

Abstract
ABHD12 is a membrane-bound hydrolytic enzyme that acts on the lyso-phosphatidylserine (lyso-PS) and lyso-phosphatidylinositol (lyso-PI) classes of immunomodulatory lipids. Human and mouse genetic studies point to a key role for the ABHD12-(lyso)-PS/PI pathway in regulating (neuro)immunological functions in both the central nervous system and periphery. Selective inhibitors of ABHD12 would offer valuable pharmacological probes to complement genetic models of ABHD12-regulated (lyso)-PS/PI metabolism and signaling. Here, we provide a detailed description of the discovery and activity-based protein profiling (ABPP)-guided optimization of reversible thiourea inhibitors of ABHD12 that culminated in the identification of DO264 as a potent, selective, and in vivo-active ABHD12 inhibitor. We also show that DO264, but not a structurally related inactive control probe (S)-DO271, augments inflammatory cytokine production from human THP-1 macrophage cells. The in vitro and in vivo properties of DO264 designate this compound as a suitable chemical probe for studying the biological functions of ABHD12-(lyso)-PS/PI pathways.
TOC Graphic
Introduction
Lysophospholipids represent an important class of signaling lipids that impact diverse physiological and disease processes1–2. Prominent lysophospholipid transmitters include lysophosphatidic acid (lyso-PA)3 and sphingosine 1-phosphate (S1P)4. These bioactive lipids have cognate receptors, mostly from the G-protein-coupled receptor (GPCR) category, and small-molecule modulators of these receptors have been clinically advanced to treat, for instance, immunological disorders4. The magnitude and duration of lysophospholipid action are controlled by specific sets of biosynthetic and degradative enzymes5–6, and these enzymes offer additional targets for pharmacological control over lysophospholipid pathways.
In recent years, other bioactive lysophospholipids, such as lysophosphatidylserine (lyso-PS) and lysophoshatidylinositol (lyso-PI), have emerged as signaling molecules that act on distinct subsets of GPCRs7–9, as well as possibly other receptor types10. Our current understanding of the physiological functions of lyso-PS and lyso-PI is limited and would benefit from selective chemical probes to perturb these lipid pathways in vivo. This challenge can be addressed, in part, by developing synthetic agonists and antagonists of the lyso-PS/PI receptors11–13. As noted above, however, an attractive and complementary strategy would be to create inhibitors of the enzymes that produce or inactivate lyso-PS/PI.
In the course of attempting to understand the biochemical basis for rare, monogenic disorders of the central nervous system (CNS) caused by deleterious mutations in enzymes from the serine hydrolase family, we discovered that ABHD12 (α/β-hydrolase domain-containing 12), loss-of-function mutations in which cause the neurological disease PHARC (Polyneuropathy, Hearing loss, Ataxia, Retinitis pigmentosa and Cataract; MIM 612674)14–15, is a principle lyso-PS/PI lipase in mammals. Mice with targeted disruption of the Abhd12 gene show elevated lyso-PS/PI, as well as increased polyunsaturated (C20:4) PS, content in the CNS16. These mice also display a subset of PHARC-like abnormalities, including auditory and motor control deficiencies, which emerge later in life (~10–18 mo) and are accompanied by brain microgliosis16, suggesting that PHARC may have an immunological underpinning. Also consistent with this premise, ABHD12 is highly expressed in innate immune cells (macrophages, microglia) and several lyso-PS receptors also show restricted expression to the immune system17–18.
To better understand the contributions of the ABHD12-(lyso)-PS/PI pathway to neuroimmunological processes, we recently reported the discovery of a selective and in vivo-active inhibitor of ABHD1219. This compound termed DO264, an N-3-pyridyl-N’−4-piperidinylthiourea, appears to act as a reversible, competitive inhibitor of ABHD12, while showing negligible interactions with other serine hydrolases as determined by activity-based protein profiling (ABPP)19. We found that DO264-treated mice displayed elevations in brain lyso-PS/PI and C20:4 PS lipids, but did not exhibit the auditory defects observed in ABHD12–/– mice, even following four weeks of inhibitor treatment. On the other hand, both DO264-treated and ABHD12–/– mice exhibited heightened immunological responses to lymphocytic choriomeningitis virus (LCMV) clone 13 infection in mice, supporting an immunomodulatory function for the ABHD12-(lyso)-PS/PI pathway.
Here, we provide a detailed account of the identification, synthesis, and structure-activity relationship (SAR) of N-3-pyridyl-N’−4-piperidinylthiourea inhibitors of ABHD12 that led to the discovery of DO264, as well as a structurally related inactive control probe (S)-DO271. We also describe the development of a tailored activity-based probe for ABHD12 – the N-hydroxyhydantoin (NHH)-carbamate JJH350 –that serves as a versatile target engagement tool for monitoring ABHD12 activity and inhibition in vivo.
Results
Exploration of NHH-carbamates furnishes JJH350 as a tailored activity-based probe for ABHD12.
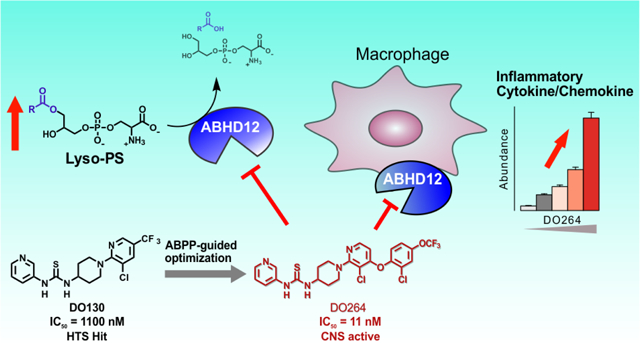
We previously described a chemical proteomic assessment of NHH-carbamates as irreversible inhibitors of serine hydrolases20. A subset of these compounds showed reactivity towards ABHD12 (e.g, JJH329 (1), IC50 = 0.32 μM, 95% CI = 0.24–0.41 μM), with a notable dependency on a key amide linker (X, Fig. 1A) appended to the NHH piperazine leaving group, without which compounds displayed negligible ABHD12 activity up to 10 μM (e.g., ABC34 (2); Figure 1A). JJH329 also showed good selectivity as assessed in mouse brain proteome by gel-based ABPP21–22 using the broad-spectrum serine hydrolase-directed probe fluorophosphonate-rhodamine (FP-Rh)23, where ABHD6 was observed as the only off-target (Figure 1B). While ABHD12 activity can be monitored by gel-based ABPP using FP-Rh in mouse brain, we found that detection of this enzyme was challenging in other mouse tissues due to overlapping signals from co-migrating serine hydrolases19. We therefore appended an alkyne handle to JJH329 to furnish an ABHD12-directed activity-based probe JJH350 (3) (Figure 1A, B; IC50 = 0.40 μM, 95% CI = 0.25–0.62 μM) which enabled direct visualization of ABHD12 by gel-based ABPP following conjugation to an azide-rhodamine reporter group24 using copper-catalyzed azide-alkyne cycloaddition (CuAAC), or click, chemistry25 (Figure 1C). ABHD12 (as well as ABHD6) could be clearly visualized in mouse brain proteome across a concentration of 0.4–10 μM of JJH350 (Figure 1C). We then used JJH350 as a target engagement probe to assess the in vivo activity of JJH329, which revealed that JJH329 (30 mg/kg, i.p.) produced only marginal inhibition of ABHD12 in mice (Figure S1). Additional optimization efforts did not lead to the identification of NHH-carbamates that showed better potency in vivo (data not shown). We therefore focused on identifying a new chemotype for ABHD12 inhibitors.
Figure 1.
Discovery of NHH-carbamate inhibitors and tailored activity-based probes for ABHD12. (A) Chemical structures and ABHD12 inhibitory activities for the indicated NHH-carbamate compounds. IC50 values were determined by gel-based competitive ABPP using the FP-Rh probe. (B) In vitro potency and selectivity of NHH-carbamate compounds in mouse brain membrane proteome as measured by gel-based competitive ABPP using the FP-Rh probe. (C) Visualization of ABHD12 in mouse brain membrane proteome using the JJH350 probe. For the gel-based ABPP assays, mouse brain membrane proteomes (1 mg/mL) were pre-incubated with NHH-carbamate compounds (45 min, 37 °C) followed by the reaction with FP-Rh probe (1 μM, 45 min, 37 °C) (A, B) or with Rh-N3 (25 μM) using CuAAC conditions (60 min, r.t.) (C).
Identification of a thiourea class of ABHD12 inhibitors by structural reassignment of the screening hit AW01275.
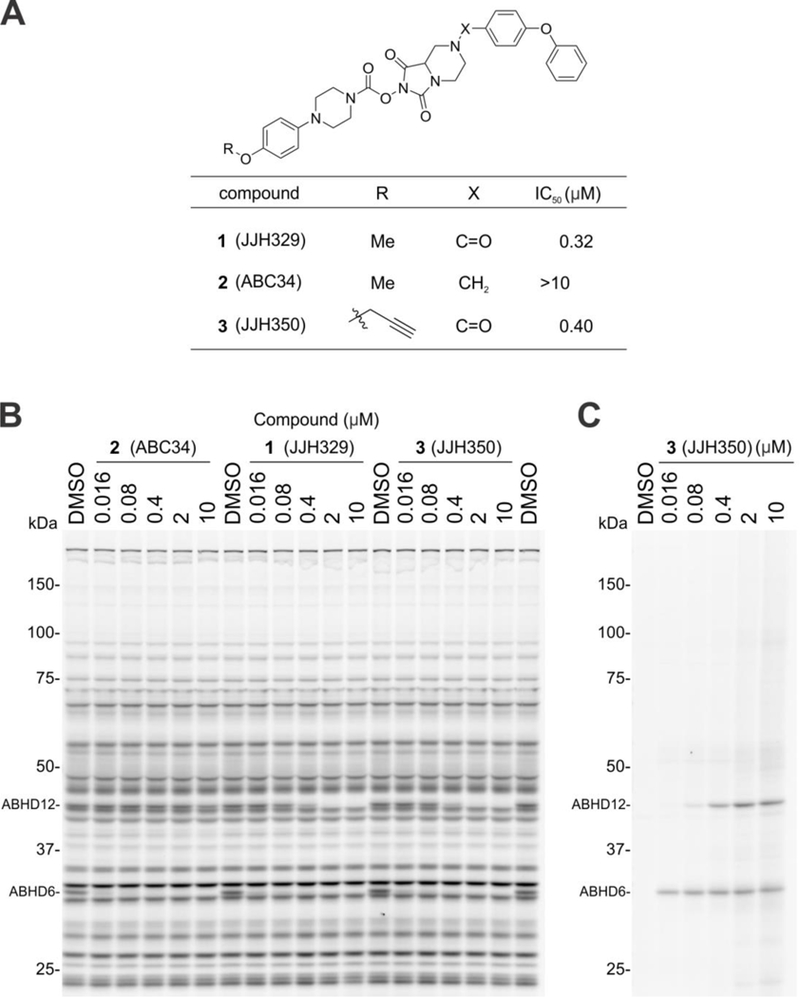
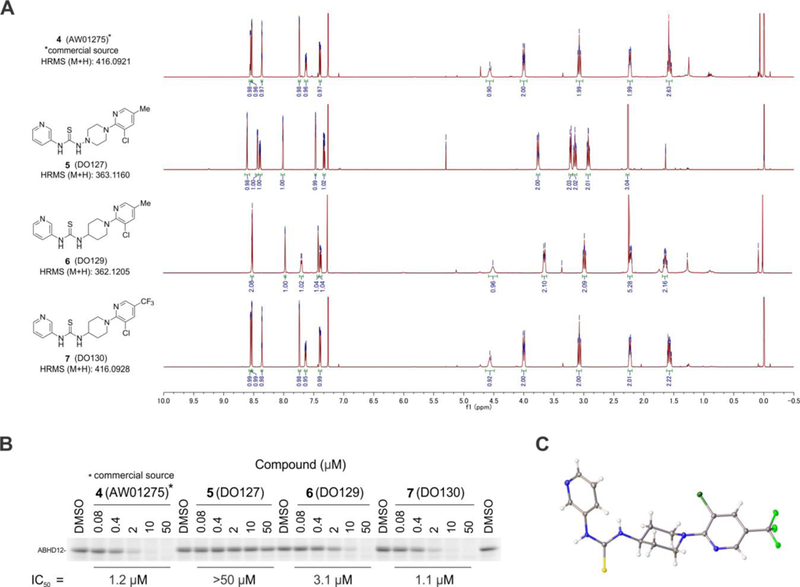
We pursued new chemotypes for ABHD12 inhibition by high-throughput screening (HTS) using a fluorescent-coupled substrate assay19, and, from the Maybridge HitFinder™ library containing ~16,000 compounds, we identified a putative hit, the thiosemicarbizide AW01275 (4, Figure 2A)19. This compound also inhibited lyso-PS hydrolysis activity of ABHD12 with an IC50 value of 1.3 μM19 and blocked the labeling of ABHD12 by JJH350 with an IC50 value of 1.2 μM (95% CI = 0.96–1.4 μM) (Figure 2B). Surprisingly, however, our chemically resynthesized stock of AW01275, termed DO127 (5) (Figure 2A), did not show any ABHD12 inhibitory activity as measured with a lyso-PS substrate assay (Figure S2) or by gel-based ABPP (Figure 2B). We found that the 1H NMR and ESI-HRMS of commercial AW01275 did not match the analytical data for DO127 (Figure 2A). In the course of exploring candidate alternative structures, we discovered that a thiourea analogue DO129 (6) (Figure 2A) exhibited ABHD12 inhibitory activity (Figure 2B) and peaks in the aliphatic region of the 1H-NMR that were similar to those of commercial AW01275 (Figure 2A). We furthermore noted that the 13C NMR spectrum for commercial AW01275 showed a distinct quadruplet (coupling constant = 270.0 Hz) peak indicative of the presence of a CF3 group (Figure S3). Based on these observations, we speculated that the correct structure of AW01275 was the thiourea DO130 (7) (Figure 2A), which we subsequently found to display identical 1H NMR, HRMS, and ABHD12 inhibitory activity as commercial AW01275 (Figure 2A and B). X-ray crystallographic analysis of commercial AW01275 confirmed its structural identity as DO130 (Figure 2C). Notably, unlike the irreversible inhibitor JJH350, DO130 did not show time-dependent inhibition of ABHD12 (Figure 3), indicating that this compound and related thioureas act as reversible inhibitors of ABHD1219.
Figure 2.
Identification and structure assignment of HTS hit AW01275 as thiourea DO130. (A) An originally assigned chemical structure and 1H-NMR of AW01275 (DO127) and its structurally related analogues, DO129 and DO130. The 1H-NMR spectra were measured in CDCl3 with D2O. (B) ABHD12 inhibitory activity of AW01275 (obtained from a commercial source), DO127, DO129, and DO130 as measured by gel-based ABPP of mouse brain membrane proteome (1 mg/mL protein) using the JJH350 probe (2 μM, 45 min, 37 oC). (C) An X-ray crystal structure of AW01275 obtained from a commercial source.
Figure 3.
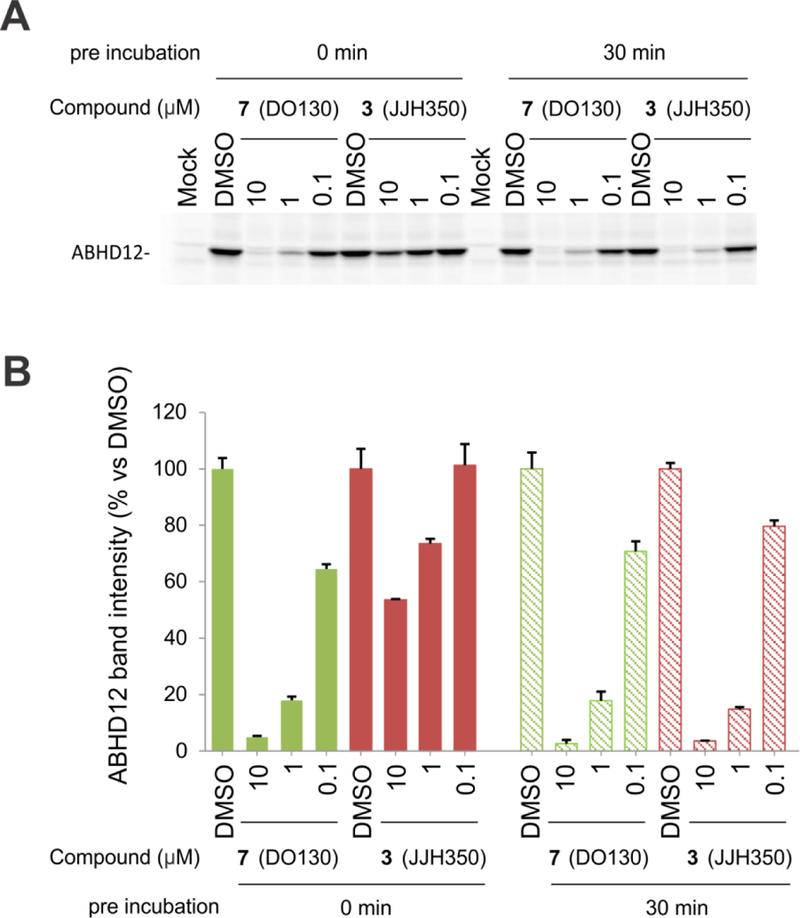
DO130 (7) does not show time-dependent inhibition of ABHD12. (A) Comparison of ABHD12 inhibitory activities of JJH350 (3) and DO130 (7) as measured by gel-based ABPP with membrane lysate of ABHD12-transfected HEK293T cells using an FP-Rh probe (1 μM, 45 min, 37 °C). A representative gel is shown. (B) Bar graph representation of ABHD12 band intensity obtained in (A). N = 3. The data represent the mean ± SD.
SAR analysis of the N-pyridyl thiourea and piperidine core of ABHD12 inhibitors.
We next set out to optimize DO130 and gain a deeper understanding of structural features that contribute to ABHD12 inhibitory activity. These initial investigations of SAR are summarized in Table 1. Given possible concerns of thioureas as potential toxicophores26, we first replaced this group with a urea (8), but this change resulted in a substantial loss in activity. Next, we explored analogs of the 3-aminopyridine group (R1), finding that methoxy-substitution (9) and conversion to 3-amino-quinoline (10) or 1,2,3,4-tetrahydroquinoline (11) also resulted in diminished potency towards ABHD12 (Table 1A). The phenyl analogue 12 retained some ABHD12 inhibitory activity, albeit slightly lower than that of DO130.
Table 1.
ABHD12 inhibitor SAR of the N-pyridyl thiourea and piperidine core.
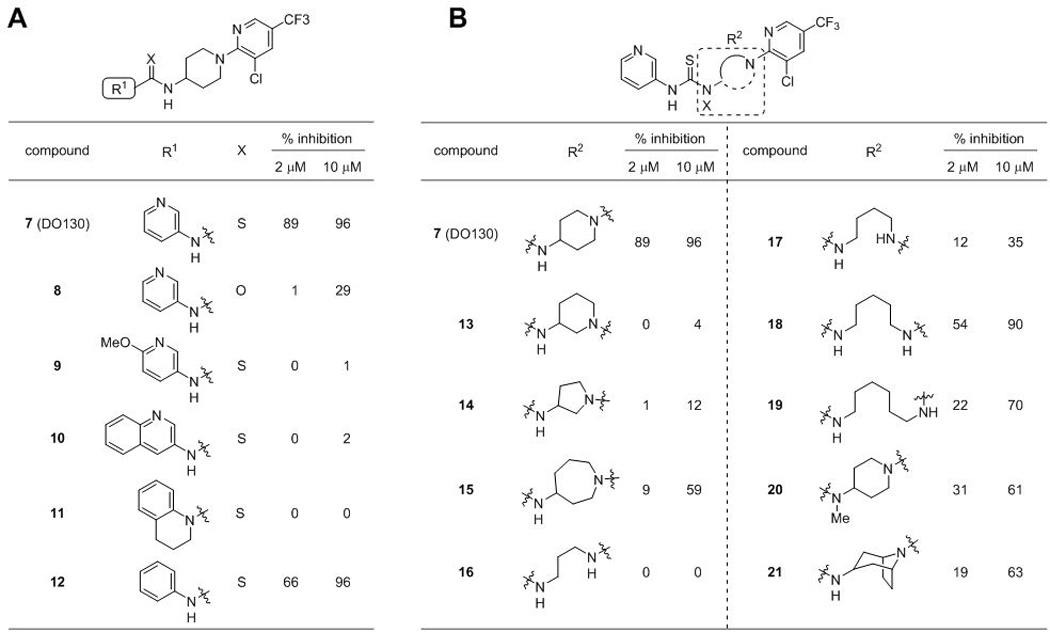 |
We next focused our attention on the SAR of the piperidine ring system (Table 1B). Repositioning the exocyclic amino group of DO130 to the 3-position (13) abolished ABHD12 inhibitory activity, while replacing the piperidine with smaller (5-membered (14)) or larger (7-membered (15)) ring systems resulted in near-complete loss (14) or substantial reductions (15) in potency. Acyclic analogs (16-19) with varying linker lengths (C3–6) were also tested and, while several displayed modest activity, none were as potent as DO130. N-methyl substitution of the exocyclic amino group (20) also weakened ABHD12 activity. Finally, we replaced the piperidine core with 8-azabiocyclo[3.2.1]octane (21) to test if conformational constraint might promote ABHD12 inhibition. Like all other deviations from the piperidine system, this too impaired ABHD12 activity relative to DO130. Together, these results demonstrated the importance of the 3-pyridylthiourea and 4-aminopiperidine moieties found in DO130 for ABHD12 activity and suggested a low tolerance to modification in these regions.
Exploration of pyridine substituent effects.
In pursuit of ABHD12 inhibitors with enhanced potency, we next explored structure-activity relationships of the distal pyridine group (Table 2). The importance of the CF3 group was apparent, as its removal (22) abolished ABHD12 activity up to 5 μM. On the otherhand, removal of the Cl group (23) resulted in only a small loss in potency. A CF3 group ring scan revealed the 4- and 6-positions (24 and 26) were favorable for ABHD12 blockade relative to 5- and 3-positions (23 and 25), leading to the first sub-micromolar inhibitors in this series. The synthetic accessibility of 4-substituted pyridines led us to further explore this position over the 6-substituted analogs, which demonstrated comparable activity. Maintaining the CF3 in the 4-position, we next scanned a Cl group across the remaining three positions of the pyridyl ring (27-29), finding 3-Cl substitution (28) to further enhanced potency (ABHD12 IC50 = 300 nM).
Table 2.
Pyridine substituent effects on ABHD12 activity.
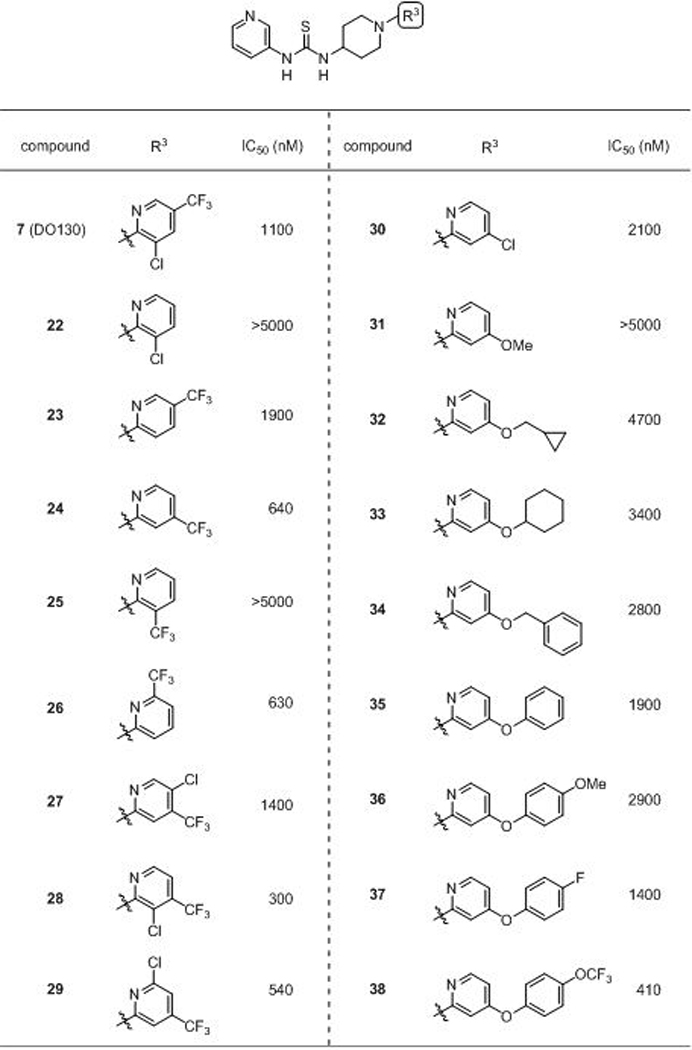 |
We then wondered if the CF3 group in the 4-position could be replaced to gain additional potency. Substituting the 4-CF3 group with a chlorine (30) modestly decreased (~3-fold) ABHD12 inhibitory activity, while conversion to a methoxy group (31) had an even more deleterious effect. Interestingly, elaborating the 4-position with alkoxyl, benzyloxy, and phenoxy groups furnished a set of compounds (32–35) with superior activity to the 5-methoxy analog (31) and revealed a scaffold trajectory that tolerated large substitutuents to potentially enhance potency. Given the superior ABHD12 inhibition of phenoxy analogue 35, we introduced substitutents to the 4-position of the distal phenyl group and observed that while a methoxy group slightly impaired activity, fluoro (37) and especially OCF3 (38) groups increased potency.
Identification of DO264 as a potent and selective ABHD12 inhibitor.
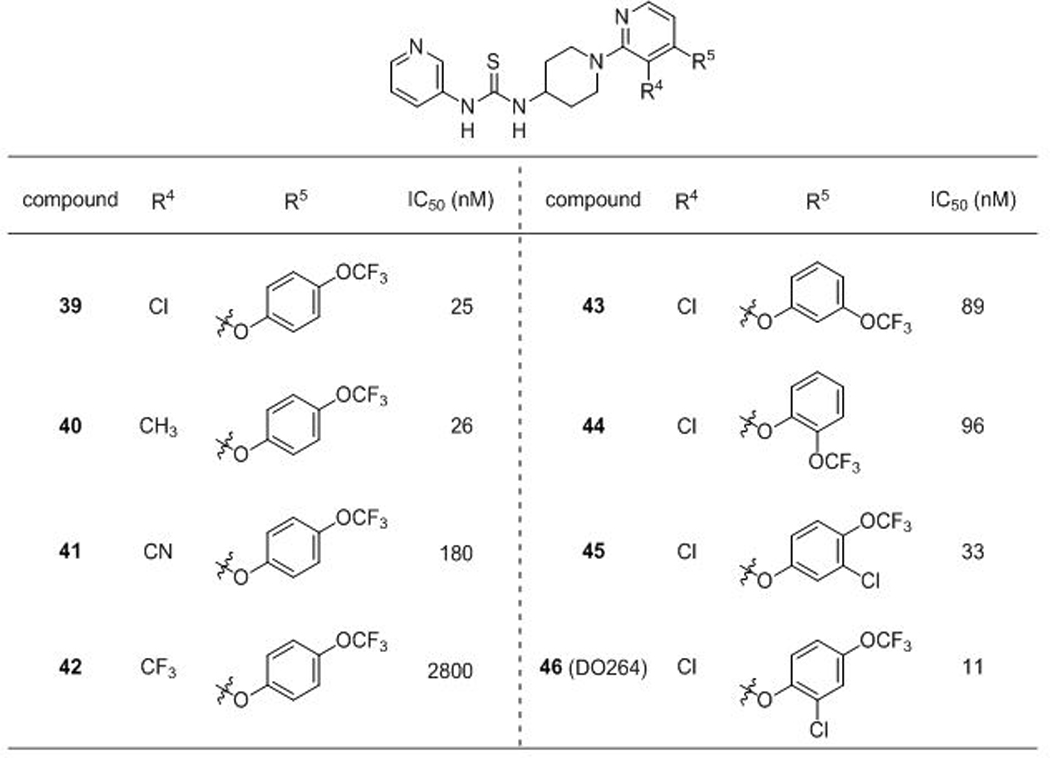
Having discovered that introducing an additional Cl group at the 3-position of the pyridine ring of 24 (28) or replacing the CF3 group of 24 with a 4-OCF3-phenyl group (38) improved ABHD12 inhibitory activity, we wondered if these positive effects would be additive when combined into a single compound. Indeed, the hybrid compound 39 displayed dramatically increased ABHD12 inhibitory activity (> 10-fold) comparing to 28 or 38 (Table 3). Conversion of the Cl group of 39 to a methyl group (40) did not alter potency, but substitution at this position with a CN (41) or CF3 (42) group reduced ABHD12 inhibitory activity, indicating that the Cl group contributed to potency primarly through a steric rather than electron withdrawing effect. The regioisomers of 39 bearing 3-OCF3 (43) or 2-OCF3 (44) groups showed ~3–4-fold lower potencies. Finally, introduction of a Cl group at the 2-position, but not the 3-position (45), of the distal phenyl provided another two-fold increase in inhibitory activity to furnish DO264 (46), a highly potent (IC50 = 11 nM) ABHD12 inhibitor
Table 3.
SAR leading to ABHD12 chemical probe DO264 (46).
 |
In the course of our SAR studies, we noted that compound 13 bearing a 3-aminopiperidine in place of the 4-aminopiperidine showed dramatically reduced ABHD12 inhibitory activity (Table 1B and Table 4). We capitalized on this observation to generate (S,R)-DO271 (47) (Table 4 and Figure 4), which showed an ~10,000-fold reduction in potency for ABHD12 inhibition compared to DO264. We found that (S)-DO271 (48) was less active than (R)-DO271 (49) (Table 4 and Figure 4), and therefore designated (S)-DO271 as an inactive control probe for biological studies. While it may seem surprising that neither enantiomer of DO271 appeared more potent than the racemate, we believe that this reflects the technical challenge of accurately measuring such weak IC50 values (> 50 μM), especially as high concentrations of compound may approach their solubility limit.
Table 4.
Identification of an inactive control probe (S)-DO271 (48).
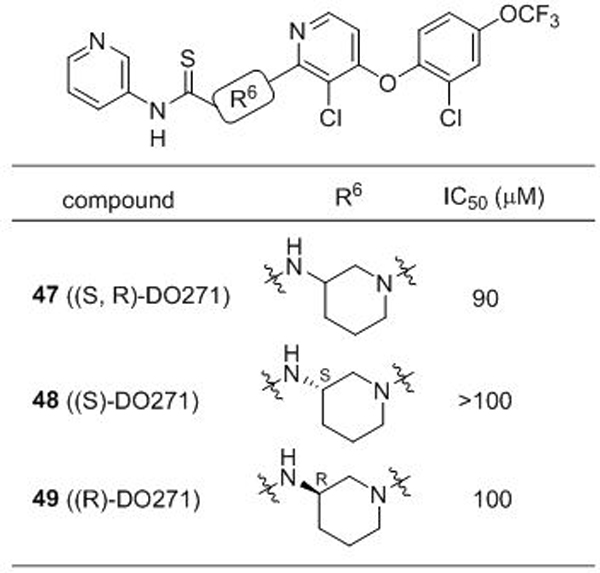 |
Figure 4.
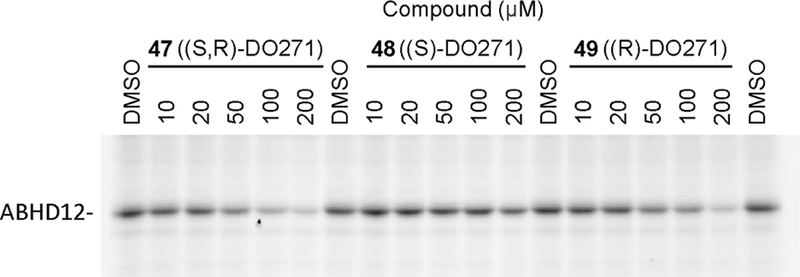
ABHD12 inhibitory activity of (S,R)-DO271 (47), (S)-DO271 (48), and (R)-DO271 (49) as measured by gel-based ABPP of mouse brain membrane proteome (1 mg/mL protein) using the JJH350 probe (2 μM, 45 min, 37 oC).
ABPP studies of DO264 (46) and other representative (thio)urea inhibitors of ABHD12 confirmed that these compounds show excellent selectivity over other serine hydrolases (Figure 5)19, including phosphatidylserine lipases, such PS-PLA127 and ABHD16A28, that contribute to lyso-PS production and the PLA1-type PI lipase DDHD1 involved in lyso-PI production29. We also confirmed the activity and selectivity of DO264 in vivo, where the compound was found to produce substantial elevations in brain lyso-PS/PI and C20:4 PS content in mice19.
Figure 5.
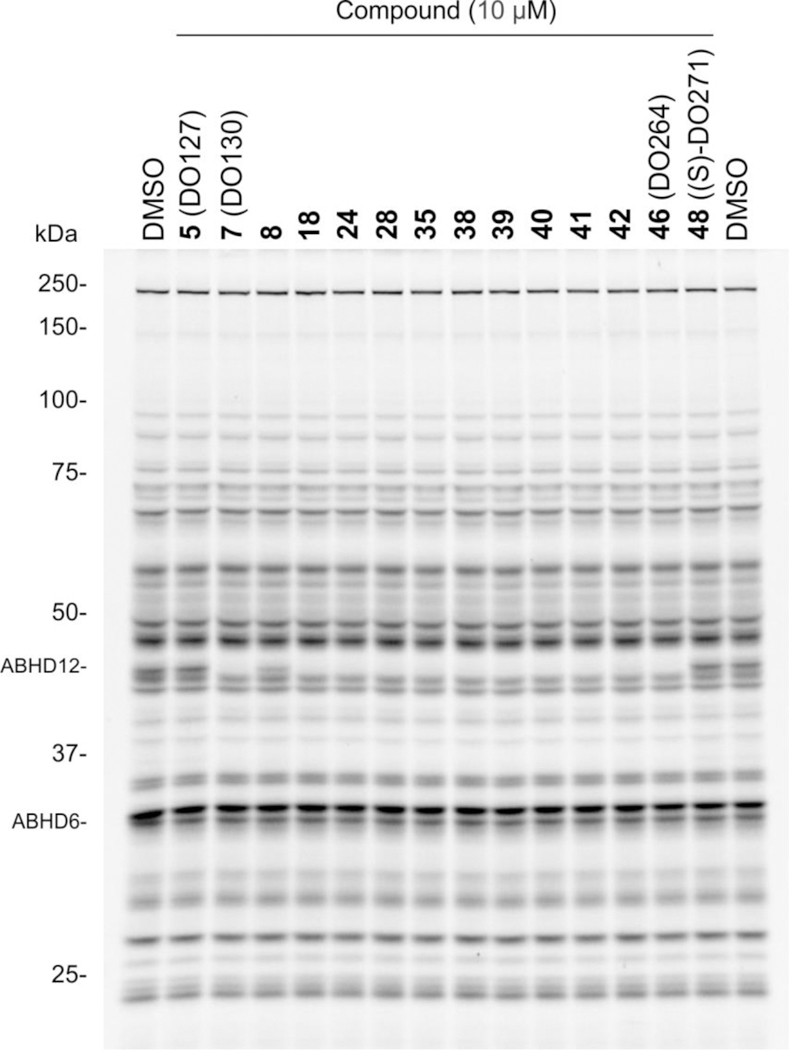
Gel-based ABPP of DO264 (46) and other representative (thio)urea inhibitors of ABHD12 in mouse brain membrane proteome using the FP-Rh probe (1 μM, 45 min, 37 °C).
ABHD12 inhibition modulates cytokine production in human THP-1 cells.
Considering that ABHD12 is highly expressed in macrophages and microglia17–18, and that genetic or pharmacological perturbation of ABHD12 produces heightened (neuro)immunological phenotypes in mice16,19, 28, we next tested whether DO264 affected cytokine production using the human monocyte THP-1 cell line as a model system. THP1 monocytes were first treated with phorbol 12-myristate 13-acetate (PMA) to produce resting macrophages characterized by changes in morphology and increased cell surface expression of CD11 and CD1430–32. Cells were allowed to rest for 24 h followed by polarization to the M1 stage with lipopolysaccharide (LPS) and IFN-γ. Each cell treatment step was performed in the presence of either DO264, DO271, or DMSO (Figure 6A). Consistent with previous studies of peritoneal macrophages from ABHD12(–/–) mice, which showed elevated TNF-α and IL-1β both basally and in response to LPS treatment28, the DO264-treated, but not DO271-treated, M1 stage THP-1 macrophages showed concentration-dependent increases in these cytokines (Figure 6B). The levels of inflammatory chemokines – CCL3 and CCL4 – were also elevated by DO264, but not DO271 (Figure 6B). Importantly, these cytokine/chemokine changes occured across a concentration range of DO264 that produced complete inhibition of ABHD12 and increases in lyso-PS/20:4 PS content in THP-1 cells. In the course of these studies, we noticed that higher concentrations of DO264 (≥ 5 μM) impaired the viability of THP1 cells (Figure S4). Since full engagement of ABHD12 is observed at lower concentrations of DO264 (1 μM)19, we believe that the cytotoxic effect is not due to inhibition of ABHD12, but rather another (as of yet) unidentified mechanism. We tested DO264 in a broader panel of cell lines and found that individual lines showed variable degrees of sensitivity to the compound, but all of the cell lines could be treated with DO264 at 1 μM without displaying cytotoxicity (Figure S4). These data indicate that cellular studies performed with DO264 should not use greater than 1 μM of the compound, which is sufficient to fully inhibit ABHD1219 without causing cytotoxicity. Finally, we should also note that the total exposure levels of DO264 can reach 3–9 μM in the periphery and brain of mice (30 mg/kg DO264, p.o. or i.p. dosing), without overt signs of toxicity, even following treatment with the compound for several weeks19. This may indicate a much lower free fraction of DO264 at pharmacologically relevant doses or that the cytotoxicity observed for this compound in culture is not representative of the compound activity in vivo.
Figure 6.
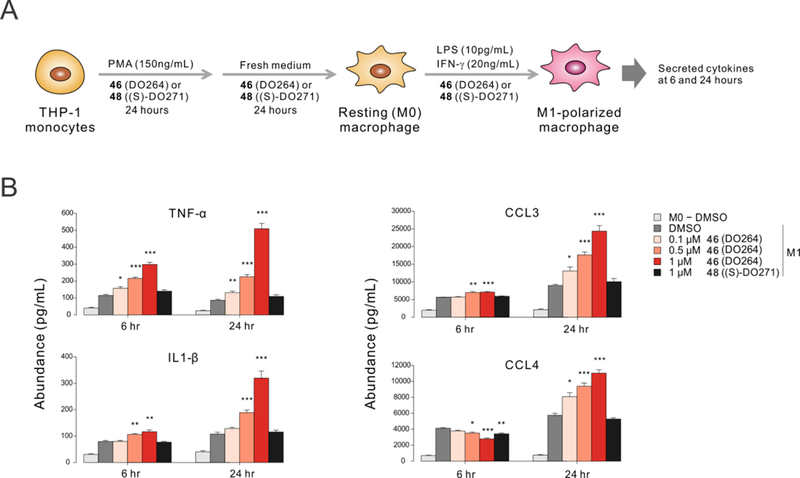
Concentration-dependent elevation of cytokines and chemokines in M1-polarized THP-1 macrophages treated with the ABHD12 inhibitor DO264. (A) PMA-differentiated THP-1 cells were polarized to M1 macrophages by stimulation with 20 ng/mL IFN-γ and 10 pg/mL LPS. (B) Cytokines and chemokines were measured 6 and 24 h after stimulation. DO264 and (S)-DO271 were included in the media at the specified concentrations throughout the experiment (A). Media samples from DMSO-treated resting (M0) macrophages were included as an additional control. Data represent mean ± SEM from four independent biological experiments. * p < 0.05; ** p < 0.01; *** p < 0.001 (Student’s t-test performed relative to M1-DMSO samples).
Conclusion
We have described herein an ABPP-guided medicinal chemistry program that culminated in the development of the first potent, selective, and in vivo-active inhibitors of ABHD12, an enzyme responsible for hydrolyzing bioactive lyso-PS/PI lipids. We have shown that this compound, termed DO264, elevates lyso-PS/PI and C20:4 PS lipids in human cells and brain tissue of treated mice19. We also found that DO264-treated mice, as well as ABHD12(–/–) mice, show exacerbated immune responses to LCMV infection that included heightened chemokine production in vivo19. Consistent with a role for the ABHD12-(lyso)-PS /PI pathway in regulating immune cell activity, we found in this current study that DO264-treated THP-1 macrophages showed heightened cytokine production. In future studies, it will be important to determine the specific (lyso)-PS/PI lipids and receptors involved in mediating the heightened immunological outcomes of ABHD12 blockade.
From a methodological perspective, the discovery of DO264 underscores the utilty of ABPP for developing not only irreversible, but also reversible inhibitors of serine hydrolases, which may be broadly underappreciated, despite past examples of success21, 33. Our program further benefited from the use of both general (FP-based) and tailored (JJH350) activity-based probes that enabled rigorous assessment of target engagement and selectivity for ABHD12 inhibitors, as well as the discovery of structurally related compounds that do not inhibit ABHD12 and can accordingly serve as inactive control probes (e.g., (S)-DO271). The toolbox of ABHD12-directed inhibitors and activity-based probes described herein should facilitate the pharmacological characterization of (lyso)-PS/PI pathways in mammalian biology and disease.
Chemistry
The synthesis of N-hydroxyhydantoin carbamates 1 and 3 from commercially available piperazine 50 has been previously described (Scheme 1, ref 19 and 20). We used a similar route to prepare carbamate 2 where the common piperazine intermediate (51) was subjected to amide coupling conditions with 4-phenoxybenzoic acid.
Scheme 1. Synthesis of 1–3a.
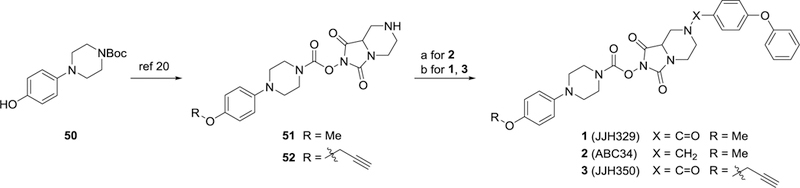
aReagents and conditions: (a) 4-phenoxybenzaldehyde, acetic acid, NaBH(OAc)3, dry THF, rt, 45% (ref 20); (b) 4-phenoxybenzoic acid, EDCI, HOBt·H2O, iPr2NEt3, rt, 74% for 2, 86% for 3.
To prepare thiocarbamates 5–7, methyl- or trifluoromethyl-chloropyridines (53 or 54, respectively) underwent 2-chloro substitution reactions with appropriate piperazines and piperidines by either a Cu-promoted amine cross-coupling (55 and 56) or SnAr reaction (57) (Scheme 2). Boc-deprotection of 56 and 57 followed by reaction of the resultant primary amines with pyridine-3-isothiocyanate gave thioureas 6 and 7. To prepare semithiocarbazide 5, the Boc group of piperazine 55 was first removed with HCl and the piperazine nitrosylated with NaNO2. Subsequent nitroso reduction of 58 furnished hydrazine 59 which was then treated with pyridine-3-isothiocyanate to provide 5 (DO127) in 55% yield over two steps.
Scheme 2. Synthesis of 5–7a.
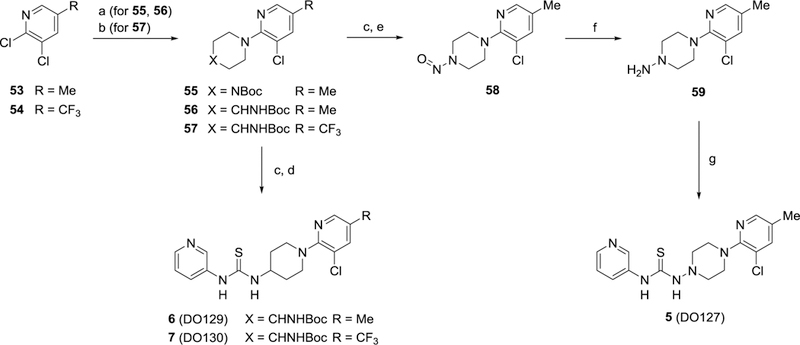
aReagents and conditions: (a) tert-butyl(piperazin-4-yl)carbamate, Cu powder, neat (for 55) or dry DMF (for 56), 120 °C, 64% for 55, 37% for 56; (b) tert-butyl(piperidin-4-yl)carbamate, K2CO3, dry DMSO, 100 °C, 91%; (c) 4N HCl in 1,4-dioxane, CH2Cl2, rt; (d) pyridine-3-isothiocyanate, iPr2NEt3, CH2Cl2, rt, 75% for 6, 94% for 7 (2 steps); (e) NaNO2, acetic acid, H2O, 0 °C to rt, 96% (2 steps); (f) Zn powder, MeOH/acetic acid, 0 °C to rt; (g) pyridine-3-isothiocyanate, iPr2NEt3, CH2Cl2, rt, 55% (2 steps).
Analogs of DO130 featuring various 3-pyridylthiourea modifications each were accessible from common piperazine intermediate 57 (Scheme 3). Following Boc-deprotection of 57, thioureas 8-11 were prepared by either treating the free amine with O-phenyl chlorothionoformate and the indicated aromatic amines or, in the case of thiourea 8, reacting the amine directly with pyridine-3-isothiocyanate. Similarly, urea 12 was synthesized through coupling deprotected 57 with pyridine-3-isocyanate.
Scheme 3. Synthesis of 8–12a.
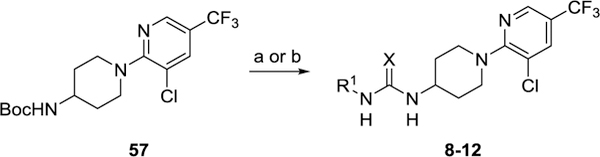
aReagents and conditions: (a) 4N HCl in 1,4-dioxane, CH2Cl2, rt then R1–NH2, phenylchlorothionoformate, iPr2NEt3, CH2Cl2, 21–45 %; (b) 4N HCl in 1,4-dioxane, CH2Cl2, rt then R1–N=C=S or R1–N=C=O, iPr2NEt3, CH2Cl2, rt, 71–100 %.
Thioureas 13-31 with varying amine cores and pendant substituted pyridines were all prepared in a similar manner outlined Scheme 4. Chloropyridines 54 and 60a–60j were coupled with the appropriate Boc-protected diamines to give intermediates 61a–61s and subsequently deprotected. As described for previous analogs, the final thioureas (13-31) were obtained through treatment of the free amines with pyridine-3-isothiocyanate. For access to 4-alkoxy or 4-phenoxy substituted pyridines 67–81, 2,4-dichloro or 2-chloro-4-fluoro pyridines were subjected to SnAr conditions in the presence of various alcohols and phenols which primarily displaced the halogen at the 4-position (Scheme 5). The remaining chloro group in the 2-position was displaced with tert-butyl(piperidin-4-yl)carbamate using similar SnAr conditions to provide 82–96. Finally, thioureas 32–46 were obtained following Boc-removal and coupling with pyridine-3-isothiocyanate. Inactive control compounds 47-49 were similarly prepared from 2-chloropyridine 79.
Scheme 4. Synthesis of 13–31a.
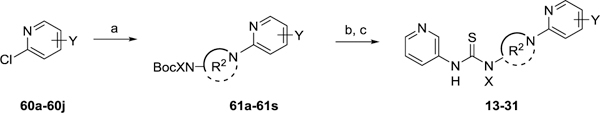
aReagents and conditions: (a) Boc-amine, K2CO3, dry DMSO, 100 °C, 19–100%; (b) 4N HCl in 1,4-dioxane, CH2Cl2, rt; (c) pyridine-3-isothiocyanate, iPr2NEt3, CH2Cl2, rt, 90–100% (2 steps)
Scheme 5. Synthesis of 32–49a.
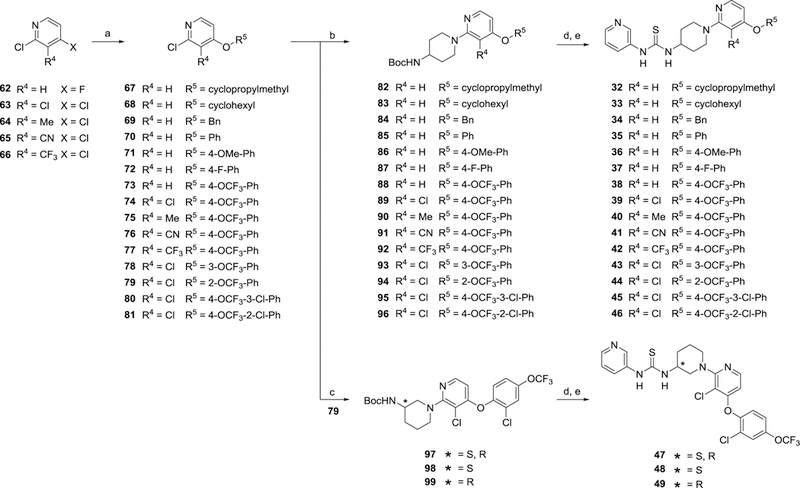
aReagents and conditions: (a) R5–OH, NaH in mineral oil, dry DMF, 0 °C then 90–100 °C, 61–100%; (b) tert-butyl(piperidin-4-yl)carbamate, K2CO3, dry DMSO, 100 °C, 20–94%; (c) tert-butyl(piperidin-3-yl)carbamate, K2CO3, dry DMSO, 100 °C, 45–51%; (d) 4N HCl in 1,4-dioxane, CH2Cl2, rt; (e) pyridine-3-isothiocyanate, iPr2NEt3, CH2Cl2, rt, 78–100% (2 steps)
Experimental Section.
Chemistry General Information.
All chemical reagents were obtained from commercial suppliers and were used without further purification. Merck silica gel TLC plates (0.25 mm, 60 F254) were used to monitor reactions. Flash chromatography was performed using SiliaFlash F60 silica gel (40–63 μm, 60 Å). NMR spectra were recorded at room temperature on Bruker DRX-600 spectrometer at 600 (1H) and 150 (13C) MHz using CDCl3 as solvent, unless stated otherwise. Chemical shifts are recorded in ppm relative to tetramethylsilane (TMS) with peaks being reported as follows: chemical shift, multiplicity (s = singlet, brs = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant (Hz). High-resolution mass spectra (HRMS) were obtained on an Agilent LC/MSD TOF mass spectrometer by electrospray ionization–time-of-flight (ESI-TOF). The single crystal X-ray diffraction studies were carried out on a Bruker Kappa APEX-II CCD diffractometer equipped with Mo Kα radiation (λ = 0.71073 Å). Purities of all final compounds were determined to be greater than 95% based on HPLC chromatograms (UV, 257 nm). Purities of all reported final compounds were determined to be greater than 95% measured by HPLC analysis (Waters Cortecs C18 column (2.1×55 mm, 1.6 mm) using a 0.1% aqueous formic acid:acetonitrile gradient (0.8 mL/min, 10–99% acetonitrile over 2.5 minutes, then 0.2 minute isocratic hold) at 35 °C. The compounds were detected using UV light (MaxPlot over 220–400 nm)). Enantiomeric excess (ee %) for compound 48 and 49 were determined to be greater than 95% ee measured by Waters UPC2 SFC with a Daicel IBN column (3 μm, 4.6×250 mm) under isocratic conditions [40% MeOH / CO2 (4 mL/min), 1600 psi backpressure] at 30 °C. The enantiomers were detected by UV light (257 nm).
1,3-Dioxo-7-(4-phenoxybenzoyl)hexahydroimidazo[1,5-a]pyrazine-2(3H)-yl 4-(4-methoxyphenyl)piperazine-1-carboxylate (1, JJH329).
1,3-dioxohexahydroimidazo[1,5-a]pyrazin-2(3H)-yl 4-(4-methoxyphenyl)piperazine-1-carboxylate·2HCl (compound 51·2HCl)20 (42 mg, 92 μmol) was dissolved in 0.5 mL DMF, and 4-phenoxy benzoic acid (24 mg, 110 μmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI·HCl) (26 mg, 140 μmol), 1-hydroxybenzotriazole monohydrate (HOBt·H2O) (21 mg, 140 μmol), N,N-diisopropylethylamine (48 μL, 270 μmol), were added. The resulting mixture was stirred at room temperature overnight and poured into saturated aqueous NaHCO3 solution. The mixture was extracted with ethyl acetate (two times). The combined organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by prep-TLC (ethyl acetate:hexane= 2:1) to afford (JJH329) (40 mg, 74%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 7.44 – 7.36 (m, 4H), 7.19 (tt, J = 7.4, 1.1 Hz, 1H), 7.09 – 7.05 (m, 2H), 7.04 – 7.00 (m, 2H), 6.92 – 6.89 (m, 2H), 6.87 – 6.82 (m, 2H), 4.21 – 4.04 (m, 2H), 3.80 (brs, 2H), 3.77 (s, 3H), 3.67 (brs, 2H), 3.11 (brs, 8H). 13C NMR (CDCl3, 150 MHz) δ 170.83, 160.09, 155.85, 154.77, 151.00, 145.19, 130.16, 129.52, 128.35, 124.56, 120.10, 119.40, 118.14, 114.68, 55.66, 54.60, 50.78, 45.42, 44.82, 39.41. HRMS calculated for C31H32N5O7 [M+H]+ 586.2302, found 586.2307.
N-3-Pyridyl-N’-[1-{3-chloro-5-(methyl)pyridin-2-yl}piperidin-4-yl]thiourea (6, DO129).
To a solution of 56 (9.7 mg, 30 μmol) in DCM (0.1 mL) was added 4N HCl in dioxane (0.1 mL) in a dropwise fashion and stirred for 2 h at room temperature. The mixture was dried under N2 stream. The residue was dissolved in DCM (0.2 mL) with iPr2NEt (21 μL, 0.12 mmol) and pyridine-3-isothiocyanate (4.5 mg, 30 μmol). The mixture was stirred for 17 h at room temperature. The mixture was diluted with DCM and washed with sat.NaHCO3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (ethylacetate only) to afford 6 (DO129) (8.1 mg, 75%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 8.56 – 8.47 (m, 2H), 7.95 (s, 1H), 7.81 (s, 1H), 7.63 (d, J = 7.5 Hz, 1H), 7.42 – 7.38 (m, 1H), 7.36 (d, 1H), 5.87 (s, 1H), 4.48 (s, 1H), 3.63 (d, 2H), 2.95 (t, 2H), 2.21 (s, 3H), 2.20 – 2.15 (m, 2H), 1.64 – 1.55 (m, 2H). 13C NMR (CDCl3, 150 MHz) δ 180.17, 156.64, 148.29, 146.62, 145.89, 139.51, 133.32, 132.71, 128.14, 124.58, 122.78, 52.68, 48.38, 31.81, 17.28. HRMS calculated for C17H21ClN5S [M+H]+ 362.1206, found 362.1205.
N-{6-(methoxy)Pyridin-3-yl}-N’-[1-{3-chloro-5-(trifluoromethyl)pyridin-2-yl}piperidin-4-yl]thiourea (9).
Step 1: To a solution of a 57 (15 mg, 39 μmol) in DCM (0.1 mL) was added 4N HCl in dioxane (0.1 mL) in a dropwise fashion and stirred for 2 h at room temperature. The mixture was dried under N2 stream. The residue was dissolved in DCM and washed with aqueous 1 N NaOH. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was used in the next step. Step 2: To a solution of an 3-amino-6-methoxypyridine (5.3 mg, 42 μmol) in DCM with iPr2NEt (27 μL, 150 μmol) was added phenylchlorothioformate (7.3 mg, 42 μmol) at 0 °C and stirred for 2 h at room temperature. The boc-deprotected amine obtained in step 1 was added and the mixture was stirred overnight at room temperature. The mixture was diluted with DCM and washed with sat.NaHCO3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by prep-TLC (ethyl acetate:hexane= 1:2) to afford 9 (5.0 mg, 29%) as a colorless amorphous. 1H NMR (CDCl3, 600 MHz) δ 8.36 (s, 1H), 8.09 – 8.07 (m, 1H), 7.74 – 7.73 (m, 1H), 7.49 (s, 1H), 7.47 – 7.43 (m, 1H), 6.82 (d, 1H), 5.62 – 5.45 (m, 1H), 4.56 (s, 1H), 4.03 – 3.94 (m, 5H), 3.06 (t, 2H), 2.26 – 2.15 (m, 2H), 1.57 –1.48 (m, 2H). 13C NMR (CDCl3, 150 MHz) δ 180.95, 163.64, 159.98, 145.41, 143.11, 137.53, 136.10, 125.85, 123.43 (q, 1JC-F = 269.9 Hz), 120.97, 120.10 (q, 2JC-F = = 33.3 Hz), 112.58, 54.12, 52.64, 47.80, 31.78. HRMS calculated for C18H20ClF3N5OS [M+H]+ 446.1029, found 446.1031.
N-3-Pyridyl-N’-[1-{3-chloro-5-(trifluoromethyl)pyridin-2-yl}piperidin-3-yl]thiourea (13).
To a solution of a 61a (20 mg, 53 μmol) in DCM (0.2 mL) was added 4N HCl in dioxane (0.2 mL) in a dropwise fashion and stirred for 2 h at room temperature. The mixture was dried under N2 stream. The residue was dissolved in DCM with iPr2NEt (37 μL, 0.21 mmol) and pyridine-3-isothiocyanate (7.9 mg, 58 μmol) was added. The mixture was stirred overnight at room temperature. The mixture was diluted with DCM and washed with sat.NaHCO3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by prep-TLC (AcOEt only) to afford 13 (24 mg, quantitative yield) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 8.54 (s, 1H), 8.46 (d, 1H), 8.32 (s, 1H), 7.81 (s, 1H), 7.69 (s, 1H), 7.67 – 7.55 (m, 2H), 7.29 (d, 1H), 4.66 (s, 1H), 3.82 – 3.62 (m, 2H), 3.30 (s, 1H), 3.15 (s, 1H), 2.32 (s, 1H), 1.95 – 1.58 (m, 3H). 13C NMR (CDCl3, 150 MHz) δ 180.16, 160.03, 147.96, 146.68, 142.74, 136.40, 133.70, 132.58, 124.43, 123.12 (q, 1JC-F = 270.2 Hz), 122.00, 120.59 (q, 2JC-F = 33.5 Hz), 120.42, 52.56, 50.99, 49.97, 28.27, 22.15. HRMS calculated for C17H18ClF3N5S [M+H]+ 416.0924, found 416.0927.
N-3-Pyridyl-N’-[1-{4-(cyclopropylmethoxy)pyridin-2-yl}piperidin-4-yl]thiourea (32).
To a solution of 82 (16 mg, 45 μmol) in DCM (0.2 mL) was added 4N HCl in dioxane (0.2 mL) in a dropwise fashion and stirred for 2 h at room temperature. The mixture was dried under N2 stream. The residue was dissolved in DCM with iPr2NEt (39 μL, 0.22 mmol) and pyridine-3-isothiocyanate (8.4 mg, 62 μmol) was added. The mixture was stirred overnight at room temperature. The mixture was diluted with DCM and washed with sat.NaHCO3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by prep-TLC (DCM/Acetone=2/3) to afford 32 (16 mg, 95%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 8.49 (s, 3H), 7.96 (d, 1H), 7.77 (s, 1H), 7.35 (s, 1H), 6.40 – 6.19 (m, 2H), 6.10 (s, 1H), 4.52 (s, 1H), 4.12 (d, 2H), 3.79 (d, 2H), 2.98 (t, 2H), 2.19 – 2.06 (m, 2H), 1.40 (s, 2H), 1.27 – 1.19 (m, 1H), 0.67 – 0.61 (m, 2H), 0.36 – 0.30 (m, 2H). 13C NMR (CDCl3, 150 MHz) δ 180.38, 166.99, 161.01, 149.09, 101.54, 93.04, 72.62, 52.60, 44.82, 31.20, 10.10, 3.36. HRMS calculated for C20H26N5OS [M+H]+ 384.1858, found 384.1861.
N-3-Pyridyl-N’-(1-[3-methyl-4-{4-(trifluoromethoxy)phenoxy}pyridine-2-yl]piperidin-4-yl)thiourea (40).
To a solution of 90 (20 mg, 43 μmol) in DCM (0.2 mL) was added 4N HCl in dioxane (0.2 mL) in a dropwise fashion and stirred for 2 h at room temperature. The mixture was dried under N2 stream. The residue was dissolved in DCM with iPr2NEt (30 μL, 0.17 mmol) and pyridine-3-isothiocyanate (6.4 mg, 47 μmol) was added. The mixture was stirred overnight at room temperature. The mixture was diluted with DCM and washed with sat.NaHCO3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by prep-TLC (DCM/Acetone=3/1) to afford 40 (25 mg, quantitative yield) as a colorless amorphous. 1H NMR (CDCl3, 600 MHz) δ 8.58 – 8.49 (m, 2H), 8.04 – 7.98 (m, 1H), 7.89 (s, 1H), 7.72 (s, 1H), 7.39 (s, 1H), 7.22 (d, 2H), 7.02 (d, 2H), 6.33 (d, 1H), 6.06 (s, 1H), 4.53 (s, 1H), 3.49 – 3.34 (m, 2H), 3.09 – 2.93 (m, 2H), 2.29 – 2.08 (m, 5H), 1.50 – 1.32 (m, 2H). 1H NMR (CDCl3, 600 MHz) δ 180.08, 163.60, 162.32, 153.96, 146.08, 144.68, 144.42, 136.39, 130.18, 123.11, 120.88, 120.09 (q, 1JC-F = 254.4 Hz), 113.87, 107.03, 50.75, 48.68, 31.17, 11.26. HRMS calculated for C24H25F3N5O2S [M+H]+ 504.1681, found 504.1677.
(R)-N-3-Pyridyl-N’-(1-[3-chloro-4-{3-chloro-4-(trifluoromethoxy)phenoxy}pyridine-2-yl]piperidin-3-yl)thiourea (49).
To a solution of 99 (20 mg, 38 μmol) in DCM (0.2 mL) was added 4N HCl in dioxane (0.2 mL) in a dropwise fashion and stirred for 2 h at room temperature. The mixture was dried under N2 stream. The residue was dissolved in DCM with iPr2NEt (27 μL, 0.15 mmol) and pyridine-3-isothiocyanate (5.7 mg, 42 μmol) was added. The mixture was stirred overnight at room temperature. The mixture was diluted with DCM and washed with sat.NaHCO3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by prep-TLC (DCM/Acetone=3/1) to afford 49 (12 mg, 57%) as a colorless amorphous. 1H NMR (CDCl3, 600 MHz) δ 8.56 (s, 1H), 8.46 (dd, J = 4.8, 1.5 Hz, 1H), 7.83 (s, 2H), 7.62 (s, 1H), 7.54 (s, 1H), 7.40 (d, J = 2.8 Hz, 1H), 7.30 (dd, J = 8.1, 4.7 Hz, 1H), 7.20 (dd, J = 8.9, 2.8 Hz, 1H), 7.13 (d, J = 8.9 Hz, 1H), 6.09 (d, J = 5.6 Hz, 1H), 4.76 – 4.06 (m, 1H), 3.79 – 2.81 (m, 4H), 2.35 (s, 1H), 1.70 (s, 3H). 13C NMR (CDCl3, 150 MHz) δ 179.99, 160.68, 160.52, 148.64, 147.93, 146.65, 146.59, 146.03, 133.71, 132.51, 127.86, 124.50, 124.07, 123.31, 121.16, 120.42 (q, 1JC-F = 257.0 Hz), 111.95, 106.05, 53.32, 51.10, 50.68, 28.23, 21.97. HRMS calculated for C23H20Cl2F3N5O2S [M+H]+ 558.0745, found 558.0755.
tert-Butyl [1-{3-chloro-5-methylpyridin-2-yl}piperidin-4-yl]carbamate (56).
A solution of 2,3-dichloro-5-(methyl)pyridine (53) (200 mg, 1.23 mmol), tert-butyl(piperidin-4-yl)carbamate (2.5 g, 12 mmol) and Cu powder (8 mg) in DMF (4 mL) was stirred for 18 h at 120 °C. The mixture was diluted with DCM and filtered through a silica pad (ethyl acetate:hexane = 1:1 as an eluent). The eluent was concentrated under reduced pressure. The residue was purified by flash column chromatography (hexane only then ethyl acetate:hexane = 1:10 to 1:3) to afford 56 (150 mg, 37%) as a yellow amorphous. 1H NMR (CDCl3, 600 MHz) δ 7.92 (d, J = 1.9 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 4.59 (d, J = 8.2 Hz, 1H), 3.65 – 3.52 (m, 3H), 2.84 (ddd, J = 13.2, 11.3, 2.5 Hz, 2H), 2.16 (s, 3H), 2.01 – 1.94 (m, 2H), 1.52 (dtd, J = 12.6, 11.0, 3.9 Hz, 2H), 1.34 (s, 9H). 13C NMR (CDCl3, 150 MHz) δ 156.82, 155.22, 145.71, 139.31, 127.76, 122.72, 79.20, 48.47, 47.83, 32.60, 28.46, 17.15. ESI-MS 326.1: ([M+H]+).
tert-Butyl [1-{3-chloro-5-(trifluoromethyl)pyridin-2-yl}piperidin-3-yl]carbamate (61a).
A solution of 2,3-dichloro-5-trifluoromethylpyridine (54) (200 mg, 0.93 mmol), tert-butyl(piperidin-3-yl)carbamate (220 mg, 1.1 mmol) and potassium carbonate (130 mg, 0.93 mmol) in dry DMF (0.5 mL) was stirred at 100 °C overnight. The mixture was diluted with DCM and filtered through a pad of silica with ethylacetate. The eluent was concentrated under reduced pressure and the residue was purified by flash column chromatography (ethyl acetate:hexane=1/8 to 1/3) to afford 61a (350 mg, 99%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 8.33 (s, 1H), 7.70 (d, J = 2.3 Hz, 1H), 5.14 – 4.79 (m, 1H), 3.89 – 3.03 (m, 5H), 1.87 – 1.74 (m, 2H), 1.74 – 1.50 (m, 2H), 1.39 (s, 9H). 13C NMR (CDCl3, 150 MHz) δ 160.52, 155.21, 142.98, 135.96, 123.35 (q, 1JC-F = 269.9 Hz), 121.47, 120.16 (q, 2JC-F = 33.2 Hz), 79.25, 53.80, 49.51, 46.52, 29.99, 28.42, 22.54. ESI-MS: 380.1 ([M+H]+).
2-Chloro-4-(cyclopropylmethoxy)pyridine (67).
To a solution of 2-cyclopropylmethanol (160 mg, 2.3 mmol) in dry DMF (1 mL) was slowly added 60% sodium hydride in mineral oil (91 mg, 2.3 mmol) at 0 °C. The mixture was warmed to room temperature and stirred for 20 min. The reaction mixture was added 4-fluoropyridine (300 mg, 2.3 mmol). The reaction mixture was stirred at 90 °C and the reaction was stirred overnight. The reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography (hexane only to ethyl acetate /hexane=1/8) to afford 67(290 mg, 70%) as a colorless oil. 1H NMR (CDCl3, 600 MHz) δ 8.09 (d, J = 5.8 Hz, 1H), 6.75 (d, J = 2.3 Hz, 1H), 6.68 (dd, J = 5.8, 2.3 Hz, 1H), 3.79 (d, J = 7.1 Hz, 2H), 1.23 – 1.16 (m, 1H), 0.64 – 0.57 (m, 2H), 0.34 – 0.27 (m, 2H). 13C NMR (CDCl3, 150 MHz) δ 166.60, 152.51, 150.18, 110.11, 109.86, 73.22, 9.75, 3.31. ESI-MS 184.1: ([M+H]+).
2-Chloro3-methyl-4-{4-(trifluoromethoxy)phenyloxy}pyridine (75).
To a solution of 4-(trifluoromethoxy)phenol (60 mg, 0.34 mmol) in dry DMF (0.2 mL) was slowly added 60% sodium hydride in mineral oil (14 mg, 0.34 mmol) at 0 °C. The mixture was warmed to room temperature and stirred for 20 min. The reaction mixture was added 2,4-dichloro-3-methylpyridine (50 mg, 0.31 mmol). The reaction mixture was stirred at 120 °C and the reaction was stirred overnight. The reaction mixture was concentrated under reduced pressure. The residue was purified by prep-TLC (ethyl acetate /hexane=1/10) to afford 75 (59 mg, 63%) as a colorless oil. 1H NMR (CDCl3, 600 MHz) δ 8.08 (d, J = 5.6 Hz, 1H), 7.31 – 7.26 (m, 2H), 7.11 – 7.06 (m, 2H), 6.56 (d, J = 5.6 Hz, 1H), 2.40 (s, 3H). 13C NMR (CDCl3, 150 MHz) δ 163.81, 153.36, 153.00, 147.60, 146.19, 123.13, 122.74, 121.55, 120.53 (q, 1JC-F = 255.5 Hz), 110.16, 12.58. ESI-MS 304.0: ([M+H]+).
tert-Butyl [1-{4-(cyclopropylmethyloxy)pyridin-2-yl}piperidin-4-yl]carbamate (82).
A solution of 67 (80 mg, 0.44 mmol), tert-butyl(piperidin-4-yl)carbamate (440 mg, 2.2 mmol) and potassium carbonate (120 mg, 0.87 mmol) in dry DMSO (1 mL) was stirred at 120 °C for 24h. The mixture was diluted with DCM and filtered through a pad of silica with ethylacetate. The eluent was concentrated under reduced pressure and the residue was purified by flash column chromatography (hexane only to hexane/ethyl acetate=1/10 to ½) to afford 82 (40 mg, 26%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 7.99 (d, J = 5.8 Hz, 1H), 6.20 (dd, J = 5.8, 2.1 Hz, 1H), 6.11 (d, J = 2.1 Hz, 1H), 4.54 – 4.42 (m, 1H), 4.14 (dt, J = 13.9, 3.6 Hz, 2H), 3.79 (d, J = 7.0 Hz, 2H), 3.72 – 3.60 (m, 1H), 2.93 (ddd, J = 13.8, 11.6, 2.7 Hz, 2H), 2.04 – 1.94 (m, 2H), 1.49 – 1.34 (m, 11H), 1.28 – 1.18 (m, 1H), 0.68 – 0.60 (m, 2H), 0.37 – 0.29 (m, 2H). 13C NMR (CDCl3, 150 MHz) δ 166.79, 161.26, 155.28, 149.22, 101.22, 92.77, 79.46, 72.48, 48.29, 44.72, 32.21, 28.52, 10.13, 3.33. ESI-MS 348.2: ([M+H]+).
tert-Butyl (1-[3-methyl-4-{4-(trifluoromethoxy)phenoxy}pyridine-2-yl]piperidin-4-yl)carbamate (90).
A solution of 75 (120 mg, 0.37 mmol), tert-butyl(piperidin-4-yl)carbamate (300 mg, 1.5 mmol) and potassium carbonate (80 mg, 0.56 mmol) in dry DMSO (0.2 mL) was stirred at 120 °C for 1h. The mixture was diluted with DCM and filtered through a pad of silica with ethylacetate. The eluent was concentrated under reduced pressure and the residue was purified by prep-TLC (ethyl acetate/hexane= 1/7) to afford 90 (64 mg, 37%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 8.01 (d, J = 5.6 Hz, 1H), 7.22 – 7.17 (m, 2H), 7.03 – 6.98 (m, 2H), 6.32 (d, J = 5.6 Hz, 1H), 4.57 (d, J = 8.1 Hz, 1H), 3.71 – 3.59 (m, 1H), 3.44 – 3.36 (m, 2H), 2.96 – 2.86 (m, 2H), 2.19 (s, 3H), 2.09 – 2.00 (m, 2H), 1.56 (dtd, J = 12.6, 10.9, 3.8 Hz, 2H), 1.44 (s, 9H). 13C NMR (CDCl3, 150 MHz) δ 164.26, 163.12, 155.32, 154.04, 146.12, 145.40, 122.86, 120.75, 120.57 (q, 1JC-F = 256.1 Hz), 114.83, 107.07, 79.41, 49.13, 47.96, 32.94, 28.39, 11.50. ESI-MS 468.1: ([M+H]+).
(R)-tert-Butyl (1-[3-chrolo-4-{2-chloro-4-(trifluoromethoxy)phenoxy}pyridine-2-yl]piperidin-3-yl)carbamate (99).
A solution of 8119 (120 mg, 0.37 mmol), (R)-tert-butyl(piperidin-3-yl)carbamate (150 mg, 0.74 mmol) and potassium carbonate (61 mg, 0.44 mmol) in dry DMSO (0.3 mL) was stirred at 100 °C for 3h. The mixture was diluted with DCM and filtered through a pad of silica with ethylacetate. The eluent was concentrated under reduced pressure and the residue was purified by prep-TLC (ethyl acetate/hexane= 1/3) to afford 99 (91 mg, 47%) as a white solid. 1H NMR (CDCl3, 600 MHz) δ 8.00 (d, J = 5.6 Hz, 1H), 7.39 (d, J = 2.8, 1H), 7.18 (dd, J = 8.9, 2.8, 1H), 7.13 (d, J = 9.0 Hz, 1H), 6.19 (d, J = 5.6 Hz, 1H), 5.33 – 4.89 (m, 1H), 3.97 – 3.65 (m, 1H), 3.51 – 3.08 (m, 4H), 1.92 – 1.59 (m, 4H), 1.44 (s, 9H). 13C NMR (CDCl3, 150 MHz) δ 161.17, 160.46, 155.40, 148.96, 146.38, 146.30, 127.78, 124.03, 123.07, 121.09, 120.42 (q, 1JC-F = 257.0 Hz), 111.94, 106.05, 79.28, 54.62, 50.43, 46.50, 29.99, 28.58, 22.51. ESI-MS 522.0: ([M+H]+).
Lyso-PS hydrolysis assay
The lyso-PS lipase activity of ABHD12 was determined as described19. Briefly, DO127 were incubated with ABHD12-overexpressing HEK293T membrane (0.25 mg/mL) in DPBS for 20 min at room temperature. 17:1 lyso-PS in DPBS was added to each reaction (100 μM final concentration) and incubated at 37 °C. After 30 min, the reaction was quenched with 400 μL of 2:1 CHCl3/MeOH (v/v) with 1 nmol 15:0 FFA as an internal standard. The mixture was vortexed and centrifuged at 1,400 x g to separate the aqueous and organic phase. The organic phase was analyzed by LC-MS (1200 LC/MSD, Agilent Technologies) using a 50 mm × 4.6 mm 5 μm Gemini C18 column (Phenomenex) coupled to a guard column (Gemini: C18: 4 × 3 mm). The LC solvents were as follows: buffer A, H2O:MeOH (95:5, v/v) with 0.1 % NH4OH (v/v); and buffer B, iPrOH:MeOH:H2O (60:35:5, v/v/v) with 0.1 % NH4OH (v/v). The LC method consisted of 0.1 mL/min 20% buffer B for 1.0 min, 0.4 ml/min isocratic mode of 100% buffer B over 7 min and equilibration with 0.5 mL/min 100% buffer A for 3 min. The MS analyses were performed using an electrospray ionization source (ESI) in negative ion mode to measure product formation. MS data were acquired in selected ion monitoring mode at m/z 267.20 for 17:1 FFA and m/z 241.20 for 15:0 FFA.
Gel-based competitive ABPP
Gel-based ABPP assays were performed as described19. Tissue proteomes (50 μL, 1 mg/mL) were treated with inhibitors for 20 min at room temperature (for (thio)urea analogues) or 45 min at 37 °C (for NHH carbamate analogues) . Then the proteomes were labeled with FP-Rh (1 μM final concentration) or JJH350 (2 μM final concentration) for 45 min at 37 °C. For FP-Rh labeled samples, the reactions were quenched by adding 20 μL of 4X SDS-PAGE loading buffer. The 30 μL of the quenched samples were loaded on gel for analysis. For JJH350-labeled samples, copper-catalyzed azide-alkyne cycloaddition (CuAAC) was used for visualizing the labeled proteins. For CuAAC, rhodamine-PEG3-N3 (1 μL/reaction, 1.25 mM in DMSO), CuSO4 (1 μL/reaction, 50 mM in H2O), TBTA (3 μL/reaction, 1.7 mM in DMSO/t-BuOH [1:4, v/v]) and tris(2-carboxyethyl)phosphine (TCEP) (1 μL/reaction, 50 mM in H2O, freshly prepared) were premixed. Then 6 μL of this click reagents mixture was immediately added to each JJH350-labeled samples (50 μL, 1 mg/mL) and incubated for 1 h at room temperature. The reactions were quenched by adding 20 μL of 4X SDS-PAGE loading buffer. The 40 μL of the quenched samples were loaded on a gel for analysis. After separation by SDS-PAGE (10% acrylamide), samples were visualized by in-gel fluorescence scanning using the ChemiDoc MP system (Bio-Rad). Band intensities were quantified using the Image Lab (5.2.1) software (Bio-Rad).
Preparation of tissue proteomes
Mouse tissues were dounce-homogenized in DPBS followed by low-speed spin (1,400 x g, 3 min, 4 °C) to remove debris. The membrane and cytosolic fractions were separated by high-speed spin (16,000 x g, 45 min, 4 °C) of the resulting homogenate lysate. After removal of the soluble supernatant, the membrane pellet was washed with cold DPBS and resuspended with cold DPBS. Total protein concentrations in membrane fractions were determined using the Bio-Rad DC protein assay kit. Samples were used immediately for the following ABPP experiments.
IC50 calculations
ABHD12 enzyme activities in mice brain membrane fraction were determined by fluorescent ABHD12 band intensity visualized by JJH350 probe followed by conjugation of rhodamine-N3 (for detail, see gel-based competitive ABPP section). The relative intensity was compared to the ABHD12 band intensity from a control-treated sample, which was set to 100%. IC50 values were determined by plotting a log(inhibitor) vs. normalized response, and the dose-response curves were generated using the Prism software (GraphPad).
Cytokine measurements in THP-1 cells
THP-1 cells (ATCC) were cultured in RPMI 1640 (Corning) with 10% heat-inactivated FBS (Omega Scientific) and 0.05mM 2-mercaptoethanol (Fisher) at a density between 2×105 to 1×106 cells/ml. On day of experiment, cells were plated into 6-well culture plates at a concentration of 2.5×106 cells/well and treated with 150ng/ml of phorbol 12-myristate 13-acetate (PMA, Sigma) for 24 hours in the presence of DO264 (1, 0.5 and 0.1μM), DO271 (1μM) or DMSO. Following PMA treatment cells were adherent and washed twice with culture media to remove PMA. Cells were allowed to rest for 24 hours to become resting macrophages in the presence of DO264, DO271 or DMSO. Resting macrophages were treated with fresh media containing 20ng/ml IFN-γ (Sigma) and 10pg/mL LPS (Sigma) for 24 hours in the presence of DO264, DO271 or DMSO to achieve M1 polarization. Aliquots of media were collected 6 and 24 hours following the addition of polarizing cytokines and selected cytokines were measured using the MSD V-Plex Plus assay.
Cell viability assay
Cells were seeded into 96-well plates at 3,000 cells per well. After 24 hours, test compounds were added to cells to final concentrations ranging from 0.1 to 10 μM. Cells were then incubated for 72 hours and cell viability was measured using CellTiter-Glo assay (Promega) following manufacturer’s instructions. Relative cell viability in the presence of test compounds was normalized to the vehicle-treated controls.
Compound treatment for in vivo studies
JJH329 was suspended in 1:1 (v/v) solution of EtOH/PEG40 (by bath sonication), and the solution was diluted with 9 volumes of DPBS followed by vortexing to obtain 1:1:18 (v/v/v) compound solution of EtOH/PEG40/DPBS. 10 μL/g mouse body weight of the freshly prepared compound solution was injected into the mouse peritoneal. Compound-treated mice were anesthetized with isoflurane and euthanized by cervical dislocation to harvest tissues. The experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Statistics
Statistical analyses were performed using the R statistical programming language or Prism. All data are shown as mean values ± SEM or SD. Two-sided Student’s t-test was used to perform statistical analyses. A p-value of < 0.05 was considered statistically significant for this study.
Supplementary Material
Acknowledgments.
We thank Micah Niphakis (Abide Therapeutics) for critical reading of the manuscript; J. Chen (Automated Synthesis Facility at Scripps Research) for measuring purity of ABHD12 inhibitors; C.E. Moore and M. Gembicky (UCSD) for X-ray crystallographic analysis of AW01275. This work was supported by the NIH (DA037660) and Abide Therapeutics.
Abbreviations Used
- ABHD12
α/β-hydrolase domain-containing 12
- PHARC
Polyneuropathy, Hearing loss, Ataxia, Retinosa pigmentosa, and Cataract
- PS
phosphatidylserine
- PI
phosphatidylinositol
- GPCR
G-protein-coupled receptor
- S1P
sphingosine 1-phosphate
- PA
phosphatidic acid
- CNS
central nervous system
- ABPP
activity-based protein profiling
- LCMV
lymphocytic choriomeningitis virus
- SAR
structure-activity relationship
- NHH
N-hydroxyhydantoin
- FP-Rh
fluorophosphonate-rhodamine
- ABHD6
α/β-hydrolase domain-containing 6
- CI
confidence interval
- CuAAC
copper-catalyzed azide-alkyne cycloaddition
- Rh-N3
rhodamine-(PEG)3-azide
- HTS
high-throughput screening
- PMA
phorbol 12-myristate 13-acetate
- LPS
lipopolysaccharide
- IFN-γ
interferon gamma
- TNF-α
tumor necrosis factor-alpha
- IL-1β
interleukin-1 beta
- CCL3
chemokine ligand 3
- CCL4
chemokine ligand 4
- SnAr
nucleophilic aromatic substitution
- EDCI
N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide
- HOBt
hydroxybenzotriazole
- FFA
free fatty acid
- TBTA
tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine
- TCEP
tris(2-carboxyethyl)phosphine
- FBS
fetal bovine serum
- SEM
standard error of the mean
- SD
standard deviation
Footnotes
Supporting Information.
Gel-based ABPP assay of JJH329-treated mice tissues, substrate assay data for DO127, 13C NMR analysis of commercial AW01275, cytotoxicity assay of DO264, additional synthetic chemistry procedures and analytical data for compound 8, 10 – 12, 14 – 31, 33 – 37, 41 – 45, 47 and X-ray crystallographic data summary for commercial AW01275 (PDF) X-ray Crystallographic Data for commercial AW01275 (CIF) Molecular formula string (CSV)
References
- 1.Grzelczyk A; Gendaszewska-Darmach E, Novel Bioactive Glycerol-Based Lysophospholipids: New Data -- New Insight into Their Function. Biochimie 2013, 95 (4), 667–679. [DOI] [PubMed] [Google Scholar]
- 2.Makide K; Kitamura H; Sato Y; Okutani M; Aoki J, Emerging Lysophospholipid Mediators, Lysophosphatidylserine, Lysophosphatidylthreonine, Lysophosphatidylethanolamine and Lysophosphatidylglycerol. Prostaglandins Other Lipid Mediat. 2009, 89 (3–4), 135–139. [DOI] [PubMed] [Google Scholar]
- 3.Yung YC; Stoddard NC; Chun J, LPA Receptor Signaling: Pharmacology, Physiology, and Pathophysiology. J. Lipid Res. 2014, 55 (7), 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosen H; Germana Sanna M; Gonzalez-Cabrera PJ; Roberts E, The Organization of the Sphingosine 1-Phosphate Signaling System. Curr. Top. Microbiol. Immunol. 2014, 378, 1–21. [DOI] [PubMed] [Google Scholar]
- 5.Adams DR; Pyne S; Pyne NJ, Sphingosine Kinases: Emerging Structure-Function Insights. Trends Biochem. Sci. 2016, 41 (5), 395–409. [DOI] [PubMed] [Google Scholar]
- 6.Castagna D; Budd DC; Macdonald SJ; Jamieson C; Watson AJ, Development of Autotaxin Inhibitors: An Overview of the Patent and Primary Literature. J. Med. Chem. 2016, 59 (12), 5604–5621. [DOI] [PubMed] [Google Scholar]
- 7.Alhouayek M; Masquelier J; Muccioli GG, Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharmacol. Sci. 2018, 39 (6), 586–604. [DOI] [PubMed] [Google Scholar]
- 8.Yamashita A; Oka S; Tanikawa T; Hayashi Y; Nemoto-Sasaki Y; Sugiura T, The Actions and Metabolism of Lysophosphatidylinositol, an Endogenous Agonist for GPR55. Prostaglandins Other Lipid Mediat. 2013, 107, 103–116. [DOI] [PubMed] [Google Scholar]
- 9.Makide K; Uwamizu A; Shinjo Y; Ishiguro J; Okutani M; Inoue A; Aoki J, Novel Lysophosphoplipid Receptors: Their Structure and Function. J. Lipid Res. 2014, 55 (10), 1986–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Kleij D; Latz E; Brouwers JF; Kruize YC; Schmitz M; Kurt-Jones EA; Espevik T; de Jong EC; Kapsenberg ML; Golenbock DT; Tielens AG; Yazdanbakhsh M, A Novel Host-Parasite Lipid Cross-Talk. Schistosomal Lyso-Phosphatidylserine Activates Toll-Like Receptor 2 and Affects Immune Polarization. J. Biol. Chem. 2002, 277 (50), 48122–48129. [DOI] [PubMed] [Google Scholar]
- 11.Ikubo M; Inoue A; Nakamura S; Jung S; Sayama M; Otani Y; Uwamizu A; Suzuki K; Kishi T; Shuto A; Ishiguro J; Okudaira M; Kano K; Makide K; Aoki J; Ohwada T, Structure-Activity Relationships of Lysophosphatidylserine Analogs as Agonists of G-Protein-Coupled Receptors GPR34, P2Y10, and GPR174. J. Med. Chem. 2015, 58 (10), 4204–4219. [DOI] [PubMed] [Google Scholar]
- 12.Kotsikorou E; Sharir H; Shore DM; Hurst DP; Lynch DL; Madrigal KE; Heynen-Genel S; Milan LB; Chung TD; Seltzman HH; Bai Y; Caron MG; Barak LS; Croatt MP; Abood ME; Reggio PH, Identification of the GPR55 Antagonist Binding Site Using a Novel Set of High-Potency GPR55 Selective Ligands. Biochemistry 2013, 52 (52), 9456–9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sayama M; Inoue A; Nakamura S; Jung S; Ikubo M; Otani Y; Uwamizu A; Kishi T; Makide K; Aoki J; Hirokawa T; Ohwada T, Probing the Hydrophobic Binding Pocket of G-Protein-Coupled Lysophosphatidylserine Receptor GPR34/LPS1 by Docking-Aided Structure-Activity Analysis. J. Med. Chem. 2017, 60 (14), 6384–6399. [DOI] [PubMed] [Google Scholar]
- 14.Chen DH; Naydenov A; Blankman JL; Mefford HC; Davis M; Sul Y; Barloon AS; Bonkowski E; Wolff J; Matsushita M; Smith C; Cravatt BF; Mackie K; Raskind WH; Stella N; Bird TD, Two Novel Mutations in ABHD12: Expansion of the Mutation Spectrum in PHARC and Assessment of Their Functional Effects. Hum. Mutat. 2013, 34 (12), 1672–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiskerstrand T; H’Mida-Ben Brahim D; Johansson S; M’Zahem A; Haukanes BI; Drouot N; Zimmermann J; Cole AJ; Vedeler C; Bredrup C; Assoum M; Tazir M; Klockgether T; Hamri A; Steen VM; Boman H; Bindoff LA; Koenig M; Knappskog PM, Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: An Inborn Error of Endocannabinoid Metabolism. Am. J. Hum. Genet. 2010, 87 (3), 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blankman JL; Long JZ; Trauger SA; Siuzdak G; Cravatt BF, ABHD12 Controls Brain Lysophosphatidylserine Pathways That Are Deregulated in a Murine Model of the Neurodegenerative Disease PHARC. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (4), 1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu C; Jin X; Tsueng G; Afrasiabi C; Su AI, BioGPS: Building Your Own Mash-up of Gene Annotations and Expression Profiles. Nucleic Acids Res. 2016, 44 (D1), D313–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viader A; Ogasawara D; Joslyn CM; Sanchez-Alavez M; Mori S; Nguyen W; Conti B; Cravatt BF, A Chemical Proteomic Atlas of Brain Serine Hydrolases Identifies Cell Type-Specific Pathways Regulating Neuroinflammation. eLife 2016, 5, e12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogasawara D; Ichu TA; Vartabedian VF; Benthuysen J; Jing H; Reed A; Ulanovskaya OA; Hulce JJ; Roberts A; Brown S; Rosen H; Teijaro JR; Cravatt BF, Selective Blockade of the Lyso-PS Lipase ABHD12 Stimulates Immune Responses In Vivo. Nat. Chem. Biol. 2018, 14 (12), 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cognetta AB 3rd; Niphakis MJ; Lee HC; Martini ML; Hulce JJ; Cravatt BF, Selective N-Hydroxyhydantoin Carbamate Inhibitors of Mammalian Serine Hydrolases. Chem. Biol. 2015, 22 (7), 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung D; Hardouin C; Boger DL; Cravatt BF, Discovering Potent and Selective Reversible Inhibitors of Enzymes in Complex Proteomes. Nat. Biotechnol. 2003, 21 (6), 687–691. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y; Patricelli MP; Cravatt BF, Activity-Based Protein Profiling: the Serine Hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (26), 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patricelli MP; Giang DK; Stamp LM; Burbaum JJ, Direct Visualization of Serine Hydrolase Activities in Complex Proteomes Using Fluorescent Active Site-Directed Probes. Proteomics 2001, 1 (9), 1067–1071. [DOI] [PubMed] [Google Scholar]
- 24.Speers AE; Adam GC; Cravatt BF, Activity-Based Protein Profiling In Vivo Using a Copper(i)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J. Am. Chem. Soc. 2003, 125 (16), 4686–4687. [DOI] [PubMed] [Google Scholar]
- 25.Rostovtsev VV; Green LG; Fokin VV; Sharpless KB, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41 (14), 2596–2599. [DOI] [PubMed] [Google Scholar]
- 26.Onderwater RC; Commandeur JN; Menge WM; Vermeulen NP, Activation of Microsomal Glutathione S-Transferase and Inhibition of Cytochrome P450 1A1 Activity as a Model System for Detecting Protein Alkylation by Thiourea-Containing Compounds in Rat Liver Microsomes. Chem. Res. Toxicol. 1999, 12 (5), 396–402. [DOI] [PubMed] [Google Scholar]
- 27.Aoki J; Nagai Y; Hosono H; Inoue K; Arai H, Structure and Function of Phosphatidylserine-Specific Phospholipase A1. Biochim. Biophys. Acta 2002, 1582 (1–3), 26–32. [DOI] [PubMed] [Google Scholar]
- 28.Kamat SS; Camara K; Parsons WH; Chen DH; Dix MM; Bird TD; Howell AR; Cravatt BF, Immunomodulatory Lysophosphatidylserines Are Regulated by ABHD16A and ABHD12 Interplay. Nat. Chem. Biol. 2015, 11 (2), 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inloes JM; Jing H; Cravatt BF, The Spastic Paraplegia-Associated Phospholipase DDHD1 Is a Primary Brain Phosphatidylinositol Lipase. Biochemistry 2018, 57 (39), 5759–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daigneault M; Preston JA; Marriott HM; Whyte MK; Dockrell DH, The Identification of Markers of Macrophage Differentiation in PMA-Stimulated THP-1 Cells and Monocyte-Derived Macrophages. PLoS One 2010, 5 (1), e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwende H; Fitzke E; Ambs P; Dieter P, Differences in the State of Differentiation of THP-1 Cells Induced by Phorbol Ester and 1,25-Dihydroxyvitamin D3. J. Leukoc. Biol. 1996, 59 (4), 555–561. [PubMed] [Google Scholar]
- 32.Tsuchiya S; Kobayashi Y; Goto Y; Okumura H; Nakae S; Konno T; Tada K, Induction of Maturation in Cultured Human Monocytic Leukemia Cells by a Phorbol Diester. Cancer Res. 1982, 42 (4), 1530–1536. [PubMed] [Google Scholar]
- 33.Lichtman AH; Leung D; Shelton CC; Saghatelian A; Hardouin C; Boger DL; Cravatt BF, Reversible Inhibitors of Fatty Acid Amide Hydrolase That Promote Analgesia: Evidence for an Unprecedented Combination of Potency and Selectivity. J. Pharmacol. Exp. Ther. 2004, 311 (2), 441–448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.