

Abstract
The margins of an expanding range are predicted to be challenging environments for adaptation. Marginal populations should often experience low effective population sizes (Ne) where genetic drift is high due to demographic expansion and/or census population size is low due to unfavorable environmental conditions. Nevertheless, invasive species demonstrate increasing evidence of rapid evolution and potential adaptation to novel environments encountered during colonization, calling into question whether significant reductions in Ne are realized during range expansions in nature. Here we report one of the first empirical tests of the joint effects of expansion dynamics and environment on effective population size variation during invasive range expansion. We estimate contemporary values of Ne using rates of linkage disequilibrium among genome-wide markers within introduced populations of the highly invasive plant Centaurea solstitialis (yellow starthistle) in North America (California, USA), and within native Eurasian populations. As predicted, we find that Ne within the invaded range is positively correlated with both expansion history (time since founding) and habitat quality (abiotic climate). History and climate had independent additive effects with similar effect sizes, indicating an important role for both factors in this invasion. These results support theoretical expectations for the population genetics of range expansion, though whether these processes can ultimately arrest the spread of an invasive species remains an unanswered question.
Keywords: Centaurea solstitialis, yellow starthistle, range expansion, ddRADseq, linkage disequilibrium Ne, climatic niche
Introduction
Adaptation is expected to be a critical component of how species respond to novel environmental conditions, such as those encountered during colonization and range expansion (Mayr 1963; Kirkpatrick & Barton 1997; Griffith & Watson 2006; Colautti & Barrett 2013; Bock et al. 2015; Hamilton et al. 2015). At the same time, it has been suggested that colonizing species might experience small population sizes that limit the ability of founding populations to respond to natural selection (Elam et al. 2007; Dlugosch et al. 2015; Gonzalez-Martinez et al. 2017; Welles and Dlugosch 2018). Small population sizes could result from both founder events and maladaptation to novel environments. A failure to adapt under these conditions could slow or limit range expansion and contribute to the formation of range limits (Bridle and Vines 2007; Eckert et al. 2008; Sexton et al. 2009; Polechová and Barton 2015; Polechova 2018). These effects are currently an active area of theoretical and experimental research (Gilbert et al. 2017; Szűcs et al. 2017a; Szűcs et al. 2017b), but empirical observations of the dynamics of population size and its influence on evolution during ongoing range expansions is scant (Ramakrishnan et al. 2010; Wootton and Pfister 2015).
Population genetic models predict that deleterious alleles may become fixed during range expansion due to the strong effects of genetic drift during colonization (Lehe et al. 2012; Peischl et al. 2013; Peischl et al. 2015), ultimately resulting in failure to adapt (Henry et al. 2015; Polechová and Barton 2015; Polechova 2018). Range expansions are expected to involve a series of founding events (repeated sampling events) as new populations establish beyond the current range boundary, resulting in reduced effective population size (Ne) and increased sampling effects as the range boundary advances (Le Corre & Kremer 1998; Excoffier 2004; Slatkin & Excoffier 2012). In particular, low Ne at the leading edge can cause random alleles, including deleterious mutations, to ‘surf’ to high frequency regardless of patterns of selection (Travis et al. 2007; Excoffier and Ray 2008; Excoffier et al. 2009; Hallatschek and Nelson 2010; Moreau et al. 2011; Peischl et al. 2013). This can create an ‘expansion load’ of deleterious alleles at the wave front, although beneficial mutations can also surf to high frequency and aid in local adaptation (Peischl et al. 2013; Peischl et al. 2015). The effects of range expansion on adaptation have been empirically observed with greatest detail in bacterial culture, where manipulative experiments have shown that the strength of genetic drift is key to determining whether allele surfing promotes or hinders adaptation (Hallatschek and Nelson 2010; Gralka et al. 2016; Bosshard et al. 2017).
Environmental conditions should also shape Ne during range expansion via their impact on population (census) size and demography. If leading edge environments are different than those experienced by source populations, then founding genotypes will not be pre-adapted and are likely to experience lower absolute fitness. Unfavorable conditions and low fitness may lead to lower abundance and/or fluctuations in population size, reducing Ne relative to larger or more stable populations (Wright 1938; Crow & Morton 1955; Kimura & Crow 1963; Frankham 1996). In a rare empirical example, Micheletti & Storfer (2015) found that streamside salamander (Ambystoma barbouri) populations on the periphery of the range were also on the margins of their climatic niche and tended toward lower Ne. Similarly, peripheral populations of the North American annual plant Arabidopsis lyrata possess greater genetic load and appear to exist at their ecological, and perhaps evolutionary limits (Willi et al. 2018). These studies address a set of long-debated hypotheses proposing that range limits form in part because they consist of ecologically and/or genetically marginal populations (Kirkpatrick & Barton 1997; Phillips 2012; Chuang & Peterson 2016), which lack the capacity to acquire adaptations that are necessary to support further expansion (i.e. the ‘central-marginal’, ‘center-periphery’ and ‘abundant center’ hypotheses: (Sagarin and Gaines 2002; Eckert et al. 2008; Pironon et al. 2015). Importantly, all of these hypotheses share the prediction that colonization will be associated with reduced response to selection for ecological reasons without requiring additional population genetic changes caused by expansion alone. The relative importance of these two factors (environment and expansion) for shaping Ne at range margins is unknown, but both have the potential to reduce opportunities for local adaptation.
Although Ne has long been used as a fundamental measure of the scale of genetic drift in populations (Wright 1931; Robertson 1960; Kimura and Crow 1963; Kimura 1964; Ohta 1992; Charlesworth 2009), little is known about how Ne changes during the process of range expansion. Most empirical population-level estimates come from the field of conservation genetics, where Ne is used to infer the potential for genetic drift to exacerbate the decline of threatened populations (Lynch et al. 1995; Frankham 1996; Sung et al. 2012). These studies have demonstrated that Ne can be highly variable within species, sensitive to local demography and modes of reproduction, and poorly predicted by census size (Frankham 1995; Turner et al. 2002; Palstra and Ruzzante 2008). For example, in recovering Chinook salmon (Orcorhynchus tshawytscha) populations, Shrimpton and Heath (2003) found up to a three-fold difference in both Ne and its ratio with census size across spawning sites. While low Ne is generally expected in declining populations, many of the same demographic factors are likely to affect Ne in founding populations (Allendorf and Lundquist 2003; Colautti et al. 2017).
Despite the potential obstacle low Ne might pose to adaptation, many species -- including large numbers of invaders -- have been successful at colonization and show evidence of adaptive evolution during range expansion (Rice and Mack 1991; Dlugosch and Parker 2008; Linnen et al. 2009; Colautti and Barrett 2013; Vandepitte et al. 2014; Colautti and Lau 2015; Li et al. 2015). Additionally, detailed studies of range expansion have found evidence of serial founding events and associated increases in genetic drift (Ramakrishnan et al. 2010; Graciá et al. 2013; White et al. 2013; Pierce et al. 2014; Peischl et al. 2018), and it is notable that few invasions appear to have expanded beyond the fundamental niches of their native range (Petitpierre et al. 2012; Tingley et al. 2014). Taken together, it appears that adaptive evolution might be achievable in many invading species, but that perhaps expansion load and ecological mismatch may act, either independently or in concert, to prevent expansion in some cases. An understanding of how founding dynamics and marginal environments shape Ne in individual wave front populations is needed to connect theoretical expectations to observed patterns of successful range expansion.
Here we estimate contemporary Ne for populations of the obligately outcrossing annual plant Centaurea solstitialis (yellow starthistle) across its invasion of California (USA) and its native range in Eurasia. In California, C. solstitialis was initially introduced in the mid 19th century into the San Francisco Bay area as a contaminant of alfalfa seed (Gerlach 1997; DiTomaso et al. 2006). Colonization by C. solstitialis resulted in a weak genetic bottleneck that is characterized by reduced private allele richness but no change in total allelic richness, nucleotide diversity, or observed heterozygosity (Barker et al. 2017). By the mid 20th century, the species was rapidly expanding through California’s Central Valley and Sierra Nevada foothill grasslands, and the current leading edge of this invasion lies above 4000 m in elevation along the west side of the Sierra Nevada Mountains (Pitcairn et al. 2006). In the North American invasion, habitat quality is often linked to the climatic environment, with warmer and drier habitats frequently supporting the densest C. solstitialis populations (Pitcairn et al. 2006; Swope and Parker 2010). During expansion, C. solstitialis has crossed climatic gradients that are largely independent in direction from the pathway of colonization (Fig. 1), allowing us to quantify the influence of both climatic environment and expansion history (time since founding) on estimates of Ne across populations.
Fig 1.

The distribution of rarefied effective population size (Ne), climatic principal component (PC) gradients, and population age (in years) across C. solstitialis populations in Eurasia and California. In all panels, circles indicate sampled populations with a diameter proportional to Ne. PC1 is positively correlated with annual temperature and temperature of the driest quarter and negatively correlated with seasonal differences in total radiation in the native (A) and invaded (C) ranges. PC2 is positively correlated with seasonal differences in temperature and negatively correlated with annual precipitation and seasonal differences in precipitation in the native (B) and invaded (D) ranges. In the California invasion, population age (E) reflects a history of expansion beginning in the San Francisco Bay area and expanding first to the North and then to the South and East of the state. Abbreviations in (E) correspond to populations in Table S1.
We used Restriction-site Associated DNA sequencing (RADseq) to estimate contemporary Ne in C. solstitialis populations sampled at a single time point. In addition to testing for the joint influence of expansion dynamics and climatic conditions on Ne in this system, we explored solutions for general problems associated with using large genome-wide marker data sets to estimate Ne. Linkage disequilibrium Ne (LD-Ne) is a powerful method for inferring contemporary Ne from single time sampled data, and does so by utilizing the frequency of statistical linkage across loci (Waples and Do 2008; Gilbert and Whitlock 2015). This method requires that loci segregate independently of each other, and while RADseq is widely used to produce population genetic datasets in non-model systems (Narum et al. 2013; Catchen et al. 2017), it is likely to violate this assumption of independence, resulting in biased calculations of Ne.
We used marker resampling and rarefaction approaches to improve inferences of variation in Ne across populations. We tested for effects of expansion history (time since founding) and habitat quality (climatic environment) on rarified Ne estimates, and compared these values to those from populations in the native range. We also explored whether estimates of genetic diversity could predict values of Ne, given that non-equilibrium population dynamics may in the short term decouple contemporary Ne from its expected long term effects on genetic variation (e.g. Nei et al. 1975; Varvio et al. 1986; Alcala et al. 2013; Epps and Keyghobadi 2015). By testing for evidence of historical and ecological effects on Ne, our goal is to shed light on the factors shaping fundamental parameters of evolution during colonization and range expansion.
Materials and Methods
Study Species
Yellow starthistle (Centaurea solstitialis) is an obligately outcrossing, diploid annual plant, native to a broad region of Eurasia. Plants grow as basal rosettes with a deep taproot, then bolt and produce up to several hundred flowering heads (capitula), which can collectively produce thousands of small (under 2mg) seeds per individual (Graebner et al. 2012; Hierro et al. 2012). Reproduction is by seed only (there is no clonal reproduction), and seeds are either unadorned (outer florets) or have a small (2mm) bristle-like pappus that appears to be better adapted for animal (including human) dispersal than for wind dispersal (Roche 1992; Gerlach 1997; Sun and Ritland 1998). Over 80% of seeds germinate within the first year, and while seeds can remain viable within the soil for up to ten years, most natural seed banks appear to be depleted in three years without new input (Joley et al. 1992; Callihan et al. 1993; Benefield et al. 2001).
Seeds of C. solstitialis were introduced to the Americas as a contaminant of alfalfa seed (Gerlach 1997), and have formed dense invading populations in mediterranean and semi arid grasslands of North and South America (DiTomaso et al. 2006). Invading populations are persistent and difficult to control (Aslan et al. 2009; Matzek and Hill 2012). Genotypes from invaded regions have evolved larger seeds, larger biomass, faster growth rates, shorter time to flowering, and greater reproductive output than those from the Eurasian native range (Eriksen et al. 2012; Widmer et al. 2007; Dlugosch et al. 2015). Invading populations in the Americas achieve densities that are more than an order of magnitude higher than those in the native range (Uygur et al. 2004; Andonian et al. 2011).
The first recorded introduction of C. solstitialis in North American occurred in the San Francisco Bay area of California, USA in 1869 (Pitcairn et al. 2006). Records indicate a subsequent expansion eastward into the Central Valley of California, then southward to San Diego, northward to southern Oregon, and further East to the Sierra Nevada mountains where the expansion remains active (Gerlach 1997; DiTomaso et al. 2006). There are also additional invading populations in the interior Pacific Northwest, but previous genetic work indicates that these are the product of separate introductions, and the California invasion is composed of a single expansion of genotypes originally from western Europe (Barker et al. 2017). Our work focuses on the California invasion.
Within California, coastal populations (closest to the initial introduction) are composed of smaller plants and reach densities that are an order of magnitude lower than those in the Central Valley and Sierra Nevadas (Swope and Parker 2010; Swope et al. 2017). Seed addition studies indicate that coastal populations are near carrying capacity despite their lower densities, while Central Valley and Sierra Nevada populations are seed limited and have the capacity to achieve higher densities (Swope and Parker 2010). Multiple biocontrol agents have been introduced to California, but have only been effective at controlling population growth in low density coastal populations, where a small decrease in vital rates has a large effect on population growth (Swope et al. 2017). In the Central Valley and Sierra Nevadas, compensatory growth and high plant densities limit the impact of biocontrol, and density dependent reproduction in C. solstitialis results in seed production that is independent of individual density across sites (Swope and Parker 2010).
Genomic Data
Genome-wide markers for C. solstitialis in this study were sampled from single nucleotide polymorphisms in double-digest RADseq (ddRADseq; (Peterson et al. 2012), previously published by Barker and colleagues (Barker et al. 2017; Dryad doi:10.5061/dryad.pf550). All sequences were obtained from C. solstitialis individuals germinated in the laboratory from wild collected seed. Seeds were sampled in 2008 from maternal plants along a linear transect in each population, with >1m separation between individuals. Populations included at least 14 individuals each grown from different maternal plants, from 12 invading populations in California and seven native populations in Europe (451 individuals total; Table S1). Sampled populations spanned the extent of the Californian invasion (Fig 1e).
Briefly, sequence data published by Barker and colleagues (2017) were generated as follows. Genomic DNA was extracted with a modified CTAB protocol (Webb and Knapp 1990) and fragmented using PstI and Mse1 restriction enzymes. Samples were individually barcoded, cleaned and size selected for fragments between 350 and 650 bp. Size selected fragments were amplified through 12 PCR cycles and sequenced on an Illumina HiSeq 2000 or 2500 platform (Illumina, Inc., San Diego, CA USA) to generate 100 bp paired-end reads. Reads were de-multiplexed with custom scripts and cleaned with the package SNOWHITE 2.0.2 (Dlugosch et al. 2013) to remove primer and adapter contaminants. Barcode and enzyme recognition sequences were removed from individual reads, and bases with phred quality scores below 20 were clipped from the 3’ end. Reads were trimmed to a uniform length of 76 base pairs. The R2 (reverse) reads from the data set were removed due to variable quality, and all analyses in this study were conducted using R1 (forward) reads only.
We used the denovo_map.pl pipeline in STACKS 1.20 (Catchen et al. 2011; Hohenlohe et al. 2011) to identify putative alleles within individuals, allowing a maximum of two nucleotide polymorphisms when merging stacks (-M parameter in STACKS), a maximum of two alleles per locus (-X), and a minimum coverage depth of five (-m). A catalog of loci and single nucleotide polymorphisms (SNPs) was generated across individuals, allowing two polymorphisms (-n) between individuals within a stack. The population.pl module in STACKS was used to calculate the population level nucleotide diversity (π) (Nei & Li 1979; Allendorf 1986). We restricted our analyses to loci that were sequenced in 80% of individuals within a population and in 90% of all populations (-r and -p parameters respectively).
Estimates of Ne
We used SNPs identified by STACKs to calculate Ne for each population using a method based on linkage disequilibrium among loci with a correction for missing data (Waples & Do 2008) implemented in the program NeEstimator v.2.01 (Do et al. 2014). This method derives estimates of Ne from the frequency of statistical linkage among loci and has been shown to be one of the best predictors of Ne (hereafter LD-Ne) for markers sampled at a single time point (Gilbert & Whitlock 2015; Wang 2016; Waples 2016). The LD-Ne method is not strongly influenced by the total genetic diversity in the sample (Charlesworth 2009; Do et al. 2014), making it particularly well suited to analyses of invading populations where low genetic diversity might arise from founder effects unrelated to the number of individuals currently reproducing in the population. We used a minimum allele frequency threshold of 0.05 for including a locus in the analyses, which was the lowest threshold that did not result in excessive loss of loci and infinite estimates of Ne at some study sites.
The ddRADseq dataset consisted of thousands of SNPs across the genome, 622 of which passed our screening requirements. Some of these loci were located in the same RAD 76bp sequence, and we expect that these and many others do not segregate independently in our data set, either due to physical proximity or the influence of selection on multi-locus allele combinations (C. solstitialis has a genome size of 850Mbp, distributed across eight chromosomes (Bancheva & Greilhuber 2005; Widmer et al. 2007). We generated an initial estimate of Ne using one randomly sampled locus from each sequence (Table S1). To minimize the likelihood of our estimates including physically linked loci, we re-sampled random sets of 20 polymorphic SNPs from unique sequences to obtain distributions of LD-Ne estimates for each population. We chose to use 20 loci because this is typical of previous studies that have estimated LD-Ne (England et al. 2006; Waples 2006 ; Waples & Do 2008; Gilbert & Whitlock 2015), and it is highly conservative relative to our genome size and chromosome number (Bancheva and Greilhuber 2006; Gaut et al. 2007; Widmer et al. 2007). Substantial increases in locus sampling would require a genetic map for C. solstitialis to ensure loci were not in physical linkage. Each population was resampled 30,000 times. Sampling distributions were generally lognormal and spanned at least four orders of magnitude (Supporting Information Fig. S1). We used median values from these distributions to identify the median estimate.
We observed a strong, positive effect of the number of individuals sampled in each population on median LD-Ne (F1,17=9.36, P=0.007). Unequal sampling has been shown to decrease the accuracy of LD-Ne estimates (England et al. 2006; Waples 2006), and NeEstimator implements a corrective algorithm to address this problem (Do et al. 2014). To account for persistent sampling effects, we produced rarefaction curves of median Ne estimated by subsampling different numbers of individuals (10 to the maximum number available per population) after marker resampling. As above, each marker resampling consisted of 30,000 Ne estimates with 20 loci. Median estimates did not asymptote at our maximum sampling effort and increased linearly (see Results). We fit a linear mixed model with random intercept and slopes implemented in the Lme4 package in R (Bates et al. 2014) to obtain population specific functions which describe the relationships between the number of individuals sampled in each population and median LD-Ne values. The estimated slope and intercepts for each population were extracted from the model and used to calculate rarefied Ne for each population at a standard value of 10 individuals (our minimum rarefaction size). We explored the relationship between rarified Ne and measures of genome-wide marker variation using nucleotide diversity (π) at variable sites, as calculated in STACKS. We used linear regression to predict π from Ne among invading populations, and among native European populations for comparison.
Effects of Expansion History and Climate on Ne
We tested for an effect of population age since founding on the rarified Ne of invading populations. We estimated the date of colonization for each population by searching the Jepson Online Herbarium (http://ucjeps.berkeley.edu/) for records of C. solstitialis in California since its first record in 1869. For each sampling location, we used the earliest date on record for the county, or for an adjacent county when the sampling location was closer to older collection records there. These dates were subtracted from the year of our seed collections (2008) to produce values of population age used in subsequent analyses. Using herbaria records to assign population ages in this manner may not represent the true population age because of the time between population founding and the first records of the population. Nevertheless, C. solstitialis has a relatively well-documented invasion history in California (858 specimens on record, 577 records with GPS data, 61 records prior to 1930 in the Jepson Herbarium), and our population age estimates are in line with historical reconstructions of a general pattern of expansion out from initial establishment in the San Francisco Bay area first to the Central Valley and then to the North, East, and South (Gerlach 1997; DiTomaso et al. 2006; Pitcairn et al. 2006).
We also tested for the influence of the climatic environment on rarefied Ne in both invading and native populations. Increasingly severe droughts reduce fecundity and density in invading C. solstitialis populations (Sheley and Larson 1994; Swope and Parker 2012), implicating a role for climatic variation in demographic performance. To quantify the climatic gradients that might be most relevant to C. solstitialis ecology, we used principal component (PC) axes of climatic variation across C. solstitialis collection sites in North America and Europe, as previously identified by Dlugosch and colleagues (2015a; Supporting Information Fig. S1). This PCA was performed on CliMond variables at 18.5 × 18.5 km resolution (Kriticos et al. 2012), extracted from a spatially thinned set of occurrence records from western North America (185 records) and Eurasia (372 records). CliMond data were chosen for this analysis because they are available worldwide, and because they include variables that vary strongly across the range of C. solstitialis (particularly solar radiation;(Dlugosch et al. 2015a)). The full CliMond dataset (35 variables) included many strongly correlated climatic variables across the range of C. solstitialis, and these were reduced to seven representative variables (Supporting Information Fig. S1). The first two PC axes explained over 72% of variation in these variables (Dlugosch et al. 2015a). Larger values along the first PC climate axis (PC1) generally indicate sites with higher temperatures and lower seasonality in total solar radiation. Larger PC2 values indicate lower annual precipitation and greater seasonality in temperature. Greater seasonal variation in temperature has been shown to be related to ecologically important traits (plant size and drought tolerance) in C. solstitialis in both the native and invaded ranges (Dlugosch et al. 2015a).
To quantify the contributions of both population age and climatic environment to variation in rarified Ne for the invaded range, we used a general linear model with Ne as the dependent variable and population age, climate PC1, climate PC2, and their interactions as explanatory variables. We constructed a separate model of rarified Ne in native range populations using only PC1 and PC2 as variables, since no information about population age is available for the native range. We used model decomposition and F-scores to identify the best fit model. To explore the relative effect of each variable and their interactions on Ne, effect sizes were calculated as partial eta-squared values, which partition the total variance in a dependent variable among all independent variables (analogous to R2 in multiple regression), using the best fit linear model with the function ‘etasq’ in the R package ‘heplots’ version 1.3–1 (Fox et al. 2008). Partial-eta squared values are standardized for differences in magnitude of the independent variables. We also tested for an overall difference in the rarified Ne of invading and native populations using a Wilcoxon signed-rank test and a Monte Carlo exact test implemented with the ‘coin’ package in R (Hothorn et al. 2006). All statistical analyses were conducted in R version 3.4.1 (R Core Team 2017).
Results
Estimates of LD-Ne varied widely depending on which set of 20 loci were subsampled (Supporting Information Fig. S2). Distributions of subsampled LD-Ne estimates spanned at least four orders of magnitude within each population. Distributions peaked strongly around median estimates (Supporting Information Fig. S2). Median estimates of LD-Ne prior to rarefaction varied from 19.5 to 38.5 across the California invasion (Supporting Information Table S1). In general, estimates were higher in central and northern California and decreased to the East and South (Fig. 1). In native populations, median Ne estimates ranged from 16.2 to 42.7, with three populations with lowest LD-Ne located on the western side of the range in Spain (Fig. 1). Median estimates were consistently lower than estimates based on all sequences, and differed in rank order among populations (Supporting Information Table S1).
We found a strong association between median LD-Ne and the number of individuals sampled across our populations (r2adj=0.32, F1,17= 9.36, P=0.007). Rarefaction sampling produced positive relationships between LD-Ne and the number of individuals resampled within each population (Fig. 2). Slopes in the linear mixed model ranged from ~0.11 to 1.19. Importantly, rarefaction removed the significant effect of sampling effort on Ne values (rarefied Ne vs. total sample size; r2adj=0.12, F1,17=3.47, P=0.08). Rarefaction fits predicted a consistent rank order of LD-Ne among populations, with differences among populations being the smallest in magnitude at our minimum sampling of 10 individuals (Supporting Information Fig S3). Therefore, we expect our rarified Ne index to be conservative for tests of relationships between Ne and explanatory variables.
Fig 2.
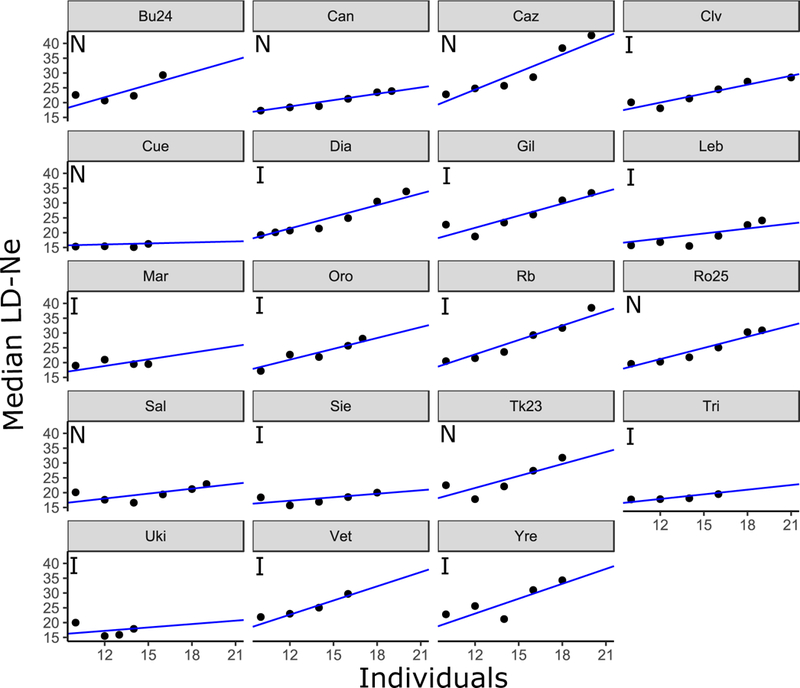
Relationships between median values of LD-Ne after locus resampling and the number of subsampled individuals for native (N) and invading (I) populations. Rarefaction was performed by linear mixed model with random slopes and intercepts.
Both climate and population age predicted rarified Ne in invading populations. The best fit linear model (Full model: r2adj=0.46, F(4,8)=4.09, P=0.0493) included significant, additive effects of population age and PC2 (Fig. 3; Table 1). Population age and PC2 were both positively correlated with rarified Ne values, indicating that Ne is largest in older populations and habitats with more temperature seasonality and lower precipitation. Age and PC2 were not significantly correlated (F1,10=2.42, P=0.15), and the model did not violate linear model assumptions of normality and no autocorrelation in the residuals (Supporting Information Fig. S4). PC2 had a greater influence on rarified Ne values than age, based on its larger standardized effect size (Table 1), although this difference was small. In contrast, rarified Ne of native range populations was not predicted by either climatic PC variable (Full model: r2adj=0.2314, F2,4=1.90, P=0.23) (Interactions: PC1: P=0.13, PC2: P=0.20).
Fig 3.
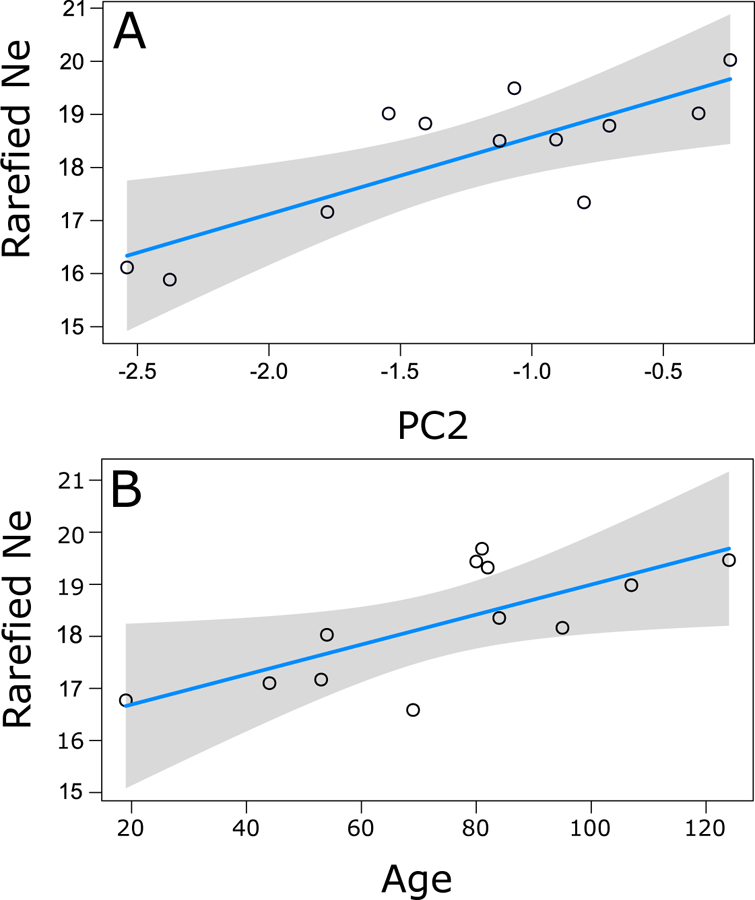
Rarefied Ne values are predicted by the second principal component (PC2) of climatic variability (A) and population age (B) in invading C. solstitialis populations. Rarefied Ne is positively correlated with PC2, for which larger values represent lower annual precipitation and greater seasonality in temperature (P=0.011), and with population age (P = 0.037). Lines show linear model fits and shading indicates the 95% confidence interval. Points represent partial residuals after accounting for other variables in the linear model.
Table 1.
Individual effects for the best fit linear model explaining rarefied effective population size (Ne) in invading populations of C. solstitialis, as a function of climatic principal component variables (PC1, PC2) and population age (Age).
| Invaded Range Populations | |||||
|---|---|---|---|---|---|
| Effect | Coefficient | Standard Error | t-value | p-value | Effect Size |
| PCA1 | 1.4503 | 0.4831 | −0.66 | 0.5278 | 0.0516 |
| PCA2 | −0.3187 | 0.4376 | 2.50 | 0.0106 | 0.5786 |
| Age | 0.0288 | 0.0115 | 3.31 | 0.0369 | 0.4387 |
Invading populations included a narrower range of rarified Ne values, nested within the distribution observed for native populations (Supporting Information Fig S5), and there was no significant difference between rarified Ne values in the native and invaded ranges (Wilcoxon signed rank test: W = 43, P = 0.97; Monte-Carlo one-way exact test: P = 0.93). Nucleotide diversity (π) also did not differ between the native and invaded ranges (r2adj=0.−0.04, F(3,15)=0.78, P=0.55; region term: t(2,15)=−1.5, P=0.15 ). There was no significant relationship between π and Ne in invading populations (r2adj=−0.037, F(1,10)=0.59, P=0.46) and there was a positive, marginally significant relationship between π and Ne in native populations (r2adj=0.43, F(1,5)=5.45, P=0.067), despite a smaller number of sampled populations from this range (Supporting Information Fig S6).
Discussion
Here we report empirical evidence for the joint effects of both range expansion and climatic environment on contemporary Ne in natural populations. We produced rarified estimates of LD-Ne across 12 populations in the invaded range of C. solstitialis and found a significant positive relationship between population age and Ne, a finding in line with theoretical expectations for the population genetics of expanding populations (Hallatschek et al. 2007; Excoffier & Ray 2008; Excoffier et al. 2009; Moreau et al. 2011; Lehe et al. 2012; Peischl et al. 2013; Peischl et al. 2015). We also found evidence that spatial variation in climatic conditions had a significant impact on Ne which was independent of population age. The effects of range expansion and climate were similar in magnitude in our study system, suggesting that both of these factors have been important for shaping evolutionary outcomes in invading populations (though their relative impact should be expected to vary across different scales of time and environment (e.g. the effect of age over time may diminish or the effect of climate may vary over both space and time) (Wegmann et al. 2006; Excoffier and Ray 2008; Gilbert et al. 2017).
We emphasize that our rarified LD-Ne values do not reflect a ‘true’ Ne value for the populations in our study. Rather, rarified estimates here represent relative values of Ne, and are useful for comparisons among populations. We expect asymptotic LD-Ne values for these populations to be larger, because we observed no asymptote with rarefaction for any of the populations in our study. However, our maximum estimates are similar to values reported in other plant and animal species using the same approach (e.g. Shrimpton & Heath 2003; Coyer et al. 2008; Wang et al. 2013; Álvarez et al. 2015). We note the LD-Ne estimation method itself also has a tendency to underestimate known values of Ne in simulations (Gilbert & Whitlock 2015), such that the true number of breeding individuals is likely higher than an asymptotic estimate.
Our resampling revealed that Ne estimates in C. solstitialis vary by at least four orders of magnitude when different sets of loci are used. This variation is expected given that particular sets of loci will capture different effects of physical linkage, history of selection, and chance sampling effects (Daly et al. 2001; Remington et al. 2001; Flint-Garcia et al. 2003). Resampling allowed us to leverage many combinations of loci across the genome to identify a well defined peak in the distribution of Ne estimates. A resampling approach is likely to be generally useful for RAD-seq and other popular methods used to generate genome-wide marker datasets, particularly where a complete reference genome is not available to determine the physical arrangement of loci.
After accounting for population and marker sampling, we found a significant effect of population age on differences in Ne across invading populations. Rarefied Ne estimates were lower in younger populations, which fits with expectations that a subset of individuals will contribute to range expansion (Excoffier & Ray 2008) and that genetic drift will be larger at the leading edge (Lehe et al. 2012; Peischl et al. 2013; Peischl et al. 2015). Estimates of contemporary Ne from C. solstitialis invading populations were within the distribution that we observed among native populations, which suggests that this species did not experience a large initial genetic bottleneck during its introduction to California, nor exceptionally low Ne during range expansion (relative to values observed in native populations). This lack of evidence for a strong genetic bottleneck is in line with models of historical demography by Barker and colleagues (2017), who inferred little reduction in Ne and maintenance of genetic diversity during the colonization of the Americas by C. solstitialis. In general, introduced species often lack strong genetic bottlenecks (Dlugosch & Parker 2008; Uller & Leimu 2011; Dlugosch et al. 2015b), and our results demonstrate that species which avoid genetic bottlenecks at introduction may still experience significant declines in Ne during range expansion. Importantly, invading populations of C. solstitialis in California are an order of magnitude higher in density than native populations (Uygur et al. 2004; Andonian et al. 2011), indicating that the fraction of the census population that is contributing to the evolutionary effective population in the invasion is much lower than in the native range.
We also observed an independent positive relationship between climatic PC2 and Ne of invading C. solstitialis populations, consistent with an impact of habitat suitability on Ne. High PC2 values reflect greater variation in seasonal temperatures and lower total annual precipitation, which typify areas of especially high C. solstitialis density in California (Dlugosch et al. 2015a). Previous studies in this system have proposed that C. solstitialis success stems from a lack of effective competitors in more drought prone habitats (Dlugosch et al. 2015a), due in part to the extensive conversion of these habitats to rangeland (Menke 1989; Stromberg and Griffin 1996). Other studies within the California invasion, however, have found that water availability (both naturally occurring and experimentally manipulated) is strongly and positively correlated with C. solstitialis density and fecundity (Enloe et al. 2004; Morghan & Rice 2006; Hulvey & Zavaleta 2012; Eskelinen & Harrison 2014), suggesting that fitness should be highest in wetter areas. Our results are most consistent with the landscape pattern of abundant C. solstitialis in drier areas, and might therefore reflect differences in human land use and the availability of native competitors across the invaded range. An underlying relationship between Ne and land use in the invasion could also explain why we did not find the same relationship with climate in the native range. Alternatively, native populations are more likely to be locally adapted, which could disrupt any relationships between climatic patterns, habitat quality, and Ne, particularly at the large geographic scale of our sampling in the native range.
Differences in rarified Ne among invading populations were not predicted by nucleotide diversity (π). Nonequilibrium populations such as the invasions here are unlikely to have had sufficient time to reach equilibrium diversity at a given Ne, and will also have been changing in size over time (Nei et al. 1975; Alcala et al. 2013; Epps & Keyghobadi 2015). Notably, we did find a marginally significant positive relationship between π and Ne in native range populations (despite a smaller population sample size), which have had more time to stabilize in population size and reach mutation-drift equilibrium. Moreover, rare alleles contribute important equilibrium genetic variation (Luikart et al. 1998) and native C. solstitialis populations have been previously shown to harbor more rare alleles than invading populations in North America (Barker et al. 2017). There is also a tendency for RAD-seq to underestimate π in more diverse genomes (Arnold et al. 2013; Cariou et al. 2016), although given the loss of rare alleles from invading populations, we might expect this to affect native populations more strongly than invading populations.
Our results support the prediction that both range expansion and habitat quality can increase the genetic drift experienced by leading edge populations. There is particular interest in whether these effects can hinder adaptation, slow further colonization, and establish static range boundaries (Bosshard et al. 2017; Lehe et al. 2012; Peischl et al. 2013; Peischl et al. 2015; Marculis et al. 2017; Birzu et al. 2018). Recent studies have demonstrated a link between differences in historical values of Ne and differences in efficacy of selection across species (e.g. (Slotte et al. 2010; Jensen & Bachtrog 2011; Strasburg et al. 2011), and both theoretical and experimental studies of bacteria have shown that the process of range expansion can reduce contemporary Ne and impose limits to adaptation and further colonization at the expansion front (Hallatschek & Nelson 2010; Lehe et al. 2012; Gralka et al. 2016; Peischl et al. 2013; Peischl et al. 2015). Natural populations of Arabidopsis lyrata demonstrate some of these effects, with greater genetic load in range edge populations associated with a lack of adaptation along an environmental cline (Willi et al. 2018). Limits to range expansion are expected to be sensitive to the specifics of evolutionary parameters in natural populations, including the magnitudes of Ne and selection, the amount and scale of gene flow across the expansion, and the genetic architecture of adaptive variation (Hallatschek & Nelson 2010; Lehe et al. 2012; Peischl et al. 2013; Peischl et al. 2015; Gralka et al. 2016).
The expansion ecology of C. solstitialis in California does not support the existence of maladapted edge populations. Populations of C. solstitialis close to the range edge can achieve higher densities than older, more interior populations (Swope et al. 2017), which runs counter to expectations of high genetic load. Additionally, evolution of increased growth and earlier flowering appears to be enhancing the invasiveness of C. solstitialis (Dlugosch et al. 2015a), suggesting that reduced Ne at the range edge has not created a barrier to adaptation and further expansion. Additional studies are needed to test for quantitative connections between expansion dynamics and the role of adaptation in this system, including detailed analyses of dispersal patterns (included biased dispersal of particular phenotypes, (Shine et al. 2011), trait and fitness differences, and demographic performance across populations. The availability of adaptive variation and the degree to which this is a limiting factor in species invasions is an active area of debate (Ellstrand & Schierenbeck 2000; Rius & Darling 2014; Bock et al. 2015), and should be particularly relevant to the colonization of habitats requiring significant niche evolution. The results reported here emphasize that an understanding of the evolutionary mechanisms that generate boundaries to range expansion in natural populations will require evaluating evidence not only for the availability of adaptive variation (Dlugosch et al. 2015a), but also for an effective response to selection.
Supplementary Material
Acknowledgements
We thank five reviewers for helpful comments on previous versions of this manuscript, and MS Barker for computational assistance. This study was supported by a USDA ELI Fellowship #2017–67011-26034 to JB, the National Institute of General Medical Sciences of the NIH under Award #K12GM000708 through the University of Arizona Center for Insect Science to BSB, USDA grant #2015–67013-23000 and NSF #1750280 to KMD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. All authors declare no conflict of interests.
Footnotes
Data Accessibility
• Genomic data used for this project are available at the NCBI sequence read archive (BioProject for C. solstitialis: PRJNA275986).
• Individual effective population size estimates from locus resampling and rarefaction runs are available at Dryad (doi:10.5061/dryad.5p26rh4).
References
- Alcala N, Streit D, Goudet J, & Vuilleumier S (2013). Peak and persistent excess of genetic diversity following an abrupt migration increase. Genetics, 193(3), 953–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf FW (1986). Genetic drift and the loss of alleles versus heterozygosity. Zoo Biology, 5(2), 181–190. [Google Scholar]
- Allendorf FW, & Lundquist LL (2003). Introduction: Population Biology, Evolution, and Control of Invasive Species. Conservation Biology, 17(1), 24–30. [Google Scholar]
- Álvarez D, Lourenço A, Oro D, & Velo-Antón G (2015). Assessment of census (N) and effective population size (Ne) reveals consistency of Ne single-sample estimators and a high Ne/N ratio in an urban and isolated population of fire salamanders. Conservation Genetics Resources, 7(3), 705–712. [Google Scholar]
- Andonian K, Hierro JL, Khetsuriani L, Becerra P, Janoyan G, Villarreal D, … Callaway RM (2011). Range-expanding populations of a globally introduced weed experience negative plant-soil feedbacks. PloS One, 6(5), e20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold B, Corbett-Detig RB, Hartl D, & Bomblies K (2013). RAD seq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Molecular Ecology, 22(11), 3179–3190. [DOI] [PubMed] [Google Scholar]
- Aslan CE, Hufford MB, Epanchin-Niell RS, Port JD, Sexton JP, & Waring TM (2009). Practical Challenges in Private Stewardship of Rangeland Ecosystems: Yellow Starthistle Control in Sierra Nevadan Foothills. Rangeland Ecology & Management, 62(1), 28–37. [Google Scholar]
- Atwater DZ, Ervine C, & Barney JN (2018). Climatic niche shifts are common in introduced plants. Nature Ecology & Evolution, 2(1), 34–43. [DOI] [PubMed] [Google Scholar]
- Bancheva S, & Greilhuber J (2006). Genome size in Bulgarian Centaurea s.l. (Asteraceae). Plant Systematics and Evolution, 257(1), 95–117. [Google Scholar]
- Barker BS, Andonian K, Swope SM, Luster DG, & Dlugosch KM (2017). Population genomic analyses reveal a history of range expansion and trait evolution across the native and invaded range of yellow starthistle (Centaurea solstitialis). Molecular Ecology, 26(4), 1131–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Maechler M, Bolker B, & Walker S (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software, 67(1), 1–48. http:doi://10.18637/jss.v067.i01 [Google Scholar]
- Benefield CB, DiTomaso JM, Kyser GB, & Tschohl A (2001). Reproductive biology of yellow starthistle: maximizing late-season control. Weed Science, 49(1), 83–90. [Google Scholar]
- Birzu G, Hallatschek O, & Korolev KS (2018).. Proceedings of the National Academy of Sciences of the United States of America, 115(16), E3645–E3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock DG, Caseys C, Cousens RD, Hahn MA, Heredia SM, Hübner S, … Rieseberg LH (2015). What we still don’t know about invasion genetics. Molecular Ecology, 24(9), 2277–2297. [DOI] [PubMed] [Google Scholar]
- Bosshard L, Dupanloup I, Tenaillon O, Bruggmann R, Ackermann M, Peischl S, & Excoffier L (2017). Accumulation of Deleterious Mutations During Bacterial Range Expansions. Genetics, 207(2), 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch J, Barker BS, Dlugosch KM, (2019) Data from: Expansion history and environmental suitability shape effective population size in a plant invasion. Dryad Digital Repository doi: 10.5061/dryad.5p26rh4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callihan RH, Prather TS, & Northam FE (1993). Longevity of Yellow Starthistle (Centaurea solstitialis) Achenes in Soil. Weed Technology, 7(1), 33–35. [Google Scholar]
- Cariou M, Duret L, & Charlat S (2016). How and how much does RAD-seq bias genetic diversity estimates? BMC Evolutionary Biology, 16(1), 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, & Postlethwait JH (2011). Stacks: building and genotyping Loci de novo from short-read sequences. G3, 1(3), 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Hohenlohe PA, Bernatchez L, Funk WC, Andrews KR, & Allendorf FW (2017). Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Molecular Ecology Resources, 17(3), 362–365. [DOI] [PubMed] [Google Scholar]
- Charlesworth B (2009). Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics, 10(3), 195–205. [DOI] [PubMed] [Google Scholar]
- Chuang A, & Peterson CR (2016). Expanding population edges: theories, traits, and trade-offs. Global Change Biology, 22(2), 494–512. [DOI] [PubMed] [Google Scholar]
- Colautti RI, Alexander JM, Dlugosch KM, Keller SR, & Sultan SE (2017). Invasions and extinctions through the looking glass of evolutionary ecology. Philosophical Transactions of the Royal Society of London. Series B Biological Sciences, 372(1712). http://doi:10.1098/rstb.2016.0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colautti RI, & Barrett SCH (2011). Population divergence along lines of genetic variance and covariance in the invasive plant Lythrum salicaria in eastern North America. Evolution, 65(9), 2514–2529. [DOI] [PubMed] [Google Scholar]
- Colautti RI, & Lau JA (2015). Contemporary evolution during invasion: evidence for differentiation, natural selection, and local adaptation. Molecular Ecology, 24(9), 1999–2017. [DOI] [PubMed] [Google Scholar]
- Coyer JA, Hoarau G, Sjøtun K, & Olsen JL (2008). Being abundant is not enough: a decrease in effective population size over eight generations in a Norwegian population of the seaweed, Fucus serratus. Biology Letters, 4(6), 755–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF, & Morton NE (1955). Measurement of Gene Frequency Drift in Small Populations. Evolution, 9(2), 202–214. [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, & Lander ES (2001). High-resolution haplotype structure in the human genome. Nature Genetics, 29, 229. [DOI] [PubMed] [Google Scholar]
- DiTomaso JM, Kyser GB, & Pitcairn MJ (2006). Yellow starthistle management guide California Invasive Plant Council Berkeley, CA. [Google Scholar]
- Dlugosch KM, Cang FA, Barker BS, Andonian K, Swope SM, & Rieseberg LH (2015a). Evolution of invasiveness through increased resource use in a vacant niche. Nature Plants, 1 http://doi:10.1038/nplants.2015.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosch KM, Anderson SR, Braasch J, Cang FA, & Gillette HD (2015b). The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Molecular Ecology, 24(9), 2095–2111. [DOI] [PubMed] [Google Scholar]
- Dlugosch KM, Lai Z, Bonin A, Hierro J, & Rieseberg LH (2013). Allele identification for transcriptome-based population genomics in the invasive plant Centaurea solstitialis. G3, 3(2), 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosch KM, & Parker IM (2008a). Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology, 17(1), 431–449. [DOI] [PubMed] [Google Scholar]
- Dlugosch KM, & Parker IM (2008b). Invading populations of an ornamental shrub show rapid life history evolution despite genetic bottlenecks. Ecology Letters, 11(7), 701–709. [DOI] [PubMed] [Google Scholar]
- Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, & Ovenden JR (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources, 14(1), 209–214. [DOI] [PubMed] [Google Scholar]
- Eckert CG, Samis KE, & Lougheed SC (2008). Genetic variation across species’ geographical ranges: the central--marginal hypothesis and beyond. Molecular Ecology, 17(5), 1170–1188. [DOI] [PubMed] [Google Scholar]
- Elam DR, Ridley CE, Goodell K, & Ellstrand NC (2007). Population size and relatedness affect fitness of a self-incompatible invasive plant. Proceedings of the National Academy of Sciences of the United States of America, 104(2), 549–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellstrand NC, & Schierenbeck KA (2000). Hybridization as a stimulus for the evolution of invasiveness in plants? Proceedings of the National Academy of Sciences of the United States of America, 97(13), 7043–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England PR, Cornuet J-M, Berthier P, Tallmon DA, & Luikart G (2006). Estimating effective population size from linkage disequilibrium: severe bias in small samples. Conservation Genetics, 7(2), 303. [Google Scholar]
- Enloe SF, DiTomaso JM, Orloff SB, & Drake DJ (2004). Soil water dynamics differ among rangeland plant communities dominated by yellow starthistle (Centaurea solstitialis), annual grasses, or perennial grasses. Weed Science, 52(6), 929–935. [Google Scholar]
- Epps CW, & Keyghobadi N (2015). Landscape genetics in a changing world: disentangling historical and contemporary influences and inferring change. Molecular Ecology, 24(24), 6021–6040. [DOI] [PubMed] [Google Scholar]
- Eriksen RL, Desronvil T, Hierro JL, & Kesseli R (2012). Morphological differentiation in a common garden experiment among native and non-native specimens of the invasive weed yellow starthistle (Centaurea solstitialis). Biological Invasions, 14(7), 1459–1467. [Google Scholar]
- Eskelinen A, & Harrison S (2014). Exotic plant invasions under enhanced rainfall are constrained by soil nutrients and competition. Ecology, 95(3), 682–692. [DOI] [PubMed] [Google Scholar]
- Excoffier L (2004). Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Molecular Ecology, 13(4), 853–864. [DOI] [PubMed] [Google Scholar]
- Excoffier L, & Ray N (2008). Surfing during population expansions promotes genetic revolutions and structuration. Trends in Ecology & Evolution, 23(7), 347–351. [DOI] [PubMed] [Google Scholar]
- Flint-Garcia SA, Thornsberry JM, & Buckler ES 4th. (2003). Structure of linkage disequilibrium in plants. Annual Review of Plant Biology, 54, 357–374. [DOI] [PubMed] [Google Scholar]
- Fox J, Friendly M, & Monette G (2008). Visualizing hypothesis tests in multivariate linear models: the heplots package for R. Computational Statistics, 24(2), 233–246. [Google Scholar]
- Frankham R (1995). Effective population size/adult population size ratios in wildlife: a review. Genetical Research, 89(5–6), 491–503. [DOI] [PubMed] [Google Scholar]
- Frankham R (1996). Relationship of Genetic Variation to Population Size in Wildlife. Conservation Biology, 10(6), 1500–1508. [Google Scholar]
- Gaut BS, Wright SI, Rizzon C, Dvorak J, & Anderson LK (2007). Recombination: an underappreciated factor in the evolution of plant genomes. Nature Reviews. Genetics, 8(1), 77–84. [DOI] [PubMed] [Google Scholar]
- Gerlach JD Jr. (1997). The introduction, dynamics of geographic range expansion, and ecosystem effects of yellow starthistle (Centaurea solstitialis). In Proceedings of the California Weed Science Society (Vol. 49, pp. 136–141). [Google Scholar]
- Gilbert KJ, Sharp NP, Angert AL, Conte GL, Draghi JA, Guillaume F, … Whitlock MC (2017). Local Adaptation Interacts with Expansion Load during Range Expansion: Maladaptation Reduces Expansion Load. The American Naturalist, 189(4), 368–380. [DOI] [PubMed] [Google Scholar]
- Gilbert KJ, & Whitlock MC (2015). Evaluating methods for estimating local effective population size with and without migration. Evolution, 69(8), 2154–2166. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Martinez SC, Ridout K, & Pannell JR (2017). Range Expansion Compromises Adaptive Evolution in an Outcrossing Plant. Current Biology, 27(16), 2544–2551. [DOI] [PubMed] [Google Scholar]
- Graciá E, Botella F, Anadón JD, Edelaar P, Harris DJ, & Giménez A (2013). Surfing in tortoises? Empirical signs of genetic structuring owing to range expansion. Biology Letters, 9(3), 20121091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralka M, Stiewe F, Farrell F, Möbius W, Waclaw B, & Hallatschek O (2016). Allele surfing promotes microbial adaptation from standing variation. Ecology Letters, 19(8), 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith TM, & Watson MA (2006). Is evolution necessary for range expansion? Manipulating reproductive timing of a weedy annual transplanted beyond its range. The American Naturalist, 167(2), 153–164. [DOI] [PubMed] [Google Scholar]
- Hallatschek O, Hersen P, Ramanathan S, & Nelson DR (2007). Genetic drift at expanding frontiers promotes gene segregation. Proceedings of the National Academy of Sciences of the United States of America, 104(50), 19926–19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallatschek O, & Nelson DR (2010). Life at the front of an expanding population. Evolution, 64(1), 193–206. [DOI] [PubMed] [Google Scholar]
- Hamilton JA, Okada M, Korves T, & Schmitt J (2015). The role of climate adaptation in colonization success in Arabidopsis thaliana. Molecular Ecology, 24(9), 2253–2263. [DOI] [PubMed] [Google Scholar]
- Henry RC, Bartoń KA, Travis JMJ (2015) Mutation accumulation and the formation of range limits. Biology letters, 11, 20140871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierro JL, Eren Ö, Villarreal D, & Chiuffo MC (2013). Non-native conditions favor non-native populations of invasive plant: demographic consequences of seed size variation? Oikos , 122(4), 583–590. [Google Scholar]
- Hohenlohe PA, Amish SJ, Catchen JM, Allendorf FW, & Luikart G (2011). Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Molecular Ecology Resources, 11(s1), 117–122. [DOI] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, van de Wiel MA, & Zeileis A (2006). A Lego System for Conditional Inference. The American Statistician, 60(3), 257–263. [Google Scholar]
- Hulvey KB, & Zavaleta ES (2012). Abundance declines of a native forb have nonlinear impacts on grassland invasion resistance. Ecology, 93(2), 378–388. [DOI] [PubMed] [Google Scholar]
- Jensen JD, & Bachtrog D (2011). Characterizing the influence of effective population size on the rate of adaptation: Gillespie’s Darwin domain. Genome Biology and Evolution, 3, 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joley DB, Maddox DM, Supkoff DM, & Mayfield A (1992). Dynamics of Yellow Starthistle (Centaurea solstitialis) Achenes in Field and Laboratory. Weed Science, 40(2), 190–194. [Google Scholar]
- Kimura M (1964). Diffusion Models in Population Genetics. Journal of Applied Probability, 1(2), 177–232. [Google Scholar]
- Kimura M, & Crow JF (1963). The Measurement of Effective Population Number. Evolution, 17(3), 279–288. [Google Scholar]
- Kirkpatrick M, & Barton NH (1997). Evolution of a species’ range. The American Naturalist, 150(1), 1–23. [DOI] [PubMed] [Google Scholar]
- Kriticos DJ, Webber BL, Leriche A, Ota N, Macadam I, Bathols J, & Scott JK (2012). CliMond: global high-resolution historical and future scenario climate surfaces for bioclimatic modelling. Methods in Ecology and Evolution, 3(1), 53–64. [Google Scholar]
- Langley CH, Tobari YN, & Kojima KI (1974). Linkage disequilibrium in natural populations of Drosophila melanogaster. Genetics, 78(3), 921–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corre V, & Kremer A (1998). Cumulative effects of founding events during colonisation on genetic diversity and differentiation in an island and stepping-stone model. Journal of Evolutionary Biology, 11(4), 495–512. [Google Scholar]
- Lehe R, Hallatschek O, & Peliti L (2012). The rate of beneficial mutations surfing on the wave of a range expansion. PLoS Computational Biology, 8(3), e1002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnen CR, Kingsley EP, Jensen JD, & Hoekstra HE (2009). On the origin and spread of an adaptive allele in deer mice. Science, 325(5944), 1095–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-M, She D-Y, Zhang D-Y, & Liao W-J (2015). Life history trait differentiation and local adaptation in invasive populations of Ambrosia artemisiifolia in China. Oecologia, 177(3), 669–677. [DOI] [PubMed] [Google Scholar]
- Luikart G, Allendorf FW, Cornuet JM, & Sherwin WB (1998). Distortion of allele frequency distributions provides a test for recent population bottlenecks. The Journal of Heredity, 89(3), 238–247. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery J, & Burger R (1995). Mutation Accumulation and the Extinction of Small Populations. The American Naturalist, 146(4), 489–518. [Google Scholar]
- Marculis NG, Lui R, & Lewis MA (2017). Neutral Genetic Patterns for Expanding Populations with Nonoverlapping Generations. Bulletin of Mathematical Biology, 79(4), 828–852. [DOI] [PubMed] [Google Scholar]
- Matzek V, & Hill S a. n. n. (2012). Response of Biomass and Seedbanks of Rangeland Functional Groups to Mechanical Control of Yellow Starthistle. Rangeland Ecology & Management, 65(1), 96–100. [Google Scholar]
- Mayr E(1963). Animal species and evolution (Vol. 797). Belknap Press of Harvard University Press Cambridge, Massachusetts. [Google Scholar]
- Menke JW (1989). Management Controls on Productivity. In Huenneke LF & Mooney HA (Eds.), Grassland structure and function: California annual grassland (pp. 173–199). Dordrecht: Springer Netherlands. [Google Scholar]
- Micheletti SJ, & Storfer A (2015). A test of the central–marginal hypothesis using population genetics and ecological niche modelling in an endemic salamander (Ambystoma barbouri). Molecular Ecology, 24(5), 967–979. [DOI] [PubMed] [Google Scholar]
- Moreau C, Bhérer C, Vézina H, Jomphe M, Labuda D, & Excoffier L (2011). Deep human genealogies reveal a selective advantage to be on an expanding wave front. Science, 334(6059), 1148–1150. [DOI] [PubMed] [Google Scholar]
- Narum SR, Buerkle CA, Davey JW, Miller MR, & Hohenlohe PA (2013). Genotyping-by-sequencing in ecological and conservation genomics. Molecular Ecology, 22(11), 2841–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Maruyama T, & Chakraborty R (1975). The bottleneck effect and genetic variability in populations. Evolution, 29(1), 1–10. [DOI] [PubMed] [Google Scholar]
- Nei M, & Li WH (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences of the United States of America, 76(10), 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, … Weigel D (2002). The extent of linkage disequilibrium in Arabidopsis thaliana. Nature Genetics, 30(2), 190–193. [DOI] [PubMed] [Google Scholar]
- Ohta T (1992). The Nearly Neutral Theory of Molecular Evolution. Annual Review of Ecology and Systematics, 23, 263–286. [Google Scholar]
- Palstra FP, & Ruzzante DE (2008). Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Molecular Ecology, 17(15), 3428–3447. [DOI] [PubMed] [Google Scholar]
- Peischl S, Dupanloup I, Kirkpatrick M, & Excoffier L (2013). On the accumulation of deleterious mutations during range expansions. Molecular Ecology, 22(24), 5972–5982. [DOI] [PubMed] [Google Scholar]
- Peischl S, Dupanloup I, Foucal A et al. (2018) Relaxed Selection During a Recent Human Expansion. Genetics, 208, 763–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peischl S, Kirkpatrick M, & Excoffier L (2015). Expansion load and the evolutionary dynamics of a species range. The American Naturalist, 185(4), E81–93. [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, & Hoekstra HE (2012). Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PloS One, 7(5), e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitpierre B, Kueffer C, Broennimann O, Randin C, Daehler C, & Guisan A (2012). Climatic niche shifts are rare among terrestrial plant invaders. Science, 335(6074), 1344–1348. [DOI] [PubMed] [Google Scholar]
- Phillips BL (2012). Range shift promotes the formation of stable range edges. Journal of Biogeography, 39(1), 153–161. [Google Scholar]
- Pierce AA, Zalucki MP, Bangura M, Udawatta M, Kronforst MR, Altizer S, … de Roode JC (2014). Serial founder effects and genetic differentiation during worldwide range expansion of monarch butterflies. Proceedings of the Royal Society of London B: Biological Sciences, 281(1797). http://doi:10.1098/rspb.2014.2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pironon S, Villellas J, Morris WF, Doak DF, & García MB (2015). Do geographic, climatic or historical ranges differentiate the performance of central versus peripheral populations? Global Ecology and Biogeography, 24(6), 611–620. [Google Scholar]
- Pitcairn M, Schoenig S, Yacoub R, & Gendron J (2006). Yellow starthistle continues its spread in California. California Agriculture, 60(2), 83–90. [Google Scholar]
- Polechova J (2018) Is the sky the limit? On the expansion threshold of a species’ range. PLoS biology, 16(6): e2005372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polechová J, Barton NH (2015) Limits to adaptation along environmental gradients. Proceedings of the National Academy of Sciences of the United States of America, 112(20), 6401–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2017. R: a language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria: http://www.r-project.org. [Google Scholar]
- Ramakrishnan AP, Musial T, & Cruzan MB (2010). Shifting dispersal modes at an expanding species’ range margin. Molecular Ecology, 19(6), 1134–1146. [DOI] [PubMed] [Google Scholar]
- Reever Morghan KJR, & Rice KJ (2006). Variation in resource availability changes the impact of invasive thistles on native bunchgrasses. Ecological Applications, 16(2), 528–539. [DOI] [PubMed] [Google Scholar]
- Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, … Buckler ES 4th. (2001). Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proceedings of the National Academy of Sciences of the United States of America, 98(20), 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice KJ, & Mack RN (1991). Ecological genetics of Bromus tectorum. Oecologia, 88(1), 91–101. [DOI] [PubMed] [Google Scholar]
- Rius M, & Darling JA (2014). How important is intraspecific genetic admixture to the success of colonising populations? Trends in Ecology & Evolution, 29(4), 233–242. [DOI] [PubMed] [Google Scholar]
- Robertson A (1960). A Theory of Limits in Artificial Selection. Proceedings of the Royal Society of London B: Biological Sciences, 153(951), 234–249. [Google Scholar]
- Roche BF Jr. (1991). Achene Dispersal in Yellow Starthistle (Centaurea solstitialis L.). Northwestern Science, 66, 62–65. [Google Scholar]
- Sagarin RD, & Gaines SD (2002). The “abundant centre” distribution: to what extent is it a biogeographical rule? Ecology Letters, 5(1), 137–147. [Google Scholar]
- Sexton JP, McIntyre PJ, Angert AL, & Rice KJ (2009). Evolution and Ecology of Species Range Limits. Annual Review of Ecology, Evolution, and Systematics, 40, 415–436. [Google Scholar]
- Sheley RL, & Larson LL (1994). Observation: Comparative Live-History of Cheat-Grass and Yellow Starthistle. Journal of Range Management, 47(6), 450–456. [Google Scholar]
- Shine R, Brown GP, & Phillips BL (2011). An evolutionary process that assembles phenotypes through space rather than through time. Proceedings of the National Academy of Sciences of the United States of America, 108(14), 5708–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimpton JM, & Heath DD (2003). Census vs. effective population size in chinook salmon: large‐and small‐scale environmental perturbation effects. Molecular Ecology, 12(10), 2571–2583. [DOI] [PubMed] [Google Scholar]
- Slatkin M, & Excoffier L (2012). Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics, 191(1), 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte T, Foxe JP, Hazzouri KM, & Wright SI (2010). Genome-wide evidence for efficient positive and purifying selection in Capsella grandiflora, a plant species with a large effective population size. Molecular Biology and Evolution, 27(8), 1813–1821. [DOI] [PubMed] [Google Scholar]
- Strasburg JL, Kane NC, Raduski AR, Bonin A, Michelmore R, & Rieseberg LH (2011). Effective population size is positively correlated with levels of adaptive divergence among annual sunflowers. Molecular Biology and Evolution, 28(5), 1569–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromberg MR, & Griffin JR (1996). Long-term patterns in coastal California grasslands in relation to cultivation, gophers, and grazing. Ecological Applications, 6(4), 1189–1211. [Google Scholar]
- Swope SM, & Parker IM (2010). Widespread seed limitation affects plant density but not population trajectory in the invasive plant Centaurea solstitialis. Oecologia, 164(1), 117–128. [DOI] [PubMed] [Google Scholar]
- Swope SM, & Parker IM (2012). Complex interactions among biocontrol agents, pollinators, and an invasive weed: a structural equation modeling approach. Ecological Applications, 22(8), 2122–2134. [DOI] [PubMed] [Google Scholar]
- Swope SM, Satterthwaite WH, & Parker IM (2017). Spatiotemporal variation in the strength of density dependence: implications for biocontrol of Centaurea solstitialis. Biological Invasions, 19(9), 2675–2691. [Google Scholar]
- Sun M, & Ritland K (1998). Mating system of yellow starthistle (Centaurea solstitialis), a successful colonizer in North America. Heredity, 80, 225. [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, & Lynch M (2012). Drift-barrier hypothesis and mutation-rate evolution. Proceedings of the National Academy of Sciences of the United States of America, 109(45), 18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szűcs M, Melbourne BA, Tuff T, Weiss-Lehman C, & Hufbauer RA (2017a). Genetic and demographic founder effects have long-term fitness consequences for colonising populations. Ecology Letters, 20(4), 436–444. [DOI] [PubMed] [Google Scholar]
- Szűcs M, Vahsen ML, Melbourne BA, Hoover C, Weiss-Lehman C, & Hufbauer RA (2017b). Rapid adaptive evolution in novel environments acts as an architect of population range expansion. Proceedings of the National Academy of Sciences of the United States of America, 114(51), 13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley R, Vallinoto M, Sequeira F, & Kearney MR (2014). Realized niche shift during a global biological invasion. Proceedings of the National Academy of Sciences of the United States of America, 111(28), 10233–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis JMJ, Münkemüller T, Burton OJ et al. (2007) Deleterious mutations can surf to high densities on the wave front of an expanding population. Molecular biology and evolution, 24(10), 2334–2343. [DOI] [PubMed] [Google Scholar]
- Turner TF, Wares JP, & Gold JR (2002). Genetic effective size is three orders of magnitude smaller than adult census size in an abundant, Estuarine-dependent marine fish (Sciaenops ocellatus). Genetics, 162(3), 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uller T, & Leimu R (2011). Founder events predict changes in genetic diversity during human-mediated range expansions. Global Change Biology, 17(11), 3478–3485. [Google Scholar]
- Uygur S, Smith L, Uygur FN, Cristofaro M, & Balciunas J (2004). Population densities of yellow starthistle (Centaurea solstitialis) in Turkey. Weed Science, 52(5), 746–753. [Google Scholar]
- Varvio SL, Chakraborty R, & Nei M (1986). Genetic variation in subdivided populations and conservation genetics. Heredity, 57, 189–198. [DOI] [PubMed] [Google Scholar]
- Vandepitte K, de Meyer T, Helsen K, van Acker K, Roldán-Ruiz I, Mergeay J, & Honnay O (2014). Rapid genetic adaptation precedes the spread of an exotic plant species. Molecular Ecology, 23(9), 2157–2164. [DOI] [PubMed] [Google Scholar]
- Wang J (2016). A comparison of single-sample estimators of effective population sizes from genetic marker data. Molecular Ecology, 25(19), 4692–4711. [DOI] [PubMed] [Google Scholar]
- Wang X-Q, Huang Y, & Long C-L (2013). Assessing the genetic consequences of flower-harvesting in Rhododendron decorum Franchet (Ericaceae) using microsatellite markers. Biochemical Systematics and Ecology, 50, 296–303. [Google Scholar]
- Waples RS (2006). A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conservation Genetics , 7(2), 167–184. [Google Scholar]
- Waples RS (2016). Making sense of genetic estimates of effective population size. Molecular Ecology, 25(19), 4689–4691. [DOI] [PubMed] [Google Scholar]
- Waples RS, & Do C (2008). ldne: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources, 8(4), 753–756. [DOI] [PubMed] [Google Scholar]
- Webb DM, & Knapp SJ (1990). DNA extraction from a previously recalcitrant plant genus. Plant Molecular Biology Reporter, 8(3), 180–185. [Google Scholar]
- Welles SR, & Dlugosch KM (2018). Population Genomics of Colonization and Invasion (Vol. 17, p. 24). Cham: Springer International Publishing. [Google Scholar]
- White TA, Perkins SE, Heckel G, & Searle JB (2013). Adaptive evolution during an ongoing range expansion: the invasive bank vole (Myodes glareolus) in Ireland. Molecular Ecology, 22(11), 2971–2985. [DOI] [PubMed] [Google Scholar]
- Widmer TL, Guermache F, Dolgovskaia MY, & Reznik SY (2007). Enhanced Growth and Seed Properties in Introduced Vs. Native Populations of Yellow Starthistle (Centaurea Solstitialis). Weed Science, 55(5), 465–473. [Google Scholar]
- Willi Y, Fracassetti M, Zoller S, & Van Buskirk J (2018). Accumulation of mutational load at the edges of a species range. Molecular Biology and Evolution doi: 10.1093/molbev/msy003 [DOI] [PubMed] [Google Scholar]
- Wootton JT, & Pfister CA (2015). Processes affecting extinction risk in the laboratory and in nature. Proceedings of the National Academy of Sciences of the United States of America, 112(44), E5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S (1931). Evolution in Mendelian Populations. Genetics, 16(2), 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S (1938). Size of population and breeding structure in relation to evolution. Science, 87(2263), 430–431. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.