

Abstract
The pathways that G protein-coupled receptor (GPCR) ligands follow as they bind to or dissociate from their receptors are largely unknown. Protease-activated receptor-1 (PAR1) is a GPCR activated by intramolecular binding of a tethered agonist peptide that is exposed by thrombin cleavage. By contrast, the PAR1 antagonist vorapaxar is a lipophilic drug that binds in a pocket almost entirely occluded from the extracellular solvent. The binding and dissociation pathway of vorapaxar is unknown. Starting with the crystal structure of vorapaxar bound to PAR1, we performed temperature-accelerated molecular dynamics simulations of ligand dissociation. In the majority of simulations, vorapaxar exited the receptor laterally into the lipid bilayer through openings in the transmembrane helix (TM) bundle. Prior to full dissociation, vorapaxar paused in metastable intermediates stabilized by interactions with the receptor and lipid headgroups. Derivatives of vorapaxar with alkyl chains predicted to extend between TM6 and TM7 into the lipid bilayer inhibited PAR1 with apparent on rates similar to that of the parent compound in cell signaling assays. These data are consistent with vorapaxar binding to PAR1 via a pathway that passes between TM6 and TM7 from the lipid bilayer, in agreement with the most consistent pathway observed by molecular dynamics. While there is some evidence of entry of the ligand into rhodopsin and lipid-activated GPCRs from the cell membrane, our study provides the first such evidence for a peptide-activated GPCR and suggests that metastable intermediates along drug binding and dissociation pathways can be stabilized by specific interactions between lipids and the ligand.
Graphical Abstract
GPCRs are among the most important classes of proteins targeted by small molecule therapeutics. A challenge for drug development has been optimizing specificity for one GPCR over related receptors.1 The explosion of high-resolution GPCR crystal structures in recent years has greatly advanced our understanding of the structural determinants of binding affinity,2 typically through analysis of ligand–receptor contacts in the orthosteric binding site.3 However, the detailed mechanisms by which GPCRs recognize and bind ligands are largely unknown. In particular, the binding and unbinding pathways by which ligands enter the orthosteric site remain largely uncharacterized except for a few exemplary cases.4,5 Knowledge of such pathways for specific GPCRs will help describe the kinetics (on and off rates) of drug binding, which is an important determinant of in vivo drug efficacy and, in some cases, even more important than the equilibrium binding affinity.6,7 In fact, binding and unbinding pathways may contribute to “kinetic selectivity” for one receptor subtype over another. An example of this phenomenon is the anticholinergic drug tiotropium, which has an off rate at the M3 muscarinic acetylcholine receptor much slower than that of the closely related M2 subtype despite comparable binding affinities.4,7 Consequently, the clinical effects of tiotropium are largely due to antagonism of the M3 receptor.8
Molecular dynamics (MD) simulations have proven to be an important tool for understanding GPCR ligand binding pathways.5,9 In some cases, the ligand is believed to visit metastable intermediates along the binding pathway. Such intermediates can be considered prebound states, where a ligand may reside for a period of time prior to either settling into the orthosteric binding site or dissociating completely from the receptor. Such states have been observed in simulations of ligands binding to and dissociating from GPCRs, and there is evidence that some metastable sites observed by MD are pharmacologically relevant.4 For example, in simulations of antagonist binding to and unbinding from muscarinic acetylcholine receptors, the observed metastable site corresponds to a well-known site for allosteric modulators.4,10,11 Metastable binding sites may occur in GPCR regions with a high degree of sequence diversity such as extracellular loops. Interactions of the drug with amino acids in these metastable binding sites may thus vary between receptor subtypes and in part determine the height of energy barriers between the free/prebound and prebound/orthosteric binding states.4,7 If this process can be understood, entry or exit paths and the novel metastable binding sites revealed might be exploited to optimize drugs for specific GPCR family members.
The orthosteric binding pocket of aminergic GPCRs such as the β-adrenergic, muscarinic cholinergic, and dopamine receptors is connected to the extracellular environment by a solvent-accessible “vestibule”12 that might host metastable binding interactions with ligands.4,5 Typical ligands for aminergic GPCRs are relatively hydrophilic and likely to enter the vestibule by diffusion from the extracellular solvent. However, not all GPCRs have vestibules providing direct solvent access to the orthosteric binding pocket. In rhodopsin, for example, the extracellular loops fold over the orthosteric binding pocket and largely shield the ligand, retinal, from the extracellular solvent.13 In such cases, the question is how the ligand gains access to the binding pocket. For rhodopsin, mutational analysis and elegant spectroscopic studies have shown that retinal likely gains access through a channel connecting the core of the receptor with the hydrophobic portion of the lipid bilayer membrane.14,15 A tunnel to the lipid membrane is also seen in sphingosine 1-phosphate receptor 1, a lipid-binding GPCR,16 and MD simulations support a pathway of inhibitor entry directly from the lipid bilayer.17 A similar binding pathway has been proposed for cannabinoid receptors.18
As a subject for the study of GPCR ligand recognition, the PAR1 receptor–vorapaxar complex provides a fascinating problem. The endogenous agonist for PAR1 is a peptide (SFLLRN) tethered to the extracellular portion of the receptor. The active form of this peptide is generated by cleavage of the PAR1 N-terminus by thrombin, which exposes the SFLLRN peptide as the new extracellular N-terminus of the receptor. 9 While structural data on the agonist peptide–PAR1 interaction are not yet available, studies of receptor mutants suggest that SFLLRN binding involves the receptor extracellular face,20,21 and recent atomic force microscopy experiments suggest that SFLLRN binds PAR1 from the extracellular solvent, because a hydrophilic PEG linker can be used to tether functional SFLLRN to the atomic force microscope tip.22 By contrast, the PAR1 antagonist vorapaxar, an antiplatelet drug, is a highly lipophilic himbacine-like molecule.23 An inactive state crystal structure of PAR1 shows that vorapaxar is buried in a pocket largely inaccessible to the solvent (Figure 1).24 Extracellular loops (ECLs) two and three fold over the vorapaxar binding site and form extensive interactions with the ligand (Figure 1A,B). Reminiscent of rhodopsin, two tunnels are observed connecting the vorapaxar binding pocket to the lipid bilayer: one between TM6 and TM7 (Figure 1C) and one between TM4 and TM5 (Figure 1D). The ethyl carbamate group of vorapaxar extends between TM6 and TM7 toward the lipid bilayer.24
Figure 1.

Tunnels into the vorapaxar binding site of PAR1. (A) Extracellular ribbon view of the PAR1–vorapaxar crystal structure (Protein Data Bank entry 3VW7) with ECL2 colored green, ECL3 colored orange, and vorapaxar rendered as spheres. TMs are labeled 1 –7. The TM4–TM5 tunnel is labeled by a single arrowhead, and the TM6–TM7 tunnel is labeled by a double arrowhead. (B) Extracellular surface rendering. (C) Surface rendering of the TM6–TM7 tunnel as seen from the plane of the plasma membrane, rotated 90° from the extracellular view. (D) Surface rendering of the TM4–TM5 tunnel as seen from the plane of the plasma membrane, rotated 115° from the view in panel C. The approximate plane of the POPC headgroups in the outer leaflet of the lipid bilayer is indicated by the dashed line.
Given the lipophilic nature of vorapaxar and its position in the PAR1 structure, we hypothesized that one or both of the tunnels described above may provide a path between the lipid bilayer and the binding pocket via which vorapaxar might enter or exit the receptor. To test this hypothesis, we performed temperature-accelerated molecular dynamics (TAMD) simulations of the dissociation of vorapaxar from PAR1. We analyzed these simulations to characterize metastable intermediates along the observed dissociation pathways. We then performed chemical biology experiments using derivatized vorapaxar compounds to provide experimental evidence that the TM6–TM7 pathway observed by TAMD is a binding pathway by which vorapaxar enters PAR1 from the lipid bilayer membrane.
MATERIALS AND METHODS
Computation
We performed all molecular dynamics simulations on Anton,25 a special-purpose computer designed to accelerate molecular dynamics simulations by orders of magnitude. All atoms, including lipids and water, were represented explicitly.
System Setup and Simulation Protocol
Simulations of PAR1 were based on the crystal structure of the PAR1–vorapaxar complex and prepared as described previously.24 Briefly, we removed T4 lysozyme from the crystallized construct, inserted the receptor into a hydrated 1-palmitoyl- 2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer, and equilibrated for 50 ns on Anton. The crystallographic sodium ion near Asp1482.50 was included in the simulations. Simulation parameters were set as in the PAR1 simulations in ref 24, except that long-range electrostatics were computed with a 64 X 64 X 64 grid.
Each production simulation was initiated from the final frame of the equilibration run using velocities sampled from the Boltzmann distribution at 310 K. Production simulations were run until a dissociation event was observed. Because vorapaxar is known experimentally to dissociate at time scales far longer than those accessible via unbiased molecular dynamics simulation, we used TAMD.26 TAMD is a method for enhancing sampling along a chosen set of collective variables (CVs); here, we use the x, y, and z coordinates of the center of mass (COM) of vorapaxar’s non-hydrogen atoms as CVs. The acceleration is achieved by harmonically tethering each CV to a fictitious particle undergoing Brownian motion at a higher temperature. With a proper choice of parameters, the sampling of the chosen CV is accelerated such that the fictitious particles still obey Boltzmann statistics at the higher, fictitious temperature, Ts, while the non-accelerated orthogonal degrees of freedom of the real system remain properly distributed at the real temperature. The spring constant tethering the COM to the fictitious particle was 100 kcal mol−1 Å−2. The friction coefficient for the fictitious particle was 100 ps kcal mol−1 Å−2, and the Ts of this fictitious particle was chosen such that kBTs was 5.0 kcal mol−1. Vorapaxar binds PAR1 very tightly and has an almost immeasurably slow off rate, and it would be expected to cross an energetic barrier over 5 kcal mol−1 upon dissociation. Simulations were run in the NPT ensemble at 310 K (37 °C) and 1 bar. The lengths of the simulations ranged from 0.9 to 7.4 μs, with ligand dissociation taking place after 0.8–7.3 μs.
Detection of Metastable States and Contact Frequency Analysis
Simulation trajectory frames were saved every 180 ps. Trajectories were visualized and analyzed using VMD27 and PyMol. To detect metastable states, we calculated displacement of the vorapaxar COM from its crystallographic position during the dissociation trajectory. Metastable states were defined as plateaus in the displacement versus time plot (±3 Å) for each vorapaxar escape event, after the ligand had moved out of the crystallographic binding pocket (see Figure 3, Figure 4, and Figure S2).
Figure 3.

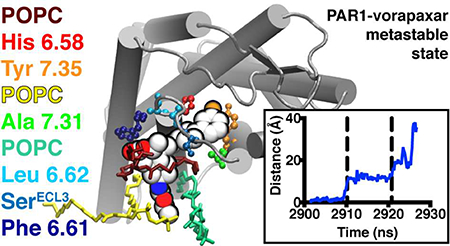
Vorapaxar interactions in the TM6–TM7 metastable state. (A) Extracellular view of the PAR1–vorapaxar crystal structure. TMs are shown as cylinders and labeled 1–7. Vorapaxar is shown as spheres, and the ethyl carbamate moiety is indicated by an arrow. (B–F) Representative poses from the five simulations in which vorapaxar dissociated from PAR1 through the TM6–TM7 tunnel. The amino acid residues and POPC molecules that interact most frequently with vorapaxar in the metastable state are colored according to a heat map, from the most frequently interacting (dark red) to the ninth most frequently interacting (dark blue). Amino acids are rendered as balls and sticks, and lipids are rendered as sticks. Ballesteros–Weinstein numbering is indicated by superscripts. Plots of the displacement of the vorapaxar COM from the crystallographic position vs simulation time are shown at the bottom right of each panel. The metastable state is indicated by vertical dashed lines.
Figure 4.

Vorapaxar interactions in the TM4–TM5 metastable state. (A) Extracellular view of the PAR1–vorapaxar crystal structure. Labeling is identical to that in Figure 3. The ethyl carbamate moiety of vorapaxar is indicated by an arrow. (B–D) Representative poses from the three simulations in which vorapaxar dissociated from PAR1 through the TM4–TM5 tunnel. The residues and lipids that interact most frequently with vorapaxar in the metastable state are colored according to a heat map, from the most frequently interacting (dark red) to the ninth most frequently interacting (dark blue). Ballesteros–Weinstein numbering is indicated by superscripts. Plots of the displacement of the vorapaxar COM from the crystallographic position vs simulation time are shown at the bottom right of each panel. The metastable state is indicated by vertical dashed lines.
To obtain a fingerprint of interactions contributing to each metastable state, we used VMD to analyze the frequency of non-hydrogen atom contacts between vorapaxar and amino acid residues in PAR1. Using a cutoff distance of 3.5 Å, we identified the residues interacting most frequently with vorapaxar in each metastable state. An identical analysis was performed to determine metastable contacts between vorapaxar and POPC molecules in the simulation.
Chemistry and Ligand Preparation
Vorapaxar was provided by Portola Pharmaceuticals in solid form. Vorapaxar derivatives were prepared as follows. First, hydrolysis of vorapaxar solid in HCl-AcOH (2:1) was performed to provide the free amine.28 The free amine is known to be an inactive metabolite of vorapaxar.29 The amine was further reacted with hexyl chloroformate or tert-butyl (6-{[(4-nitrophenoxy)- carbonyl]oxy}hexyl)carbamate30 to yield hexyl vorapaxar or N-boc-hexyl vorapaxar, respectively (Scheme 1). The crude product was purified by reverse phase high-performance liquid chromatography and confirmed by mass spectrometry.
Scheme 1. Chemical Synthesis of Vorapaxar Derivatives (see Materials and Methods)a.

aVorapaxar consists of three pharmacophores: an ethyl carbamate (curly brace), a tricyclic group that contains a methylfuranone ring (asterisk), and a fluorophenyl-pyridine group (R2).
To prepare ligands for use with cellular assays, vorapaxar derivatives were dissolved to a concentration of 10 mM in dimethyl sulfoxide (DMSO) and then rapidly diluted 100-fold into a solution such that its final composition was 100 μM ligand, 10 mM 2-hydroxypropyl-β-cyclodextrin (HPβCD), 10% DMSO, and 90% water. The solution was rapidly vortexed and typically appeared turbid at this point. The solution was nutated at room temperature for 48 h with protection from light and then centrifuged at 17000g for 30 min. The clarified supernatant was removed to a clean tube, and the soluble vorapaxar derivative concentration was estimated by ultraviolet (UV) absorbance using an ε305 of 19.3 mM−1 cm−1 (see below). A sample including HPβCD and DMSO but no vorapaxar derivative was prepared in parallel using the same method and served as a vehicle control in cell experiments.
Cell Culture and Signaling Assays
Cell-based assays were performed in Rat1 fibroblasts stably transfected with N-terminally FLAG-tagged human PAR1. Generation of this cell line has been previously described.31 Untransfected Rat1 fibroblasts served as a control. To confirm the cell surface expression of FLAG-PAR1 under typical culture conditions, confluent cells were washed and fixed with paraformaldehyde. The primary antibody was anti-FLAG M1 (Sigma) used at a final concentration of 3 μg/mL. The secondary antibody was Alexa488 goat anti-mouse IgG (Molecular Probes) used at a 1:500 dilution. For microscopy, cells were grown to confluency on coverslip chambers (MatTek) and images were taken on a Zeiss LSM 700 microscope.
Phosphoinositide hydrolysis assays were performed as previously described.21,24 For calcium flux assays, Rat1 cells were plated at a density of 20000 cells/well in 96-well blackwalled fluorescent microplates (Corning Costar) and grown in Dulbecco’s modified Eagle’s medium (DMEM) High Glucose with 10% fetal bovine serum for 48 h until the cells were confluent. At that time, the medium was exchanged for serum-free medium [DMEM High Glucose, 20 mM HEPES, and 0.1% bovine serum albumin (BSA)] and incubated for 4 h prior to fluorescent labeling.
For fluorescent labeling,32 probenecid (Sigma, used as an organic anion transporter inhibitor to prevent dye efflux) was freshly prepared as a 100 mM stock in 1 M NaOH. The assay medium was prepared by diluting probenecid to a concentration of 2.5 mM in Hank’s balanced salt solution with 20 mM HEPES (no BSA or phenol red) and adjusting the pH to 7.4. Fluo4-AM (Molecular Probes) was dissolved to a concentration of 4 mM in DMSO, mixed at a 1:1 ratio with the surfactant Pluronic F-127 (20%), and diluted 1:1000 into assay medium. Serum-free medium was aspirated, and cells were labeled with Fluo4-AM (final concentration of 2 μM) for 45 min at room temperature. Fluo4-AM-containing medium was aspirated off and exchanged for fresh assay medium prior to calcium flux assays. For inhibitor experiments, vorapaxar derivatives were solubilized with HPβCD as described above, diluted into Hank’s balanced salt solution, and incubated with the cells for 10–90 min at room temperature. Fluo4 fluorescence was recorded using a Tecan M-1000 plate reader with excitation at 485 nm and emission at 525 nm. The assay was initiated by rapid addition of PAR1 agonist peptide SFLLRN (American Peptide Co.) using an automatic eight-channel pipettor (Ramin E4 XLS), and calcium transients were recorded. The final concentration of SFLLRN was 3 μM in a working volume of 210 μL per 96-well plate. Data from pharmacologic experiments were analyzed, and curve fitting was performed using Prism 7.0 (GraphPad).
RESULTS AND DISCUSSION
Temperature-Accelerated Molecular Dynamics Simulations of the Vorapaxar–PAR1 Complex
To probe the dissociation of vorapaxar from PAR1, we performed atomistic MD simulations of the PAR1–vorapaxar complex embedded in a POPC lipid bilayer. In previous work with the PAR1–vorapaxar crystal structure, unbiased MD simulations ≤10 μs in length did not result in spontaneous dissociation of vorapaxar.24 This is consistent with the slow experimental off rate of vorapaxar (on the order of 20 h).23 To observe vorapaxar dissociation on a computationally accessible time scale, we ran TAMD simulations in which the rate of thermal motion of vorapaxar was increased to accelerate its dissociation (see Materials and Methods). Thirteen TAMD simulations ranging in length from 0.9 to 7.4 μs with a median of 1.7 μs were performed. Of the 13 simulations, vorapaxar dissociated from the lipid bilayer eight times and from the extracellular face of the receptor between ECL2 and ECL3 five times (Figure 2). Of the lipid bilayer exits that were observed, vorapaxar passed between TM4 and TM5 (Figure 2A) in three of the simulations and between TM6 and TM7 in five of the simulations (Figure 2C).
Figure 2.

Ligand dissociation trajectories observed in TAMD simulations of vorapaxar dissociating from PAR1. (A) Ligand dissociation pathway between TM4 and TM5, observed in three simulations. (B) Ligand dissociation pathway into the extracellular solvent, observed in five simulations. (C) Ligand dissociation pathway between TM6 and TM7, observed in five simulations. In panels A–C, the PAR1 backbone is shown as light brown ribbons and vorapaxar atoms are shown as spheres. The crystallographic pose of vorapaxar is colored red, as well as representative poses from intermediate (pink) and unbound states (blue) observed in the simulations. (D) Overlay of the COM of vorapaxar (small spheres) during dissociation from PAR1 (light brown ribbons) between TM6 and TM7 in five different simulations. Simulation time progresses from red to white to blue in each simulation, and the time step is 180 ps. The metastable state is indicated by a dashed circle. The direction of vorapaxar motion is indicated by arrows.
Notably, in all of the trajectories where vorapaxar exited into the lipid bilayer, the drug paused for a period of time in a pocket outside of the crystallographic binding site, with vorapaxar forming a set of interactions with PAR1 different from that seen in the crystal structure. We will refer to such positions of the ligand as “metastable states” along the dissociation pathway, and they are characterized in more detail below. The metastable state observed during exits between TM6 and TM7 was reproducible, occurring each time after the vorapaxar COM had moved 11 ± 1a Å laterally out of the crystallographic binding site toward the TM6–TM7 tunnel. The consistency of this metastable state can be appreciated by the dense cluster of spheres representing the ligand COM at evenly spaced time points in all the TM6–TM7 escape trajectories (Figure 2D). Metastable states were also observed for vorapaxar exits between TM4 and TM5, but they were more heterogeneous (Figure S1A) and occurred closer to the crystallographic position (7.6 ± 0.3 Å measured from the vorapaxar COM).
The metastable states observed in the five extracellular exit trajectories were the most heterogeneous (Figures S1B and S2). In one trajectory, vorapaxar dissociates without visiting a metastable state (not shown). A second trajectory exhibits a very short (540 ps) metastable state formed by interactions exclusively with ECL residues (Figure S2B). A third trajectory shows a metastable state with vorapaxar lying between ECL3 and the TM4–TM5 exit tunnel (Figure S2C), thus having features of both TM4–TM5 and TM6–TM7 exits before dissociating into the extracellular solvent. In the fourth trajectory, vorapaxar visits a metastable intermediate similar to that seen during TM6–TM7 exit (Figure S2E and Movie S1), and in the fifth trajectory, it visits a metastable intermediate similar to that seen during TM4–TM5 exit (Figure S2F). Given the heterogeneity of the extracellular exits, we did not investigate the amino acid contacts made with the ligand beyond what is shown in Figure S2.
Detailed Analysis of Vorapaxar in the TM6–TM7 Metastable State
Vorapaxar is a roughly T-shaped molecule made of (i) a core tricyclic group that includes a methylfuranone moiety, (ii) a fluorophenyl-pyridine group, and (iii) an ethyl carbamate group (Scheme 1). In the PAR1–vorapaxar crystal structure, the ethyl carbamate group protrudes through the tunnel between TM6 and TM7 and points toward the lipid bilayer, while the fluorophenyl-pyridine group points down (toward cytosol) within the binding pocket toward TM4. In all of the trajectories in which vorapaxar exited between TM6 and TM7, the ethyl carbamate group led the way out of the binding pocket and into the outer leaflet of the lipid bilayer (Figure 3, arrow, and Movie S2). This observation formed the basis for our chemical biology experiments described below, in which the ethyl carbamate was derivatized with long alkyl groups.
The remainder of the ligand followed the ethyl carbamate as vorapaxar tilted out of the PAR1 binding pocket. The metastable state along this pathway was consistently found at a point where the fluorophenyl-pyridine group rests between TM6 and TM7, with the ligand mostly out of the binding pocket and protruding through the TM6–TM7 tunnel into the lipid bilayer (Figure 3). The metastable state was visually similar in all five simulations that captured a TM6–TM7 exit and featured interactions of vorapaxar with both PAR1 residues and POPC molecules (Figure 3B–F). To better compare the similarities and differences between the metastable states observed in the five trajectories, we quantified the frequency of non-hydrogen atom contacts formed by vorapaxar with PAR1 residues and POPC molecules, using a distance cutoff of 3.5 Å The qualitative results of this analysis are shown in Figure 3, with the top nine interacting residues (or lipids) color-coded according to a heat map (from dark red, which indicates the most frequently interacting residue, to dark blue, which indicates the ninth most frequently interacting residue).
In the TM6–TM7 metastable states detected, the most consistent interactions observed were between the fluorophenyl-pyridine moiety of vorapaxar (−R2, Scheme 1) and aromatic amino acids in TM6 and TM7. In particular, Tyr3537.35 and His3366.58 interacted with the fluorophenyl ring in every TM6–TM7 exit trajectory (Figure S3). Additional interactions between Leu3326.54 and His342ECL3 and the vorapaxar fluorophenyl ring (Figure 3B,E,F), as well as between Ala3497.31 and the pyridine ring (Figure 3B,D,F), were seen in some trajectories. As one moves out along the vorapaxar skeleton, the methylfuranone ring of the tricyclic group tended to exit PAR1 in the plane of the lipid–water interface and interacted with polar residues in ECL3 [particularly Ser344 (see Figure 3C–E)] as well as lipid headgroups and bulk water.
Most strikingly, all of the TM6–TM7 metastable states revealed that lipids frequently interacted with vorapaxar, particularly with the himbacine-like tricyclic ring and the ethyl carbamate moiety (Figure S4). Lipids made both hydrophobic and polar interactions with vorapaxar, and in every simulation, a POPC molecule interacted with vorapaxar more frequently than the first or second most frequently interacting amino acid residue (dark red or red in Figure 3).
Detailed Analysis of Vorapaxar in the TM4–TM5 Metastable State
Compared with the TAMD trajectories in which vorapaxar exited PAR1 through the TM6–TM7 trajectory described above, the predominant metastable state observed with TM4–TM5 exits occurred after a smaller displacement from the crystallographic binding site (Figure 4). This occurred at a position where the vorapaxar ethyl carbamate had slid from the TM6–TM7 tunnel in toward the center of the seven-TM bundle (arrows in Figure 4 and Movie S3). Deep in this metastable binding pocket, the fluorophenyl-pyridine moiety of vorapaxar interacted with hydrophobic residues in TM3 and TM5, namely, Gly2334.56 (Figure 4B–D), Tyr1873.37 (Figure 4C,D), and one or more of Leu2294.52, Phe2715.39, Phe2745.42, or Phe2785.46. The methylfuranone ring and tricyclic group rested in the upper (toward extracellular) portion of the binding pocket and interacted with ECL2 residues (namely Thr261 and Leu262). The part of the metastable binding pocket facing the center of PAR1 was consistently formed by His33 6 6.58, Tyr3376.59, and Tyr3537.35, which interacted with the ethyl carbamate. POPC molecules formed frequent interactions in only one of the three metastable states observed during TM4–TM5 exits (Figure 4B).
Preparation of Alkylated Derivatives of Vorapaxar
On the basis of the consistency of the metastable state and lipid interactions associated with the TM6–TM7 exit pathway and the principle of microscopic reversibility, which implies that ensembles of binding and dissociation pathways should be identical at equilibrium,33 we hypothesized that the TM6–TM7 pathway may be a path by which vorapaxar enters the PAR1 binding pocket.34 Vorapaxar binds to PAR1 slowly, with full inhibition of PAR1 function requiring up to 30 min even at high drug concentrations. We reasoned that if vorapaxar indeed enters PAR1 from the lipid bilayer via the TM6–TM7 path, derivatizing the ethyl carbamate position of vorapaxar (arrows in Figure 3) with long chain alkyl “tails” that would be able to follow the remainder of the vorapaxar molecule into the binding pocket and remain protruding through the TM6–TM7 tunnel (Figure 1C) would not substantially disrupt the already slow entry of the drug and its antagonist function. To test this prediction, we first converted vorapaxar back to the free amine by removing the ethyl carbamate and then converted the free amine to either a hexyl carbamate group or an N-boc-hexyl carbamate (Scheme 1 and Figure 5). The former compound (hexyl vorapaxar) effectively extends the ethyl carbamate alkyl chain that protrudes between TM6 and TM7 by four carbons, while the latter (N-boc-hexyl vorapaxar) extends the chain by four carbons and a bulky N-boc group (Figure 5).
Figure 5.

Kinetics of PAR1 inhibition by vorapaxar derivatives. Time course of inhibition of the calcium flux response of Rati fibroblasts stably expressing human PAR1 by (A) vorapaxar, (B) hexyl vorapaxar, and (C) N-boc-hexyl vorapaxar. After incubation with 1 μM vorapaxar derivatives for the indicated time, cells were stimulated with 3 μM agonist peptide SFLLRN. The response is expressed as the percent obtained after incubation with vehicle alone in the absence of an inhibitor. Data are representative of three independent experiments (see also Figure S10), and each data point represents the mean ± the standard deviation (error bars) of three to six individual 96-well plates. The pooled data from all experiments were subjected to curve fitting (Table 1). At the right are chemical structures of vorapaxar derivatives. An increasing alkyl chain length and bulk predicted to extend through the TM6–TM7 exit tunnel is indicated by the arrow.
Vorapaxar and its alkylated derivatives are very hydrophobic,35 which presented a challenge to delivery of well- defined concentrations to cells (a requirement for precise kinetic experiments). Vorapaxar has an absorbance band in the UV, likely arising from the fluorophenyl-pyridine moiety.36 We determined the extinction coefficient of vorapaxar to be 19.3 ± 0.6 mM−1 cm−1 at 305 nm in DMSO (Figure S5A,B). Hexyl and N-boc-hexyl vorapaxar had identical absorption spectra and gave similar extinction coefficients at 305 nm (derivatization of the ethyl carbamate position should have no effect on the fluorophenyl-pyridine chromophore). We took advantage of this absorption band to characterize the solubility of the vorapaxar derivatives in various carrier solutions (Figure S5C). After screening several cyclodextrins commonly used to enhance the solubility of hydrophobic drugs,37 we found HPβCD to be most effective. The solubility of the derivatives in 10% DMSO and 10 mM HPβCD was 89 ± 6 μM; that of N-boc-hexyl vorapaxar was 14.4 ± 0.2 μM, and that of hexyl vorapaxar was 7.8 ± 1.0 μM [n = 3 determinations (Figure S5D)].
Cell Signaling Assays
We first tested hexyl vorapaxar for activity in a human PAR1-dependent SFLLRN- or thrombin- triggered inositol trisphosphate (IP3) accumulation assay and found it to be equally efficacious to vorapaxar (Figure S6; antagonists were added at a concentration of 100 nM for 1 h at 37 °C prior to agonist addition). Encouraged by this preserved activity, we went on to test the kinetics of inhibition of human PAR1 by the vorapaxar derivatives as assessed by inhibition of SFLLRN-triggered increases in the intracellular calcium concentration in Rat1 fibroblasts stably expressing human PAR1 (hPAR1) and loaded with Fluo4. This cell line showed uniform membrane expression of hPAR1 (Figure S7) and rapid agonist-triggered intracellular calcium flux upon addition of SFLLRN (Figure S8A and Movie S4). Untransfected Rat1 fibroblasts showed a minimal response to SFLLRN (Figure S8B). The EC50 for the SFLLRN response in this assay was 265 nM [210–330 nM, 95% confidence interval (Figure S8C)], and we chose 3 μM SFLLRN as a near-saturating concentration of the agonist for inhibition assays.
For inhibition assays (Figure S8D), hPAR1-expressing cells were incubated for 10–75 min with vorapaxar derivatives or with vehicle controls containing HPβCD and DMSO at matched concentrations. A vehicle containing <2 mM HPβCD had no significant effect in these experiments, suggesting that cholesterol depletion of membranes by cyclodextrin, if it occurred under these conditions, did not have a substantial effect on PAR1 function. HPβCD concentrations of >2 mM caused loss of loaded Fluo4 and toxicity (Figure S9C,D) and were avoided; 3% DMSO alone had no effect in this assay (Figure S9A,B), but the DMSO concentration was maintained at <2% in inhibition assays.
Vorapaxar, hexyl vorapaxar, and N-boc-hexyl vorapaxar were all effective hPAR1 antagonists. As previously shown, the free amine derivative of vorapaxar (which lacks the ethyl carbamate group) is inactive as an inhibitor of human PAR1.29 In calcium flux assays, incubation with 1 μM vorapaxar, hexyl vorapaxar, or N-boc-hexyl vorapaxar for 45 min at 25 °C produced nearly complete inhibition of SFLLRN responses (Figure 5 and Figure S10A,B). At 500 nM, inhibition was complete by 60–80 min at 25 °C (Figure S10C,D). Under these conditions, the HPβCD vehicle caused no more than 30% decay in the response over the duration of the assay. The 1 μM inhibition curves were well fit by a first-order exponential, and there was a trend toward slower inhibition kinetics with an increasing alkyl chain length (Table 1). However, the observed first-order rate constants (kobs) differed by only ~3-fold between the fastest (vorapaxar) and slowest (N-boc-hexyl vorapaxar) binders. We conclude that derivatizing vorapaxar with bulky alkyl chains predicted to extend through the TM6–TM7 solvent tunnel in PAR1 (Figure 1C) does not prevent antagonist function and has a relatively small effect on the kinetics of inhibition (Figure 5).
Table 1.
Results of First-Order Exponential Fits to the Time Course of Inhibition for Vorapaxar Derivativesa
| inhibitor | kobs (min−1) | R2 | n (of 96 wells) |
|---|---|---|---|
| vorapaxar | 0.112 ± 0.005 | 0.62 | 80 |
| hexyl vorapaxar | 0.054 ± 0.004 | 0.85 | 29 |
| N-boc-hexyl vorapaxar | 0.036 ± 0.003 | 0.79 | 53 |
kobs is reported as the mean ± the standard error. A statistically significant difference was found in kobs between all groups (p < 0.0001; extra sum of squares F test).
Reconciling Experimental and TAMD Results
We hypothesized that vorapaxar can enter and exit PAR1 from the plasma membrane via a path between TM6 and TM7. This hypothesis was based on the hydrophobic nature of vorapaxar and previous observations suggesting that lipid ligands for GPCRs may “tunnel” into the receptor from the plasma membrane14–18 together with the position of vorapaxar in the PAR1 co-crystal structure,24 in which the ethyl carbamate “tail” of vorapaxar extends between TM6 and TM7 toward the lipid bilayer. The TAMD studies support this hypothesis. The TM6–TM7 pathway of vorapaxar dissociating from PAR1, in which the vorapaxar COM “followed” the ethyl carbamate tail out of the binding pocket, was the mechanism of exit most consistently observed in TAMD simulations, with reproducible amino acid contacts detected in a metastable state along the path (Figure 3). To experimentally test the TM6–TM7 entry/ exit pathway, we used a chemical biology approach, again inspired by the position of the ethyl carbamate group in the crystal structure and the path observed in TAMD simulations. The hexyl and N-boc-hexyl “tails” that replaced the ethyl carbamate in the vorapaxar derivatives tested were envisioned to extend through the TM6–TM7 tunnel if the derivatized molecule adopted the same position as the parent in the vorapaxar–PAR1 co-crystal (Figure 1). We expected that these longer and bulkier groups would sterically interfere with vorapaxar’s reaching the crystallographic pose and impede the already slow onset of antagonist function unless entry occurred by the TM6–TM7 pathway. Addition of these large alkyl groups to vorapaxar did not decrease its effectiveness as an antagonist and did not slow the onset of antagonism by >2–3- fold (Figure 5 and Table 1). Thus, taken as a whole, the simulation and experimental data suggest that vorapaxar can enter and exit PAR1 via the TM6–TM7 tunnel.
This study has several limitations. We did not observe vorapaxar binding to PAR1 on the time scale of the MD simulations and were therefore unable to simulate the effects of derivatizing vorapaxar on binding via the different unbinding pathways observed. Our inability to observe repeated binding and unbinding also prevented estimates of the relative likelihood of the different pathways. TAMD does add energy to the system; the unbinding pathways observed in TAMD simulations may differ from those that would have been observed in extremely long unbiased MD simulations, and the potential energy functions (force fields) that all MD simulations employ are inherently approximate.
Because of difficulty in solubilizing the vorapaxar derivatives and the requirement for cyclodextrin vehicles, we were unable to perform direct binding assays to precisely determine the kinetics of hexyl vorapaxar and N-boc-hexyl vorapaxar binding relative to the parent compound. Instead, we relied on functional assays to demonstrate the efficacy of all three inhibitors on a similar time scale (Figure 5 and Figure S6) to infer that their on rates were similar. The limitations of TAMD/MD and binding analyses prevent the establishment of a precise relationship between the kinetics observed in the TAMD simulations and our cell culture studies, and while our data are consistent with the notion that vorapaxar can enter or exit PAR1 via the TM6–TM7 pathway and the lipid bilayer, we cannot exclude the possibility that vorapaxar can also enter or exit PAR1 via the TM4–TM5 or ECL pathways or others not detected in these studies.
We did not perform site-directed mutagenesis to perturb the major interactions seen in the TM6–TM7 metastable state. A Tyr3537.35Ala mutation resulted in a loss of PAR1 surface expression (data not shown). While it would be of interest to mutate other key residues interacting in the metastable state (such as His3366.58), we felt that it would be difficult to distinguish subtle effects on inhibition kinetics from changes in receptor structure and binding affinity. Thus, we used a stably transfected cell line known to have robust surface expression of wild-type human PAR1 (Figure S7) to compare the function of vorapaxar derivatives.
Lastly, one of our most interesting observations is the finding that interactions between vorapaxar and lipid molecules appear to contribute to the metastable state along the TM6–TM7 pathway. As is convention in MD studies, our studies employed lipid bilayers comprised of hydrated POPC. Phosphatidylcholine is the most abundant phospholipid species in mammalian lipid bilayers and is enriched in the outer leaflet, where the interactions of interest in our study occurred. How inclusion of additional lipids would affect the MD simulations is unknown.
CONCLUSIONS
Using a combination of MD simulation, kinetic cell signaling experiments, and a novel panel of inhibitors, we provide evidence that the small molecule antagonist vorapaxar can bind and unbind from PAR1 via the lipid bilayer membrane. While lipid bilayer entry pathways have been described for rhodopsin/retinal and sphingosine 1-phosphate receptor 1,14,17 which have natural small molecule lipid ligands, this work provides evidence that hydrophobic small molecules can bind to a peptide-activated GPCR in this manner. Our results suggest that vorapaxar can dissociate through the tunnel between TM6 and TM7 into the lipid bilayer (Figure 1C). Along this route, we reproducibly detected a metastable binding site (Figures 2D and 3) featuring interactions of vorapaxar with GPCR residues (Figure 3 and Figure S3) and lipids in the outer leaflet of the lipid bilayer membrane (Figure 3 and Figure S4).
This study has several broader implications for the design and optimization of GPCR therapeutics. First, our data suggest that a small molecule antagonist can enter a peptide-activated GPCR from the lipid bilayer. Given the soluble nature of most endogenous peptide hormones, it is expected that the native ligands for these receptors bind from the extracellular solvent. Our work suggests that tunnels facing the lipid bilayer should not be neglected as potential targets for structure-guided drug design, particularly as more high-resolution crystal structures of peptide-activated GPCRs are determined. Second, the contribution of membrane lipids to a metastable binding intermediate could be exploited to modulate drug properties. Modifying interactions between GPCR ligands and lipid headgroups, as observed between the ethyl carbamate and methylfuranone moieties of vorapaxar and POPC headgroups during MD simulations (Figure S4), could allow tuning of on and off rates, or even selectivity for membrane subdomains with dissimilar lipid compositions.
Despite dramatic recent advances in GPCR crystallography, no structure has yet trapped a ligand in a metastable binding intermediate. Given the overall importance of kinetics to understanding receptor–drug interactions and in vivo efficacy,7 MD represents an important tool for probing pathways of ligand binding and unbinding. Chemical biology experiments using modified ligands as kinetic probes, in addition to dynamic high-resolution techniques such as nuclear magnetic resonance and electron paramagnetic resonance, may synergize with MD to help clarify these crucial mechanisms of drug recognition by GPCRs.
Supplementary Material
ACKNOWLEDGMENTS
Vorapaxar was a kind gift of Portola Pharmaceuticals.
Funding
M.P.B. acknowledges Research Scholars funding from the University of California, San Francisco’s, Department of Anesthesia and Perioperative Care. S.R.C. acknowledges support from National Institutes of Health Grant R35 HL135755.
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- PAR1
protease-activated receptor-1
- TM
transmembrane helix
- MD
molecular dynamics
- ECL
extracellular loop
- TAMD
temperature- accelerated molecular dynamics
- POPC
1-palmitoyl-2-oleoyl- sn-glycero-3-phosphocholine
- CV
collective variable
- COM
center of mass
- DMSO
dimethyl sulfoxide
- HPβCD
2- hydroxypropyl-β-cyclodextrin
- DMEM
Dulbecco’s modified Eagle’s medium
- BSA
bovine serum albumin
- IP3
inositol trisphosphate
Footnotes
The authors declare no competing financial interest.
All measurements are reported as means ± the standard deviation unless otherwise noted.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.8b00577.
Figures and captions showing additional trajectories of the vorapaxar COM during MD simulations, details of the vorapaxar metastable state prior to extracellular exit, interactions of vorapaxar with PAR1 and lipids in the TM6–TM7 metastable state, absorption spectroscopy of vorapaxar, phosphoinositide accumulation assay for inhibition of PAR1 by vorapaxar and hexyl vorapaxar, cell surface expression of human PAR1 in Rat1 fibroblasts, calcium flux inhibition assay for PAR1, vehicle controls for the calcium assay, inhibition of calcium flux by vorapaxar derivatives, and captions for the movies (PDF)
Molecular dynamics simulation of the exit of vorapaxar from PAR1 (MPG)
Molecular dynamics simulation of the exit of vorapaxar from PAR1 (MPG)
Molecular dynamics simulation of the exit of vorapaxar from PAR1 (MPG)
Calcium flux assay in Rat1 fibroblasts (AVI)
REFERENCES
- (1).Baker JG (2005) The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharmacol 144, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, and Kobilka BK (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273. [DOI] [PubMed] [Google Scholar]
- (3).Katritch V, Cherezov V, and Stevens RC (2013) Structure- function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol 53, 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, and Kobilka BK (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, and Shaw DE (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A 108, 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Guo D, Mulder-Krieger T, IJzerman AP, and Heitman LH (2012) Functional efficacy of adenosine A(2)A receptor agonists is positively correlated to their receptor residence time. Br. J. Pharmacol 166, 1846–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pan AC, Borhani DW, Dror RO, and Shaw DE (2013) Molecular determinants of drug-receptor binding kinetics. Drug Discovery Today 18, 667–673. [DOI] [PubMed] [Google Scholar]
- (8).Barnes PJ (2000) The pharmacological properties of tiotropium. Chest 117, 63S–66S. [DOI] [PubMed] [Google Scholar]
- (9).Gonzalez A, Perez-Acle T, Pardo L, and Deupi X (2011) Molecular basis of ligand dissociation in beta-adrenergic receptors. PLoS One 6, e23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Redka DS, Pisterzi LF, and Wells JW (2008) Binding of orthosteric ligands to the allosteric site of the M(2) muscarinic cholinergic receptor. Mol. Pharmacol 74, 834–843. [DOI] [PubMed] [Google Scholar]
- (11).Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, and Kobayashi T (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chien EY, Liu W, Zhao Q, Katritch V, Won Han G, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, and Stevens RC (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, and Miyano M (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- (14).Piechnick R, Ritter E, Hildebrand PW, Ernst OP, Scheerer P, Hofmann KP, and Heck M (2012) Effect of channel mutations on the uptake and release of the retinal ligand in opsin. Proc. Natl. Acad. Sci U. S. A 109, 5247–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hildebrand PW, Scheerer P, Park JH, Choe HW, Piechnick R, Ernst OP, Hofmann KP, and Heck M (2009) A ligand channel through the G protein coupled receptor opsin. PLoS One 4, e4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, and Stevens RC (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Stanley N, Pardo L, and Fabritiis GD (2016) The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep 6, 22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, and Reggio PH (2010) A lipid pathway for ligand binding is necessary for a cannabinoid G proteincoupled receptor. J. Biol. Chem 285, 17954–17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vu TK, Hung DT, Wheaton VI, and Coughlin SR (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068. [DOI] [PubMed] [Google Scholar]
- (20).Gerszten RE, Chen J, Ishli M, Ishil K, Wang L, Nanevicz T, Turck CW, Vu TK, and Coughlin SR (1994) Specificity of the thrombin receptor for agonist peptide is defined by its extracellular surface. Nature 368, 648–651. [DOI] [PubMed] [Google Scholar]
- (21).Nanevicz T, Wang L, Chen M, Ishii M, and Coughlin SR (1996) Thrombin receptor activating mutations. Alteration of an extracellular agonist recognition domain causes constitutive signaling. J. Biol. Chem 271, 702–706. [DOI] [PubMed] [Google Scholar]
- (22).Pfreundschuh M, Alsteens D, Wieneke R, Zhang C, Coughlin SR, Tampe R, Kobilka BK, and Muller DJ (2015) Identifying and quantifying two ligand-binding sites while imaging native human membrane receptors by AFM. Nat. Commun 6, 8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, and Chintala M (2008) Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J. Med. Chem 51, 3061–3064. [DOI] [PubMed] [Google Scholar]
- (24).Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, and Kobilka BK (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492, 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA, Young C, Deneroff MM, Batson B, Bowers KJ, Chow E, Eastwood MP, Ierardi DJ, Klepeis JL, Kuskin JS, Larson RH, Lindorff-Larsen K, Maragakis P, Moraes MA, Piana S, Shan Y, and Towles B (2009) Millisecond- scale molecular dynamics simulations on Anton. In Proceedings of the Conference on High Performance Computing, Networking, Storage and Analysis (SC09), ACM, Portland, OR. [Google Scholar]
- (26).Maragliano L, and Vanden-Eijnden E (2006) A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett 426, 168–175. [Google Scholar]
- (27).Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (28).Voronin KN, and Penner NA (2013) Active metabolite of a thrombin receptor antagonist. U.S. Patent 8,575,351 B2 Merck Sharp & Dohme Corp.
- (29).Ghosal A, Lu X, Penner N, Gao L, Ramanathan R, Chowdhury SK, Kishnani NS, and Alton KB (2011) Identification of human liver cytochrome P450 enzymes involved in the metabolism of SCH 530348 (Vorapaxar), a potent oral thrombin protease-activated receptor 1 antagonist. Drug Metab. Dispos 39, 30–38. [DOI] [PubMed] [Google Scholar]
- (30).Christensen MK, and Bjorkling F (2012) Squaric acid derivatives as inhibitors of the nicotinamide. Patent WO 2009156421A1.
- (31).Hung DT, Wong YH, Vu TK, and Coughlin SR (1992) The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. J. Biol. Chem 267, 20831–20834. [PubMed] [Google Scholar]
- (32).Emkey R, and Rankl NB (2009) Screening G protein-coupled receptors: measurement of intracellular calcium using the fluorometric imaging plate reader. Methods Mol. Biol 565, 145–158. [DOI] [PubMed] [Google Scholar]
- (33).Astumian RD (2012) Microscopic reversibility as the organizing principle of molecular machines. Nat. Nanotechnol 7, 684–688. [DOI] [PubMed] [Google Scholar]
- (34).Lewis GN (1925) A New Principle of Equilibrium. Proc. Natl. Acad. Sci U. S. A 11, 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Xia Y, Chackalamannil S, Greenlee WJ, Wang Y, Hu Z, Root Y, Wong J, Kong J, Ahn HS, Boykow G, Hsieh Y, Kurowski S, and Chintala M (2010) Discovery of a vorapaxar analog with increased aqueous solubility. Bioorg. Med. Chem. Lett 20, 6676–6679. [DOI] [PubMed] [Google Scholar]
- (36).Krumholz P (1951) Structural studies on polynuclear pyridine compounds. J. Am. Chem. Soc 73, 3487–3492. [Google Scholar]
- (37).Ahuja A, Baboota S, Ali J, and Mustafa G (2011) Cyclodextrins as potential excipients in pharmaceutical formulations: solubilizing and stabilizing effects In Cyclodextrins in pharmaceutics, cosmetics, and biomedicine: Current and future industrial applications (Bilensoy E, Ed.) 1st ed., pp 19–43, John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.