

Abstract
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV), including granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), are autoimmune conditions associated with small vessel inflammation. Earlier studies indicate that complement activation via the alternative pathway plays a major role in the pathogenesis. In this study we have investigated if ANCA-activation of neutrophils from AAV patients leads to activation of the alternative complement pathway. C5a-primed neutrophils (PMN) from 10 AAV patients and 10 healthy controls (HC) were stimulated with PMA or IgG purified from PR3-ANCA positive patients (ANCA IgG). The supernatants were analyzed for release of complement proteins and markers of different granules by ELISA, and release of microparticles (MP) by flow cytometry. The ability of the supernatants to activate the alternative complement pathway was determined by incubation with normal serum and C3bBbP and C5a were measured by ELISA. MP were analyzed by flow cytometry and removed by centrifugation. The supernatants from the AAV patients’ neutrophils produced significantly more C3bBbP compared with HCs (p = 0.0001). C3bBbP levels correlated with the number of MP. After removal of MP from the supernatants, alternative pathway activation was significantly lower. This study shows that primed and ANCA-stimulated neutrophils from AAV patients have a greater ability to activate the alternative complement pathway compared to primed neutrophils from healthy controls. This finding emphasizes the role of complement in the pathogenesis of AAV - underlining the therapeutic potential of C5a and other complement blockade.
Introduction
Primary systemic vasculitis is characterized by relapsing-remitting inflammation and necrosis of blood vessel walls and sometimes granuloma formation. Small-vessel vasculitis lesions with little or no immune complex deposition (pauci-immune) in conjunction with anti-neutrophil cytoplasmic autoantibodies (ANCA) characterize ANCA-associated vasculitides (AAV). AAV affect small vessels in various organs, such as the kidneys, and include granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA). ANCA are in most cases directed against proteinase 3 (PR3) or myeloperoxidase (MPO), two important enzymes in the host defense against bacteria which are located in the granules of neutrophils and monocytes [1].
ANCA can activate primed neutrophils (PMN) to release their granular content, produce reactive oxygen species and mediate the release of microparticles (MP) from neutrophils as previously described by our group and others [2, 3]. In AAV, immune complexes and complement were previously considered not to be involved in the pathogenesis, since depositions detected by immunofluorescence are generally absent or scanty in the lesions, which differs from immune complex-mediated and anti-GBM glomerulonephritis [4]. However, low levels of immune complexes and complement do exist at sites of vascular inflammation and necrosis. Haas and Eustace performed studies on 126 renal biopsies from patients with crescentic glomerulonephritis associated with positive ANCA serology and/or necrotizing small vessel arteritis and found immune complex deposits in 54% of these [5]. In the majority of these cases immunofluorescence was positive for Ig and/or C3.
Activation of neutrophils is thought to play a major role in the pathogenesis and this was demonstrated in vivo when a murine disease model of MPO-ANCA vasculitis was established [6]. This opened new avenues for experiments that also suggested a critical role for complement activation in AAV, via the alternative and terminal pathways and that intervening in complement activation can prevent disease progression [7–9]. Anti-MPO IgG was induced in MPO-deficient mice and transferred into wild-type mice, resulting in crescentic glomerulonephritis. When the recipient mice were deficient in C5 or factor B of the alternative pathway no disease developed. Mice deficient in C4 developed glomerulonephritis to the same extent, indicating that there was no involvement of the classical or lectin pathways. When mice were pre-treated with a C5-inhibiting monoclonal antibody the lesions could be prevented.
After release from the bone marrow into the circulation, neutrophils can be primed by pro-inflammatory mediators, e.g. TNF-α and C5a and become attached to locally activated endothelium. ANCA can then activate these attached neutrophils. By mechanisms that are still unclear, the alternative pathway is activated, leading to generation of C5a which primes surrounding neutrophils by binding to C5a receptors. C5a recruits more neutrophils to the site through chemotaxis and creates an inflammatory amplification loop that finally results in necrotizing vascular injury [10]. Patients with ANCA disease produce higher plasma levels of complement factors C3a, C5a, soluble C5b-9, and Bb in active disease than in remission while no difference was reported in plasma C4d [8]. These data support the hypothesis that ANCA-induced neutrophil activation activates the alternative complement pathway.
Animal and in vitro studies have demonstrated a pivotal role of C5a and its neutrophil receptor C5aR (CD88). In a murine model of MPO-ANCA vasculitis, C5aR-deficient mice injected with anti-MPO IgG were protected from disease to a higher degree than wild type mice (5 out of 6) [11]. Further studies on blockade of the C5aR confirmed the central role in the pathogenesis of AAV and a clinical trial of a small-molecule C5aR antagonist is ongoing [12]. Less is known about the functional role of the second receptor of C5a, C5L2, and studies have shown contradictory results with anti- or pro-inflammatory effects in different disease settings [13]. The role of C5L2 in the pathogenesis of AAV was recently investigated in a study by Hao et al. Neutrophils where pre-incubated with a C5L2 blocking antibody, before C5a priming and subsequent stimulation with ANCA, leading to less release of granular proteins and less membrane expression of PR3—suggesting a pro-inflammatory role of C5L2 [14].
Clinical and experimental data strongly indicate activation of the alternative complement pathway, but the mechanisms by which complement activation occurs in AAV remain unclear. The current study investigated if supernatants from ANCA-activated AAV patient neutrophils activate the alternative complement pathway in normal serum and explored the routes of activation by investigating the expression of C5a receptors and the role of MP.
Materials and methods
Blood samples and patients
PMN were isolated from 10 PR3-ANCA positive patients in stable clinical remission, as assessed by Birmingham Vasculitis Activity Score (BVAS) 0 [15], and 10 healthy controls (HC). Table 1.
Table 1. PMN donor characteristics.
| Age Median (IQR) |
Sex | Diagnosis | BVAS Median (IQR) |
Corticosteroids (mg/day) Median (IQR) |
ANCA | |
|---|---|---|---|---|---|---|
|
AAV (n = 10) |
68 (15) | 6/10 Male | 10/10 GPA | 0 (0) | 5 (2.5) | 10/10 PR3 |
|
HC (n = 10) |
52 (32) | 3/10 Male | - | - | - | - |
PMN = polymorphonuclear neutrophils, AAV = ANCA-associated vasculitis, HC = healthy controls, IQR = inter quartile range, BVAS = Birmingham vasculitis activity score, GPA = granulomatosis with polyangiitis, ANCA = anti-neutrophil cytoplasmic antibodies.
Serum samples for IgG purification were obtained from AAV patients with PR3–ANCA specificity. The ANCA IgGs that we use have been well characterized before by toxicity index, epitope mapping and affinity [2]. HC IgG was used as negative control in some assays.
Written informed consent was retrieved from all donors and the Helsinki declaration was followed. These studies were conducted with permission from the Regional Ethical Review Board, Lund, Sweden, DNR 110/2008.
IgG purification
IgG was purified using NAb protein G spin columns (Thermo Scientific, Rockford, IL, USA) and concentrated to 2 mg/mL, as previously described [2].
PMN purification
Heparinized blood was collected from 10 patients and 10 HCs and the PMN were purified by density gradient centrifugation as previously described [2].
Degranulation assay
We performed two series of experiments in which PMN were primed with TNF-α in one arm and with C5a in the other. PMN primed with TNF-α were stimulated with 10 different ANCA IgG as previously described by our group [2]. PMN primed with C5a were stimulated with 4 different ANCA IgG. After optimization experiments a C5a concentration of 60 ng/mL was chosen.
Purified PMN were adjusted to 5 x 106 cells/mL in KRG (Krebs–Ringer buffer with glucose: 130 mM NaCl, 5 mM KCl, 1·27 mM MgSO4, 0·95 mM CaCl2, 10mM NaH2PO4/Na2HPO4, pH 7·4, 5 mM glucose). PMN were exposed to cytochalasin B (CLB, Sigma-Aldrich, Steinheim, Germany, 5 μg/ml) for 5 min followed by priming with TNF-α (R&D systems, Abingdon, UK, 2 ng/ml) or C5a (R&D Systems, Abingdon, UK, 60 ng/mL) for 15 min. The cells were then stimulated with IgG (200 μg/ml) for 15 min. All stimulations were performed at 37°C, with gentle shaking. As controls, cells stimulated with PMA (4 μg/ml), HC IgG or buffer only were used. Subsequent steps were performed as previously described [2].
Degranulation ELISA
Supernatants and pellets from the degranulation assay were run through 5 different ELISAs to analyze the degree of degranulation, as previously described [16]. Albumin was used to detect release from secretory vesicles, gelatinase for gelatinase granules, lactoferrin for specific granules, myeloperoxidase for azurophil granules. The other main ANCA autoantigen PR3 was also analyzed. Subcellular fractionation of neutrophils has shown that PR3 can be detected mainly in azurophil granules, but also secretory vesicles and specific granules [17].
We also studied the release and total contents of properdin. Properdin was analyzed by the commercial Hycult Properdin ELISA kit from Nordic Biosite, with a detection rate of 0.31–20 ng/ml. Supernatants were diluted 1:25 and pellets 1:50.
Release was calculated as the amount of protein in the supernatant divided with the total amount of protein in the supernatant and pellet all together.
ROS assay
Purified PMN were adjusted to 2x106 cells/ml in Hanks’ balanced salt solution (HBSS++) (Hyclone) and primed with C5a 60 ng/mL for 15 min at 37°C followed by Cytochalasin B (Sigma-Aldrich, Steinheim, Germany; 5 μg/ml) exposure for 5 min at 37°C. PMN were then stimulated with either purified ANCA IgG (200 μg/ml), HC IgG or a monoclonal anti PR3 antibody (a kind gift from Dr Zhao, Beijing, 5 μg/ml), phorbol 12-myristate 13-acetate (PMA) (Glycotope Biotechnology, Heidelberg, Germany; 100 ng/ml) or HBSS at 37°C. This was performed in duplicate samples in white 96-well polystyrene plates (Thermo Scientific) in the presence of luminol (Sigma-Aldrich, Fluka; 1 μg/ml) and the scavenger superoxide dismutase (SOD) (Sigma-Aldrich; 5 U/ml) and catalase (Sigma-Aldrich; 2400 U/ml) to detect intracellular ROS, or isoluminol (Sigma-Aldrich; 1 μg/ml) alone to detect extracellular ROS. PMN were stimulated for a total of 60 min and luminescence, as a measure of ROS production, was monitored in a microplate reader (Tristar LB941; Berthold Technologies, Bad Wildbad, Germany) every 10 min. Each well was measured for 0·1 s with filter. The software MicroWin 2000 was used for analysis. We chose to report the value after 20 min of stimulation, as in most cases this time point corresponds to maximum stimulation.
Complement activation assays by ELISA
The complement activation assays were performed on the PMN supernatants generated in the degranulation assay.
C3bBbP complex and C5a formation
100 μL of all supernatants from the degranulation assay were put on microdialysis against Veronal buffered saline (pH 7.4), MWCO 6–8000 at 4° overnight. Then 90 μL of microdialysis sample was added to 90 μL of HC serum on ice, with gentle shaking at 37°. 50 μL samples were removed at 0,1 and 4 hours and 5 μL 0.5 M EDTA was added. Samples were immediately put in the freezer at -80°C.
C3bBbP ELISA assay
Polystyrene 96-well plates (Maxisorp; Nunc, Roskilde, Denmark) were coated with anti-properdin IgG (IgG fraction of rabbit antiserum against human properdin, a kind gift from dr K Persson, dept. of Virology, Malmö) diluted 1/500 in PBS overnight at 4°. Plates were washed three times with ELISA wash (0.9% NaCl, 0.05% Tween 20) between each of the following incubation steps. ELISA plates were blocked with incubation buffer (PBS 1% BSA) while gently shaking for 2 hours at room temperature. Samples (1/10 in PBS-T (0.05% Tween 20)) were added for 2 hours at room temperature. The detection antibody a-C3c (IgG fraction of rabbit antiserum against human C3c, Dako A0062, Dako, Glostrup, Denmark) was conjugated with alkaline phosphatase and incubated at dilution 1/500 in PBS-T for 2 hours at room temperature [18].
C5a ELISA assay
C5a was measured in all samples (1/200) according to manufacturer’s instructions using a commercial C5a ELISA kit (A025 MicroVue, Quidel, San Diego, CA, USA).
Detection, quantification and removal of MP
MP released from the stimulated cells were measured in the supernatants by flow cytometry (BD Accuri C6), as previously described [19]. Supernatants from the degranulation assay were used. In short, 100 μL supernatant was mixed with an equal volume of binding buffer and then incubated with 7 μL of CD66-PE (551480 BD Pharmingen) and 5 μL of Annexin V-FITC (556419 BD Bioscience) for 20 min at room temperature. The MP gate was determined by 0.8–1 μM fluorescent beads, as previously described. All CD66/Annexin V positive events identified by size were considered to be MP. Negative (50 μM EDTA in PBS) and positive (supernatants from PMN stimulated with 100 ng/ ml TNF-α) controls were included. MP were removed from the supernatants by centrifugation at 21 300 g, 30 min.
Expression of C5aR1 and C5L2
Isolated and unstimulated PMN from 5 patients and 5 HCs were tested for C5aR and C5L2 surface expression by FACS. They were stained with monoclonal antibodies against C5aR, APC anti-human CD88 (C5aR) (BioLegend, San Diego, CA, USA) and C5L2, PE anti-human C5L2 (BioLegend, SanDiego, CA, USA) and analyzed using a fluorescence activated cell sorter (FACS) Canto II (BD Biosciences, Stockholm, Sweden). The samples were blocked using human IgG (0,5 mg/ml) in PBS containing 5% mouse serum and 1% BSA (Sigma Chemical Co., St Louis, MO, USA). The neutrophil population was identified by forward and side scatter gating. Mean fluorescence intensity (MFI) and percentage of C5aR and C5L2-positive cells were recorded.
The effects of priming with either TNF-α or C5a with or without pre priming with CLB were studied. A negative control was kept at 4°C. Samples were analyzed at 5, 10, 15, 30 and 60 min.
Further experiments were performed to test surface expression of C5aR and C5L2 at different time points on stimulated neutrophils from 2 patients and 2 HCs. PMN were primed with either CLB and TNF-α or CLB and C5a. Each sample was made in duplicates, one with only priming and one with the addition of a monoclonal
PR3 antibody (a kind gift from Dr Zhao, Beijing, 5μg/ml) at 15 min. Samples were then analyzed at 30 and 60 min.
Statistical analysis
All statistical analyses were performed on GraphPad Prism 7.0 software (GraphPad Software, San Diego, CA, USA). Correlations were determined by Spearman’s correlation test. Mann-Whitney U-test was used for two group comparisons. All p-values were considered significant at p < 0.05.
Results
PMN stimulation
PMN from AAV patients and HC degranulated to a similar extent upon ANCA-stimulation after priming with TNF-α, as it has been shown previously [2] and the same was seen after priming with C5a. C5a priming, however, generated a significantly higher degree of degranulation, especially of PR3 and MPO, compared to TNF-α priming (45–50% versus 5–10%), Table 2. In order to obtain an extra effect, on top of the priming effect, with ANCA stimulation, the PMN had to be pre-incubated with CLB and there was a variation in the effect of individual ANCA IgGs. However, there was no significant difference when comparing to what degree the individual IgGs stimulated PMN from HC or from AAV patients. HC IgG also stimulated degranulation to some extent, Table 3.
Table 2. Degranulation (% release, median (IQR)).
| PMN/Priming | HSA | MMP-9 | LF | MPO | PR3 | P |
|---|---|---|---|---|---|---|
|
AAV/TNF-α (n = 50) |
62 (9) | 38 (22) | 12 (12) | 5 (5) | 4 (4) | 34 (22) |
|
AAV/C5a (n = 20) |
75 (8) p < 0.001 |
55 (21) p < 0.01 |
37 (15) p < 0.0001 |
44 (35) p < 0.0001 |
31 (18) p < 0.0001 |
44 (18) p = ns |
|
HC/TNF-α (n = 50) |
62 (13) | 46 (17) | 13 (6) | 4 (3) | 4 (3) | 42 (6) |
|
HC/C5a (n = 20) |
75 (7) p < 0.001 |
55 (20) p < 0.01 |
40 (9) p < 0.0001 |
47 (14) p < 0.0001 |
42 (13) p < 0.0001 |
46 (12) p = ns |
IQR = inter quartile range. PMN = polymorphonuclear neutrophils. HSA = human serum albumin, MMP-9 = matrix metalloproteinase 9 (gelatinase), LF = lactoferrin, MPO = myeloperoxidase, Pr3 = proteinase 3, P = properdin. AAV = ANCA-associated vasculitis. HC = healthy control. N = 50 AAV PMN samples (5 PMN donors, 10 different ANCA IgG stimuli) and 50 HC PMN samples (5 PMN donors, 10 different ANCA IgG stimuli) respectively. P-values representing significance of the differences between TNF-α priming and C5a priming. Statistical method: Mann Whitney.
Table 3. Degranulation controls (% release, median (IQR)).
| Stimuli | HSA | MMP-9 | LF | MPO | PR3 |
|---|---|---|---|---|---|
| HC IgG (n = 20) | 49 (8) | 25 (6) | 2 (3.4) | 0.6 (0.5) | 2 (1.6) |
|
PMA (n = 10) |
75 (5) | 85 (22) | 48 (9) | 14 (29) | 10 (17) |
|
PBS (n = 10) |
29 (5) | 6 (2) | 3 (3) | 3 (3) | 3 (3) |
IQR = inter quartile range. PMN = polymorphonuclear neutrophils. HSA = human serum albumin, MMP-9 = matrix metalloproteinase 9 (gelatinase), LF = lactoferrin, MPO = myeloperoxidase, PR3 = proteinase 3, P = properdin. AAV = ANCA-associated vasculitis. HC = healthy control. PMA = phorbol myristate acetate. PBS = phosphate buffered saline. N = 20 samples = 10 PMN donors (5 AAV and 5 HC) and 2 HC IgG stimuli. N = 10 samples = 10 PMN donors (5 AAV and 5 HC).
Intracellular ROS production was measured by stimulating primed PMN with ANCA IgG in the presence of luminol together with the scavengers SOD and catalase. Extracellular ROS was measured using isoluminol instead, as it is not able to cross membranes. There was a substantial variation in the amount of intracellular and extracellular ROS obtained with different stimulating IgGs. Priming with C5a before ANCA IgG stimulation, generated increased intracellular ROS in AAV PMN compared to HC PMN (p<0,001), Table 4.
Table 4. Oxidative burst after ANCA stimulation.
| PMN/Priming | IC ROS (RLU) median (IQR) p = AAV vs HC |
EC ROS (RLU) median (IQR) p = AAV vs HC |
|---|---|---|
|
AAV/TNF-α (n = 50) |
10872 (5581) p = ns |
1920 (1306) p = ns |
|
HC/TNF-α (n = 50) |
9589 (4405) | 1942 (1211) |
|
AAV/C5a (n = 20) |
13080 (3440) p < 0.001 |
1580 (2819) p = ns |
|
HC/C5a (n = 20) |
7770 (2540) | 980 (1963) |
| PMN/Controls | ||
|
AAV/HBSS (n = 10) |
0 (143) p = ns |
0 (44) p = ns |
|
HC/HBSS (n = 10) |
0 (0) | 0 (6) |
|
AAV/PMA (n = 10) |
566 (9880) p = ns |
336 (1374) p = ns |
|
HC/PMA (n = 10) |
8732 (5677) | 956 (594) |
PMN = polymorphonuclear neutrophils, IC ROS = intra cellular reactive oxygen species, EC = extra cellular, IQR = inter quartile range, AAV = ANCA associated vasculitis, HC = healthy controls, HBSS = buffer/negative control, PMA = phorbol 12-myristate 13-acetate, n = number of samples. P-values representing significance of the differences between AAV patients and HC. Statistical method: Mann Whitney.
Complement activation and microparticle release
Alternative complement pathway activation was measured in the supernatants of stimulated AAV PMN and HC PMN. C3bBbP-formation as well as C5a levels were measured over time. C3bBbP-levels peaked at 60 minutes and C5a at 240 minutes and therefore those time points were chosen for further analyses.
C3bBbP levels generated by incubation of the PMN supernatants with serum were significantly higher for AAV patients than for healthy controls, p = 0.01, Fig 1. There was a substantial variation in the amount of generated C3bBbP, depending on which ANCA IgG that had been used for PMN stimulation, Fig 2. Two of the ANCA IgGs having greatest impact (IgG4 and IgG7) had a significantly greater effect on AAV PMN compared to HC PMN, p < 0.001. Both Fig 1 and Fig 2 show results from PMN primed with TNF-α. Priming with C5a gave similar results, but with non-significant difference between patients and controls, possibly due to the strong impact by C5a itself on PMN and less additional effect of ANCA IgG stimulation.
Fig 1. Increased C3bBbP levels in supernatants from AAV PMN.
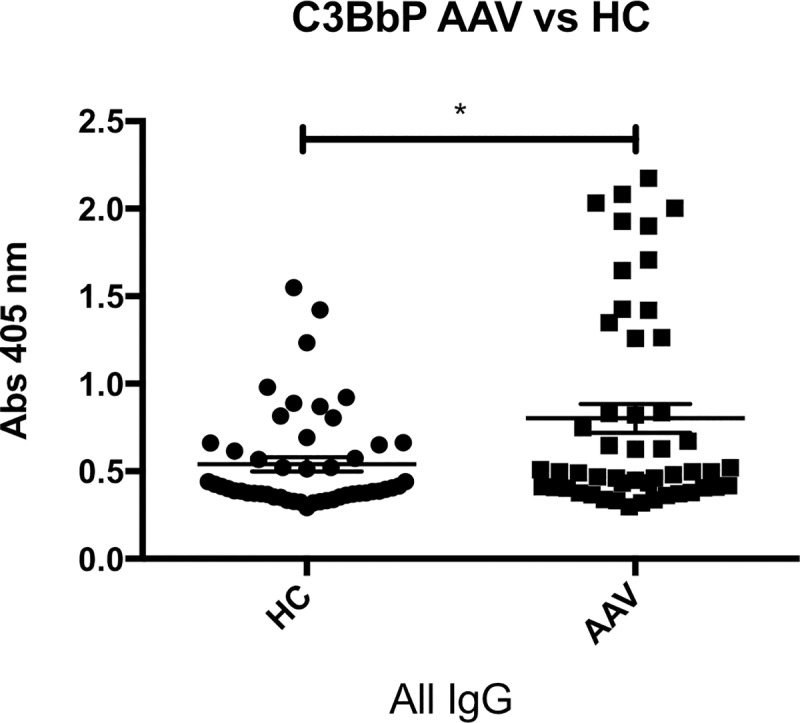
C3bBbP (stable alternative pathway C3 convertase, a sign of alternative pathway activation) formed in normal serum after incubation with supernatants from AAV PMN (neutrophils from vasculitis patients) or HC PMN (neutrophils from healthy controls), that have been primed with TNF-α and then stimulated with ANCA IgG (immunoglobulin G fraction from PR3-ANCA positive patients). Mean and SEM are shown. N = 50 AAV PMN samples (5 PMN donors, 10 different ANCA IgG stimuli) and 50 HC PMN samples (5 PMN donors, 10 different ANCA IgG stimuli). Statistical analysis: Mann Whitney, p = 0.01.
Fig 2. Activation with different ANCA IgG leads to varying degree of C3bBbP formation.

C3bBbP (stable alternative pathway C3 convertase) formed in normal serum after incubation with supernatants from five AAV PMN (neutrophils from vasculitis patients) and five HC PMN (neutrophils from healthy controls) primed with TNF-α and then stimulated with 10 different ANCA IgGs (immunoglobulin G fraction from PR3-ANCA positive patients) and one HC IgG (immunoglobulin G fraction from a healthy blood donor). PMA (phorbol 12-myristate 13-acetate) was used as positive control and PBS as negative control. Number of neutrophil supernatants tested = 5 in each bar. Means and SEMs are shown. Statistical analysis: Mann Whitney, p < 0.001.
There was a great variation in the amount of C5a generated with different stimulating ANCA IgGs, (Fig 3). One of the ANCA IgGs having greatest impact (IgG7) had a significantly greater effect on AAV PMN compared to HC PMN when priming with TNF-α, but no significant difference was seen between AAV patients and healthy controls when priming with C5a. C5a in the supernatants correlated with MP release (r = 0.3, p < 0.05).
Fig 3. Activation with different ANCA IgG leads to varying degree of C5a formation.

C5a measured in normal serum after incubation with supernatants from five AAV PMN (neutrophils from vasculitis patients) and five HC PMN (neutrophils from healthy controls) primed with TNF-α and then stimulated with 10 different ANCA IgGs (immunoglobulin G fraction from PR3-ANCA positive patients) and one HC IgG (immunoglobulin G fraction from a healthy blood donor). PMA (phorbol 12-myristate 13-acetate) was used as positive control and PBS as negative control. Number of neutrophil supernatants tested = 5 in each bar. Means and SEM are shown. Statistical analysis: Mann Whitney, p = 0.01.
Neither the neutrophil contents of properdin (median 15 ng/ml IQR 8 ng/ml, n = 10, vs median 17 ng/ml IQR 4 ng/ml, n = 10), nor the release upon neutrophil stimulation differed significantly between AAV patients and HC, Table 2. C3bBbP-generation correlated with C5a generation in both groups of PMN donors (r = 0,70 p<0,001) and with intracellular ROS in the AAV patients (r = 0.9 p < 0.001).
PMN microparticles were measured in the supernatants of the stimulated PMN. The released amount, after stimulation with ANCA IgG, was significantly higher in AAV samples compared with HC, Fig 4. There was a substantial variation in the amount of microparticles released, with different stimulating ANCA IgG. IgG7 had a significantly greater impact on MP generation from AAV PMN compared to HC PMN, p = 0.01. Supernatants from PMN only incubated with PBS contained very low MP levels (mean 662, SEM 79), whereas supernatants from PMN incubated with HC IgG contained slightly higher MP numbers (mean 1671, SEM 135). C3bBbP levels in the PMN supernatants correlated with the number of MP, regardless of priming with TNF-α (Fig 5) or C5a (r = 0.4, p < 0.001), and C3bBbP generation was significantly reduced when microparticles had been removed from the supernatants before incubation with healthy serum, Fig 6.
Fig 4. Increased microparticle release from AAV PMN.
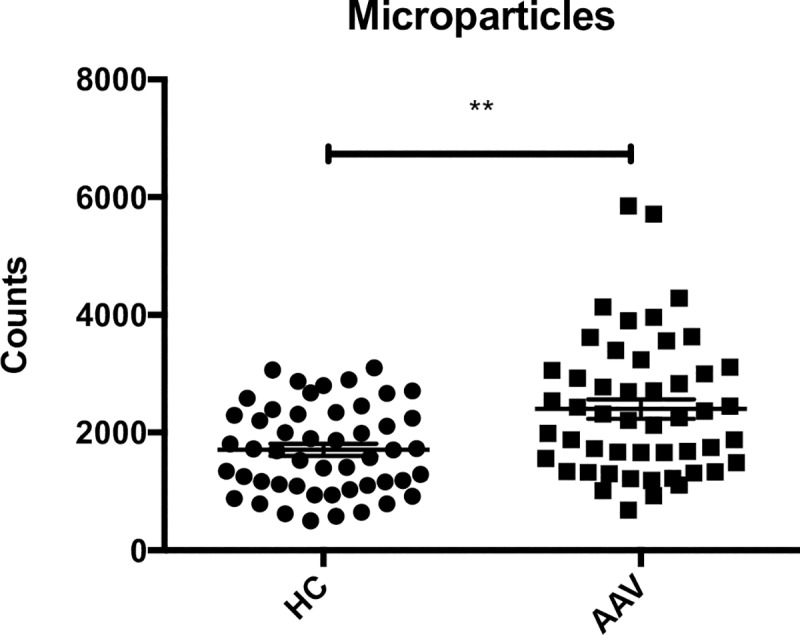
Release of CD66+ microparticles from five AAV PMN (neutrophils from PR3-ANCA positive vasculitis patients) and five HC PMN (neutrophils from healthy controls), after ANCA IgG (immunoglobulin G fraction from PR3-ANCA positive patients) stimulation, with 10 different ANCA IgGs. Means and SEM are shown. N = 50 AAV PMN samples and 50 HC PMN samples. Statistical analysis: Mann Whitney, p-value = 0.001.
Fig 5. C3bBbP formation correlates with microparticle release.
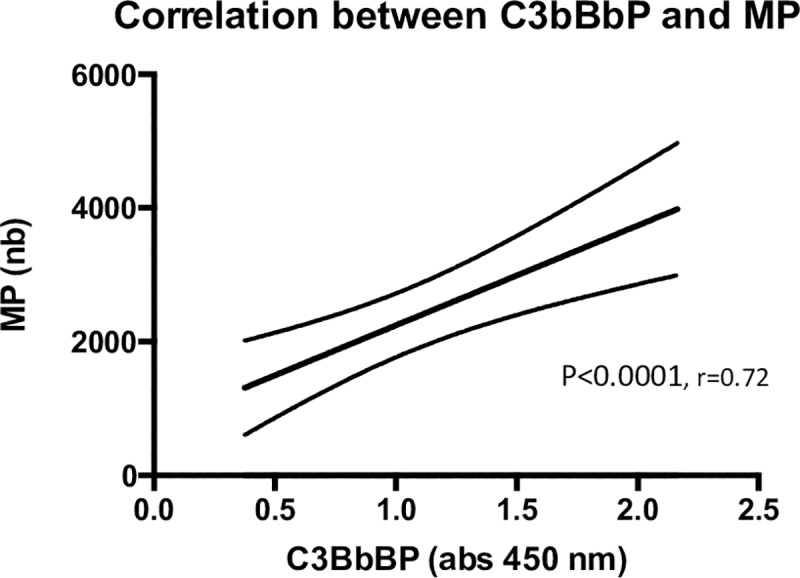
Correlation analysis of the number of CD66+ microparticles in PMN supernatants, and C3bBbP formation in normal serum, after incubation with the same supernatants. PMN supernatants were collected from 10 AAV PMN (neutrophils from 10 different PR3-ANCA positive vasculitis patients), after ANCA IgG stimulation, using two different ANCA IgG (IgG4 and IgG7 in Fig 1B), number of samples = 20, r = 0.72, p < 0,0001. Statistical analysis: Spearman.
Fig 6. Less C3bBbP formation after microparticle removal.

C3bBbP formation in normal serum after incubation with supernatants from seven AAV PMN (neutrophils from vasculitis patients) after ANCA IgG stimulation, using two different ANCA IgG (IgG4 and IgG7 in Fig 1B), before and after removal of CD66+ microparticles from the supernatants by centrifugation. Means and SEM are shown. Number of samples in each group = 14, p < 0,0001. Statistical analysis: Wilcoxon matched pairs signed rank test.
C5a receptor cell surface expression
Since CLB was needed in order to get a significant effect of ANCA stimulation on degranulation and ROS-production after PMN priming with either TNF-α or C5a, we studied the effects of CLB and priming on PMN cell surface C5a receptor expression. Resting AAV and HC PMN displayed the expected C5aR/C5L2 distribution on their membrane and there was no difference between AAV patients and HC (data not shown).
TNF-α itself did not seem to have any significant impact on C5a receptor expression. C5a seemed to induced temporary CD88 down regulation/internalization. CLB pre priming increased and prolonged CD88 down regulation and led to reciprocal C5L2 up regulation, Fig 7.
Fig 7. Effects of priming on membrane C5aR expression.

Effects of pre-priming with CLB (cytochalasin B) or not, in combination with TNF-α or C5a priming, on membrane C5aR expression on HC PMN (neutrophils from healthy controls). Percentages of cells expressing CD88 and C5L2 are shown. Number of experiments = 2, showing representative values.
We also studied the additional effect of adding ANCA IgG, Fig 8. Stimulation of PMN from AAV patients and HC with ANCA IgG, after priming with either TNF-α or C5a was performed. AAV PMN tended to downregulate CD88 less efficiently than HC PMN, however the experiments were only done on PMN from 2 AAV patients and 2 HC.
Fig 8. Effects of priming and ANCA IgG stimulation on membrane C5aR expression.

Effects of ANCA IgG stimulation of primed AAV PMN (neutrophils from two different vasculitis patients) on membrane C5aR expression are displayed to the left and HC PMN (neutrophils from two different healthy controls) are displayed to the right. Percentages of cells expressing CD88 and C5L2 are shown. Number of experiments = 2, showing representative values.
Discussion
The alternative complement pathway has received increasing attention in the context of AAV during the last 10 years. Both animal studies and a clinical trial on C5aR blockade have given us evidence of its importance in these traditionally pauci immune diseases. [9, 11–13] Previous studies have predominantly focused on MPO-ANCA-associated vasculitis and stimulation with MPO-ANCA, probably since the existing animal models are in MPO-ANCA associated disease. In this study we studied patients with PR3-ANCA-associated vasculitis and PR3-ANCA IgG preparations. Due to results from other studies, we focused on the potential role of the alternative pathway although our findings do not exclude involvement of other complement activation pathways. We could demonstrate increased C3bBbP generation in supernatants from AAV PMN compared with healthy controls. This effect correlated with intracellular ROS production and microparticle release, whereas no correlation could be seen neither with simply the degree of degranulation, nor with release of properdin or PMN contents of properdin. These findings imply a loss of inhibition in AAV PMN. Deficient function of factor H has recently been described in AAV by Chen et al, leading to decreased alternative pathway regulation [20–22]. Decreased regulation of the alternative pathway in the fluid phase would be expected to lead to increased production of anaphylatoxins, creating a vicious circle of increased complement activation and increased PMN activation, already suggested in the pathogenesis of AAV[23]. In our degranulation assay, priming with C5a instead of TNF-α resulted in a 10-fold increased release of the ANCA antigens PR3 and MPO, implying an increased exposure and thereby possibly an increased risk of autoimmunity in C5a-driven inflammation.
Intracellular ROS production was found to be increased in AAV PMN, which is consistent with our previous results–and this has also recently been confirmed by Nishide et al, describing a possible relationship with low levels of membrane bound SEMA4D, leading to loss of inhibition.[24] Interestingly, we as well as others, have seen that the results of in vitro stimulation of PMN with ANCA, are largely dependent on the use of CLB, a compound known to inhibit actin polymerization, and thereby affecting several processes such as phagocytosis, pinocytosis, membrane transport of glucose and nucleosides, and perhaps also the distribution of available receptors on the cell surface [25–28]. The C5a receptor C5L2 is normally internalized by clathrin-mediated endocytosis, but after CLB treatment C5L2 expression on the cell surface increases, Fig 7. When exposing the PMN for C5a, CD88 is internalized as expected, however the effect is enhanced and prolonged by CLB pretreatment. Is there a circulating factor in vivo, affecting the PMN in a fashion similar to CLB? One candidate is Sphingosin-1 phosphate, which has some similar effects and has been implicated to play a role in AAV, and is known to have a role in the CD88/C5L2 balance [29, 30]. The C5L2 function has been debated, but there is increasing evidence of a pro inflammatory function and Hao et al could demonstrate reduced PMN priming for oxidative burst and degranulation by ANCA stimulation, after C5L2 blockade [14, 31]. Increased C5L2 expression has been seen in renal biopsies from AAV patients. [32] There is an ongoing clinical trial on C5aR blockade in the treatment of AAV [12]. The compound under study is selective for CD88 and it is unclear how selective CD88 blockade will effect C5L2 expression, which thus would be interesting to study.
Camous et al demonstrated that PMN-derived microparticles could activate the alternative complement pathway. [33] Our results point in the same direction, given the correlation between microparticle release and C3bBbP formation, and the demonstration of increased microparticle release from AAV PMN compared to HC PMN, after stimulation. In addition, AAV PMN seem to respond to stimulation in a way leading to more alternative pathway activation, compared to HC PMN. The mechanisms behind these findings need to be further explored. A role for properdin in the complement activation, as was hypothesized also in the article by Camous, cannot be excluded although we found no difference in neutrophil properdin content or expression between cells from patients and controls. Expression of properdin and other complement proteins on neutrophil-derived microparticles would be an important focus of further studies. A possible limitation to our study is the definition of microparticles within 0.8–1 μm, excluding smaller microparticles that might be biologically important [34], and this will be taken into consideration in future studies.
Conclusions
This study shows that supernatants from stimulated AAV PMN have a greater ability to activate the alternative complement pathway compared to HC PMN. This increased ability could be caused by increased release of microparticles. Our findings emphasize the role of complement in the pathogenesis in AAV - underlining the therapeutic potential of C5a and other complement blockade.
Acknowledgments
Some of the data presented in this paper have been discussed at the 10 th International congress on Autoimmunity (Leipzig, Germany, 2016) and the 17th International Vasculitis and ANCA Workshop (London, UK, 2015) and published in abstract form.
Abbreviations
- ANCA
anti neutrophil cytoplasmic antibodies
- AAV
ANCA-associated vasculitis
- PR3
proteinase 3
- MPO
myeloperoxidase
- GPA
granulomatosis with polyangiitis
- HC
healthy controls
- PMN
neutrophil granulocytes
- MP
microparticles
- MPA
microscopic polyangiitis
- ELISA
enzyme-linked immunosorbent assay
- PMA
phorbol myristate acetate
- IgG
immunoglobulin G
- C5a
complement factor 5a
- TNF-α
tumor necrosis factor alpha
- FACS
fluorescence activated cell sorting
- C5aR
complement factor 5a receptor, CD88
- C5L2
complement factor 5a receptor 2
- BVAS
Birmingham vasculitis activity score
- ANCA IgG
Immunoglobulin G fraction from PR3-ANCA positive patients
- CLB
cytochalasin B
Data Availability
All relevant data are available at DOI: 10.5281/zenodo.3236852.
Funding Statement
This work was funded by the Skåane University Hospital research funds, the Swedish Rheumatism Association, the Foundation of the National Board of Health and Welfare, and the Swedish Research Council 65x-15152. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. There was no commercial company funding.
References
- 1.Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: immunodiagnostic and pathogenetic aspects. Clin Exp Immunol. 1993;91(1):1–12. 10.1111/j.1365-2249.1993.tb03345.x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohlsson SM, Ohlsson S, Soderberg D, Gunnarsson L, Pettersson A, Segelmark M, et al. Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti-neutrophil cytoplasmic antibodies. Clin Exp Immunol. 2014;176(3):363–72. Epub 2014/03/29. 10.1111/cei.12301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(11):4115–9. Epub 1990/06/01. 10.1073/pnas.87.11.4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charles Jennette J, Xiao H, Hu P. Complement in ANCA-associated vasculitis. Semin Nephrol. 2013;33(6):557–64. Epub 2013/10/29. S0270-9295(13)00134-4 [pii] 10.1016/j.semnephrol.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haas M, Eustace JA. Immune complex deposits in ANCA-associated crescentic glomerulonephritis: a study of 126 cases. Kidney Int. 2004;65(6):2145–52. Epub 2004/05/20. 10.1111/j.1523-1755.2004.00632.x S0085-2538(15)49959-4 [pii]. . [DOI] [PubMed] [Google Scholar]
- 6.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110(7):955–63. 10.1172/JCI15918 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170(1):52–64. 10.2353/ajpath.2007.060573 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83(1):129–37. Epub 2012/08/24. S0085-2538(15)55692-5 [pii] 10.1038/ki.2012.313 . [DOI] [PubMed] [Google Scholar]
- 9.Huugen D, van Esch A, Xiao H, Peutz-Kootstra CJ, Buurman WA, Tervaert JW, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71(7):646–54. Epub 2007/02/15. S0085-2538(15)52429-0 [pii] 10.1038/sj.ki.5002103 . [DOI] [PubMed] [Google Scholar]
- 10.Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34(3):J276–86. Epub 2009/12/17. S0896-8411(09)00154-1 [pii] 10.1016/j.jaut.2009.11.014 . [DOI] [PubMed] [Google Scholar]
- 11.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol. 2014;25(2):225–31. 10.1681/ASN.2013020143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J Am Soc Nephrol. 2017;28(9):2756–67. Epub 2017/04/13. 10.1681/ASN.2016111179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48(14):1631–42. 10.1016/j.molimm.2011.04.014 . [DOI] [PubMed] [Google Scholar]
- 14.Hao J, Wang C, Yuan J, Chen M, Zhao MH. A pro-inflammatory role of C5L2 in C5a-primed neutrophils for ANCA-induced activation. PLoS One. 2013;8(6):e66305 Epub 2013/06/21. 10.1371/journal.pone.0066305 PONE-D-13-08877 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87(11):671–8. . [PubMed] [Google Scholar]
- 16.Udby L, Calafat J, Sorensen OE, Borregaard N, Kjeldsen L. Identification of human cysteine-rich secretory protein 3 (CRISP-3) as a matrix protein in a subset of peroxidase-negative granules of neutrophils and in the granules of eosinophils. Journal of leukocyte biology. 2002;72(3):462–9. Epub 2002/09/12. [PubMed] [Google Scholar]
- 17.Witko-Sarsat V, Cramer EM, Hieblot C, Guichard J, Nusbaum P, Lopez S, et al. Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood. 1999;94(7):2487–96. . [PubMed] [Google Scholar]
- 18.Voller A, Bidwell DE, Bartlett A. Enzyme immunoassays in diagnostic medicine. Theory and practice. Bull World Health Organ. 1976;53(1):55–65. [PMC free article] [PubMed] [Google Scholar]
- 19.Stahl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111(11):5307–15. 10.1182/blood-2007-08-106153 . [DOI] [PubMed] [Google Scholar]
- 20.Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH. The functional activities of complement factor H are impaired in patients with ANCA-positive vasculitis. Clinical immunology (Orlando, Fla). 2017;175:41–50. Epub 2016/12/13. 10.1016/j.clim.2016.11.013 . [DOI] [PubMed] [Google Scholar]
- 21.Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH. Complement Factor H Inhibits Anti-Neutrophil Cytoplasmic Autoantibody-Induced Neutrophil Activation by Interacting With Neutrophils. Frontiers in immunology. 2018;9:559 Epub 2018/04/05. 10.3389/fimmu.2018.00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH. Myeloperoxidase influences the complement regulatory activity of complement factor H. Rheumatology (Oxford, England). 2018. Epub 2018/02/23. 10.1093/rheumatology/kex529 . [DOI] [PubMed] [Google Scholar]
- 23.Flint SM, McKinney EF, Smith KG. Emerging concepts in the pathogenesis of antineutrophil cytoplasmic antibody-associated vasculitis. Current opinion in rheumatology. 2015;27(2):197–203. Epub 2015/01/30. 10.1097/BOR.0000000000000145 . [DOI] [PubMed] [Google Scholar]
- 24.Nishide M, Nojima S, Ito D, Takamatsu H, Koyama S, Kang S, et al. Semaphorin 4D inhibits neutrophil activation and is involved in the pathogenesis of neutrophil-mediated autoimmune vasculitis. Annals of the rheumatic diseases. 2017;76(8):1440–8. Epub 2017/04/19. 10.1136/annrheumdis-2016-210706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein I, Hoffstein S, Gallin J, Weissmann G. Mechanisms of lysosomal enzyme release from human leukocytes: microtubule assembly and membrane fusion induced by a component of complement. Proceedings of the National Academy of Sciences of the United States of America. 1973;70(10):2916–20. Epub 1973/10/01. 10.1073/pnas.70.10.2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bengtsson T, Dahlgren C, Stendahl O, Andersson T. Actin assembly and regulation of neutrophil function: effects of cytochalasin B and tetracaine on chemotactic peptide-induced O2- production and degranulation. Journal of leukocyte biology. 1991;49(3):236–44. Epub 1991/03/01. 10.1002/jlb.49.3.236 . [DOI] [PubMed] [Google Scholar]
- 27.Radford DJ, Lord JM, Savage CO. The activation of the neutrophil respiratory burst by anti-neutrophil cytoplasm autoantibody (ANCA) from patients with systemic vasculitis requires tyrosine kinases and protein kinase C activation. Clin Exp Immunol. 1999;118(1):171–9. Epub 1999/10/30. 10.1046/j.1365-2249.1999.01043.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malawista SE, Gee JB, Bensch KG. Cytochalasin B reversibly inhibits phagocytosis: functional, metabolic, and ultrastructural effects in human blood leukocytes and rabbit alveolar macrophages. The Yale journal of biology and medicine. 1971;44(3):286–300. Epub 1971/12/01. [PMC free article] [PubMed] [Google Scholar]
- 29.Sadahira Y, Ruan F, Hakomori S, Igarashi Y. Sphingosine 1-phosphate, a specific endogenous signaling molecule controlling cell motility and tumor cell invasiveness. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(20):9686–90. Epub 1992/10/15. 10.1073/pnas.89.20.9686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao J, Huang YM, Zhao MH, Chen M. The interaction between C5a and sphingosine-1-phosphate in neutrophils for antineutrophil cytoplasmic antibody mediated activation. Arthritis Res Ther. 2014;16(4):R142 Epub 2014/07/09. 10.1186/ar4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang T, Garstka MA, Li K. The Controversial C5a Receptor C5aR2: Its Role in Health and Disease. Journal of immunology research. 2017;2017:8193932 Epub 2017/07/15. 10.1155/2017/8193932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan J, Gou SJ, Huang J, Hao J, Chen M, Zhao MH. C5a and its receptors in human anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res Ther. 2012;14(3):R140 Epub 2012/06/14. 10.1186/ar3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux-Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117(4):1340–9. Epub 2010/11/11. 10.1182/blood-2010-05-283564 . [DOI] [PubMed] [Google Scholar]
- 34.Cointe S, Judicone C, Robert S, Mooberry MJ, Poncelet P, Wauben M, et al. Standardization of microparticle enumeration across different flow cytometry platforms: results of a multicenter collaborative workshop. Journal of thrombosis and haemostasis: JTH. 2017;15(1):187–93. Epub 2016/09/24. 10.1111/jth.13514 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are available at DOI: 10.5281/zenodo.3236852.