

Abstract
While highly active antiretroviral therapy (HAART) has significantly reduced mortality rates in patients with human immunodeficiency virus type 1 (HIV-1), its efficacy may be impeded by emergence of drug resistance caused by lack of patient adherence. A therapeutic strategy that requires infrequent drug administration as a result of sustained release of antiretroviral drugs would put less burden on the patient. Long-acting antiretroviral prodrugs for HIV therapy were synthesized through modification of the active drugs, emtricitabine (FTC) and elvitegravir (EVG), with docosahexaenoic acid (DHA) in one-step, one-pot, high-yielding reactions. The in vitro drug release profiles of these synthetic conjugates demonstrated sustained and controlled release of the active drug over a period of 3–4weeks attributable to the hydrolysis of the chemical linker in conjunction with the hydrophilicity of the parent drug. Both conjugates exhibited superior antiviral activities in tissue culture models of HIV replication as compared to those of the free drugs, strengthening their role as potent prodrugs for HIV therapy. Pharmacokinetic analysis in CD1 mice further confirmed the long-acting aspect of these conjugates with released drug concentrations in plasma detected at their respective IC90/IC95 values over a period of 2 weeks and discernable amounts of active drug even at 6weeks. Our findings suggest that the injectable small molecule conjugates could be used as long-acting controlled release of FTC and EVG in attempts to mitigate adherence-related HIV resistance.
Keywords: Emtricitabine, Elvitegravir, Sustained release formulations, Antiretroviral therapy, Lipid drug conjugate
1. Introduction
Approximately 36.7 million people worldwide are affected by human immunodeficiency virus type 1 (HIV-1), of which 20.9 million receive highly active antiretroviral therapy (HAART; World Health Organization) (HIV/AIDS). Since the first antiretroviral, zidovudine, hit the market 32 years ago, there has been a steady growth of available treatment options (~ 35 drugs) across seven distinct drug-classes, based on their molecular mechanism and resistance profiles (HIV/AIDS). Due to the increased number of currently available therapies, it has become even more imperative to tailor a treatment plan that addresses tolerability, safety, and convenience. Conventional antiretroviral therapy regimen requires daily oral intake of a combination of at least three anti-HIV drugs that have the potency to suppress the viral replication and prevent development of drug resistance (Clinical Guidelines). While HAART is not a cure for HIV, it prolongs and improves the quality of life, and reduces the risk of HIV transmission (Cohen et al., 2011).
With more anti-HIV FDA-approved drugs currently on the market, there has been tremendous progress in improving access of these drugs to high-risk individuals in low-and middle-income countries. However, one of the primary challenges of HAART is poor adherence (50–75%) to medications, which is extremely common in patients taking oral treatment for chronic diseases (Rajoli et al., 2015; Coleman et al., 2012; Kim et al., 2018). Such noncompliance renders the drug combination ineffective against suppressing HIV replication and can lead to the emergence of drug resistance (Paydary et al., 2013; Gross et al., 2001; Genberg et al., 2012). In resource-constrained environments, especially with limited drug options, it becomes increasingly difficult to effectively address the development of acquired resistance and switch regimens in a timely manner (World Health Organization, 2010). All these challenges underscore the need for long-acting therapeutics that can replace the daily pill burden by being administered on a more in-frequent basis (Boffito et al., 2014). Simplification of HIV therapeutic regimens can lead to improvement in patient adherence and make lifelong HIV management easier for the patients (Kerrigan et al., 2018).
As an alternative to the more conventional oral HAART regimen, long-acting injectables (LAIs) can be administered on a more infrequent basis, reduce patient non-compliance, and potentially minimize the development of drug resistance (Owen and Rannard, 2016). Although HIV-targeted injectable crystalline nanoparticle formulations have progressed to clinical trials, some concerns include prolonged systemic exposure and challenges in removing drug in the event of toxicity (Spreen et al., 2013). Other drug nanoparticle formulations required multiple doses to achieve an enhanced pharmacokinetic (PK) effect over the corresponding soluble drugs (Gautam et al., 2013). Attempts at encapsulating the active drugs into carriers have been limited by poor drug loading (Mandal et al., 2017) and “leaky” particles resulting from passive diffusion (Mandal et al., 2017; Tshweu et al., 2014). Our lab has previously developed an injectable biopolymeric microconfetti delivery system for one week controlled release of saquinavir from a single injection (Collier et al., 2016). However, a more sustained drug release profile is required to achieve fewer administrations to address the previously mentioned practical challenges faced in HIV therapy.
Direct chemical modification of an antiretroviral drug that transforms them into a LAI could circumvent the barriers that exist with the aforementioned injectable systems. To this end, we identified two current drugs with reactive functional moieties that can be used as chemical handles for further modification: emtricitabine (FTC) and elvitegravir (EVG). FTC, a nucleoside reverse transcriptase inhibitor (NRTI), was approved by the FDA in 2003 for the prevention and treatment of HIV infection in adults and children (Saag, 2006). EVG, belonging to a different HIV drug class, integrase inhibitors, was FDA-approved in 2014 as a single pill formulation. EVG prevents HIV transmission by blocking an essential HIV enzyme required by the virus for integration of its genetic code into the host DNA (Unger et al., 2016). Due to their high potencies, both drugs are part of several combination therapies that are currently being used worldwide by patients. However, their long-term efficacy is limited by daily administration (Truong et al., 2015), poor bioavailability (Bastiaans et al., 2014), and metabolism by cytochrome P450 3A4 (CYP3A4) (Klibanov, 2009). LAI versions of these drugs could improve their systemic circulation and reduce the daily intake requirement, ultimately reducing the overall drug cost (Barnhart, 2017). To realize this goal, we sought to develop two standalone LAI-based derivatives of antiretrovirals (FTC and EVG), with the potential for fewer drug administrations compared to traditional HAART therapy.
Herein, we report the synthesis of polyunsaturated fatty-acid (PUFA) modified-FTC and EVG as long-acting derivatives of the parent drugs. Success of this approach is dependent upon the cleavage rate of the chemical bond between the parent drug and the derivatizing moiety. We evaluated their antiviral activities against HIV-infected cells and compared to those of the free drugs. In vitro drug release profiles were generated in attempts to demonstrate sustained release of the active antiretrovirals. Pharmacokinetic analyses were also conducted in mice to discern the long-acting aspect of these drug conjugates.
2. Materials and methods
2.1. General considerations
Deuterated solvents were purchased from Cambridge Isotope Laboratories (Andover, MA, USA) and used without further purification. Compounds FTC-DHA and EVG-DHA were prepared according to literature procedures, with slight modifications. (Agarwal et al., 2013) All reagents were purchased from Sigma Aldrich (St. Louis, MO, USA) and used without further modification, unless otherwise indicated. Emtricitabine (FTC) was purchased from 1 ClickChemistry, Inc. (Kendall Park, NJ, USA). Elvitegravir (EVG) was purchased from Pharma-Block, Inc. (Sunnyvale, CA, USA). Reactions were performed under a dry nitrogen atmosphere, unless otherwise noted. All flash chromatography was carried out using a 75-mm inner diameter column containing 100-mm length of silica gel under a positive pressure of laboratory air. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1NL4–3 Infectious Molecular Clone (pNL4–3) from Dr. Malcolm Martin, TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc.
2.2. Instrumentation
1H and 13C NMR spectra were recorded on an Inova 400 FT-NMR spectrometer (400 MHz for 1H NMR, 100 MHz for 13C NMR). 1H NMR data are reported as follows: chemical shift (multiplicity (b = broad, s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, and m = multiplet) and peak assignments). 1H and 13C chemical shifts are reported in ppm downfield from tetramethylsilane (TMS). Electrospray-ionization mass spectrometric (ESIMS) data was obtained on a Q Exactive™ HF-X Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (ThermoFisher Scientific, Waltham, MA, USA). UV-vis absorption spectra for all samples were obtained on a SpectraMax M2 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA).
2.3. Synthesis of ((2R,5S)-5-(4-amino-5-fluoro-2-oxopyrimidin-1(2H)-yl)-1,3-oxathiolan-2-yl)methyl (4E,7E,10E,13E,16E,19E)-docosa-4,7,10,13,16,19-hexaenoate (FTC-DHA)
FTC (800 mg, 3.24 mmol), docosahexanoic acid (DHA) (2.13 gm, 6.47 mmol), and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, O-(Benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HBTU) (2.45 gm, 6.47 mmol) were dissolved in dry dimethylformamide (DMF, 20 mL) in a 100 mL round-bottom flask. Diisopropylethylamine (DIPEA) (18.6 mL, 0.11 mol) was added to the reaction mixture via a gas tight syringe, and the reaction was allowed to proceed for 18 h at room temperature. The reaction mixture was then concentrated under reduced pressure and purified by flash chromatography (50:50 v/v ethyl acetate/hexanes) to afford compound FTC-DHA as a colorless oil (1.18 gm, 65% yield). 1H NMR (Fig. S1 in ESI†, CDCl3, 400 MHz, δ ppm): 8.17 (d, 1H, CFCH), 6.22 (s, 2H, NH2), 5.18–5.68 (m, 1H, NCHCH2, 1H, OCHCH2, 12H, CH vinylic protons), 4.38 (dd, 2H, CH2OCO), 3.19 (dd, 2H, NCHCH2S), 2.69–2.87 (m, 10H, CH2 vinylic protons), 2.31–2.47 (m, 4H, OCOCH2CH2), 1.95–2.07 (m, 2H, CH2CH3), 0.92 (t, 3H, CH3). 13C NMR (Fig. S2 in ESI†, CDCl3, 100 MHz, δ ppm): 172.33 (C=OO), 158.76 (CNH2), 152.15 (C=O), 131.98 (CF), 126.97 (CFCH), 127.29–129.72 (11C, CH, vinylic carbons), 87.54 (NCHO), 85.66 (OCHS), 62.88 (CH2OC=O), 39.23 (NCHCH2S), 33.75 (CH2C=OO), 25.53–25.71 (5C, CH2 vinylic carbons), 22.07 (CH2CH2C=OO), 20.56 (CH2CH3), 14.23 (CH3). ESIMS (Fig. S3 in ESI†): m/z calculated for C30H40FN3O4S (M +H)+: 558.73. Found: 558.28.
2.4. Synthesis of 6-(3-chloro-2-fluorobenzyl)-1-(1-(((4Z,7E,10E,13E,16E,19E)-docosa-4,7,10,13,16,19-hexaenoyl)oxy)-3-methylbutan-2-yl)-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (EVG-DHA)
EVG (650 mg, 1.45 mmol), DHA (958 mg, 2.9 mmol), and HBTU (1.1 gm, 2.9 mmol) were dissolved in dry DMF (20 mL) in a 100 mL round-bottom flask. DIPEA (8.3 mL, 47.9 mmol) was added to the reaction mixture via a gas tight syringe, and the reaction was allowed to proceed for 18 h at room temperature. The reaction mixture was then concentrated under reduced pressure and purified by flash chromatography (50:50 v/v ethyl acetate/hexanes) to afford EVG-DHA as a pale yellow oil (1 gm, 91% yield). 1H NMR (Fig. S4 in ESI†, CDCl3, 400 MHz, δ ppm): 8.72 (s, 1H, COOHCH), 8.19 (s, 1H, aromatic proton), 7.28–7.37 (m, 1H, aromatic proton), 7.13–7.24 (m, 1H, aromatic proton), 6.86–7.04 (m, 1H, aromatic proton), 5.12–5.44 (12H, CH vinylic protons), 4.67 (dd, 2H, CH2OCO), 4.03 (s, 2H, CFCCH2), 3.94 (s, 3H, OCH3), 2.68–2.84 (m, 10H, CH2 vinylic protons), 2.57–2.67 (m, 1H, NCHCH2), 2.07–2.21 (m, 4H, OCOCH2CH2), 1.89–2.04 (m, 2H, CH2CH3), 1.07–1.24 (m, 1H, CH(CH3)2), 0.89 (m, 6H, CH(CH3)2), 0.79 (t, 3H, CH3). 13C NMR (Fig. S5 in ESI†, CDCl3, 100 MHz, δ ppm): 177.03 (C = O, aromatic carbon), 175.95 (C = OO), 167.54 (COOH), 165.82 (COCH3), 162.59 (CF), 144.05 (CHCHN), 142.33 (CN, aromatic carbon), 131.98 (CHCH2CH3, vinylic carbon), 129.61 (CFC), 129.18 (CH2CCH, aromatic carbon), 127.67–129.18 (11C, CH, vinylic carbons), 127.03 (CHCHCH), 124.44 (CHCHCH), 120.99 (CCl), 119.69 (CC=O, aromatic carbon), 116.68 (CH2C, aromatic carbon), 106.98 (CCOOH), 95.78 (CHCOCH3), 75.30 (NCH), 64.31 (CH2OC=O), 56.12 (OCH3), 33.71 (5C, CH2, vinylic carbons), 30.47 (CH2C = OO), 28.97 (CH(CH3)2), 25.52 (CH2CH3), 22.28 (CH2CH2C=OO), 19.48–19.69 (CH (CH3)2), 14.31 (CH3). ESIMS (Fig. S6 in ESI†): m/z calculated for C45H53ClFNO6 (M + H) + : 758.37. Found: 758.36.
2.5. Hydrolytic release curves
The hydrolysis of FTC-DHA and EVG-DHA were studied at pH 7.4 (0.1 M phosphate buffered saline). All experiments were performed in triplicate. Each drug conjugate (1 mg/mL) was stirred continuously while being maintained at 37 °C. At appropriate time intervals (1, 3, 7, 10, 14, 17, 21, 24, 28, 31, 35, and 42 days) samples were withdrawn and UV absorbance was recorded (300 nm for free FTC released from FTC-DHA and 320 nm for free EVG released from EVG-DHA).
2.6. Antiviral activity in vitro
HIV-1NL-43 was prepared by transfecting 293 T cells using ExpressFect (Denville Scientific) with NL4.3 provirus plasmid. (Kulkarni et al., 2017; Adachi et al., 1986) Transfected cells were incubated for 48 h at 37 °C and the supernatant was collected, centrifuged at 400 × g for 5 min, and passed through a 0.45 μm filter. 1 × 106 HeLa-derived human TZM-bl cells, (Wei et al., 2002; Platt et al., 2009, 1998; Takeuchi et al., 2008; Derdeyn et al., 2000) which contain a luciferase gene under the control of a HIV-1 promoter, were plated in each well of a 96 well plate and were incubated with 50 μL HIV-1NL-43 along with serial dilutions of each compound. After 48 h at 37 °C, culture supernatants were removed and cells were lysed using 100 μL of 1X Passive Lysis Buffer (Promega, USA) overnight at — 80 °C and three subsequent freeze-thaw cycles. Tat-induced luciferase reporter gene expression was measured in TZM-bl cells by luminescence produced via reaction with Bright-Glo™ reagent (Promega, USA) quantified on a Spectramax i3X (Molecular Devices, USA). Cells which have been successfully infected with HIV-1NL-43 produce luciferase enzyme that reacts with Bright-Glo™ reagent, and therefore, viral infectivity correlates directly to luminescence.
2.7. Pharmacokinetic evaluation of FTC-DHA and EVG-DHA conjugates
CD-1 mice (female, 10–11 weeks old) were purchased from Charles River Laboratories (Wilmington, MA, USA). Free FTC drug solution (30 μL; 1 g/ml in PBS; 1000 mg/kg) was injected intramuscularly into the right leg of each mouse (n = 2) with collection time points of 1, 6, 12, 24, 36, 48, 72, 168, 240, and 336 h. For the PK study using FTC-DHA, drug conjugate solution (22 μL; 1g/mL in ethanol; 750 mg/kg) was injected intramuscularly into the right leg of each mouse (n = 3). Collection time points were 1, 6, 12, 24 h, 3, 7, 14, 21, 28, 35, and 42 days post injection.
Free EVG drug solution (25 μL; 1 g/ml in ethanol; 1000 mg/kg) was injected intramuscularly into the right leg of each mouse (n = 1) with collection time points of 1, 6, 12, 24, 36, 48, 72, 168, and 336 h. For the PK study using EVG-DHA, drug conjugate solution (19 μL; 1 g/mL in ethanol; 750 mg/kg) was injected intramuscularly into the right leg of each mouse (n = 1). Collection time points were 6, 24, 48 h, 7, 14, 22, 28, 35, and 42 days post injection. At each time point for all the in vivo studies, whole blood was collected by submandibular bleeds followed by euthanasia of mice via CO2.
Plasma from both studies was obtained by centrifuging the whole blood samples in Greiner Bio-One MiniCollect capillary blood collection system tubes (Fisher Scientific, Hampton, NH, USA) for 10 min at 4000 × g in a Thermo Scientific Sorvall Legend Micro 21 microcentrifuge (Hampton, NH, USA) and stored at — 80 °C until LC/MS analysis.
2.8. LC/MS quantification of plasma samples
FTC and EVG were extracted from mouse plasma samples using a protein precipitation with isotopically-labeled internal standard (Emtricitabine 13C, 15N and Elvitegravir-d6, respectively). FTC was eluted from a Waters Atlantis T3 (2.1 × 100 mm, 3 μm particle size) analytical column using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B). EVG was eluted from a Waters Atlantis T3 (2.1 × 50 mm, 3 μm particle size) analytical column using 10 mM ammonium acetate (mobile phase A) and acetonitrile (mobile phase B). Both analytes were detected on an AB Sciex API-5000™ triple quadrupole mass spectrometer. Standards and quality controls (QC) were prepared in duplicate, and a calibration curve was generated using a weighted linear regression of analyte:internal standard peak area ratio vs. concentration. FTC and EVG concentrations in QCs and study samples were calculated from this calibration curve using Sciex Analyst Chromatography Software. The acceptance criteria of the assay was ± 20% of nominal concentration for standards and QCs. The quantifiable ranges were 2–20,000 ng/mL (FTC) and 12.5–10,000 ng/mL (EVG).
Plotting and non-compartmental analysis (NCA) were performed using Phoenix WinNonlin v8.0 software. The sparse sampling function was applied for a composite NCA of concentration data from all animals. The linear trapezoidal rule was used to calculate AUC0–42 d. The terminal elimination rate constant (kel) was calculated by fitting a linear regression line to the observed terminal concentration versus time data on a semi-log plot. All values below the limit of quantification were imputed at half the lower limit of quantification (2 ng/mL) for analysis purposes. Steady-state PK parameter estimates included kel, t½ (half-life), Cmax (maximal or peak concentration), and AUC0–42 d (area under the steady-state plasma concentration-time curve over 42 days).
3. Results and discussion
3.1. Chemistry
FTC-and EVG-modified docosahexanoic ester conjugates were synthesized in simple one-step, one-pot, and high-yielding reactions (Scheme 1). Although similar chemistry has been previously reported with anti-HIV drugs lamivudine (Guo et al., 2017); FTC (Agarwal et al., 2013), and abacavir (Singh et al., 2016) conjugated to myristic acid, multi-step syntheses with lower overall yields were required to obtain the desired compounds. Our strategy eliminated the need for additional synthetic manipulations and had improved yields. Taking advantage of the inherent chemical moieties in the antivirals and modifying them to yield prodrugs allows for complete drug incorporation when compared to methods that encapsulate drugs in carriers through physical loading (Ogunwuyi et al., 2016). In designing these conjugates, there were two major components that required careful consideration: chemical linker and choice of PUFA. While there is a broad range of linkers to choose from, carboxylic ester-based ones are convenient since the precursors are widely applicable across many therapeutic platforms and do not need prior chemical modification (Nobs et al., 2004). The ester bond is known to undergo slow hydrolysis when subjected to enzymatic cleavage under physiological conditions in vivo (Fukami and Yokoi, 2012); facilitating extended release of the active antiretroviral from the synthesized prodrugs.
Scheme 1.
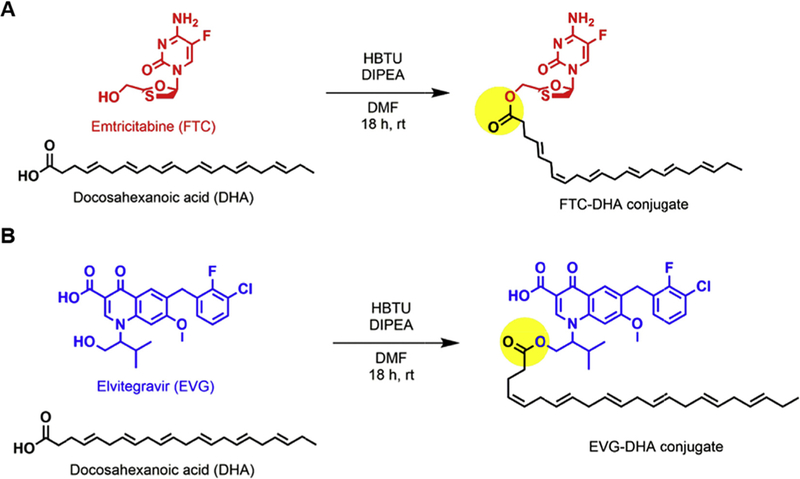
Synthetic schemes of (a) FTC-DHA and (b) EVG-DHA conjugates. The yellow circle highlights the ester bond, allowing for the potential prolonged release of the active drug when subjected to physiological conditions. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
One of the major complications in HAART is that some anti-retrovirals have limited bioavailability due to metabolism via the cytochrome P450 (CYP) CYP3A4 in the intestine and the liver (Klibanov, 2009). As a result, they typically require the drug to be co-administered with pharmacokinetic enhancers (Walubo, 2007; Ramanathan et al., 2007). To minimize drug-drug interactions while maintaining therapeutic drug concentrations in plasma, we directed our efforts in identifying relevant candidates that allow the therapeutics to remain in the bloodstream for a longer time. PUFAs have been shown to inhibit drug-metabolizing CYP enzymes (Yao et al., 2006), which can reduce the rate of metabolism of the active antiretrovirals. Of the available PUFA options, docosahexaenoic acid (DHA) was chosen because it demonstrated greater inhibitory potency against CYP3A4 (Yao et al., 2006). Such competitive inhibition of the antiretroviral-CYP3A4 enzyme interaction could potentially boost the drug’s concentration and bioavailability, resulting in an improved PK profile.
An added advantage of conjugating long fatty chains to the anti-retrovirals is the increased partition coefficients (log P) compared to those of the unmodified drugs, leading to an enhanced lipophilicity of the resultant conjugate (Agarwal et al., 2013). Through increased lipophilicity (log P > 5), the DHA conjugate can improve cell uptake (Irby et al., 2017). Our calculations also confirmed that the fatty chainmodified conjugates have higher log P values (> 5) compared to their respective unconjugated drugs (< 5) (Table 1).
Table 1.
Calculated (calc.) partition coefficients for the free drugs FTC and EVG; free DHA; synthesized FTC-DHA and EVG-DHA conjugates. Chem3D 15.1 software was used for generating log P values.
| Compound | Log P (calc.) |
|---|---|
| FTC (Emtricitabine) | –1.29 |
| EVG (Elvitegravir) | 4.56 |
| DHA (Docosahexaenoic acid) | 7.48 |
| FTC-DHA | 7.29 |
| EVG-DHA | 13.14 |
3.2. Cell-based antiretroviral activity
To evaluate the antiretroviral efficacy of the synthesized conjugates, TZM-bl cells were treated with indicated doses of free FTC, EVG, DHA, FTC-DHA, or EVG-DHA conjugates. PUFA-modified anti-HIV drugs exhibited higher activity against cell-associated virus than their corresponding parent drugs (Fig. 1). FTC-DHA (IC50 = 1.39 μM) was ~12 times more potent than free FTC (IC50 = 17.2 μM) alone, while EVG-DHA (IC50 = 0.14 μM) was ~220 times more potent than free EVG (IC50 = 30.9 μM). Previously published myristoylated FTC conjugate has also been shown to have a higher anti-HIV activity (24 times) when compared to that of FTC alone, indicating that modification of the parent drug generated a more potent prodrug (Agarwal et al., 2013). Another NRTI-lipid conjugate, abacavir-myristic acid, had comparable antiviral activity with its native form against monocyte-derived macrophages (Singh et al., 2016). Our results clearly demonstrate that the PUFA derivatives of the antiretrovirals reported herein have enhanced antiviral activity as compared to that of the parent drugs.
Fig. 1.
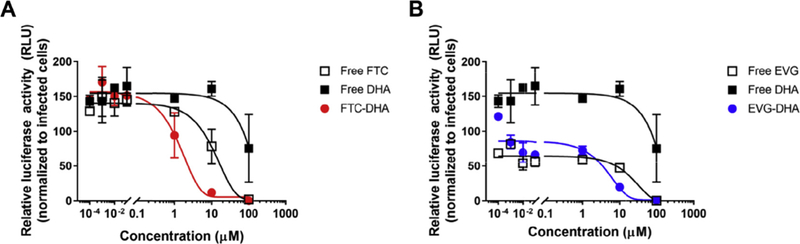
Antiviral activities of conjugates against HIV-infected cells. TZM-bl cells were treated for 48 h with μM doses of (a) free FTC, free DHA, and FTC-DHA conjugate; (b) free EVG, free DHA, and EVG-DHA conjugate and evaluated for intracellular viral activity, as measured by luciferase assay. Data are presented as mean ± standard deviation (n = 2).
3.3. Drug release profile
Drug release kinetics were evaluated for the synthesized FTC-DHA and EVG-DHA conjugates. As mentioned previously, ester bonds are known to degrade over a longer period of time, preventing burst release of the antiretroviral (Acton et al., 2013). Using the ester bond chemistry for our conjugates, we also demonstrated that FTC-DHA conjugate had a gradual and sustained drug release profile over a period of 21 days, while EVG-DHA demonstrated a slower drug release over a period of 30 days (Fig. 2). We hypothesize that due to the hydrophobicity of the EVG drug compared to the more hydrophilic FTC (Table 1), it may be releasing at a much slower rate (Fu and Kao, 2010). At the terminal time point, the remaining aggregate was subjected to 1H NMR analysis to determine if any drug remained. The NMR spectrum displayed peaks pertinent to only DHA, indicating 100% of the drug had been released (Figs. S7 and S8 in ESI†). The slow and sustained release over several days that we observed with our conjugates is in contrast to entecavir (an antiretroviral used to treat hepatitis B virus (HBV) infection) fatty acid conjugate achieved through similar chemistry where a significant amount of active antiretroviral (15–41%) was released within the first hour (Ho et al., 2018).
Fig. 2.
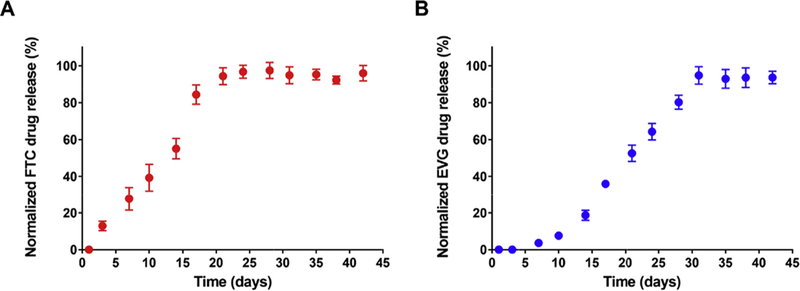
In vitro release profiles of free FTC and EVG from (a) FTC-DHA and (b) EVG-DHA conjugates, respectively. Drug release was performed at pH 7.4 in PBS at 37 °C and drug concentration was determined using UV-vis spectroscopy. Data are presented as mean ± standard deviation (n = 3).
3.4. Pharmacokinetics
In vivo experiments were conducted to determine the pharmacokinetic profiles of the unmodified drugs and the free drug released from the synthesized conjugates in CD1 mice, when given as an intramuscular injection (Fig. 3). The absence of any observable absorption phase in the pharmacokinetic profiles could be attributed to the rapid diffusion that small molecules are known to undergo. Absorption of the free drugs and their conjugates could have occurred prior to the first drug-plasma concentration detection time point (6 h). All treatment groups peaked at the initial time points followed by steady decline of the release rate of the active antiretrovirals in plasma over time for both the FTC-DHA and EVG-DHA conjugates. The drug concentrations sufficient to provide 90% inhibition of viral replication (IC90) for FTC in plasma is 14 ng/mL (Molina et al., 2004), whereas the IC95 for EVG in plasma has been reported to be 45 ng/mL (Podany et al., 2017), As is evident in Fig. 3, both free versions of the drugs were cleared within 48 h post injection with an unexpected rise in plasma concentration for free FTC at day 7. We speculate that rates of drug absorption could vary from different parts of the muscle, leading to slightly higher levels of drug being detected in plasma at later time points. Despite this observed variation in drug-plasma concentration, all subsequent time points have drug concentrations below their respective IC90/IC95 values. In stark contrast, the plasma concentrations at 2 weeks of FTC and EVG released from the synthesized conjugates exceeded their respective IC90 and IC95 antiviral activities. Analysis of plasma samples even at 6 weeks illustrated detectable amounts of active drug that had been released from the conjugates. Presumably, even higher plasma concentrations of the drugs could be achieved in humans due to the faster systemic circulation clearance rates in mice (Lin et al., 1999).
Fig. 3.
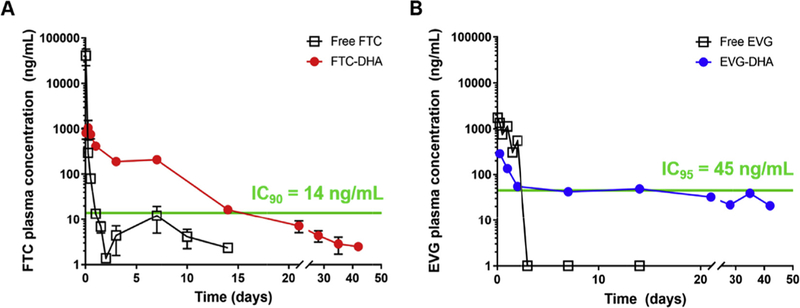
Plasma concentrations of (a) FTC and (b) EVG following in vivo release from intramuscular injections of FTC-and EVG-DHA conjugates, respectively. Data for FTC-DHA are presented as mean ± standard deviation (n = 3); EVG-DHA data are presented as absolute values (n = 1).
Pharmacokinetic parameters kel (elimination rate constant) and t½ (half-life) are indicative of the rate at which the active drug is cleared from the blood. Controlled release of the active drug from the FTC-DHA and EVG-DHA conjugates led to ~ 4-fold and ~ 65-fold improvements in the t½ over their corresponding free drugs, respectively (Table 2). As the conjugates exhibit longer half-lives, their rates of elimination are significantly diminished when compared to their free drug counterparts. A higher Cmax (maximum plasma concentration) value correlates with the rate of drug absorption. Both of the free drugs have much higher Cmax values when compared to those of their conjugates with a rapid decline in drug concentrations, indicating that they are absorbed quicker than their modified analogs. Rapid absorption and clearance of the free drugs renders them ineffective for long-term therapy, as it would require frequent dosing to maintain therapeutic concentrations. Our approach maintains consistent drug-plasma concentrations above the IC90 or IC95 attributable to gradual release of the active drug which could enable infrequent dosing, ultimately leading to improved patient adherence and clinical outcomes. Considering the long-term implications of HAART therapy (Dakin et al., 2006; Chariot et al., 1999; Ofotokun and Weitzmann, 2010), long-acting injectables could have substantial benefits for HIV+ individuals.
Table 2.
Comparison of the plasma pharmacokinetic parameters of the free versions of FTC and EVG to their PUFA-modified counterparts. Parameters include elimination rate constant (kel), half-life (t½), maximum plasma concentration (Cmax), and area under the curve for the plasma concentration over 42 days (AUC0–42 d).
| Parameter | Free FTC | FTC-DHA | Free EVG | EVG-DHA |
|---|---|---|---|---|
| kel(h–1) | 0.013 | 0.003 | 0.071 | 0.0011 |
| t½(h) | 54.7 | 228.06 | 9.75 | 643.44 |
| Cmax (ng/mL) | 41,050 | 1,054 | 1,750 | 285 |
| AUC0–42 d (h× ng/mL) | 127,462 | 74,252 | 48,344 | 39,177 |
4. Conclusion
We have reported the synthesis of injectable long-acting antiretrovirals to address the emergence of resistance due to patient non-compliance. The one-step, one-pot chemistry is modular and has been used to modify anti-HIV drugs from two different classes with DHA to obtain lipid drug conjugates. These prodrugs demonstrated a sustained drug release profile in vitro and in vivo due to the slow hydrolysis of the ester bond, which could lead to reductions in dose and frequency of administration. Additionally, the collaborative effort of DHA could also contribute to longer circulation, potentially improving therapeutic efficacy. This strategy transformed short-acting unmodified drugs with half-lives of a few hours to extended-release prodrugs with improved PK profiles. Due to the ease of chemical conjugation and sustained drug concentrations in plasma over at least two weeks, this approach is promising in its ability to deliver therapeutic doses of FTC and EVG for an extended period of time.
Supplementary Material
Acknowledgements
We would like to acknowledge the University of North Carolina Chapel Hill Department of Chemistry Chemical Research Instrumentation, Teaching, and Core Laboratories (CRITCL) Mass Spectrometry Core Laboratory for allowing us to use the ESI-MS equipment used within this manuscript. Additionally, we would like to acknowledge the Eshelman Institute for Innovation for providing the financial support for this research.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpharm.2018.10.017.
References
- Acton AL, Fante C, Flatley B, Burattini S, Hamley IW, Wang Z, Greco F, Hayes W, 2013. Janus PEG-based dendrimers for use in combination therapy: controlled multi-drug loading and sequential release. Biomacromolecules 14 (2), 564–574. [DOI] [PubMed] [Google Scholar]
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA, 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59 (2), 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal HK, Chhikara BS, Bhavaraju S, Mandal D, Doncel GF, Parang K, 2013. Emtricitabine prodrugs with improved anti-HIV activity and cellular uptake. Mol. Pharm. 10 (2), 467–476. [DOI] [PubMed] [Google Scholar]
- Antiretroviral Therapy for HIV Infection in Adults and Adolescents. World Health Organization: 2010. [PubMed] [Google Scholar]
- Barnhart M, 2017. Long-acting HIV treatment and prevention: closer to the threshold. Glob. Health Sci. Pract. 5 (2), 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaans DE, Cressey TR, Vromans H, Burger DM, 2014. The role of formulation on the pharmacokinetics of antiretroviral drugs. Expert Opin. Drug Metab. Toxicol. 10 (7), 1019–1037. [DOI] [PubMed] [Google Scholar]
- Boffito M, Jackson A, Owen A, Becker S, 2014. New approaches to antiretroviral drug delivery: challenges and opportunities associated with the use of long-acting injectable agents. Drugs 74 (1), 7–13. [DOI] [PubMed] [Google Scholar]
- Chariot P, Drogou I, de Lacroix-Szmania I, Eliezer-Vanerot MC, Chazaud B, Lombes A, Schaeffer A, Zafrani ES, 1999. Zidovudine-induced mitochondrial disorder with massive liver steatosis, myopathy, lactic acidosis, and mitochondrial DNA depletion. J. Hepatol. 30 (1), 156–160. [DOI] [PubMed] [Google Scholar]
- Clinical Guidelines: Offering Information on HIV/AIDS Treatment, Prevention, and Research. https://aidsinfo.nih.gov/guidelines/.
- Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, Hakim JG, Kumwenda J, Grinsztejn B, Pilotto JH, Godbole SV, Mehendale S, Chariyalertsak S, Santos BR, Mayer KH, Hoffman IF, Eshleman SH, Piwowar-Manning E, Wang L, Makhema J, Mills LA, de Bruyn G, Sanne I, Eron J, Gallant J, Havlir D, Swindells S, Ribaudo H, Elharrar V, Burns D, Taha TE, Nielsen-Saines K, Celentano D, Essex M, Fleming TR, 2011. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 365 (6), 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman CI, Roberts MS, Sobieraj DM, Lee S, Alam T, Kaur R, 2012. Effect of dosing frequency on chronic cardiovascular disease medication adherence. Curr. Med. Res. Opin. 28 (5), 669–680. [DOI] [PubMed] [Google Scholar]
- Collier MA, Gallovic MD, Bachelder EM, Sykes CD, Kashuba A, Ainslie KM, 2016. Saquinavir loaded acetalated dextran microconfetti -a long acting protease inhibitor injectable. Pharm. Res. 33 (8), 1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakin CL, O’Connor CA, Patsdaughter CA, 2006. HAART to heart: HIV-related cardiomyopathy and other cardiovascular complications. AACN Clin. Issues 17 (1), 18–29. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E, 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74 (18), 8358–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Kao WJ, 2010. Drug release kinetics and transport mechanisms of non-degrad-able and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 7 (4), 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami T, Yokoi T, 2012. The emerging role of human esterases. Drug Metab. Pharmacokinet. 27 (5), 466–477. [DOI] [PubMed] [Google Scholar]
- Gautam N, Roy U, Balkundi S, Puligujja P, Guo D, Smith N, Liu XM, Lamberty B, Morsey B, Fox HS, McMillan J, Gendelman HE, Alnouti Y, 2013. Preclinical pharmacokinetics and tissue distribution of long-acting nanoformulated antiretroviral therapy. Antimicrob. Agents Chemother. 57 (7), 3110–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genberg BL, Wilson IB, Bangsberg DR, Arnsten J, Goggin K, Remien RH, Simoni J, Gross R, Reynolds N, Rosen M, Liu H, 2012. Patterns of antiretroviral therapy adherence and impact on HIV RNA among patients in North America. AIDS 26 (11), 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross R, Bilker WB, Friedman HM, Strom BL, 2001. Effect of adherence to newly initiated antiretroviral therapy on plasma viral load. AIDS 15 (16), 2109–2117. [DOI] [PubMed] [Google Scholar]
- Guo D, Zhou T, Arainga M, Palandri D, Gautam N, Bronich T, Alnouti Y, McMillan J, Edagwa B, Gendelman HE, 2017. Creation of a long-acting nano-formulated 2’,3’-Dideoxy-3’-thiacytidine. J. Acquir. Immune Defic. Syndr. 74 (3), e75–e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIV/AIDS: Data and Statistics. http://www.who.int/hiv/data/en/.
- Ho MJ, Lee DR, Im SH, Yoon JA, Shin CY, Kim HJ, Jang SW, Choi YW, Han YT, Kang MJ, 2018. Microsuspension of fatty acid esters of entecavir for parenteral sustained delivery. Int. J. Pharm. 543 (1–2), 52–59. [DOI] [PubMed] [Google Scholar]
- Irby D, Du C, Li F, 2017. Lipid-drug conjugate for enhancing drug delivery. Mol. Pharm. 14 (5), 1325–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrigan D, Mantsios A, Gorgolas M, Montes ML, Pulido F, Brinson C, deVente J, Richmond GJ, Beckham SW, Hammond P, Margolis D, Murray M, 2018. Experiences with long acting injectable ART: A qualitative study among PLHIV participating in a Phase II study of cabotegravir + rilpivirine (LATTE-2) in the United States and Spain. PLoS One 13 (1), e0190487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee E, Park BJ, Bang JH, Lee JY, 2018. Adherence to antiretroviral therapy and factors affecting low medication adherence among incident HIV-infected individuals during 2009–2016: a nationwide study. Sci. Rep. 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klibanov OM, 2009. Elvitegravir, an oral HIV integrase inhibitor, for the potential treatment of HIV infection. Curr. Opin. Investig. Drugs 10 (2), 190–200. [PubMed] [Google Scholar]
- Kulkarni MM, Ratcliff AN, Bhat M, Alwarawrah Y, Hughes P, Arcos J, Loiselle D, Torrelles JB, Funderburg NT, Haystead TA, Kwiek JJ, 2017. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology 14 (1), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YS, Nguyen C, Mendoza JL, Escandon E, Fei D, Meng YG, Modi NB, 1999. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J. Pharmacol. Exp. Ther. 288 (1), 371–378. [PubMed] [Google Scholar]
- Mandal S, Belshan M, Holec A, Zhou Y, Destache CJ, 2017. An enhanced emtricitabine-loaded long-acting nanoformulation for prevention or treatment of HIV infection. Antimicrob. Agents Chemother. 61, (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JM, Peytavin G, Perusat S, Lascoux-Combes C, Sereni D, Rozenbaum W, Chene G, 2004. Pharmacokinetics of emtricitabine, didanosine and efavirenz administered once-daily for the treatment of HIV-infected adults (pharmacokinetic substudy of the ANRS 091 trial). HIV Med. 5 (2), 99–104. [DOI] [PubMed] [Google Scholar]
- Nobs L, Buchegger F, Gurny R, Allemann E, 2004. Current methods for attaching targeting ligands to liposomes and nanoparticles. J. Pharm. Sci. 93 (8), 1980–1992. [DOI] [PubMed] [Google Scholar]
- Ofotokun I, Weitzmann MN, 2010. HIV-1 infection and antiretroviral therapies: risk factors for osteoporosis and bone fracture. Curr. Opin. Endocrinol. Diabetes Obes. 17 (6), 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunwuyi O, Kumari N, Smith KA, Bolshakov O, Adesina S, Gugssa A, Anderson WA, Nekhai S, Akala EO, 2016. Antiretroviral drugs-loaded nanoparticles fabricated by dispersion polymerization with potential for HIV/AIDS treatment. Infect. Dis. 9, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen A, Rannard S, 2016. Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: insights for applications in HIV therapy. Adv. Drug Deliv. Rev. 103, 144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paydary K, Khaghani P, Emamzadeh-Fard S, Alinaghi SAS, Baesi K, Zandie S, 2013. The emergence of drug resistant HIV variants and novel anti-retroviral therapy. Asian Pac. J. Trop. Biomed. 3, 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D, 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72 (4), 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC, 2009. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J. Virol. 83 (16), 8289–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podany AT, Scarsi KK, Fletcher CV, 2017. Comparative clinical pharmacokinetics and pharmacodynamics of HIV-1 integrase strand transfer inhibitors. Clin. Pharmacokinet. 56 (1), 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajoli RK, Back DJ, Rannard S, Freel Meyers CL, Flexner C, Owen A, Siccardi M, 2015. Physiologically based pharmacokinetic modelling to inform development of intramuscular long-acting nanoformulations for HIV. Clin. Pharmacokinet. 54 (6), 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan S, Shen G, Cheng A, Kearney BP, 2007. Pharmacokinetics of emtricitabine, tenofovir, and GS-9137 following coadministration of emtricitabine/tenofovir disoproxil fumarate and ritonavir-boosted GS-9137. J. Acquir. Immune Defic. Syndr. 45 (3), 274–279. [DOI] [PubMed] [Google Scholar]
- Saag MS, 2006. Emtricitabine, a new antiretroviral agent with activity against HIV and hepatitis B virus. Clin. Infect. Dis. 42 (1), 126–131. [DOI] [PubMed] [Google Scholar]
- Singh D, McMillan J, Hilaire J, Gautam N, Palandri D, Alnouti Y, Gendelman HE, Edagwa B, 2016. Development and characterization of a long-acting nano-formulated abacavir prodrug. Nanomedicine 11 (15), 1913–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreen WR, Margolis DA, Pottage JC Jr., 2013. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr. Opin. HIV AIDS 8 (6), 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, McClure MO, Pizzato M, 2008. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research∇. J. Virol. 82, 12585–12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong WR, Schafer JJ, Short WR, 2015. Once-Daily, Single-Tablet Regimens For the Treatment of HIV-1 Infection. Pharm. Ther. 40 (1), 44–55. [PMC free article] [PubMed] [Google Scholar]
- Tshweu L, Katata L, Kalombo L, Chiappetta DA, Hocht C, Sosnik A, Swai H, 2014. Enhanced oral bioavailability of the antiretroviral efavirenz encapsulated in poly(epsilon-caprolactone) nanoparticles by a spray-drying method. Nanomedicine 9 (12), 1821–1833. [DOI] [PubMed] [Google Scholar]
- Unger NR, Worley MV, Kisgen JJ, Sherman EM, Childs-Kean LM, 2016. Elvitegravir for the treatment of HIV. Expert Opin. Pharmacother. 17 (17), 2359–2370. [DOI] [PubMed] [Google Scholar]
- Walubo A, 2007. The role of cytochrome P450 in antiretroviral drug interactions. Expert Opin. Drug Metab. Toxicol. 3 (4), 583–598. [DOI] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC, 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46 (6), 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HT, Chang YW, Lan SJ, Chen CT, Hsu JT, Yeh TK, 2006. The inhibitory effect of polyunsaturated fatty acids on human CYP enzymes. Life Sci. 79 (26), 2432–2440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.