

Abstract
Duchenne muscular dystrophy (DMD) is a common and devastating genetic disease primarily caused by exon deletions that create a genetic frameshift in dystrophin. Exon skipping therapy seeks to correct this by masking an exon during the mRNA maturation process, restoring dystrophin expression, but creating an edited protein missing both the original defect and the therapeutically skipped region. Crucially, it is possible to correct many defects in alternative ways, by skipping an exon either before or after the patient’s defect. This results in alternatively edited, hybrid proteins that might have different properties and therapeutic consequences. We examined three such dystrophin exon-skipped edits, Δe45–53, Δe46–54, and Δe47–55, comprising two pairs of alternative repairs of Δe46–53 and Δe47–54 DMD defects. We found that in both cases, Δe46–54 was the more stable repair as determined by a variety of thermodynamic and biochemical measurements. We also examined the origin of these differences with molecular dynamics simulations, which showed that these stability differences were the result of different types of structural perturbations. For example, in one edit there was partial unfolding at the edit site that caused domain-localized perturbations while in another there was unfolding at the protein domain junctions distal to the edit site that increased molecular flexibility. These results demonstrate that alternative exon skip repairs of the same underlying defect can have very different consequences at the level of protein structure and stability and furthermore that these can arise by different mechanisms, either locally or by more subtle long-range perturbations.
Graphical Abstract
Duchenne muscular dystrophy (DMD) is a severe and unfortunately prevalent pediatric genetic disease that affects approximately one in 5000 male births and is a result of the loss of dystrophin proteins from muscle cells.1–3 The prognosis for those with this condition is bleak: progressive muscle wasting leading to loss of ambulation in the second decade of life, with death typically in the second or third decade of life. Exon skipping is the first, and so far, only curative therapy for this condition and offers the promise of a significantly better life to these patients and their families.
The most common type of genetic defect underlying DMD is a large, kilobase scale deletion of one or more exons. In a protein as large as dystrophin, this often results in the juxtaposition of exons of incompatible reading frames and also inevitably introduces a new stop codon that truncates the protein. The nonsense-mediated decay system generally flags and destroys these defective transcripts,4,5 so that the truncated protein is expressed far below normal levels, resulting in dystrophin typically being undetectable in DMD cells.
Exon skipping seeks to address this deficiency by masking additional exons to correct the reading frame and restore dystrophin expression. Clinically, this is done with antisense oligonucleotide analogues (AONs), which are similar to short DNA primers but with modified backbones to improve their pharmacological properties.6,7 This aims to restore expression by correcting the damaged reading frame, with the clinical aim of slowing or reversing muscle deterioration. However, although reading frame correction restores protein expression, the proteins produced lack the regions corresponding to the patient’s original defect as well as the therapeutically skipped exon, thus altering the protein’s structure. While deleting a region corresponding to several exons from a protein’s structure and reattaching the remaining regions are drastic alterations, especially because these newly attached regions may not be natively proximal and so have not evolved to be structurally compatible, we have shown that in some cases stable structures can form.8
Dystrophin acts largely as a mechanical stabilizing agent9,10 and is thought to work as a molecular shock absorber, so changes in its structure and stability likely alter those functions. In addition, dystrophin also contains a variety of binding sites for other proteins, most significantly neuronal nitric oxide synthetase11 and phospholipids,12 and edits may disrupt and modify those interactions.13 How edits impact the structure and function of dystrophin is an open question and presumably of clinical importance. Insight into this is provided by Becker muscular dystrophy (BMD), which is caused by naturally occurring in-frame defects in dystrophin. BMD is a milder but more heterogeneous condition, varying from being nearly as severe as DMD to nearly benign, with the suspicion that some defects might be so benign as to be subclinical.14,15 Exon skipping essentially aims to convert DMD to therapeutically induced BMD. However, due to the complexity of the dystrophin gene, there are a very large number of defects known. While in aggregate many edits have been identified, many individual defects are known only from a few cases or are unknown. It thus becomes difficult to accurately assess clinical impact, as patients with the same defect can progress very differently. However, by pooling different types of defects, several studies have shown a clear and significant relationship between edit type and clinical severity,16–18 although the statistics do not currently allow for the comparison of arbitrary pairs of defects in most cases.
The comparison of edit types to clinical severity is becoming increasingly important as additional exon skipping compounds become available. In many cases, a patient’s underlying defect can be treated in different ways, skipping different exons, to create alternative repairs with potentially different properties and clinical outcomes. Currently, there is only one choice for exon skipping, eteplirsen (Exondys51), which targets exon 51.19 However, clinical trials for compounds targeting exons 44, 45, and 53 are underway (e.g., NCT02329769, NCT02500381, and NCT01957059, respectively, in the United States and other programs worldwide7,20,21), and preclinical programs exist for other exons. If development of the therapies targeting these new exons is successful, the prospect of patients and their physicians facing a choice about which to use will become a reality. Because different therapies will result in the expression of distinct dystrophin structures, a rational, structure-based basis for making this choice is needed.
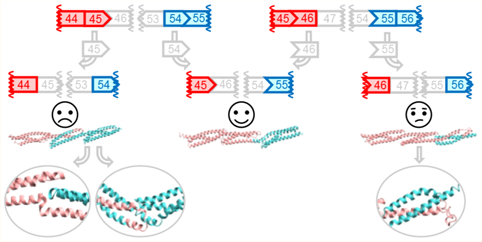
DMD-causing mutations are most commonly found in two hot spot regions of the gene where exons of different starting and ending reading frames are clustered. These occur in the central rod region of the protein that consists of 24 copies of the spectrin-type repeat (STR) rod domain.22,23 The largest and most significant of these is the hot spot 2 region, spanning exons 43–59, which corresponds to STRs D16–D24 (Figure 1). Within this region there are 64 possible exon-edited proteins. While the structures of all of these edited proteins can be applied to DMD therapy, here we are most concerned with pairs of edits that may be end points of alternative exon skip repairs of the same underlying DMD mutation. These cases represent those in which a choice of therapy exists and are more urgently needed in the near term as exon skipping choices become available. The most common type of choice occurs when the exon immediately before or after the original deletion is targeted. For instance, patients with an out-of-frame exon 52 defect could be treated to either skip exon 51 with eteplirsen to create an in-frame Δe51–52 skip or a potential exon 53 skip reagent creating Δe52–53. Not every defect has such simple alternatives, but many do: such pairs lie adjacent on a diagonal from the top left to bottom right in Figure 1, and there are 31 such edits. More possibilities arise with multiexon skipping, which is a more demanding task but has been studied in cell culture and animal models. Furthermore, in general terms, the question of “what edit to make” is of even greater importance with more flexible techniques, such as CRISPR gene editing, which although not yet in the clinic is widely viewed with great promise and potential.
Figure 1.

All possible exon edits in the hot spot 2 region, exons 42–60. Viable exon edits result in in-frame fusion if the ending phase of the 5′ remaining exon is compatible with the starting phase of the 3′ remaining exon. There are three possible reading frames for each exon boundary {<, |, >} corresponding to the protein reading frame at the exon junctions {−1 or one base before the end of a codon, 0 or at the codon boundary, 1 or one base into a codon}. The approximate STR boundaries are also shown in alternating green and yellow, although it must be emphasized that STR boundaries do not exactly correspond with exon boundaries. There are 64 in-frame fusions (f45>55 is a deletion of 46–54 fusing exons 45 and 55 at a phase 1 boundary). Blank cells are unviable, out-of-frame DMD defects. Alternative repairs occur when skipping of either the 5′ or the 3′ exon converts a DMD defect into an in-frame edit. These lie in pairs along a diagonal as shown. Of the 64 possible edits, 31 share this property. We selected three edits that comprise two alternative repairs: f44|54, f44>55, and f45|65, shown in bold and more conventionally referred to by the exons deleted, i.e., Δe45–53, Δe46–54, and Δe47–55, respectively.
Because there are a wide array of possible edits when both the entire dystrophin gene and future editing modalities such as CRISPR are considered, we are conducting studies to determine not only “which edit is best” but also the basic principles that make an edit better or worse. This involves studies at a range of scales from atomic to organismal. Here, we focus on the impact of the protein structure, because it is the dystrophin protein that performs the crucial muscle stabilizing function. We have selected one such set of three edits, Δe45–53, Δe46–54, and Δe47–55, encompassing alternative exon skipping repairs of two frameshifting DMD-type deletions, Δe46–53 and Δe47–54 (Table 1), and demonstrated that one of these, Δe46–54, produced a protein that is significantly more stable than the other two. Furthermore, we conducted molecular modeling and molecular dynamics (MD) simulations on these edits to elucidate the molecular basis of these differences. We found that the destabilizing edits did not appear to cause structural perturbations proximal to the edit site but rather function distally at STR junctions that became “non-wild-type” when regions away from the edit site that are not normally in contact are spatially juxtaposed. This provides the first molecular level understanding of these stability differences in exon-edited rods.
Table 1.
Exon Repairs in This Studya
| DMD-type defect (out of frame) | exon skip target | in-frame repair edit studied | |
|---|---|---|---|
| Δe46–53 | exon 45 | Δe45–53 | |
| exon 54 | Δe46–54 | ||
| Δe47–54 | exon 46 | Δe47–55 | |
| exon 55 |
Edits in this study were chosen to be alternative repairs of specific DMD-type defects.
METHODS
Concise methods are given, with complete details available in the Supporting Information. The dystrophin protein studied has UniProt entry P11532.
Target Selection.
To study the local perturbations introduced by exon skipping, we produced exon-edited targets in the context of a region that was as small as possible and has nativelike dystrophin rod ends.24,25 This entailed identification of biophysical domain boundaries of the dystrophin rod to locate appropriate termini. In previous work,26 we determined that D16 and D17 form a thermodynamically cooperating domain, as do D20–D22. On the basis of that, the boundaries of a minimal rod containing these edits were set at the N-terminus of D16 and the C-terminus of D22 (Figure 2). However, one of our edits, Δe47–55, removes large portions of D22 (53 of the 117 amino acids, 45%) leaving behind a fractional STR at the C-terminus. To ensure well-formed rod ends, we utilized a second cohort of experimental targets with a C-terminal boundary at D24. As such, we produced two series of exon-edited proteins, in both D16:22 and D16:24 parent molecules.
Figure 2.

Cloning strategy. Target proteins were expressed in both D16:22 and D16:24 parent molecules. At the top, the exon structure is shown in alternating pink and blue bars, aligned with the STR structure in this region. There are two alternative alignments describing the end points of dystrophin STRs as described in the text, KK and WGK. These differ by at most eight amino acids, so we utilized end points that are eight amino acids larger than the KK alignment to be compatible with both, as in our previous work on wild-type and exon-edited STRs. Polymerase chain reaction and Gibson assembly were used to achieve the exon edits (indicated by the dashed lines) to add glutathione S-transferase (GST) and His9 affinity tags.
Cloning and Protein Production.
Proteins were cloned, expressed, and purified as previously described.13 Briefly, plasmids containing the genes encoding the target proteins were assembled by the Gibson protocol27 using unedited dystrophin gene fragments previously studied and transformed into Escherichia coli NEB express, which is a commercially modified DH5α strain optimized for protein expression. Concordance of all constructed targets with the intended sequences as indicated in Figure 2 was confirmed by direct DNA sequencing over the entire dystrophin region. Proteins were expressed in a double affinity-tagged form utilizing an N-terminal GST (glutathione S-transferase) domain and a C-terminal His9 tag. Only the full-length, undegraded protein was selected by both affinity protocols. To remove any remaining non-full-length species, a final ion exchange purification step was used. All proteins were purified to a single band of >98% at the appropriate molecular weight as assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
Biophysical Characterization.
Proteins were characterized as previously described.13 Because STRs are triple-α-helical motifs, total helicity was measured by circular dichroism (CD) to assess how well-folded they were initially, which is reported as both mean residue ellipticity at 222 nm (φ222) and fractional helicity as determined by a three-basis set model28 that has been extensively used by us and others29–31 for STR helicity. To measure overall thermodynamic stability, we performed thermal denaturation as followed by CD, which provided the melting temperature, Tm, as well as the enthalpy of unfolding, ΔH. Here, we used the signal at 222 nm, φ222, which was collected every 0.2 °C and fast Fourier transform (FFT) filter smoothed (lowpass cutoff of 0.2 °C), and then the derivative, dφ222/dT, was fit to the John–Weeks equation to yield ΔH and Tm.32 Because unstructured regions are invisible to denaturation sensitive assays, we used sensitivity to protease challenge to assess how much and the extent to which the target protein contained poorly folded regions.33,34 To avoid primary sequence bias, we use a highly nonspecific protease, proteinase K (PK). The reported measure is the concentration of PK that achieved 50% degradation in a defined assay (30 min at 37 °C in PBS buffer), the PK50 value. This assay has been applied by us and others35 to a wide range of native and exon-edited dystrophin rods.
Our standard of reproducibility was that all data were acquired from at least three independent experiments on at least two independent batches of protein. Our standard of statistical significance is P < 0.05 (significant) or P < 0.005 (highly significant) using a two tailed Student’s t test as well as an effect size d of >2, where d is Cohen’s d = (μ1 –μ2)/s.
Computational Modeling.
Initial Model Development.
Because empirical studies of the three target edits produced similar results in both the D16:22 and D16:24 contexts, only the D16:22 case was pushed forward to computational studies due to its smaller size. The primary sequences of three targets in the D16:22 family were submitted to the Robetta automated structure prediction server.36 The output models were ranked by a combination of ProQ2 score37 and TM score,38 but all were similar in creating rodlike, STR-containing structures.
Implicit Solvent Molecular Dynamics Simulations.
The top model for each target was chosen for further computational study. These structures were processed to remove hydrogens in VMD,39 and then the topology and coordinate files were developed using the LEaP package of the AMBER software suite.40 Structures were minimized and equilibrated through a multistage protocol (more details can be found in the Supporting Information). We conducted six independent MD production runs (with independent equilibration), three of 2 μs and three shorter runs of 0.5 μs, with the first 250 μs discarded as further equilibration. The fact that 250 μs was sufficient equilibration time was justified by examining both the total helicity and backbone root-mean-aquare deviation (RMSD) (Figure S1) for lack of system drift after that time. Conformations after that were saved every 50 ps, for a total of 120000 coordinate sets for each target, which was the standard implicit data set. All implicit solvent calculations utilized the “GB-Neck2” solvation model and parameters developed by Nguyen, Roe, and Simmerling, which was enabled with the igb = 8 AMBER configuration keyword and the mbondi3 radii set.41 The SHAKE algorithm was used with a 2 fs time step and the AMBER ff14SB force field,42 along with the GPU-accelerated version of PMEMD.43 Simulations were performed on local resources as well as the Extreme Science and Engineering Development Environment (XSEDE).44
Explicit Solvent Molecular Dynamics Simulations.
These initial data sets were developed using an implicit solvent model, which is a trade-off between physical concordance and computational tractability. To examine the effect of this and provide reassurance that solvation effects were not unduly perturbing our data, an additional set of simulations was performed in an explicit solvent environment. As starting points for explicit simulation, the implicit standard data set for each target was clustered using the hierarchical agglomerative algorithm45 with average linkage. Representative structures from all clusters with >1.5% abundance (four for Δe45–53 and Δe46–54 and three for Δe47–55) were solvated at 150 mM NaCl and pH 7, relaxed by a multistep protocol, and subjected to three 250 ns explicit MD simulations. Only the last 150 ns was used for analysis (i.e., 450 ns for the three runs), and conformations were once again saved every 50 ps, for a total of 9000 coordinate sets for these three runs from each starting structure. This yielded a standard explicit data set of 36000 coordinate sets for the four-cluster run of Δe45–53 and Δe46–54 and 27000 for coordinate sets for the three-cluster run of Δe47–55.
Computational Analysis.
Standard data sets obtained as described above were analyzed for helicity using cpptraj,46 which itself uses the DSSP algorithm that detects an (i, i + 4) hydrogen bond.47 Inter-STR bending was assessed by defining an STR vector from the positions of conserved heptad hydrophobic residues (see Figure 6 and Figure S2 and the Supporting Information) in each STR, and then the angle between vectors of adjacent STRs was calculated with cpptraj. Pairwise intraresidue interaction energies within each STR were calculated by the Molecular Mechanics/Generalized Born Surface Area (MMGBSA) protocol,48 with the Generalized Born implicit solvent model49 using the MMPBSA.py script.50 Coordinated motions indicative of structural interactions were assessed by general motional correlation.51 Visualization was performed in VMD and modified in GIMP,52 and data plots were created by OriginLab and Matplotlib.53
Figure 6.

Initial models. All targets were modeled by Robetta to produce typical STR-like triple-α-helical bundles. Shown are Δe45–53 (top), Δe46–54 (middle), and Δe47–55 (bottom). The location of the edit site is at the transition from pink to blue. Junction sites J1 (left) and J2 (right) are colored darker pink and blue, respectively. The triads of internal hydrophobic sites (see also Figure S2) selected to define each “STR vector” are colored brown.
RESULTS
Experimental Section.
Secondary Structure Determined by CD.
All proteins exhibited circular dichroism (CD) spectra with double minima at ~208 and ~222 nm, which is characteristic of highly α-helical proteins (Figure 3). However, they varied significantly in either φ222 or the fractional α-helix content, fα, with Δe46–54 having the highest values, Δe47–55 close behind, and Δe45–53 having the lowest. These were consistent in both D16:22 and D16:24 parents. Crystal structures of non-dystrophin STRs show helicity values in the range of 85–90%, and spectroscopic studies of unskipped dystrophin 2-STR rods in this region show values in the range of 65–87%26 (Figure 4). By this measure, both Δe46–54 and Δe47–55 exhibit unremarkable helicity values consistent with typical STRs. On the other hand, Δe45–53 exhibits significantly lower helicity values, 36% in D16:22 and 49% in D16:24, that are not consistent with a well-formed STR-type structure and suggest a significant disruption in this edit’s structure.
Figure 3.

CD structure and stability. CD and thermal denaturation yield secondary structure and unfolding energies as measures of stability. The left panels show CD spectra at 25 °C, with cyan showing D16:22 and blue D16:24 parents. The right panels show raw thermal unfolding profiles and the derivative plots for each system for the temperature range of 0.5–98.5 °C. Fit curves in pink (D16:22) and red (D16:24) show the three-component secondary structure analysis of the CD spectra. Fit curves in the raw thermal denaturation profiles (center) indicate the FFT-filtered data, and the derivative plot (right) shows the thermodynamic fit used to determine Tm and ΔH (which is inversely proportional to peak width). The gray vertical line is at the physiologically relevant temperature of 37 °C. This shows the greater stability of Δe46–54 with respect to the other edits, especially with regard to ΔH as shown by the very sharp transition. Indeed, the other edits are beginning to unfold at physiological temperatures.
Figure 4.

Concordance of experimental stability measures. Various measures of good stable structures are compared for the three target edits in the D16:22 (cyan) and D16:24 (blue) contexts. In all cases, the Δe46–54 edit is more stable and shows better structure than either Δe45–53 or Δe47–55. All data shown are means of n > 3 experiments from at least two independently produced batches of protein, and error bars reflect one standard deviation. Thick lines above indicate statistically significant differences at the P < 0.005 level (two-tailed), and thin lines at the P < 0.05 level (all with effect size d > 2). Complete P and d values are listed in Table S1. For comparison, values for wild-type 2STR constructs in the region being studied are shown by gray dashed lines, as described in the text. Only the more stable Δe46–54 lies within the range for all measures.
Thermodynamic Stability Determined by Thermal Denaturation.
Structural stability was also probed by thermal denaturation as monitored by CD. The three edits all displayed typical sigmoidal unfolding transitions, in the range of 50–70 °C (Figure 3). By taking the derivative of the signal with respect to temperature, we can obtain thermodynamic properties of these transitions more accurately and, crucially, in a manner largely independent of spectroscopic parameters.32 This yielded melting temperatures in the range of 62–64 °C for all edits, which is in the range typically observed for STRs of 55–70 °C. Once again, Δe46–54 was the most stable, but by only a few degrees (Figure 4).
Although the melting temperatures were similar for all systems, the sharpness of the denaturation curves displayed large differences. This manifests in differences in the width of the peaks in the derivative plots and in the slopes of the transitions in the raw CD traces. Underlying this, the slope of the transition is proportional to the enthalpy of unfolding, ΔH, via the van’t Hoff equation [(d ln K)/dT = –ΔH/(RT2)]. The Δe46–54 edit has a transition significantly narrower than that of either Δe45–53 or Δe47–55 and yielded significantly higher ΔH values upon analysis [565 and 624 kJ/mol for D16:22 and D16:24 contexts, respectively (see Table 2)], which is consistent with, and even slightly larger than, unedited STR values in this region that vary from 304 to 539 kJ/mol26 (Figure 4). The other two edits, Δe47–55 and Δe45–53, exhibit broad low-enthalpy transitions, in the range of 163–215 kJ/mol, which is less than the values for typical STRs in that region.
Table 2.
Experimental Measures of Stability and Well-Foldednessa
| parent:edit | fa | φ222 (mdeg dmol−1 cm−2) | ΔH (kj/mol) | Tm (°C) | PK50 (μg) |
|---|---|---|---|---|---|
| D16:22:Δe45–53 | 0.36 ± 0.03 | 17.0 ± 0.9 | 164 ± 40 | 62.9 ± 0.9 | 1.8 ± 0.8 |
| D16:22: Δe46–54 | 0.93 ± 0.01 | 32.4 ± 0.5 | 565 ± 95 | 68.5 ± 0.6 | 9.5 ± 2.4 |
| D16:22: Δe47–55 | 0.80 ± 0.03 | 28.7 ± 0.6 | 220 ± 23 | 62.7 ± 0.6 | 1.5 ± 0.1 |
| D16:24: Δe45–53 | 0.49 ± 0.08 | 19.4 ± 2.0 | 215 ± 14 | 62.8 ± 0.4 | 0.4 ± 0.2 |
| D16:24: Δe46–54 | 0.88 ± 0.04 | 30.5 ± 1.5 | 624 ± 63 | 64.6 ± 0.3 | 3.6 ± 0.5 |
| D16:24: Δe47–55 | 0.72 ± 0.04 | 25.8 ± 1.1 | 194 ± 10 | 61.6 ± 0.5 | 1.0 ± 0.2 |
Error ranges are given as one standard deviation.
Importantly, with exon skipping therapy patients do not have the option to move back to the wild type but rather can choose between two alternative edits. As such, the most relevant comparison is to compare alternatives to each other, not to the wild type. In all measurements, Δe46–54 was significantly different from the other edits (P < 0.005) and more stable in terms of both Tm and enthalpy as shown in Figure 4. Conversely, no differences were seen between identical edits in the two expression contexts, D16:22 versus D16:24.
Global Folding Structure Determined by Protease Challenge.
The presence of regions so disrupted that they might be unfolded even at low temperatures is not detected by thermal denaturation techniques as they do not unfold with an increase in temperature. Such regions are, however, suggested by differences in helical content. To confirm the presence of these possibly unfolded regions, protease challenge was conducted, because proteolytic susceptibility is correlated with flexible and disordered backbone regions. Once again, Δe46–54 is the most well folded with PK50 values [proteinase K (PK) giving 50% degradation under our standard assay condition] of 13 ng in D16:22 and 5.1 ng in D16:24 (Figure 5). For comparison, the PK50 values of unskipped 2-STR rods in this region range from 8 to 22 ng (see Figure 4), suggesting that this motif is folded in a manner similar to that of unedited rods. The slightly lower value in the D16:24 context may arise from the D22:D23 junction that is not present in the D16:22 series. Mapping studies showed an apparent noncooperative junction between these two STRs.26 However, Δe47–55 and Δe45–53 both display significantly reduced PK50 values in both contexts, in the range of 0.6–2.3 ng. These values are all significantly different than that of Δe46–54 and well below these unedited rod PK50 values, consistent with significant destabilization of these two edits.
Figure 5.

Protease challenge. Target proteins were exposed to various concentrations of proteinase K under standard conditions and analyzed by SDS–PAGE. Molecular mass standards (leftmost or rightmost lane) are at 80, 60, 40, 30, 20, and 10 kDa. In the rest of the gel lanes, the level of proteinase K varies from high to low from left to right, respectively, as shown in the graphs. The amount of full-length protein remaining was assessed by densitometry and fit as described to an exponential decay, yielding a half-life value, PK50. Once again, Δe46–54 was the most stable as assessed by its resistance to proteolysis.
Various measures of stability and well-foldedness are listed together in Table 2 and are concordant as shown in Figure 4. In all cases, Δe46–54 is more stable and well-folded than Δe45–53 and Δe47–55 regardless of whether it was expressed in a D16:22 or D16:24 context. Once again, we judged the statistical relevance with P < 0.005 thresholds or in some cases as noted in Figure 4 (P < 0.05). Via comparison of the less stable edits, Δe45–53 was less stable than Δe47–55, with the exception of the ΔH of unfolding between these two in both parents and the PK50 in the D16:22 context, for which no statistically significant conclusion could be drawn. This strong agreement between such disparate techniques, as well as the agreement between parent molecules, provides confidence that the differences measured reflect underlying differences in the biophysical consequences of these edits on the dystrophin rod.
Computational Section.
Initial Models and Helicity.
The models produced by Robetta were all triple-helical STR-like structures (Figure 6). STR proteins are dominated by their three-bundle α-helices, so the simplest metric we examined both experimentally and computationally was total helicity. The experimental data showed significant differences in total helical content as assessed by CD, and our models reproduced the experimental rank order of Δe46–54 > Δe47–55 > Δe45–53, with helicities of 82.8, 81.7, and 80.7, respectively. However, while this order matches (see Figure S3) and is in general agreement for Δe46–54 and Δe47–55, these computational helicity differences are not statistically significant, in contrast to the experimental differences (Figure 4). Furthermore, the Δe45–53 value is well above the very low experimental value of 36% seen in the D16:22 context, which as previously noted is not compatible with a typical STR structure. In homology modeling in general, output structures are based on known, well-folded, stable, and unedited proteins; therefore, it is not surprising that we obtained such well-folded structures in all cases. Modeling methods thus may bias partially disordered proteins toward their most structured conformer, even while the dynamic behavior of these proteins may result in a lower degree of structure when assessed in a time-averaged fashion.
To assess the dynamic behavior of these models, a series of MD simulations were performed. Here we observed a loss of helicity. This was marginal in terms of the overall helicity, but an examination of this on a residue by residue basis showed that certain regions of the molecule exhibited a high degree of local unfolding (Figure 7). This primarily occurred in the junction regions between the STRs, which is where the third helix of the leading STR propagated into the first helix of the next STR. In the homology model structures, this junction consists of a continuous α-helix that propagates from the third helix of one STR propagates directly into the first helix of the subsequent STR without a break. This long continuous inter-STR helix is also seen in many crystallographic structures of multi-STR rods from homologous proteins,54–56 and this linkage is thought to contribute to the thermodynamic cooperativity seen in some, but not all, tandem STRs. It is also thought from prior computational studies that this local unwinding of the helix at the junction is an initial event in a force-related extension of STR rods, related to their mechanical stabilization role.57,58
Figure 7.

Initial and dynamic helicity during MD runs. Shown are the average helicity values at each residue during the three long 1.75 μs MD runs (red, green, and blue plots) for each target protein, compared to the initial model starting structure of each model. The edit site, ES, and Junction regions are colored yellow. This shows dynamic unfolding of the junction regions, but to different extents in the three edits studied.
Because the edit site is of particular interest and may result in a “scar” in the edited proteins, we also assessed helical unfolding in a 10-residue window centered on the edit sites. The full distribution of unwinding is shown in Figure 8 and summarized in Table 3 as the average number of residues that unfold (mean nonhelical gap size) and the fraction of frames with a gap.
Figure 8.

Helicity of the edit site, ES, and junction regions J1 and J2. The fraction of frames with gaps of nonhelical amino acids of various sizes in the J1, J1, and ES regions is shown. All three edit sites (ES) are largely helical and well-formed, whereas Δe45–53 has both J1 and J2 junctions with significant unfolding, in fact typically more than a full turn (3.6 residues) of an α-helix. This illustrates that a large portion of the effect of these edits is felt not locally, at ES, but distal to the specific edit site at the junctions. For Δe46–54, only junction J2, which partially overlaps with ES, was significantly perturbed. However, the low degree of ES unfolding shows this unfolding is skewed to the junction, not the edit site. Finally, Δe46–54 had junction sites and an edit site that remained largely helical. Junctions are classified as wild-type (wt) or non-wild-type (nonwt) as described in the text, and the nonwt junctions show larger losses of helicity.
Table 3.
Junction and Edit Site Unfolding Metricsa
| mean nonhelical gap size | fraction of frames with a nonhelical gap | |||||
|---|---|---|---|---|---|---|
| ES | Jl | J2 | ES | Jl | J2 | |
| Δe4S–53 | 0.01 | 2.22 | 1.77 | 0.01 | 0.74 | 0.78 |
| Δe46–54 | 0.12 | 0.59 | 2.59 | 0.06 | 0.26 | 0.89 |
| Δe47–55 | 0.46 | 0.50 | 0.55 | 0.21 | 0.25 | 0.30 |
Non-wild-type junctions, as described in the text and Figure 8, are highlighted in bold.
During simulations, the junction regions exhibited an extensive loss of helicity, especially in the least empirically stable edit, Δe45–53. For this edit, the average gap was ~2 residues for both J1 and J2, with larger gaps not being uncommon. Furthermore, the presence of a gap was a dominant structural feature, with ~75% of frames having a gap. This unfolding was heterogeneous and occurred at various spots in this junction region, with many residues adopting either a helical or unfolded conformation at different times (Figure S4). In addition, in each of the three runs there were differences in the helicity of J1, and to a lesser extent J2, of Δe45–53 as seen by nonoverlapping lines in Figure 7. In contrast, the most empirically stable edit, Δe46–54, displayed a well-structured J1, with a mean gap size of 0.5 and only 35% of frames showing a break. However, in J2, there was a mean gap of 2.6 residues present in 90% of the frames. For Δe47–55, which was empirically of intermediate stability, we observed well-structured J1 and J2 regions with low, <0.5 mean gap sizes and only a minority of frames showing a break.
In contrast, the edit site (ES) was comparatively well structured in all cases with <0.5 residue being nonhelical and only infrequently exhibited any nonhelical structure. In fact, the least stable protein empirically, Δe45–53, showed almost no ES perturbation, with only 1% of frames having a gap, and a mean gap of only 0.01 residue. This strongly suggests that, at least for some exon skip events, the impact of the edit on protein structure does not occur locally. The empirically most stable target, Δe46–54, showed a greater degree of ES helicity loss, at 6% of frames and a mean gap of 0.2 residue, which is a comparatively minor perturbation and smaller than the loss of helicity at the junction sites. Only for Δe47–55 was significant perturbation observed, where 21% of frames had some gap, with a mean gap size of 0.46. While this does not approach the size of the gap seen for the perturbed junctions (either junction of Δe45–53 or J2 of Δe46–54), it is comparable to the relatively unperturbed junctions of this protein and is a disturbance that is larger than that seen for nonedit regions in the three helices of each STR.
Rod Bending.
We then sought to understand the impact this loss of junction structure has on the overall rod shape. Because these rods link and stabilize various cellular components during muscle cell functions, their rigidity and shape directly impact their function. Therefore, a continuous inter-STR helix should enforce the classic linear STR rod seen in many crystal structures of non-dystrophin STRs (there are no known multi-STR atomic resolution structures from dystrophin, only one single STR dystrophin structure59), whereas a loss of helicity to a more random coil-type junction may allow for bending motion. This may result in an entropic spring-type action60 or could be a prelude to further unfolding under stress, in a more classical Hookian spring mechanism. To assess this bending, for each STR an “STR vector” was defined on the basis of hydrophobic residues in the amphipathic AbcDefg heptad located at the ends of the STR hydrophobic core (Figure 6, brown residues and S2 heptad), and the angle between adjacent STR vectors was measured.
This loss of junction helicity is directly associated with increased rod flexibility (Figure 9), and the least stable edit, Δe45–53, is significantly more flexible as a result of this unfolding. Correlations of the inter-STR angles angle 1 (A1) and angle 2 (A2) show that for all edits there is a preferred conformer with modest inter-STR angles in the range of 40–60°. However, for Δe45–53, there is a distinct second population with a highly bent A1 of >90°. This highly bent A1 conformer occurs with significant J1 unfolding of only >4 residues (but not J2) (Figure 9, A1 vs J1 map). Loss of helicity is thus seen to allow, but not create, a highly bent structure. For Δe46–54, no distinct highly bent population is observed, although there are rare conformers with highly bent A1 that are also correlated with loss of J1 helicity. For Δe47–55, there are few conformers with large bends of >90° (<0.1%), in keeping with the very low degree of unfolding in this edit. This illustrates that edit-induced junction perturbation impacts the global properties of the rod, such as its rigidity.
Figure 9.

STR bending in relation to junction helicity. The left side shows the correlation between A1 and A2 within each edit, and the right side the correlation between angle bending and loss of J1 or J2 helicity. Δe43–55 show two distinct populations based upon bending of angle A2. We note that in increases in A1 are correlated with an increased level of unfolding at J1 (but not J2), suggesting that loss of helicity here is contributing to the increased flexibility of this edit. For A2, both Δe45–53 and Δe46–54 have a broad distribution centered around ~45° but extending beyond 90°; these are also both of the edits that exhibit significant J2 unwinding (8). In contrast, Δe47–55 showed almost no loss of J2 helicity [<30% of frames with any unfolding and a mean gap size of 0.55 (see Table 3)] and angle 2 distributed in a lower and more narrow range centered around 15°.
Motional Correlation Analysis.
In two cases, “hybrid STRs” consist of parts of two different naturally occurring STRs occur: Δe45–53 where the second STR consists of a portion of D17 and portion of D21 and Δe47–55 where the third STR consists of D18 and D22 (see Figures 2 and 6). To assess how this impacts internal STR dynamics, motional correlation analysis was performed (Figure 10). Well-formed and tightly interacting domains are expected to exhibit a high degree of correlation, whereas looser or disturbed domains less correlation. Interactions between adjacent secondary structural elements can be seen, and the triple α-helical bundle STR structure provides a unique signature, with three well-defined helix-bundling interactions seen: the two antiparallel AB and CB linkages and the parallel AC linkage producing a characteristic “STR box” signature (see Figure S5 for a guide to interpreting STR correlation maps). This pattern is present in most of the correlation maps, in particular those of Δe45–53 and Δe46–54, indicating a high degree of interaction and tight helix–helix interactions.
Figure 10.

Correlation analysis of residue motion for each edit. Locations of ES are highlighted with blue lines. Intra-STR interactions are seen by a characteristic STR box motif (see Figure S5 for interpretation). We see this very easily in Δe45–53 and Δe46–54 and even in the second and somewhat in the first STR of Δe47–55. However, the third STR of Δe47–55 is significantly disrupted, with long helical correlation breaking up into much shorter-range interactions. We note that this is not a general consequence of edit site (ES) location, as ES in Δe45–53 is squarely in the middle of STR2, which is not perturbed. Off-diagonal inter-STR interactions are seen strongly for J1 but are absent for J2 of Δe46–54, in agreement with the increased level of unfolding of J2 but not J1 for this edit (as seen in Figure 8).
In contrast, in the Δe47–55 correlation map, this box signature is present in only the first and second STRs, while the third, which contains the ES in this target, is highly disrupted. Rather than three long individual, and highly correlated, regions, there is a scattering of smaller, local regions with less long-range motional correlation in all three helices, indicating a loss of long-range internal interactions. The Δe45–53 target also contains a hybrid STR with an internal ES; however, in this case, the long-range correlations are retained, and this edit does not appear to disrupt global STR interactions. In the case of Δe46–54, the edit site lies in the second STR but relatively close to the STR2–STR3 junction and also appears not to perturb long-range interactions.
Finally, the overall level of correlation is higher for Δe45–53 than for the other proteins, while that of Δe47–55 is the lowest. This is a result of the increased flexibility of Δe45–53 (and decreased flexibility of Δe47–55) as demonstrated by, and corroborating, the STR angle analysis. This results in correlated motion of all atoms in each STR as they bend relative to the rod as a whole. This is confirmed as this excess correlation disappears when the correlation is calculated only within individual STRs (Figure S6).
Energetics of STR Motifs.
The internal energetics of each STR were estimated using an MMGBSA energy analysis of each STR in each target protein. This is not a true measure of folding stability, which is the difference in energy between the folded and unfolded states, as the unfolded state is not sampled in these simulations. However, the total internal energy can give an indication of the thermodynamic forces involved in maintaining the STR structure and how the edits may perturb them. Here, all molecules have similar internal energies for their first STR (Figure 11A). This is sensible, because the first STR in all cases is a wild-type STR, the 16th in native dystrophin, and has an identical amino acid sequence in all targets. However, the second STR has a significantly lower stabilizing energy in Δe45–53, and the third STR has a lower stabilizing energy in Δe47–55. These two are precisely the STRs where the edit site is located and therefore are non-wild-type, hybrid STRs consisting of sections of two different wild-type STRs: a hybrid D17/21 in the case of Δe45–53 and a hybrid D18/22 in the case of Δe47–55. Therefore, while these hybrid STRs can form, they appear to have reduced stability, the extent of which may vary depending on the specific nature of the STRs hybridized. This is in accord with the loss of internal STR structure seen in the correlation matrices for these STRs. Once again, only Δe46–54 escapes this, with none of its three STRs energetically perturbed. In that case, the edit occurs near the end of D18, which is largely intact, with only a comparatively small section of D21 (approximately five amino acids) fused to it. In this sense, STR2 of Δe46–54 is “almost” wild-type.
Figure 11.

MMGBSA energy of interaction. (A) Total pairwise interaction for all residues within each STR of each edit. Values shown are averages calculated over the three 1.75 μs runs, with error bars of one standard deviation. Statistical significance at the criteria specified [P < 0.005; d > 2 (all values listed in Table S2)] is indicated by bars near the top. STR1 is a wild-type (wt) STR, does not contain any edit sites, and has the same primary sequence for all targets; the total STR energies are quite similar. The edit site in Δe45–53 occurs in STR2, which is a hybrid (hyb) STR in this case, consisting of part of wild-type D17 and D21; it exhibits an energy that is lower than that of STR2 of Δe46–54 or Δe47–55. Similarly, STR3 of Δe47–55 is a hybrid STR in this target; it too exhibits an energy that is lower than that of STR3 of the other targets. For Δe46–54, the edit site occurs near the end of STR2, very close to its junction with STR3, and STR2 is very nearly wild-type, indicated as wt*. (B) Individual pairwise interaction energies for the W277 and W284 heptad triads of Δe45–53 and Δe46–54 and the homologous V270 triad of Δe47–55 (see Figure S2 for the position of these in the alignment and Figure 6 for a view of these positions in the models). We note that when the edit replaced this W with a smaller and less hydrophobic V, the interaction energy is reduced and is an example of how the overall destabilization occurs for these edited STRs.
We also identified specific amino acid level interactions that are perturbed in the Δe47–55 case. We noticed that the region near the C-terminus of STR3—the perturbed, hybrid STR in this target—was particularly destabilized and appeared to partially unbundle, up to the first hydrophobic triad (Figure S7). In the other edits, this site is a tryptophan, but it has been replaced with a much smaller and less hydrophobic valine in Δe47–55. Tryptophan residues are particularly well conserved in STR motifs and have been proposed to strongly contribute to their stability.61 We thus examined the energetic interactions between these triad partners and found that they were significantly weakened (Figure 11B). While the reduction is only a modest fraction of the reduction seen in the overall STR energy, it shows how perturbation of these heptad triads can be significant.
Explicit Solvent Simulations.
The majority of our simulations were performed using an implicit solvent model as a trade-off between physical reality and computational feasibility. Implicit solvent models are less physically realistic and may skew results due to the solvation effect; however, explicit solvent models suffer from slower simulation speeds and therefore lower rates of sampling, thereby introducing a different source of error. To understand whether solvent effect biases impacted our results, additional explicit solvent simulations were performed starting from conformers derived from a clustering analysis of the implicit solvent standard data set. If these conformers were nonphysical as a result of differences between the implicit and explicit solvent force field, then we would expect that those conformers would drift from their initial cluster when subjected to explicit MD simulations. This did not happen, as shown by the RMSD analysis in Table 4. All runs stayed within their starting cluster on average, with the sole exception of the third cluster of Δe46–54, which was a minor conformer representing only 2.2% of the implicit solvent data set. An evolution of this RMSD distance over time is shown in Figure S8.
Table 4.
Explicit MD Movementa
| rmsd from | ||||||
|---|---|---|---|---|---|---|
| cluster | abundance | size | 1 | 2 | 3 | 4 |
| Δe45–53 | ||||||
| 1 | 0.62 | 21.1 | 13.5 | 27.0 | 21.3 | 23.2 |
| 2 | 0.17 | 19.7 | 30.6 | 20.9 | 26.2 | 35.8 |
| 3 | 0.06 | 18.0 | 23.5 | 24.3 | 17.2 | 29.9 |
| 4 | 0.06 | 19.6 | 20.5 | 32.5 | 27.6 | 20.5 |
| Δe46–54 | ||||||
| 1 | 0.88 | 20.6 | 13.9 | 24.7 | 19.8 | 22.6 |
| 2 | 0.03 | 21.4 | 25.8 | 16.1 | 28.4 | 31.7 |
| 3 | 0.02 | 18.7 | 22.1 | 29.4 | 25.0 | 14.8 |
| 4 | 0.02 | 19.4 | 21.3 | 28.7 | 24.5 | 14.3 |
| Δe47–55 | ||||||
| 1 | 0.92 | 19.1 | 12.0 | 19.6 | 20.0 | |
| 2 | 0.03 | 18.8 | 18.3 | 17.9 | 24.7 | |
| 3 | 0.02 | 20.4 | 18.4 | 25.1 | 16.0 | |
Explicit MD runs were begun from conformation points produced by cluster analysis of the implicit standard data set. Clusters with >1.5% abundance were used (four for Δe45–53 and Δe46–54 and three for Δe47–55). The abundance and size (rmsd) of each cluster are indicated. We then monitored the average rmsd of each trajectory from the representative structures of each of the other clusters. If solvation differences between implicit and explicit runs perturbed the energetics, then some clusters would be expected to rapidly evolve away from their starting points toward another cluster. The lowest-rmsd (closest) cluster is highlighted in bold. In fact, most stayed closest to their starting structure and within the size of the starting cluster. The sole exceptions are the lowest-abundance cluster in Δe45–53, which appeared to evolve to a point midway between cluster 1 and cluster 4, and the third cluster of Δe46–54 which appears to have evolved into another very low abundance (2%) cluster 4. This suggests that solvation model difference effects are not extreme.
The fact that none of these conformers was rapidly perturbed (i.e., within the three 250 ns simulations per system) when moved to an explicit solvent model suggests that the differences between these solvation models are not severe and do not greatly impact the conformers obtained. While all conformers are in equilibrium and interconvertible during simulations, either explicit or implicit, the longer more rapid pace of implicit simulations allowed us to more fully sample the conformational space. We also examined the conformations of all edits during the explicit runs, redoing the helicity and bending analysis, and no great differences were observed. This is shown in dynamic per residue helicity (Figure S9, homologous to Figure 6), junction and edit site helicity loss (Figure S10, homologous to Figure 8), and the overall rod bending (Figure S11, homologous to Figure 9).
DISCUSSION
Our results indicate that Δe46–54 is more stable than and structurally more similar to unskipped dystrophin rods than the other two edits studied, with Δe47–55 being intermediate and Δe45–53 being the least stable and well-formed. Consistent results were obtained across several empirical stability measures, regardless of the parent rod in which it was expressed, D16:22 or D16:24. Helicity, PK50, and thermal denaturation measurements, which probe the order, disorder, and stabilization energy of the folded form, respectively, give the same overall order: Δe46–54 > Δe47–55 > Δe45–53. This consistency between the parent rod and measurement types provides confidence that these differences are intrinsic to the edit and not some peculiarity of the context in which the edit is studied or the techniques used to probe it. Altogether, we obtain the following empirical picture of these edits. (1) Δe45–53 is an edit that produces a perturbed structure of low helicity inconsistent with STR structure and has significant disorder and significantly reduced stabilization enthalpy. (2) Δe46–54 is an edit that produces a typical STR-type structure that is well-formed with few disordered regions, high helicity, and typical unfolding thermodynamics. (3) Δe47–55 is also a perturbative edit resulting in a significant disorder, but while the structure has reduced stability when thermally challenged, it does have appropriate helicity at low temperatures and so might be considered to be a marginal edit.
MD To Understand the Molecular Origins of Experimental Effects.
However, as much as we can determine the phenomenological properties of these edits by this type of characterization, we cannot understand at a molecular level why some edits seem to produce viable structures or, conversely, why some do not. Unfortunately, there are no high-resolution structures of dystrophin rods in this region to help understand how such edits might manifest at an atomic level, although there are many known homologous STR structures from other proteins. To work around this, we developed homology-based models and used molecular dynamics simulations to help understand the molecular basis of these differences.
The models produced were consistent with STR structures seen in other proteins and were grossly consistent with the experimental evidence that these are structures dominated by α-helices. However, they were more helical than our experimentally produced structures as assessed by CD. We believe this is partially due to a bias in many homology algorithms that may favor highly structured outputs. This idea is supported by our MD simulations, which showed that helicity dropped somewhat once the molecules were equilibrated to 300 K and further reduced by local unfolding in a dynamic fashion during long runs. While we did come closer to the helicity values for the least empirically structured motifs, we did not obtain full agreement. However, we were able to simulate only on the order of 2 μs, which is very short compared to the time scale of the in vitro experiments (minutes to hours).
Nonetheless, our computational results did reproduce the experimentally observed stability and have the virtue of being able to provide insights into why and how this instability arises in some edits but not others. The most important conclusion is that in some cases, the exon editing has impacts well away from the edit site. In our least stable motif, Δe45–53, the edit site was in fact very well structured. This edit site occurs in the middle of the second STR in the target and is almost completely helical even in dynamic fashion during runs. However, the edit induced changes in the STR junction regions. With the juxtaposition of new residues that are not normally in contact with one another, these junctions are destabilized and begin to unfold, and MD runs show a high degree of loss of helicity at both junctions for this edit.
Junctions can be classified as either wild type, consisting of only native-type contacts, or non-wild type, containing some non-native contacts. Each STR consists of three helices, A, B, and C, connected by A–B and B–C loops. At each junction, the A–B loop of the first STR interacts with the junction-spanning helix, as does the B–C loop of the following STR. In some cases, the two loops also interact directly. We see that for J1 of Δe46–54 and Δe47–55, these elements (A–B loop, junction-spanning helix, and B–C loop) all lie on the same side of the edit, colored pink in Figure 6, and thus are unchanged from those of wild-type unedited dystrophin. However, for J1 of Δe45–53, the B–C loop comes from the region after the edit, blue, which creates a non-wild-type, non-native interaction. A similar look at J2 shows that all J2’s juxtaposed the region from the other side of the edit site and thus are all non-wild type. In Table 3, we see that wild-type junctions (J1 of Δe46–54 and Δe47–55) are strongly helical, with a low frequency of gap formation and in small gap sizes when formed. In contrast, the non-wild-type junctions (J1 of Δe45–53 and J2 of all targets) are less helical with both more frequent breaks and larger gap sizes. This demonstrates that the impact of edits may occur well away from the edit site by inducing unfolding at non-native junctions.
So far, for two targets, Δe45–53 (our least empirically stable) and Δe46–54 (most stable), MD has provided a reasonable, if somewhat surprising, narrative for how these edits impact structure and stability: they do not act directly on the edit site but rather work distally by destabilizing the STR junctions. This leads to junction unfolding and presumably instability.
Unfortunately, Δe47–55 does not conform to this explanation. Its junction regions show the least loss of helicity of any target, in both time and extent, and its ES region shows the greatest perturbation, albeit modest compared to those of junction regions. This perturbation is seen as a dip in the helicity half of the second helix of STR3 of Δe47–55 near residues 260–280 in the per-residue helicity plot (Figure 7). A much smaller dip is seen near ES of Δe46–54. For this edit, MD shows that the experimental destabilization occurs directly due to ES site perturbation. Correlation analysis demonstrated loss of long-range interaction, and energy analysis showed a reduced overall inter-residue interaction energy within the edit containing STR. This paints a complex picture; in some cases, the edits act locally, whereas in others, they are locally accommodated in the STR structure but their effects propagate to STR junctions where they have a large impact on structure.
Relevance to Exon Skipping Therapy.
As exon skipping therapy has been developed, most of the effort has gone into simply getting it to work: finding a pharmacologically acceptable compound that is effective in inducing some exon skipping in enough tissue to provide a clinically meaningful benefit. With the recent approval of eteplirsen, a milestone has been reached, although there is certainly a long way to go in improving both delivery and skipping efficiency and in expanding the range of exons targeted to bring this treatment option to more patients. We are now at the stage where skipping for many, if not all, exons is scientifically within reach.6,7,62 However, even in the limit of perfect exon skipping, the protein that is created will have some edit and may therefore be functionally different from wild-type dystrophin. This results in DMD patients essentially becoming therapeutically created BMD patients, and in many cases, patients and clinicians may have choices in determining which form of BMD to aspire to.
Retrospective studies of BMD patients provide some guidance on how to choose but also demonstrate great heterogeneity and uncertainty. There are many thousands of known mutations for DMD and BMD, meaning that many specific edits are rare. Given these small sample sizes and the fact that patients with the same underlying exon deletion can progress at highly different rates,17,18,25 it is challenging in most cases to compare one edit against another. To get around this sampling problem, the various BMD defects have typically been analyzed in structurally related groups to increase sample size and statistical power. This has demonstrated correlations with the underlying structure of the protein on a general level.18 However, this grouping may obscure the underlying physical causes of an ailment if dystrophin molecules with diverse tertiary structures are analyzed together.
Clinical relevance has been most clearly demonstrated in a relationship between BMD defects and dilated cardiomyopathy63 (DCM), a leading cause of mortality in BMD. In that study, to achieve statistical power, many edits were grouped on the basis of their features in relation to the STR structure of the rod, and certain groups were associated with a large delay in the onset of DCM. “In-phase” edits that were expected to have less perturbed STR structures progressed more rapidly, with an 11-year difference in age of DCM diagnosis than “out-of-phase” edits that were expected to have more perturbed STR structures. While this study showed a link between DCM severity and edit structure, it also emphasized that patients with the same underlying defect can progress at very different rates. For instance, the most rapidly progressing patient in the more favorable in-phase cohort (patient 109; DCM at 15 years of age, Figure 3A in that paper) was more severely afflicted than any of the supposedly worse-off out-of-phase group. However, in aggregate, this clinical evidence shows that the underlying protein structure is important (i.e., the significant delay in DCM) even if variability of progression may be an inherent part of BMD.
We believe that factors relating to structural characteristics of the edit at the protein level are what underlie this clinically significant correlation. Dystrophin is a structural protein whose primary function is to provide a mechanical linkage in muscle cells. It also has multiple scaffolding and organizational functions, e.g., organizing the DGC and NOS.11,64,65 All of these rely on the proper folding, structure, and stability of this protein and might easily be perturbed in edited proteins, either natural, as in BMD, or therapeutically produced, as in exon skipping or other therapies.
Relation to Clinically Observed Cases.
We note that two of the edits corresponding to the ones studied here have been clinically observed, Δe45–53 and Δe46–54, and are found 95 and 4 times, respectively, in the public Leiden database (ref 66, September 2017 update). Δe47–55 is not found. The underlying DMD-type deletions that might be repaired by exon skipping to these edits have also been clinically observed: Δe46–53 is noted 25 times, and Δe47–54 is found five times. Of course, this database is not an unbiased survey of the human population but is composed of only clinically observed cases and therefore is biased toward those seeking medical attention. All DMD patients require medical attention, and many BMD patients do, as well, as they progress. However, whether there is a cohort of subclinical BMD-type deletions out there who are unknown to the scientific community because they are so subclinical that they do not seek help is an open question. Such subclinical edits are the “holey grail” of exon skipping but are likely under-represented in this database.
The creation frequency of deletions is related to two factors: size of the regions in which the breakpoints must lie (i.e., the size of the flanking introns) and the length of the deletions, with the length dependence scaling with an exponent of −1.5.67 Using this, we might thus calculate a cross section for each exon edit from the known sizes of dystrophin exons and introns; this is shown in Table 5. Other factors such as chromatin structure and CpG islands are also important but appear to make only a minor contribution to mutational hot spots.68 This explains the observed prevalence somewhat, as we can see that intron 46i is quite small, reducing the observed prevalence of Δe47–54 and Δe47–55, whereas intron 44i is quite large, increasing the prevalence of Δe45–53.
Table 5.
Observed Frequencies of Relevant Edits and Breakpoint Intron Sizesa
| size | length | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| deletion | 5’ intron | 3’ intron | maximum | minimum | cross section | prevalence | ratio | ||
| DMD | |||||||||
| Δe46–53 | 45i | 36111 | 53i | 21230 | 310195 | 252852 | 5.1 | 25 | 4.9 |
| Δe47–54 | 46i | 2334 | 54i | 30127 | 304218 | 271755 | 0.5 | 5 | 11.0 |
| BMD | |||||||||
| Δe45–53 | 44i | 248401 | 53i | 21230 | 558772 | 289139 | 19.1 | 95 | 5.0 |
| Δe46–54 | 45i | 36111 | 54i | 30127 | 340477 | 274237 | 6.4 | 4 | 0.6 |
| Δe47–55 | 46i | 2334 | 55i | 120219 | 424627 | 302072 | 1.3 | 0 | 0.0 |
The cross section is calculated form intron sizes and overall length as described in the text, and the prevalence is as observed in the Leiden clinical database at the time of writing as described in the text.
Selection is also important. The DMD alleles are under extremely strong X-linked selection, because virtually all males with DMD do not reproduce. As expected, this 100% culling of males results in a mutation-selection equilibrium in which one-third of cases are de novo mutations, with the rest being created in female carriers in the prior few generations and little long-term inheritance.69 However, for BMD-type edits, this is not the case, with a much greater carrier incidence observed, ~90%,70 demonstrating that BMD alleles overall are under lower selection pressure, which should increase their incidence relative to that of DMD. When examining the ratio of database incidence to this cross section, we find the opposite: the DMD-type deletions and one of our BMD-type edits are represented at a similar frequency, and two edits in particular seem underrepresented (Δe46–54 and Δe47–55). The evidence for the abnormally low prevalence of Δe47–55 is weak, because none are reported whereas only approximately six might be expected using a prevalence to cross section ratio of 5. However, for Δe46–54, the evidence is stronger: four cases have been reported, whereas 32 might have been expected. Interestingly, Δe46–54, based on the biophysical evidence, is the edit most concordant with wild-type properties. Conversely, Δe45–53 is significantly destabilized and is also observed in the same ratio to its expected frequency as DMD defects in this region, suggesting that it is severe enough that these patients seek medical attention at rates similar to those of severely affected DMD patients. We have identified one report of this edit in the literature that has clinically meaningful progression data. In that case, the Δe45–53 patient has an age of DCM onset of 36 years,18 significantly below the median of 43 years in the relevant cohort in that study, making it a comparatively severe case as expected from the analysis described above.
Consequences.
Empirically, this study shows that alternative exon skip repairs can result in repaired proteins of very different stabilities. In this case, we found that the Δe46–54 repair is significantly more stable than the Δe45–53 or Δe47–55 repair. This means that DMD defects Δe46–53 or Δe47–54 can be repaired in two different ways, with different consequences with respect to the nature of the protein produced. While the experimental evidence is clear on which edit is more stable, it does not shed light on the molecular details of why this is. Computational studies suggest that this can arise in different ways. Perturbation of the edit site is the most obvious way and is the case for Δe47–55, where most of the disrupted residues are in the ES region. Juxtaposing amino acids that did not evolve to be in the proximity might quite naturally be expected to produce suboptimal interactions and reduced stability. However, a subtler effect is seen in Δe45–53, which has the largest experimentally observed destabilization. In this case, the edit produces changes in the global tertiary structure that destabilize regions distal to the edit site, in particular the junction regions between adjacent STRs. In all known high-resolution structures of multi-STR rods, the last helix of the leading STR propagates directly into the first helix of the next. Furthermore, the loop regions of the other helices (A–B loop of the leading STR, B–C loop of the following STR) are in the proximity of and interact with these junction regions. This then provides a model for how these edits perturb these structures: by the placement of different and unnatural combinations of these two loops and the junction helix together at each junction, essential stabilizing interactions that are present in naturally evolved tandem STR junctions are lost. In some cases, this may fortuitously result in productive interactions (i.e., in our most stable edit, Δe46–54), whereas in others, it might be destabilizing. This shows the idiosyncratic nature of exon editing and the value of both experimental and computational analysis of each edit in identifying such compatible and incompatible junctions. Overall, our MD results suggest that the consequences of an exon edit can impact structure in a number of distinct ways.
This heterogeneity may be less than satisfying in terms of providing a simple or uniform explanation for exon edit stability and suggests that a simple heuristic understanding of the exon edit impact at the protein level may not be possible. However, it illuminates possible origins of these differences and emphasizes that each repair should be understood on a case by case basis. This highlights the need for a thorough analysis of each edit and suggests that a combined experimental and computational approach can provide meaningful comparisons in cases in which alternative exon skipping repair is possible.
This is most crucial in cases in which a clinical choice may be made. Currently, there is only one choice for exon skipping, eteplirsen (Exondys51), which targets exon 51. However, clinical trials for compounds targeting exons 44, 45, and 53 are underway (i.e., NCT02329769, NCT02500381, and NCT01957059, respectively), and preclinical programs exist for many other exons. If development of the therapies targeting these new exons is successful (and there is no reason to think they will pose any greater regulatory hurdles than exon 51), the prospect of patients and their physicians facing a choice about which to use will become a reality. Because the disease is slowly progressive, it seems likely that the consequences of this choice will manifest only after years of therapy. Some serious symptomology such as DCM generally develops only in the second or later decades of life and may be exacerbated, or ameliorated, by the choice of therapy made decades earlier, and careful consideration of this choice may be important for optimal clinical outcomes.
Supplementary Material
ACKNOWLEDGMENTS
The computational aspects of this work were supported by the National Institute of General Medical Sciences of the National Institutes of Health (Grant 1R35GM119647). This work used the Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation Grant ACI-1053575.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00062.
(PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, Weber DR, and DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17, 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Falzarano MS, Scotton C, Passarelli C, and Ferlini A (2015) Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 20, 18168–18184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hamuro L, Chan P, Tirucherai G, and AbuTarif M (2017) Developing a Natural History Progression Model for Duchenne Muscular Dystrophy Using the Six-Minute Walk Test. CPT: Pharmacometrics Syst. Pharmacol. 6, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Khajavi M, Inoue K, and Lupski JR (2006) Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet 14, 1074. [DOI] [PubMed] [Google Scholar]
- (5).Linde L, and Kerem B (2008) Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 24, 552–563. [DOI] [PubMed] [Google Scholar]
- (6).Aartsma-Rus A, Straub V, Hemmings R, Haas M, Schlosser-Weber G, Stoyanova-Beninska V, Mercuri E, Muntoni F, Sepodes B, Vroom E, and Balabanov P (2017) Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 27, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Nakamura A (2017) Moving towards successful exon-skipping therapy for Duchenne muscular dystrophy. J. Hum. Genet 62, 871–876. [DOI] [PubMed] [Google Scholar]
- (8).Menhart N (2006) Hybrid spectrin type repeats produced by exon-skipping in dystrophin. Biochim. Biophys. Acta, Proteins Proteomics 1764, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, and Chamberlain JS (2002) Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med 8, 253–261. [DOI] [PubMed] [Google Scholar]
- (10).Le S, Yu M, Hovan L, Zhao Z, Ervasti J, and Yan J (2018) Dystrophin As a Molecular Shock Absorber. ACS Nano 12, 12140–12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, and Duan D (2009) Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest 119, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sarkis J, Hubert J-F, Legrand B, Robert E, Chéron A, Jardin J, Hitti E, Le Rumeur E, and Vié V (2011) Spectrin-like repeats 11–15 of human dystrophin show adaptations to a lipidic environment. J. Biol. Chem 286, 30481–30491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sahni N, Mangat K, Le Rumeur E, and Menhart N (2012) Exon edited dystrophin rods in the hinge 3 region. Biochim. Biophys. Acta, Proteins Proteomics 1824, 1080–1089. [DOI] [PubMed] [Google Scholar]
- (14).Le Rumeur E (2015) Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosnian J. Basic Med. Sci 15, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zimowski JG, Pilch J, Pawelec M, Purzycka JK, Kubalska J, Ziora-Jakutowicz K, Dudziñska M, and Zaremba J (2017) A rare subclinical or mild type of Becker muscular dystrophy caused by a single exon 48 deletion of the dystrophin gene. J. Appl. Genet 58, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Anthony K, Arechavala-Gomeza V, Ricotti V, Torelli S, Feng L, Janghra N, Tasca G, Guglieri M, Barresi R, Armaroli A, Ferlini A, Bushby K, Straub V, Ricci E, Sewry C, Morgan J, and Muntoni F (2014) Biochemical Characterization of Patients With In-Frame or Out-of-Frame DMD Deletions Pertinent to Exon 44 or 45 Skipping. JAMA Neurol 71, 32–40. [DOI] [PubMed] [Google Scholar]
- (17).Findlay AR, Wein N, Kaminoh Y, Taylor LE, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Howard MT, Day JW, McDonald C, Nicolas A, Le Rumeur E, Weiss RB, Flanigan KM, and United Dystrophinopathy Project (2015) Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol 77, 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kaspar RW, Allen HD, Ray WC, Alvarez CE, Kissel JT, Pestronk A, Weiss RB, Flanigan KM, Mendell JR, and Montanaro F (2009) Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ.: Cardiovasc. Genet 2, 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aartsma-Rus A, and Krieg AM (2017) FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 27, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Komaki H, Nagata T, Saito T, Masuda S, Takeshita E, Sasaki M, Tachimori H, Nakamura H, Aoki Y, and Takeda S (2018) Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med 10, eaan0713. [DOI] [PubMed] [Google Scholar]
- (21).Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S, and Aoki Y (2016) Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am. J. Transl. Res 8, 2471–2489. [PMC free article] [PubMed] [Google Scholar]
- (22).Djinovic-Carugo K, Gautel M, Ylänne J, and Young P (2002) The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett. 513, 119–123. [DOI] [PubMed] [Google Scholar]
- (23).Nicolas A, Delalande O, Hubert J-F, and Le Rumeur E (2014) The spectrin family of proteins: a unique coiled-coil fold for various molecular surface properties. J. Struct. Biol 186, 392–401. [DOI] [PubMed] [Google Scholar]
- (24).Findlay AR, Wein N, Kaminoh Y, Taylor LE, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Howard MT, Day JW, McDonald C, Nicolas A, Le Rumeur E, Weiss RB, Flanigan KM, and United Dystrophinopathy Project (2015) Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol 77, 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nicolas A, Raguénès-Nicol C, Ben Yaou R, Ameziane-Le Hir S, Chéron A, Vié V, Claustres M, Leturcq F, Delalande O, Hubert J-F, Tuffery-Giraud S, Giudice E, Le Rumeur E, and French Network of Clinical Reference Centres for Neuromuscular Diseases (CORNEMUS) (2015) Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet 24, 1267–1279. [DOI] [PubMed] [Google Scholar]
- (26).Mirza A, Sagathevan M, Sahni N, Choi L, and Menhart N (2010) A biophysical map of the dystrophin rod. Biochim. Biophys. Acta, Proteins Proteomics 1804, 1796–1809. [DOI] [PubMed] [Google Scholar]
- (27).Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang R-Y, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM, Merryman C, Vashee S, Krishnakumar R, Assad-Garcia N, Andrews-Pfannkoch C, Denisova EA, Young L, Qi Z-Q, Segall-Shapiro TH, Calvey CH, Parmar PP, Hutchison CA, Smith HO, and Venter JC (2010) Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56. [DOI] [PubMed] [Google Scholar]
- (28).Greenfield N, and Fasman GD (1969) Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8, 4108–4116. [DOI] [PubMed] [Google Scholar]
- (29).Calvert R, Kahana E, and Gratzer WB (1996) Stability of the dystrophin rod domain fold: evidence for nested repeating units. Biophys. J 71, 1605–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ipsaro JJ, Huang L, Gutierrez L, and MacDonald RI (2008) Molecular epitopes of the ankyrin-spectrin interaction. Biochemistry 47, 7452–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wolny M, Grzybek M, Bok E, Chorzalska A, Lenoir M, Czogalla A, Adamczyk K, Kolondra A, Diakowski W, Overduin M, and Sikorski AF (2011) Key amino acid residues of ankyrin-sensitive phosphatidylethanolamine/phosphatidylcholine-lipid binding site of βI-spectrin. PLoS One 6, No. e21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).John DM, and Weeks KM (2000) van’t Hoff enthalpies without baselines. Protein Sci. 9, 1416–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fontana A, Polverino de Laureto P, De Filippis V, Scaramella E, and Zambonin M (1997) Probing the partly folded states of proteins by limited proteolysis. Folding Des. 2, R17–26. [DOI] [PubMed] [Google Scholar]
- (34).Hubbard SJ (1998) The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1382, 191–206. [DOI] [PubMed] [Google Scholar]
- (35).McCourt JL, Rhett KK, Jaeger MA, Belanto JJ, Talsness DM, and Ervasti JM (2015) In vitro stability of therapeutically relevant, internally truncated dystrophins. Skeletal Muscle 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kim DE, Chivian D, and Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ray A, Lindahl E, and Wallner B (2012) Improved model quality assessment using ProQ2. BMC Bioinf. 13, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang Y, and Skolnick J (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710. [DOI] [PubMed] [Google Scholar]
- (39).Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33. [DOI] [PubMed] [Google Scholar]
- (40).Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, and Woods RJ (2005) The Amber biomolecular simulation programs. J. Comput. Chem 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nguyen H, Roe DR, and Simmerling C (2013) Improved Generalized Born Solvent Model Parameters for Protein Simulations.J. Chem. Theory Comput 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, and Walker RC (2012) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD, Roskies R, Scott JR, and Wilkens-Diehr N (2014) XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng 16, 62–74. [Google Scholar]
- (45).Shao J, Tanner SW, Thompson N, and Cheatham TE (2007) Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput 3, 2312–2334. [DOI] [PubMed] [Google Scholar]
- (46).Roe DR, and Cheatham TE (2013) PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (47).Kabsch W, and Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (48).Chowdhury R, Rasheed M, Keidel D, Moussalem M, Olson A, Sanner M, and Bajaj C (2013) Protein-protein docking with F(2)Dock 2.0 and GB-rerank. PLoS One 8, No. e51307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mongan J, Simmerling C, McCammon JA, Case DA, and Onufriev A (2007) Generalized Born Model with a Simple, Robust Molecular Volume Correction. J. Chem. Theory Comput 3, 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, and Roitberg AE (2012) MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput 8, 3314–3321. [DOI] [PubMed] [Google Scholar]
- (51).Lange OF, and Grubmüller H (2006) Generalized correlation for biomolecular dynamics. Proteins: Struct., Funct., Genet 62, 1053–1061. [DOI] [PubMed] [Google Scholar]
- (52).Peck A (2008) Beginning GIMP: from novice to professional, 2nd ed., Apress, Distributed to the Book trade worldwide by Springer-Verlag, Berkeley, CA. [Google Scholar]
- (53).Hunter JD (2007) Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 9, 90–95. [Google Scholar]
- (54).Djinović-Carugo K, Young P, Gautel M, and Saraste M (1999) Structure of the alpha-actinin rod: molecular basis for cross-linking of actin filaments. Cell 98, 537–546. [DOI] [PubMed] [Google Scholar]
- (55).Kusunoki H, Minasov G, Macdonald RI, and Mondragón A (2004) Independent movement, dimerization and stability of tandem repeats of chicken brain alpha-spectrin. J. Mol. Biol 344, 495–511. [DOI] [PubMed] [Google Scholar]
- (56).Ortega E, Manso JA, Buey RM, Carballido AM, Carabias A, Sonnenberg A, and de Pereda JM (2016) The Structure of the Plakin Domain of Plectin Reveals an Extended Rod-like Shape. J. Biol. Chem 291, 18643–18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Mirijanian DT, and Voth GA (2008) Unique elastic properties of the spectrin tetramer as revealed by multiscale coarse-grained modeling. Proc. Natl. Acad. Sci. U. S. A 105, 1204–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Paramore S, Ayton GS, and Voth GA (2006) Extending a Spectrin Repeat Unit. II: Rupture Behavior. Biophys. J 90, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Muthu M, Richardson KA, and Sutherland-Smith AJ (2012) The crystal structures of dystrophin and utrophin spectrin repeats: implications for domain boundaries. PLoS One 7, No. e40066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Heinrich V, Ritchie K, Mohandas N, and Evans E (2001) Elastic thickness compressibilty of the red cell membrane. Biophys. J 81, 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Pantazatos DP, and MacDonald RI (1997) Site-directed mutagenesis of either the highly conserved Trp-22 or the moderately conserved Trp-95 to a large, hydrophobic residue reduces the thermodynamic stability of a spectrin repeating unit. J. Biol. Chem 272, 21052–21059. [DOI] [PubMed] [Google Scholar]
- (62).Niks EH, and Aartsma-Rus A (2017) Exon skipping: a first in class strategy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther 17, 225–236. [DOI] [PubMed] [Google Scholar]
- (63).Kaspar RW, Allen HD, and Montanaro F (2009) Current understanding and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J. Am. Acad. Nurse Pract 21, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Greenberg DS, Sunada Y, Campbell KP, Yaffe D, and Nudel U (1994) Exogenous Dp71 restores the levels of dystrophin associated proteins but does not alleviate muscle damage in mdx mice. Nat. Genet 8, 340–344. [DOI] [PubMed] [Google Scholar]
- (65).Judge LM, Arnett ALH, Banks GB, and Chamberlain JS (2011) Expression of the dystrophin isoform Dp116 preserves functional muscle mass and extends lifespan without preventing dystrophy in severely dystrophic mice. Hum. Mol. Genet 20, 4978–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).White SJ, and den Dunnen JT (2006) Copy number variation in the genome; the human DMD gene as an example. Cytogenet. Genome Res 115, 240–246. [DOI] [PubMed] [Google Scholar]
- (67).Zhang Z, and Gerstein M (2003) Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 31, 5338–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kondrashov AS (2003) Direct estimates of human per nucleotide mutation rates at 20 loci causing mendelian diseases. Hum. Mutat 21, 12–27. [DOI] [PubMed] [Google Scholar]
- (69).Grimm T, Kress W, Meng G, and Müller CR (2012) Risk assessment and genetic counseling in families with Duchenne muscular dystrophy. Acta Myol. 31, 179–183. [PMC free article] [PubMed] [Google Scholar]
- (70).Lee T, Takeshima Y, Kusunoki N, Awano H, Yagi M, Matsuo M, and Iijima K (2014) Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J. Hum. Genet 59, 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.