pixels was taken as too small to be a mitochondrion and was discarded from further analysis. After this, the single molecule localization data of mitochondrial CII was mapped onto the binary image of TOM20 taken of the same area. A MATLAB program then counted the number of single molecule events localized in each mitochondrion. Six images were taken of each sample (at least n = 3), and the data were pooled to gather statistics on the average number of single molecule events per unit area of mitochondrion. Thus, the number of single molecule localizations was used as a proxy for the amount of mitochondrial complex II in the mitochondria.
MRI Imaging and Analysis
Calculating the volume of the striatum, hippocampus, cerebellum, cortex, from control and HdhQ(150/150) mice was performed on Amira software (Thermo Scientific). Dimensions of the fields were set to 256 × 256 × 100, and the voxel size to 117 × 117 × 122. Based on the Allen mouse Brain Atlas (Lein et al., 2007), the right ventricle of the brain (in striatum), positioning it in the center of the view. A lighter surrounding area is visualized and traced along that border, marking the striatum. The traces are checked for accuracy in the coronal and sagittal views. Because of mouse positioning during MRI, the brain can appear larger on one half of the view, and the brightness and contrast have to be optimized to minimize this apparent size difference. The brain volume was calculated using the “Segmentation Editor” subprogram on the Amira Software. The pixel values were converted to cubic millimeters.
Seahorse Assay for Maximum Reserve Capacity (MRC)
Pelleted synaptosomes or astrocytes in a Seahorse XFe96 well plate (Agilent 102601–100) were gently layered with pre-warmed Reserve Capacity buffer (XF Assay media (Agilent 102365–100), 15mM D-glucose, 70mM sucrose, 200mM D-Mannitol, 2mM HEPES, 1mM EGTA, 0.20% BSA (Fatty acid free, Sigma A6003), 5mM L-Malate, 4mM ADP, 20mM sodium pyruvate, pH 7.2). The plate was transferred to the XF flux analyzer for respiration measurement. The order of additions for the assay protocol was 1) Oligomycin (inhibitor of ATP synthase/CV) (10μg/mL), 2) FCCP (an ETC decoupler) (8mM), 3) a mixture of Rotenone (CI inhibitor) (4μM) and Antimycin A (CIII inhibitor) (2μg/mL), and 4) a mixture of Ascorbate (10μM) and N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD) (10μM) (substrates for CIV).
Metabolomic Network Analysis for Tissue from WT and HD Brain Regions
Following animal decapitation, the whole brain was placed on an ice bed and each brain region (striatum, hippocampus, and cerebellum) was immediately isolated and snap-frozen in liquid nitrogen, lyophilized, and stored at −80°C until analysis (Lee et al., 2013). The freeze-dried tissue was mechanically ground with a single 5 mm diameter steel ball followed by the addition of 0.75 mL extraction solvent of isopropanol:methanol:water (3:3:2), vortexed, and sonicated (2 min). After 5 min centrifugation (16,100 rcf 5 min), 0.70 mL supernatant was split and transferred to new vials and concentrated to dryness for GC-MS and LC-MS analysis. For GC-MS analysis, the dried extracts were mixed with 10μL of the first derivatization solution comprising 40 mg/mL of 98% methoxyamine hydrochloride (CAS No. 593–56–6, Sigma, St. Louis, MO) in pyridine (silylation grade, Pierce, Rockford, IL), and incubated at 30°C, 800 rcf for 90 min (ThermoStat plus, Eppendorf North America Inc, San Diego, CA). The mixtures were then incubated with 90 μL of MSTFA containing 1% TMCS (1mL bottles, Pierce, Rockford, IL) and a mixture of retention index markers (C8-C30) at 37°C for 60 min. The derivatives (0.5 μL) were injected using Gerstel automatic liner exchange system and cold injection system (Baltimore, MD), and separated by an Agilent 6890 gas chromatograph (Santa Clara, CA) equipped with an RTX-5Sil MS column (Restek, Gellefonte, PA). Mass spectrometric analysis was conducted on a Leco Pegasus III time of flight mass spectrometer and result files were preprocessed after data acquisition and stored as ChromaTOF-specific *.peg files and as generic *.txt result files for post-processing. Compound identification was performed based on the Binbase algorithm including chromatogram validation, RT index marker finding, and spectra matching against the Fiehn mass spectral library (Fiehn, 2016). Identified metabolites were reported if present within at least 50% of the samples per study design group (Lee et al., 2013). Quantitative values were calculated based on peak height of single quantum mass. Missing value replacement was done based on the Binbase algorithm, which processes raw data for post-matching and replacement (Fiehn, 2016). The post-processed data were sample-wise normalized by the sum of the peak intensities of all structurally identified compounds (total useful MS) for statistical analysis.
Reverse phase LC-MS analysis was performed on Agilent 1200 capillary LC system coupled online with Agilent 6520 dual-ESI-Q-TOF mass spectrometer (positive ionization mode). Chromatographic separation was done with an Acquity UPLC BEH C18 column (150 mm x 2.1 mm, 1.7 μm particles) (Waters Corporation, Milford, MA, USA) at a flow rate of 20 μL/min (buffer A: H2O with 0.1% formic acid; buffer B: acetonitrile with 0.2% ammonium acetate). LC condition was as follows: equilibration in 3% buffer B for 3 min, 3–50% buffer B gradient over 5 min, 50–99% buffer B gradient over 30 min, 99% buffer B for 10 min, and reequilibration in 3% buffer B for 10 minute injection volume of sample was 4 μL for scan only MS profiling (MS1) and 10 μL for MS/MS analysis. MS/MS analysis was conducted based on target list specified from the analysis of profiling data (collision energy, 10–20 eV). All raw data were converted to mzXML files using Trapper (Institute for Systems Biology, Seattle, WA, USA). The MZmine software (version 2) was used for data processes such as mass detection, chromatogram builder, chromatogram deconvolution (local minimum search), isotopic peaks grouper, peak alignment (join aligner) and gap filling. The processed data was exported as .csv file format with the information of the mass over charge (m/z) ratio, a retention time (RT), and intensity prior to normalization by the sum of total useful MS (preprocessed m/z-RT list).
Univariate statistics was evaluated based on the two-tailed Student’s t-test using the Statistica software (ver. 8.0, StatSoft, Tulsa, OK). Significance Analysis of Microarray (SAM) was applied to address the False Discovery rate (FDR) problem. The delta value was set to 1, which led to resultant FDR <0.056 and q value <0.052 as cut-off criteria. Pathway over-representation analysis was conducted on the lists of metabolites with significant up-regulation and down-regulation, respectively. The results were presented based on significance (Global test) and pathway impact (Relative-betweeness centrality) implemented in MetaboAnalyst 4.0 (http://www.metaboanalyst.ca/MetaboAnalyst/faces/home.xhtml). Multivariate statistics by orthogonal partial least squares discriminant analysis (OPLS-DA) was performed in SIMCA 14 (Umetrics AB, Umea, Sweden). Metabolic network maps (MetaMapp) were constructed as previously reported (Lee et al., 2013). Structural similarity was computed based on Tanimoto score that was calculated from the Structure Clustering module of PubChem Compound database. A Tanimoto score was set to 0.7 for cut-off value for metabolite pair with structural similarity. In addition, connection of metabolite pair coupling by an enzymatic transformation (Kegg reaction pair) was integrated into the structural similarity matrix and the double-layered connectivity matrix was exported into the Cytoscape interface where edge information was stored as simple interaction format (.SIF file). Detailed node information was stored as separated node attribute file (.txt file). The imported single graph in Cytoscape interface was visualized in an organic layout with some modifications for visual clarity. Node colors indicate significant changes of metabolite levels (p < 0.05) in HdhQ(150/150) brains compared to controls (red = elevated metabolites, blue = decreased metabolites in HdhQ(150/150) brains. Node size represents the fold change of the differentially-expressed metabolites in HD brain compared to the control.
β-oxidation Activity of Peroxisomes and Mitochondria of Post-nuclear Supernatants and Astroglia
Mice were sacrificed by guillotine and their brains were immediately dissected and placed on ice in cold PBS. Brain regions from two animals were combined, weighed, and placed in 10 volumes of cold isolation buffer for homogenization (0.225M sucrose, 75mM mannitol, 5mM Tris pH7.5, 1mM EGTA). Tissues were homogenized on ice for 10 passes with the tight Dounce pestle (size B). This homogenate was centrifuged for 3 min at 1300 rcf at 4°C and the post-nuclear supernatant (Kristian, 2010) was retained on ice until assay. Post-nuclear supernatants were assayed for mitochondrial β-oxidation capacity using [1-14C]palmitic acid (C16) and for peroxisomal β-oxidation using [1−14C]lignoceric acid (C24) (both with final fatty acid concentration of 20μM)(both labeled FAs are from Moravek Biochemicals, Brea, CA) as described (Watkins et al., 1991) with quantification of water soluble metabolites. The palmitic acid assay was run for 30 min and the lignoceric acid for 1 hr. During water soluble metabolite isolation, the Folch extraction was replaced with the addition of 20 μL of 20% BSA just before stopping the reaction, followed by the usual perchloric acid precipitation of the fatty acids. After overnight refrigeration of the stopped assays, they were centrifuged at 9000 rcf for 12 min and 200μL of the supernatant was counted for 14C activity. In half of the studies, a second palmitic acid assay with mitochondrial β-oxidation inhibitors was added. These post-nuclear supernatant assays were preincubated at room temperature for 15 minutes with the mitochondrial inhibitors, +/− etomoxir (CPT1 inhibitor, Sigma) and Antimycin A (CIII inhibitor, Sigma) prior to the assay, to determine background levels. FAO by astrocyte cell cultures was measured in flasks as previously described (Lee et al., 2015; Watkins et al., 1991). Astrocyte cells were plated in T25 flasks with DMEM supplemented with 10% FBS and 1% penicillin/streptomycin and grown to 70% confluence. The cells were washed with 13 × PBS and the reaction mixture (DMEM supplemented with 0.1 μCi of [1-14C] oleate, 100μM L-carnitine hydrochloride, and 0.2% BSA) or the reaction mixture containing 100μM etomoxir was added to cells. The flasks were sealed with a rubber stopper containing a hanging well filled with filter paper and incubated in a 37°C incubator for 4 hr. Carbon dioxide was trapped by adding 150μL of 1M NaOH to the filter paper in the center well and 200μL of 1M perchloric acid to the reaction mixture. Then the samples were incubated at 55°C for 1 hr and the filter paper was placed in scintillation fluid and counted.
Quantification of Intracellular Mitochondrial ROS Levels Using MitoSOX
Cultured primary astrocytes were washed and incubated in HBSS (with Mg++ and Ca++) in the presence of 1mM sodium pyruvate and 5μM MitoSOX for 10 min in the dark at 37°C and 5% CO2. Mitochondrial ROS was quantified using the MitoSOX reagent (Molecular Probes M36008). MitoSOX reagent (a derivative of hydroethidine) is a blue fluorescent molecule that is converted intracellularly, via a process involving superoxide, to a fluorescent molecule (2-OH-Mito-E+) with an emission maximum of ~560nm (red) (Zielonka and Kalyanaraman, 2010). An alternative intracellular (superoxide independent) pathway forms (Mito-E+) (an analog of ethidium) which has an emission maximum of ~590nm (yellow) (Zhao et al., 2005). Both molecules are excited at 488nm and have overlapping emissions (Zhao et al., 2005), and intercalate into DNA (Zielonka and Kalyanaraman, 2010). The cells were washed with growth medium and immediately imaged live at 37°C and 5% CO2 using a Zeiss 710 confocal microscope and a 488nm excitation laser and using a 100x N/A=1.4 oil lens, with imaging optical section set at 1 A.U. and keeping the laser intensity low. Image analysis of ROI was performed using ImageJ:Fiji (Schneider et al., 2012). We selected two wave-length ranges of minimal overlap (R1 = 523–543nm) (R2 = 600–640nm) to interrogate superoxide levels in cells. R2 is seen in the nucleus and R1 is observed faintly inside the nucleus and most intensely in the perinuclear space. R1 is visible as groups of foci that co-localize with MitoTracker™ Green and are considered to be in mitochondria. The level of mitochondrial superoxide was calculated using the intensity of R1 in the perinuclear space, excluding signal inside the nucleus (ie. excluding signal that co-localizes with R2).
Seahorse Analysis for Mitochondrial FA Import in Astrocytes
To test whether FA import into MT was altered in HdhQ(150/150) astrocytes, we inhibited carnitine palmitoyltransferase-1 (CPT-I) using etomoxir (Pike et al., 2011; Xu et al., 2003), an irreversible inhibitor, before or after uncoupling with FCCP. Astrocytes were gently layered in a Seahorse XFe96 well plate (Agilent 102601–100) with pre-warmed buffer (XF Base media (Agilent 102353–100), 0.22mM palmitate-BSA, 5mM HEPES, 1mM EGTA, 0.5mM L-glucose, 0.5mM L-carnitine, 0.20% BSA (Fatty acid free, Sigma A6003), 2mM L-Malate, and pH 7.2). The plate was pre-incubated at 37°C for 1hr and transferred to the XF flux analyzer for respiration measurement. The order of additions for the assay was FCCP (8μM), and Etomoxir (20μM).
Metabolite Analysis of Plasma from 20-Week WT and HdhQ(150/150) Mice
Blood was collected via syringe heart puncture and plasma was collected after centrifugation and removal of erythrocytes. The plasma samples were lyophilized and analyzed at the NIH-supported West Coast Metabolomics Center at UC Davis. Samples were analyzed for ‘untargeted” analysis of primary metabolites using GC-TOF MS.
Oxygen Consumption Rate (OCR) in Fatty Acid and Glucose Media
For some assays, primary astrocytes were cultured in media designed to mimic the striatal metabolic pools in normal and HdhQ(150/150) animal brains. A media with high levels of glucose and amino acids mimics the striatum of normal animals (WT, C57BL/6) (composition: DMEM:F12 (Hyclone SH30261.01), 20% FBS (JRS 43640), 5 or 25mM L-glucose, 1× non-essential amino acids (NEAA) (Quality Biologicals 116–078–721), 1mM sodium pyruvate (total), 2.5mM L-glutamate (total), 1× antibiotic/antimycotic (Gibco 15240062)). A media with low levels of glucose and added fatty acids (FA) (composition: DMEM:F12 (Hyclone SH30261.01), 20% FBS (JRS 43640), 1mM L-glucose, 1x fatty acid supplement (Sigma F7050), 1mM sodium pyruvate (total), 2.5mM L-glutamate (total), 1x antibiotic/antimycotic (Gibco 15240062)) mimics the metabolic content of HdhQ(150/150) animals striatum. The most significant forms in the MS profiles were medium chain FAs, of the size that directly undergoes β-oxidation in MT. Thus, the FAs mix from Sigma and the FA in natural tissue did not exclude any size range of FAs. Prior to the assay, astrocytes were grown in the relevant media at 37°C, 5% CO2 and the media was replaced every 1–2 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n, the definition of center, dispersion and precision measures (mean ± SE) and statistical significance are reported in the Figures and Figure Legends. SE analysis was performed in Microsoft Excel. Data or in GraphPad Prism. A two-tailed Student’s t-test was used for pairwise comparisons, whereas multiple comparisons were analyzed with one-way analysis of variance (ANOVA) and Bonferroni’s post hoc test. A two-way analysis of variance (ANOVA) was used to measure the mean differences in astrocyte cell lines that differed in brain region and genotype. Data were judged as statistically significant when p < 0.05 by two-tailed Student’s t test. In figures, asterisks denote statistical significance as calculated by two-tailed Student’s t-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). Box and Whisker plots are defined by the values in percentage above and below the mean. We did not include additional statistical tests for data distributions.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-βII spectrin | BD | 612562/RRID:AB_399853 |
| Rabbit anti-TOM20 | Santa Cruz | sc-11415/RRID:AB_2207533 |
| Mouse anti-mitochondrial complex II | Invitrogen | 459230/RRID:AB_2532233 |
| Anti-mouse-Alexa Flour 647 | Jackson | A21236/RRID:2535805 |
| Anti-rabbit-Alexa Flour 647 | Jackson | A21245/RRID:2535813 |
| Anti-rabbit-Alexa Flour 555 | Jackson | A21249/RRID:2535817 |
| Rabbit polyclonal anti-53BP1 | Bethyl Labs | A300–272A/RRID:AB_185520 |
| Mouse monoclonal anti-8-oxo-dG | Trevigen | 4354-mc-050/RRID:AB_1857195 |
| Mouse monoclonal anti-Complex I (subunit DNUFB8) | Invitrogen | 459210/RRID:AB_2532232 |
| Rabbit polyclonal anti-Complex I (subunit DNUFS1) | ThermoFisher | PA5–22309/RRID:AB_1115187 |
| Mouse monoclonal anti-Complex II (subunit 30kDa Ip) | Invitrogen | 459230/RRID:AB_2532233 |
| Mouse monoclonal anti-Complex III (core2, UQCRC2) | Invitrogen | 459220 |
| Mouse monoclonal anti-complex IV (OxPhos c-IV subunit alpha) | Invitrogen | 459600/RRID:AB_2532240 |
| Mouse monoclonal anti-Complex V (ATP synthase, subunit alpha) | Invitrogen | 459240/RRID:AB_2532234 |
| Mouse monoclonal anti-GFAP Cy3 conjugate | Abeam | Ab49874/RRID:AB_880203 |
| Rabbit polyclonal anti-GFAP | Invitrogen | PA5–16291/RRID:AB_1098076 |
| Rabbit monoclonal anti-H3 (phosphor-Histone H3/Ser10) | Millipore | 04–817/RRID:AB_1163440 |
| Mouse monoclonal anti-Htt (181–810aa) | Millipore | MAB-2166/RRID:AB_1121314 |
| Mouse monoclonal anti-Htt (2146–254aa) | Millipore | MAB-2168/RRID:AB_94595 |
| Rabbit polyclonal anti-Iba1 | Wako | 019–19741/RRID:AB_839504 |
| Mouse monoclonal anti-Neun Alexa Fluor 488 conjugate | Millipore | MAB377X/RRID:AB_2149209 |
| Mouse monoclonal anti-Neun Alexa Fluor 555 conjugate | Millipore | MAB377A5 |
| Mouse monoclonal anti-tubulin | Sigma | T7451/RRID:AB_609894 |
| Rabbit polyclonal anti-ubiquitin | DAKO | Z0458/RRID:AB_2315524 |
| Goat anti-mouse IgG(H+L) Alexa Fluor 488 conjugate | Invitrogen | A31620 |
| Goat anti-mouse IgG(H+L) Alexa Fluor 568 conjugate | Invitrogen | A11019/RRID:AB_143162 |
| Goat anti-rabbit IgG(H+L) Alexa Fluor 488 conjugate | Invitrogen | A11070/RRID:AB_2534114 |
| Goat anti-rabbit IgG(H+L) Alexa Fluor 555 conjugate | Invitrogen | A31630 |
| Rabbit anti-mouse IgG (H+L) HRP | Sigma-Aldrich | A9044/RRI D:AB_258431 |
| Goat anti-rabbit IgG (H+L) HRP | Sigma-Aldrich | A6154/RRID:AB_258284 |
| Biological Samples | ||
| Astrocytes | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formaldehyde | EMS | 15714 |
| Bovine serum albumin | Sigma LS | A3059–100G |
| Triton-X 100 | Sigma LS | T8787–100ML |
| Saponin | Sigma LS | 54521–10G |
| PBS | Corning Cellgro | 21–040-CV |
| Tris-HCI | Corning | 46–030-CM |
| Cysteamine | Sigma LS | 30070–10G |
| Glucose | Sigma LS | G7528–250G |
| Glucose oxidase | Sigma LS | G2133–250KU |
| Catalase | Sigma LS | C30–500MG |
| (+)-Sodium L-Ascorbate | Sigma-Aldrich | A4034 |
| 14C-Carnitine | Amersham | CFA 668 |
| 1-butanol | Fisher scientific | A399–4 |
| Antibiotic-Antimycotic | Life Technologies | 15240062 |
| Antimycin A | Sigma-Aldrich | A8674 |
| ATP | Sigma Aldrich | A9062 |
| Bovine Serum Albumin (Fatty acid free) | Sigma-Aldrich | A6003 |
| Calcium Chloride, CaCl2 | Sigma-Aldrich | C5080 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 |
| D-Glucose | Sigma-Aldrich | G5767 |
| DMEM Glutamax, high glucose | Life Technologies | 10569044 |
| EGTA (Ethylene glycol-bis(2-aminoethylether)-N,N,N,N-tetraacetic acid) | Sigma-Aldrich | E33889 |
| Fatty acid supplement | Sigma-Aldrich | F7050 |
| FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) | Sigma-Aldrich | C2920 |
| GlutaMax 1 (CTS), 100x | ThermoFisher | A1286001 |
| Glutathione | Sigma Aldrich | G6529 |
| Halt Protease Inhibitor Cocktail (100X) | Life Technologies | 78430 |
| HEPES (1M solution) | Sigma-Aldrich | H4034 |
| Hydrochloric acid | Mallinkrodt | 5587 |
| L(−) Malic acid | Sigma-Aldrich | M6413 |
| L-Carnitine | Sigma Aldrich | C-7518 |
| L-Carnitine hydrochloride | Sigma-Aldrich | C0283 |
| Magnesium chloride | Fisher Scientific | M33500 |
| Magnesium Sulfate, MgSO4 | Sigma-Aldrich | |
| Malonyl-CoA | Sigma Aldrich | M4263 |
| MEM Non-Essential Amino Acids | VWR | 10128–762 |
| MES (2-[N-morpholino] ethanesulfonic acid) | Sigma Aldrich | M5287 |
| n-octyl-b-D-glucopyranoside | Sigma Aldrich | O8001 |
| Normocure broad spectrum antibacterial reagent | Invivogen | Ant-noc |
| Palmitic Acid | Sigma-Aldrich | P5585 |
| Palmitoyl-CoA | Sigma Aldrich | P9276 |
| Phosphate buffered saline | Sigma Aldrich | P4417 |
| Ponceau S (0.1 % solution) | Sigma-Aldrich | P7170 |
| Potassium Chloride, KCI | Sigma-Aldrich | P9541 |
| Potassium cyanide | Sigma Aldrich | 207810 |
| Potassium Phosphate, KH2PO4 | Sigma-Aldrich | P0662 |
| Rotenone | Sigma Aldrich | R8875 |
| Sodium Chloride, NaCl | Sigma-Aldrich | S9888 |
| Sodium Pyruvate (100mM) | ThermoFisher | 11360070 |
| Sodium Succinate (dibasic hexahydrate) | Sigma-Aldrich | S5047 |
| Sodium Sulfate, Na2SO4 | Sigma-Aldrich | 23859 |
| Sucrose | Sigma-Aldrich | S03898 |
| Sudan Black B | Sigma-Aldrich | 199664 |
| TMPD (N,N,N,N-Tetramethyl-p-phenylenediamine dihydrochloride) | Caymen | 70455 |
| Trizma | Sigma Aldrich | T4003 |
| Trypsin (0.25%) EDTA | Life Technologies | 25200056 |
| True Black Lipofuscin Autofluorescence Quencher | Biotium | 23007 |
| XF Assay Medium (contains 2mM GlutaMax) | Seahorse/Agilent | 102365–100 |
| XF Base Assay Medium | Seahorse/Agilent | 102353–100 |
| XJB-5–131 | P. Wipf laboratories | N/A |
| Critical Commercial Assays | ||
| Complex I Enzyme Activity Microplate Assay Kit (Colorimetric) | Abeam | Ab109721 |
| Complex II Enzyme Activity Microplate Assay Kit (Colorimetric) | Abeam | Ab109908 |
| Complex IV Enzyme Activity Microplate Assay Kit (Colorimetric) | Abeam | Ab109911 |
| NuPage NOVEX 4–12% Tris-Bis SDS premade gels (1.5mm × 15 wells) | Life Technologies | NP0336BOX |
| MitoSOX™ Red Mitochondrial Superoxide Indicator | Invitrogen | M36008 |
| MitoTracker Deep Red FM | Invitrogen | M22426 |
| MitoTracker Green FM | Invitrogen | M7514 |
| MitoTracker Red CMXRos | Invitrogen | M7512 |
| NAD/NADH assay kit (colorimetric) | Abeam | Ab65348 |
| Invitrogen Novex NuPAGE MOPS SDS Running Buffer (20X) | Life Technologies | NP0001 |
| Pierce 660nm Protein Assay Reagent | Life Technologies | 22660 |
| Nissl Stain (Cresyl violet, Nova Ultra) | IHCWorld | 1W-3007A |
| TrueBlack Lipofuscin Autofluorescence Quencher | Biotium | 23007 |
| XF Plasma Permeabilizer | Seahorse | 102504–100 |
| XF Cell Mito Stress Kit | Seahorse/Agilent | 103015–100 |
| Experimental Models: Cell Lines | ||
| Primary mouse astrocytes, P1–3, C57BL/6J | This paper | N/A |
| Primary mouse astrocytes, P1–3, HdhQ150(150/150) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Jackson Labs | 000664 |
| HdhQ150 (B6.129P2-Htttm2Detl/150J) | Jackson Labs | 004595 |
| Software and Algorithms | ||
| Insight 3D single molecule localization | Huang et al., 2008; Xu et al., 2013 | Insight 3D single molecule localization |
| Quantity One | Biorad | N/A |
| Seahorse Wave Controller software | Seahorse/Agilent | N/A |
| Globals for Images - SimFCS 4.0 | Globals Software | N/A |
| Python & Seaborn | PyData, Python.org | N/A |
| Zen 2012 (black edition) | Carl Zeiss Microscopy GmbH | N/A |
| BinBase | Fiehn, 2016; Skogerson et al., 2011 | https://c0de.g00gle.c0m/archive/p/binbase/ |
| MZmine 2 | Pluskal et al., 2010 | http://mzmine.github.io/ |
| Significant analysis of microarray | Multiple Experiment Viewer | http://mev.tm4.org |
| SIMCA 14 | Umetrics | |
| Cytoscape 3.4.0 | Shannon et al., 2003 | https://cytoscape.org/ |
| MetaboAnalyst 4.0 | Chong et al., 2018 | https://www.metaboanalyst.ca/ |
| GC | Agilent | 6890 |
| LC | Agilent | 1200 |
| Time-of-flight mass spectrometer | Leco | Pegasus III |
| Q-TOF mass spectrometer | Agilent | 6520 |
| Other | ||
| Flome-built STORM microscope | Huang et al., 2008; Rust et al., 2004 | |
| Eclipse Ti-E inverted optical microscope | Nikon | |
| Oil immersion objective CFI Plan Apochromat λ 100x, N/A=1.45 | Nikon | |
| 647 nm laser | MPB Communications | |
| 560 nm laser | MPB Communications | |
Highlights.
Region-specific neuronal toxicity arises from metabolic reprogramming in astrocytes
Glucose is low in the Huntington striatum but neuronal mitochondria are unaffected
Astrocytes compensate by fuel switching to fatty acids but increase oxygen damage
Toxicity occurs when oxygen damage exceeds the energy benefits of fatty acid oxidation
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health grants NS060115 and GM119161 (to CTM); the Pew Biomedical Scholars Award (to K.X.); National Institutes of Health grants NIH P41-GM103540 and NIH P50-GM076516 (to E.G.); the basic science research program through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (NRF-2016R1C1B2007982) (to D.Y.L); the Evelyn Willing Bromley Chair funds at Children’s Hospital of Philadelphia (to M.J.B); and NIH DK78775 (to J.V.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2019.03.004.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, and Walter P (2008). Molecular Biology of the Cell, Fifth Edition (Garland Publishing Science; ). [Google Scholar]
- Arun S, Liu L, and Donmez G (2016). Mitochondrial biology and neurological diseases. Curr. Neuropharmacol 14, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barupal DK, Haldiya PK, Wohlgemuth G, Kind T, Kothari SL, Pinkerton KE, and Fiehn O (2012). MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinformatics 13, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, and Magistretti PJ (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. [DOI] [PubMed] [Google Scholar]
- Bender A, Auer DP, Merl T, Reilmann R, Saemann P, Yassouridis A, Bender J, Weindl A, Dose M, Gasser T, et al. (2005). Creatine supplementation lowers brain glutamate levels in Huntington’s disease. J. Neurol 252, 36–41. [DOI] [PubMed] [Google Scholar]
- Besson MT, Alegría K, Garrido-Gerter P, Barros LF, and Liévens J-C (2015). Enhanced neuronal glucose transporter expression reveals metabolic choice in a HD Drosophila model. PLoS One 10, e0118765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvento G, Valette J, Flament J, Mochel F, and Brouillet E (2017). Imaging and spectroscopic approaches to probe brain energy metabolism dysregulation in neurodegenerative diseases. J. Cereb. Blood Flow Metab. 37, 1927–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussicault L, Hérard AS, Calingasan N, Petit F, Malgorn C, Merienne N, Jan C, Gaillard MC, Lerchundi R, Barros LF, et al. (2014). Impaired brain energy metabolism in the BACHD mouse model of Huntington’s disease: critical role of astrocyte–neuron interactions. J. Cereb. Blood Flow Metab. 34, 1500–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E (2014). The 3-NP model of striatal neurodegeneration. Curr. Protoc. Neurosci 67, 9.48.1–9.4814. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N (2016). Mutant huntingtin and elusive defects in oxidative metabolism and mitochondrial calcium handling. Mol. Neurobiol 53, 2944–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, and Dubinsky JM (2003). Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci 23, 4858–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, and Dubinsky JM (2005). Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s disease. J. Neurochem 93, 1361–1370. [DOI] [PubMed] [Google Scholar]
- Carroll JB, Deik A, Fossale E, Weston RM, Guide JR, Arjomand J, Kwak S, Clish CB, and MacDonald ME (2015). HdhQ111 mice exhibit tissue specific metabolite profiles that Include Striatal Lipid Accumulation. PLoS One 10, e0134465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho C, Correia SC, Cardoso S, Plácido AI, Candeias E, Duarte AI, and Moreira PI (2015). The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev. Neurother 15, 867–884. [DOI] [PubMed] [Google Scholar]
- Choi SW, Gerencser AA, and Nicholls DG (2009). Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem 109, 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, and Xia J (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury KR, Das S, and Bhattacharyya NP (2016). Differential proteomic and genomic profiling of mouse striatal cell model of Huntington’s disease and control; probable implications to the disease biology. J. Proteom 132, 155–166. [DOI] [PubMed] [Google Scholar]
- Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, and Squitieri F (2006). Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J. Nucl. Med 47, 215–222. [PubMed] [Google Scholar]
- Covarrubias-Pinto A, Moll P, Solís-Maldonado M, Acuña AI, Riveros A, Miró MP, Papic E, Beltrán FA, Cepeda C, Concha II, et al. (2015). Beyond the redox imbalance: oxidative stress contributes to an impaired GLUT3 modulation in Huntington’s disease. Free Radic. Biol. Med 89, 1085–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Diguet E, Malgorn C, D’Aurelio M, Galvan L, Petit F, Benhaim L, Guillermier M, Houitte D, Dufour N, et al. (2013). A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum. Mol. Genet 22, 3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta R, Alfonso-García A, Cinco R, and Gratton E (2015). Fluorescence lifetime imaging of endogenous biomarker of oxidative stress. Sci. Rep 5, 9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Caiolfa VR, Zamai M, and Gratton E (2008). The phasor approach to fluorescence lifetime imaging analysis. Biophys. J 94, L14–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O (2016). Metabolomics by gas chromatography-mass spectrometry: combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol 114, 30.4.1–30.4.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O, Barupal DK, and Kind T (2011). Extending biochemical databases by metabolomic surveys. J. Biol. Chem 286, 23637–23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gårseth M, Sonnewald U, White LR, Rød M, Zwart JA, Nygaard O, and Aasly J (2000). Proton magnetic resonance spectroscopy of cerebrospinal fluid in neurodegenerative disease: indication of glial energy impairment in Huntington chorea, but not Parkinson disease. J. Neurosci. Res 60, 779–782. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, and Zeviani M (2012). Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv. Exp. Med. Biol 748, 65–106. [DOI] [PubMed] [Google Scholar]
- Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, Wheeler VC, Persichetti F, and MacDonald ME (2003). Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet 12, 497–508. [DOI] [PubMed] [Google Scholar]
- Gouarné C, Tardif G, Tracz J, Latyszenok V, Michaud M, Clemens LE, Yu-Taeger L, Nguyen HP, Bordet T, and Pruss RM (2013). Early deficits in glycolysis are specific to striatal neurons from a rat model of Huntington disease. PLoS One 8, e81528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J, Pellman JJ, Brustovetsky T, Harris RA, and Brustovetsky N (2015). Oxidative metabolism in YAC128 mouse model of Huntington’s disease. Hum. Mol. Genet 24, 4862–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J, Pellman JJ, Brustovetsky T, Harris RA, and Brustovetsky N (2016). Oxidative metabolism and Ca2+handling in isolated brain mitochondria and striatal neurons from R6/2 mice, a model of Huntington’s disease. Hum. Mol. Genet 25, 2762–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley RR, Reid SJ, Patassini S, Rudiger SR, Obolonkin V, McLaughlan CJ, Jacobsen JC, Gusella JF, MacDonald ME, Waldvogel HJ, et al. (2016). Metabolic disruption identified in the Huntington’s disease transgenic sheep model. Sci. Rep 6, 20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, and Bolaños JP (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 11, 747–752. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Rodriguez-Rodriguez R, Gaebler A, Casals N, Scheller A, and Kuerschner L (2017). Astrocytes and oligodendrocytes in grey and white matter regions of the brain metabolize fatty acids. Sci. Rep 7, 10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, and Zhuang X (2008). Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Disatnik MH, and Mochly-Rosen D (2015). Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol. Med 7, 1307–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen AHP, van Hal M, op den Kelder IC, Meier RT, de Ruiter AA, Schut MH, Smith DL, Grit C, Brouwer N, Kamphuis W, et al. (2017). Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 65, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Looger LL, and Khakh BS (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristian T (2010). Isolation of mitochondria from the CNS. Curr. Protoc. Neurosci 52, 7.22.1–7.22.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR, Szmacinski H, Nowaczyk K, and Johnson ML (1992). Fluorescence lifetime imaging of free and protein-bound NADH. Proc. Natl. Acad. Sci. USA 89, 1271–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Xun Z, Platt V, Budworth H, Canaria CA, and McMurray CT (2013). Distinct pools of non-glycolytic substrates differentiate brain regions and prime region-specific responses of mitochondria. PLoS One 8, e68831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ellis JM, and Wolfgang MJ (2015). Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 10, 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legan SK, Rebrin I, Mockett RJ, Radyuk SN, Klichko VI, Sohal RS, and Orr WC (2008). Overexpression of glucose-6-phosphate dehydrogenase Extends the Life Span ofDrosophila melanogaster. J. Biol. Chem 283, 32492–32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, and Detloff PJ (2001). Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet 10, 137–144. [DOI] [PubMed] [Google Scholar]
- Liot G, Valette J, Pépin J, Flament J, and Brouillet E (2017). Energy defects in Huntington’s disease: why “in vivo” evidence matters. Biochem. Biophys. Res. Commun 483, 1084–1095. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. (2015). Glial lipid droplets and ROS Induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Mora DA, Camacho V, Pérez-Pérez J, Martínez-Horta S, Fernández A, Sampedro F, Montes A, Lozano-Martínez GA, GómezAnson B, Kulisevsky J, et al. (2016). Striatal hypometabolism in premanifest and manifest Huntington’s disease patients. Eur. J. Nucl. Med. Mol. Imaging 43, 2183–2189. [DOI] [PubMed] [Google Scholar]
- Mochel F, and Haller RG (2011). Energy deficit in Huntington disease: why it matters. J. Clin. Invest 121, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Itakura M, Kubo T, Kaneshige A, Harada N, Izawa T, Azuma YT, Kuwamura M, Yamaji R, and Takeuchi T (2017). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) aggregation causes mitochondrial dysfunction during oxidative stress-induced cell death. J. Biol. Chem 292, 4727–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambron R, Silajd zic E, Kalliolia E, Ottolenghi C, Hindmarsh P, Hill NR, Costelloe SJ, Martin NG, Positano V, Watt HC, et al. (2016). A metabolic study of Huntington’s disease. PLoS One 11, e0146480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oláh J, Klivényi P, Gardián G, Vécsei L, Orosz F, Kovacs GG, Westerhoff HV, and Ovádi J (2008). Increased glucose metabolism and ATP level in brain tissue of Huntington’s disease transgenic mice. FEBS J. 275, 4740–4755. [DOI] [PubMed] [Google Scholar]
- Oliveira JMA, Jekabsons MB, Chen S, Lin A, Rego AC, Gonçalves J, Ellerby LM, and Nicholls DG (2007). Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem 101, 241–249. [DOI] [PubMed] [Google Scholar]
- Osellame LD, Blacker TS, and Duchen MR (2012). Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab 26, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, and Greenamyre JT (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci 5, 731–736. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, Messing A, Steinhäuser C, Lee JM, Parpura V, Hol EM, Sofroniew MV, and Verkhratsky A (2016). Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345. [DOI] [PubMed] [Google Scholar]
- Pike LS, Smift AL, Croteau NJ, Ferrick DA, and Wu M (2011). Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 1807, 726–734. [DOI] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, and Oresic M (2010). MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyzos A, Holt A, Brown C, Cosme C, Wipf P, Gomez-Marin A, Castro MR, Ayala-Peña S, and McMurray CT (2016). Mitochondrial targeting of XJB-5–131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum. Mol. Genet 25, 1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyzos AA, and McMurray CT (2017). The chicken or the egg: mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech. Ageing Dev. 161, 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Jin YN, von Bernhardi R, and Johnson GVW (2013). Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol. Neurodegener 8, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, and Young AB (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA 85, 5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds NC Jr., Prost RW, and Mark LP (2005). Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington’s disease. Brain Res. 1031, 82–89. [DOI] [PubMed] [Google Scholar]
- Ross CA, Kronenbuerger M, Duan W, and Margolis RL (2017). Mechanisms underlying neurodegeneration in Huntington disease: applications to novel disease-modifying therapies. Handb. Clin. Neurol 144, 15–28. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Malgat M, Mazat JP, and Letellier T (1999). Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem 274, 33426–33432. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, and Zhuang X (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (Storm). Nat. Methods 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Lakadamyali M, Zhang F, and Zhuang X (2004). Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol 11, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameni S, Syed A, Marsh JL, and Digman MA (2016). The phasor-FLIM fingerprints reveal shifts from OXPHOS to enhanced glycolysis in Huntington disease. Sci. Rep 6, 34755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer J, Jackson S, Taroni F, Swift P, and Turnbull DM (1997). Characterisation of carnitine palmitoyltransferases in patients with a carnitine palmitoyltransferase deficiency: implications for diagnosis and therapy. J. Neurol. Neurosurg. Psychiatry 62, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld P, and Reiser G (2013). Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 33, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skogerson K, Wohlgemuth G, Barupal DK, and Fiehn O (2011). The volatile compound BinBase mass spectral database. BMC Bioinformatics 12, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari C, Nourse JL, Flanagan LA, and Gratton E (2012). Phasor fluorescence lifetime microscopy of free and protein-bound NADH reveals neural stem cell differentiation potential. PLoS One 7, e48014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supplie LM, Dűking T, Campbell G, Diaz F, Moraes CT, Götz M, Hamprecht B, Boretius S, Mahad D, and Nave KA (2017). Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci 37, 4231–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza M, Marullo M, Di Paolo E, Cesana E, Zuccato C, Biella G, and Cattaneo E (2015). Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 22, 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Bahney J, and Herculano-Houzel S (2016). The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J. Comp. Neurol 524, 3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, and Richardson EP (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol 44, 559–577. [DOI] [PubMed] [Google Scholar]
- Watkins PA, Ferrell EV, Pedersen JI, and Hoefler G (1991). Peroxisomal fatty acid b-oxidation in HepG2 cells. Arch. Biochem. Biophys 289, 329–336. [DOI] [PubMed] [Google Scholar]
- Xu FY, Taylor WA, Hurd JA, and Hatch GM (2003). Etomoxir mediates differential metabolic channeling of fatty acid and glycerol precursors into cardiolipin in H9c2 cells. J. Lipid Res. 44, 415–423. [DOI] [PubMed] [Google Scholar]
- Xu K, Zhong G, and Zhuang X (2013). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun Z, Rivera-Sánchez S, Ayala-Peña S, Lim J, Budworth H, Skoda EM, Robbins PD, Niedernhofer LJ, Wipf P, and McMurray CT (2012). Targeting of XJB-5–131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s disease. Cell Rep. 2, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, and Kalyanaraman B (2005). Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. USA 102, 5727–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Clabough EBD, Sarkar S, Futter M, Rubinsztein DC, and Zeitlin SO (2010). Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 6, e1000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, and Kalyanaraman B (2010). Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic. Biol. Med 48, 983–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, and Sollott SJ (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS Release. Physiol. Rev 94, 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.