

Abstract
Ewing sarcoma (ES) are aggressive pediatric bone and soft tissue tumors driven by EWS-ETS fusion oncogenes, most commonly EWS-FLI1. Treatment of ES patients consists of up to 9 months of alternating courses of 2 chemotherapeutic regimens. Furthermore, EWS-ETS-targeted therapies have yet to demonstrate clinical benefit, thereby emphasizing a clinical responsibility to search for new therapeutic approaches. Our previous in silico drug screening identified entinostat as a drug hit that was predicted to reverse the ES disease signatures and EWS-FLI1- mediated gene signatures. Here, we establish preclinical proof of principle by investigating the in vitro and in vivo efficacy of entinostat in preclinical ES models, as well as characterizing the mechanisms of action and in vivo pharmacokinetics of entinostat. ES cells are preferentially sensitive to entinostat in an EWS-FLI1 or EWS-ERG- dependent manner. Entinostat induces apoptosis of ES cells through G0/G1 cell cycle arrest, intracellular reactive oxygen species (ROS) elevation, DNA damage, homologous recombination (HR) repair impairment and caspase activation. Mechanistically, we demonstrate for the first time that HDAC3 is a transcriptional target of EWS-FLI1 and that entinostat inhibits growth of ES cells through suppressing a previously unexplored EWS-FLI1/HDAC3/HSP90 signaling axis. Importantly, entinostat significantly reduces tumor burden by 97.4% (89.5 mm3 vs. 3,397.3 mm3 of vehicle, p < 0.001) and prolongs the median survival of mice (15.5 days vs. 8.5 days of vehicle, p < 0.001), in two independent ES xenograft mouse models, respectively. Overall, our studies demonstrate promising activity of entinostat against ES, and support the clinical development of the entinostat-based therapies for children and young adults with metastatic/relapsed ES.
Keywords: Ewing sarcoma, entinostat, histone deacetylase inhibitor, EWS-FLI1, HDAC3
Introduction
Ewing sarcoma (ES), the second most common bone and soft tissue cancer in pediatric patients in the United States [1], is a highly lethal malignancy. The 5-year event-free survival for localized ES is between 60% to 80% [2], but this falls to less than < 20% in patients with metastatic or relapsed disease [3]. Current chemotherapy regimens incorporate vincristine, doxorubicin, cyclophosphamide, ifosfamide and etoposide in compressed cycles [4]. These drugs are conventional non-targeted cytotoxic agents and their use is often limited by severe side effects as well as serious late effects in survivors that can include secondary leukemias, renal toxicity, and cardiac failure [5]. Thus, new targeted therapeutic approaches are needed to expand treatment options and to improve the outcomes for patients with ES, especially for those with metastatic or recurrent disease.
More than 85% of ES cases are characterized by the chromosomal translocation t(11;22)(q24;q12) leading to the EWS-FLI1 fusion gene [6]. Other translocations, which fuse EWS gene to different ETS family members (e.g., ERG, ETV1, E1AF, FEV), have also been described in ES and account for 5–15% of all cases. EWS-FLI1 functions as an aberrant transcription factor that drives the malignant transformation and disease progression [6, 7]. Currently, only one small molecule compound, TK216 (Oncternal Therapeutics, Inc) that inhibits the biological activity of the ETS-family transcription factor oncoproteins in a variety of tumor types is in Phase 1 clinical trial (NCT02657005) for patients with relapsed or refractory ES. Several other compounds which indirectly inhibit EWS-FLI1 through alteration of upstream/downstream pathways have demonstrated to date limited clinical benefit [8–12]. Furthermore, developing drugs that specifically target the family of EWS-ETS oncoproteins, such as EWS-FLI1, has proven very challenging due to the lack of the enzymatic activity and its biochemical makeup as an intrinsically disordered protein [13]. Therefore, as an alternative approach, we employed integrated bioinformatics combined with high-throughput screening to search for clinically relevant and well tolerated drugs (FDA-approved or in clinical trials) that can reverse ES disease signatures and/or EWS-FLI1 -mediated gene signatures on a genome-wide scale as novel chemotherapeutic options for the treatment of ES [14].
Among the drug hits identified from in silico screening, entinostat (MS-275, SNDX-275) was predicted to reverse the ES disease signatures and EWS-FLI1-mediated gene signatures. Entinostat is an oral class I histone deacetylase (HDAC) inhibitor developed by Syndax Pharmaceuticals, which selectively inhibits HDAC1 and HDAC3. Entinostat has shown marked anti-tumor activity against a variety of solid and hematological malignancies in preclinical models [15–17]. More importantly, considerable data have demonstrated that entinostat has a reasonable safety profile and promising efficacy in patients with leukemia, lymphoma, melanoma, prostate cancer, renal cancer, non-small cell lung cancer and breast cancer, either alone or in combination with other therapies [15, 18–20]. Relevant to our studies, a recent Phase 1 study of entinostat as a monotherapy to establish dosing and safety is being evaluated in pediatric patients with recurrent or refractory solid tumors (NCT02780804), including central nervous system (CNS) tumor and lymphoma, but not for ES or other pediatric sarcoma patients.
To date, the regulation of entinostat on the oncogenic driver EWS-FLI1 and the downstream targets/effectors of EWS-FLI1 is unknown, and the significance of entinostat’s targets (e.g.,HDAC3) in the pathogenesis of ES has not been explored. Previously, Jaboin et al. [21] and Sonnemann et al. [22] reported the in vitro and in vivo anti-tumor activity of entinostat against ES, which is mediated through DNA synthesis inhibition, cell cycle arrest, increases in the expression of p21, TGF-βRII and c-myc, as well as the induction of apoptosis. To answer the unsolved questions, in the present study, we defined the molecular mechanisms by which entinostat potently suppressed ES tumor growth, uncovered a new role for EWS-FLI1/HDAC3/HSP90 signaling axis in maintaining the oncogenic phenotype of ES, and identify HDAC3, one of the target proteins of entinostat, as a novel transcriptional target of EWS-FLI1 and an essential effector that promotes ES cell viability and genomic stability. We also established the preclinical proof of principle of entinostat in the treatment of ES by characterizing the in vitro and in vivo efficacy, as well as the pharmacokinetics of entinostat using mouse models. Together, our findings delineate a new oncogenic pathway with pharmacologically targetable components (e.g., HDAC3 and HSP90) in ES and demonstrate the therapeutic potential of entinostat in treating ES.
Materials and Methods
Cell culture and chemicals
Hs 919.T., Hs 822.T., Hs 863.T., RD-ES, SK-ES-1 and HEK-293T cells were purchased from ATCC. TC-71, CHLA-258 and COG-E-352 cells were obtained from Children’s Oncology Group Cell Culture and Xenograft Repository. A673 cells were kindly provided by Dr. Mizuki Azuma from University of Kansas. All ES cell lines were maintained as previously described [14] and HEK-293T cells were cultured in DMEM with 10% FBS. Cell identities were confirmed by short tandem repeat profiling at the University of Kansas Cancer Center Clinical Molecular Oncology Laboratory. Cells were cultured for no more than 2 months for all experiments and routine mycoplasma testing was performed by using LookOut® Mycoplasma qPCR Detection Kit (Sigma-Aldrich).
YK-4-279 was made by Albany Medical Research Labs (AMRI, Albany, NY). For in vitro studies, entinostat was purchased from Selleckchem. For in vivo studies, entinostat was purchased from LC Laboratories. All other reagents were obtained from Thermo Fisher Scientific unless otherwise specified.
Cell viability assays
Cells were treated with drugs or vehicle for 72 h, followed by cell viability assessment using CellTiter-Blue reagent (Promega). Fluorescence was read by Infinite® M200 Pro plate reader (Tecan) at the excitation/emission wavelengths of 544 nm/590 nm. The IC50 value was determined by non-linear regression analysis to fit the data to the log (inhibitor) versus normalized response with variable slope model in GraphPad Prism 5.0 software.
Cell growth rate and doubling Time
Doubling time was determined by counting cell numbers from triplicate 25 cm3 flasks every 2–3 days. Cells were stained by using Muse® Count &Viability Assay Kit and cell numbers were counted by using the Muse® Cell Analyzer (EMD Millipore, Merck KGaA, Darmstadt, Germany). Cell growth rate can be calculated by using the formula growth rate = ln(N(t) / N(0)) * t−1, where N(t) is the number of cells at time t, N(0) is the number of cells at time 0, and t is time in hours. The doubling time calculation formula is doubling time = ln(2) * growth rate-1.
siRNA transfection assays
Cells were transfected with 50 nM of siRNAs by using DharmaFECT 1 (Dharmacon) or Lipofectamine® RNAiMAX (Thermo Fisher Scientific) Transfection Reagent following the manufacturer’s instructions. The siRNAs used in the study are detailed in Supplementary Table S1. Seventy-two hours post-transfection, cells were collected for Western blot analysis. For drug sensitivity assays, cells were transfected with indicated siRNAs first. Twenty-four hours posttransfection, cells were subjected to entinostat treatment and cell viability was determined 72 h post drug treatment as described above.
Cell cycle analysis
After drug treatment, cells were harvested and fixed in 70% ethanol overnight and then stained with FxCycle™ PI/RNase Staining Solution (Life Technologies). DNA content was measured using the Attune™ NxT flow cytometer (Invitrogen). Cells were gated based on vehicle treatment of each cell line. Data was analyzed using FlowJo software.
ROS measurement
Cells were treated with entinostat in 96-well black-walled clear-bottom plates. After treatment, cells were washed with PBS and incubated with 10 μM 2′7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Molecular Probes) at 37 °C for 15 minutes. The dye was removed, and cells were washed with PBS and scanned by Infinite® M200 Pro plate reader (Tecan) at the excitation/emission wavelengths of 485 nm/535 nm.
Caspase3/7 activity assay
Cells were treated with vehicle or drugs in 96-well black-walled clear-bottom plates. After treatment, the CellTiter-Blue reagent (Promega) was added and plates were further incubated for 3 h before the cell viability was measured as mentioned above. The Caspase-Glo® 3/7 Reagent (Promega) was then added in each well and cells were further incubated for 1 h. Luminescence was then measured as per the manufacturer’s instructions. The caspase-3/7 activities were normalized to the cell viability and expressed as the percentages relative to the vehicle control of each cell line.
Co-immunoprecipitation
After drug treatment, whole cell lysates were collected using the Universal Magnetic Co-IP Kit (Active Motif). Protein concentration was determined using the Bradford Protein Assay Kit (BioRad) and lysate was incubated with 5 μg of mouse anti-HSP90 antibody (Santa Cruz, sc-13119) or mouse anti-IgG antibody (Cell Signaling, #5415) overnight at 4 °C on a rotator. Magnetic beads were then added to the lysates and tumbled for 2 hours on a rotator at room temperature. Immunoprecipitated proteins were resolved on the 4–20% Mini-PROTEAN TGX™ Precast Gels (BioRad) followed by Western blot analysis.
Western blot analysis
Proteins were separated on the 4–20% Mini-PROTEAN TGX™ Precast Gels (Bio-Rad) and transferred onto PVDF membranes (Bio-Rad). Blots were probed with primary antibodies followed by secondary antibodies. Independent experiments were performed at least twice. All antibodies with catalog numbers and their blotting conditions are listed in the Supplementary Table S2. Immunoreactive bands were visualized by using ECL 2 Western Blotting Substrate and the densitometric analysis was performed by using ImageJ software (NIH, version 1.50i) followed by β-actin normalization.
Chromatin immunoprecipitation (ChIP) - polymerase chain reaction (PCR) assay
The chromatin immunoprecipitation experiments were performed by using SimpleChIP® Plus Enzymatic Chromatin IP Kit (magnetic beads) (Cell Signaling) according to the manufacturer’s protocol. After formaldehyde fixation, nuclease digestion and sonication, DNA-protein complexes were immunoprecipitated with ChIP grade anti-FLI1 antibody (Abcam, ab15289) or normal rabbit IgG (Cell signaling, #2729). DNA-protein complexes were then reverse cross-linked and the immunoprecipitated DNA was analyzed by PCR. Primer sequences and PCR products’ sizes are listed in Supplementary Table S3. PCR products were submitted for Sanger sequencing (Genewiz, South Plainfield, NJ).
Luciferase reporter assays
The LightSwitch™ Promoter Reporter GoClone plasmid DNA construct for HDAC3 gene was purchase from Active Motif (product ID S720148). HEK-293T cells were plated in white 96-well plates and transfected with an EWS-FLI1 (type I fusion) expression plasmid or an empty vector along with an HDAC3 promoter-reporter plasmid using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. After 48 hours, LightSwitch™ Luciferase Assay Reagent (Active Motif) was added and plates were incubated at room temperature for 30 minutes before collecting the luciferase reporter signal in a luminometer (Infinite® M200 Pro plate reader, Tecan).
Pharmacokinetic in vivo studies
For details, see Supplementary materials and methods.
In vivo xenograft mouse models
All in vivo efficacy studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Kansas Medical Center (KUMC) (IACUC# 2017–2387). Four-week old female athymic nude mice (Foxnlnu/Foxnlnu) were purchased from The Jackson Laboratory (Bar Harbor, ME) and inoculated subcutaneously in the right flanks with a suspension of TC-71 cells (1 × 106) or CHLA-258 cells (5 × 106) mixed with an equal volume of ice-cold Matrigel (BD Biosciences). After tumors were established, mice were randomized into two treatment groups and treated as follows: 1) vehicle control; equivalent dose of entinostat vehicle, oral gavage, once daily and 2) entinostat; 25 mg/kg of entinostat in 2% DMSO and 30% PEG300 (pH 2.1–2.5), oral gavage, once daily. Tumor volume and body weight were measured at least twice per week. Tumor volumes were measured with calipers and calculated using the following formula: volume (mm3) = length × (width)2/2. Mice were humanely euthanized and gross necropsies were performed when tumor volumes exceeded 4,000 mm3 (TC-71 study) or after 36 days of treatment (CHLA-258 study).
Statistical analysis
GraphPad Prism 5.0 software was used to generate data plots and for most statistical analyses. Data are presented as mean ± SD or mean ± SEM as indicated. Comparisons of two groups were carried out using two-tailed Student’s t test. For the TC-71 xenograft mouse study, the effect of drug treatments on mice survival was evaluated by the Kaplan-Meier survival curve and log-rank test, and tumor growth rates were estimated by fitting a linear mixed model with random intercept and time slope in SAS 9.4. P values less than 0.05 were considered statistically significant.
Results
Entinostat is preferentially toxic to ES cells and EWS-FLI1 and EWS-ERG confer sensitivity to entinostat treatment
We first confirmed the expression of the EWS-FLI1 and EWS-ERG oncoproteins in a panel of six ES cell lines (A673, TC-71, RD-ES, SK-ES-1, CHLA-258 and COG-E-352). Three control cell lines, including one benign osteoid osteoma cell line (Hs 919.T) and two non-tumorigenic cell lines of human bone cells origin (Hs 822.T and Hs 863.T), are negative for an EWS rearrangement (Figure 1A). Meanwhile, three EWS-FLI1 in-frame isoforms fusing EWS to different exons of FLI1 and one EWS-ERG fusion protein were detected in the ES cells (Figure 1A). The chromosomal translocations in all tested cell lines were further validated by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) and fluorescence in situ hybridization (FISH) analysis (Supplementary Figure S1). The EWS-FLI1 or EWS-ERG fusion characteristics of the ES cell lines used in the study are listed in Supplementary Table S3.
Figure 1. Entinostat is preferentially toxic to EWS-rearranged ES cells and EWS-FLI1 and EWS-ERG confer sensitivity to the treatment of entinostat.
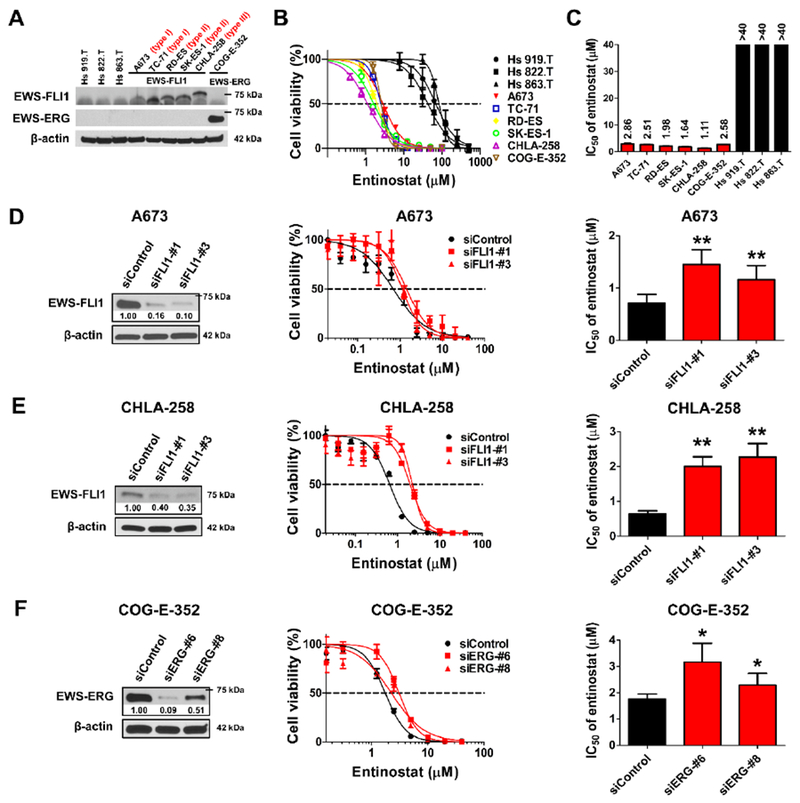
A, Western blot analysis of the expression of EWS-FLI1 and EWS-ERG fusion proteins in non-EWS-rearranged benign cells (Hs 919.T, Hs 822.T and Hs 863.T) and EWS-rearranged ES cells (A673, TC-71, RD-ES, SK-ES-1, CHLA-258 and COG-E-352). Three molecular subtypes (type I, II and III) of EWS-FLI1 fusion transcript are labeled. B, Dose-response curves of entinostat across non-EWS-rearranged benign cells (black) and EWS-rearranged ES cells (red, blue, yellow, green, purple and brown). Cell viability is normalized to the DMSO-treated controls and shown as mean ± SD of two to five independent experiments, each performed in triplicate. C, IC50 values of entinostat across non-EWS-rearranged benign cells (black) and EWS-rearranged ES cells (red). Data are mean ± SEM and detailed in Supplementary Table S5. Mean values are indicated above the bars. D, Representative Western blot image shows the downregulated expression of EWS-FLI1 in A673 cells 72 h post siRNA transfection. Cell viability was assessed in A673 cells treated by siControl, siFLI1-#1 or siFLI1-#3 for 24 h then followed by the incubation of entinostat for 72 h, and was presented as mean ± SD of two independent experiments (n = 3). IC50 values of entinostat in A673 cells were expressed as mean ± SEM. E, Representative Western blot image shows the downregulated expression of EWS-FLI1 in CHLA-258 cells 72 h post siRNA transfection. Cell viability was assessed in CHLA-258 cells treated by siControl, siFLI1-#1 or siFLI1 -#3 for 24 h then followed by the incubation of entinostat for 72 h, and was presented as mean ± SD of two independent experiments (n = 3). IC50 values of entinostat in CHLA-258 cells were expressed as mean ± SEM. F, Representative Western blot image shows the downregulated expression of EWS-ERG in COG-E-352 cells 72 h post siRNA transfection. Cell viability was assessed in COG-E-352 cells treated by siControl, siERG-#6 or siERG-#8 for 24 h then followed by the incubation of entinostat for 72 h, and was presented as mean ± SD of two independent experiments (n = 3). IC50 values of entinostat in COG-E-352 cells were expressed as mean ± SEM. β-actin served as a loading control. The relative intensities of protein bands are shown under the immunoblots after normalization for the levels of β-actin. * p < 0.05, ** p < 0.01 vs. siControl by Student’s t test.
We then assessed the toxicity of entinostat on ES and control cells. Entinostat is selectively toxic to ES cells as compared with benign cells (Figure 1B). The IC50 values of entinostat in ES cell lines range from 1.11 to 2.86 μM, which are at least 15-fold lower than their counterparts ranging from 43.18 to 83.86 μM in benign cells (Figure 1C and Supplementary Table S5). We also noticed that all six ES cell lines displayed comparable sensitivity to entinostat, even though their doubling time ranged from 24 hours to 93 hours (Supplementary Table S5).
In light of the exceptional sensitivity of ES cells with either EWS-FLI1 or EWS-ERG fusion oncogenes to entinostat, we hypothesized that EWS-FLI1 or EWS-ERG imparts a selective vulnerability to entinostat treatment in ES cells. To test this hypothesis, we knocked down EWS-FLI1 or EWS-ERG using different siRNAs. Our data showed that silencing of EWS-FLI1 or EWS-ERG rendered A673, CHLA-258 and COG-E-352 cells less sensitive to entinostat treatment. The range of the IC50 values of entinostat in siFLI1- or siERG-transfected cells increased by 1.3- to 3.5- fold, when compared with non-targeting siControl-transfected cells (Figure 1D, 1E & 1F).
Entinostat treatment induces cell cycle arrest, increases ROS levels and promotes apoptosis in ES cells
To investigate the mechanisms by which entinostat inhibited the growth of ES cells, we first examined the effects of entinostat on cell cycle distribution, intracellular ROS levels and apoptosis in vitro. In TC-71 cells, treatment with entinostat for 24 h inhibited cell cycle progression and induced G0/G1 arrest (Figure 2A). The G0/G1 cell accumulation was then followed by a significant increase of hypodiploid cells in the sub-G0 phase after 48 h exposure to entinostat, which indicates cell death (Figure 2A). Consistently, Western blot analysis showed that entinostat treatment substantially increased the expression of G1 gatekeeper p21Waf1/Cip1, and led to decreased expression of cyclin D1 and EWS-FLI1 (Figure 2B). Interestingly, we did not observe significant cell cycle disturbance in CHLA-258 cells except for the accumulation of cells in the sub-G0 phase 24 h and 48 h after entinostat treatment (Figure 2A). A modest increase in p21Waf1/Cip1 expression and marked decreases in cyclin D1 and EWS-FLI1 expression were detected 24 h post treatment in CHLA-258 cells (Figure 2B).
Figure 2. Entinostat causes cell cycle arrest, increases ROS levels and induces apoptosis in ES cells.
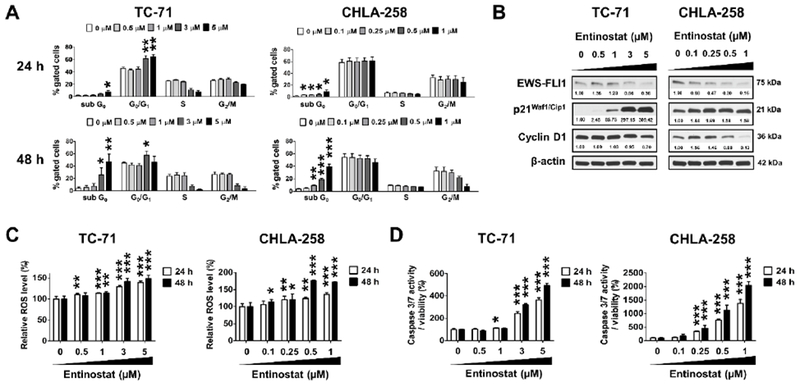
A, Cell cycle analysis in TC-71 and CHLA-258 cells treated with entinostat at the indicated concentrations for 24 h and 48 h, respectively. B, Western blot images showing the down-regulation of EWS-FLI1 and cyclin D1 expression, as well as the up-regulation of p21waf1/Cip1 expression 24 hours post entinostat treatment in TC-71 and CHLA-258 cells. β-actin served as a loading control. The relative intensities of protein bands are shown under the immunoblots after normalization for the levels of β-actin. C, ROS levels in TC-71 and CHLA-258 cells following the treatment of entinostat at the indicated concentrations for 24 h and 48 h, respectively. D, Caspase 3/7 activities of TC-71 and CHLA-258 cells treated with entinostat at the indicated concentrations for 24 h and 48 h, respectively. Data are presented as the mean ± SD (n = 3 – 6). * p < 0.05, ** p < 0.01, ***p < 0.001 vs. 0 μM at respective cell phase or time point by Student’s t test. H, hours.
It has been reported that HDAC inhibitors induce ROS accumulation [23], thus leading to DNA damage and apoptosis. Treatment with entinostat significantly increased the ROS levels dose- and time-dependently in TC-71 and CHLA-258 cells (Figure 2C). Consequently, entinostat profoundly induced caspase 3/7 activity in both cell lines (Figure 2D).
Entinostat suppresses the EWS-FLI1/HDAC3/HSP90 signaling
Our Western blot data showed that exposure to entinostat resulted in the decreased expression of EWS-FLI1 in ES cells (Figure 2B). Since prior publications demonstrated that EWS-FLI1 is a client protein of the chaperone HSP90 [24] and its degradation is primarily through the ubiquitin/proteasome pathway [25], we investigated that possibility. We hypothesized that entinostat could lead to persistent acetylation of HSP90, impairing its chaperone function, followed by depletion of HSP90 client proteins including EWS-FLI1. Supporting this hypothesis, treatment with entinostat for 6 h induced hyperacetylation of HSP90 in both TC-71 and CHLA-258 cells (Figure 3A). After 48 h treatment, EWS-FLI1 expression was reduced in a dose-dependent fashion in TC-71, SK-ES-1 and CHLA-258 cells (Figure 3B). Similarly, EWS-ERG expression was also reduced dose-dependently in COG-E-352 cells following entinostat treatment (Figure 3B).
Figure 3. Entinostat suppresses the EWS-FLI1/HDAC3/HSP90 signaling axis in ES cells.
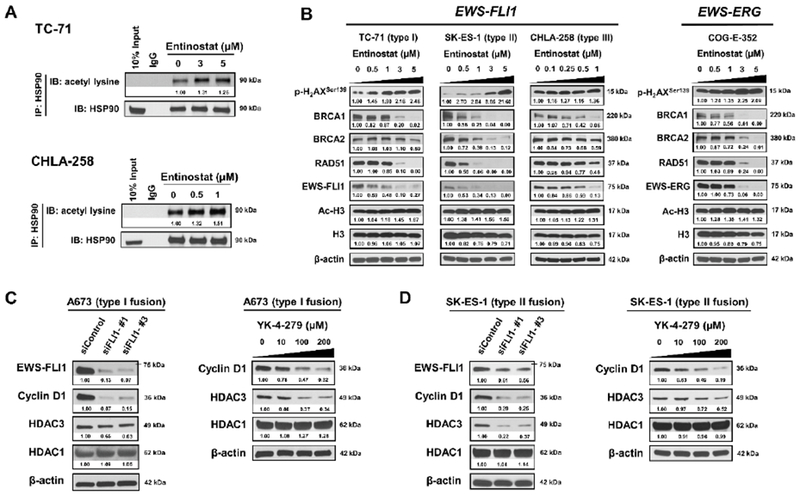
A, Entinostat induced hyperacetylation of HSP90 in both TC-71 and CHLA-258 cells 6 hours post treatment. IgG, immunoglobulin G. B, Representative Western blots showing changes in protein expression in TC-71, SK-ES-1, CHLA-258 and COG-E-352 cells after 48 h incubation with entinostat. Cell lysates were probed with the indicated antibodies. C, Knockdown of EWS-FLI1 leads to the downregulation of HDAC3 expression in A673 cells at 72 h post transfection. Meanwhile, YK-4–279 treatment reduced the expression of HDAC3 dose-dependently in A673 cells at 12 h post treatment. β-actin served as a loading control. D, Knockdown of EWS-FLI1 leads to the downregulation of HDAC3 expression in SK-ES-1 cells at 72 h post transfection. Meanwhile, YK-4–279 treatment reduced the expression of HDAC3 dose-dependently in SK-ES-1 cells at 12 h post treatment. Cyclin D1 is a validated transcriptional target of EWS-FLI1 and served as a positive control. β-actin served as a loading control. The relative intensities of protein bands are shown under the immunoblots after normalization for the levels of β-actin.
Many major components of the homologous recombination (HR) (e.g., BRCA1, BRCA2 and RAD51) and non-homologous end joining (NHEJ) (e.g., Ku70, Ku80 and DNA-PKcs) DNA double-strand breaks (DSBs) repair pathways have been demonstrated to be the clients of HSP90 [26, 27]. To further determine whether the expression of other HSP90 client proteins was regulated by entinostat, we detected the protein expression of BRCA1, BRCA2, RAD51, Ku70, Ku80 and DNA-PKcs in ES cells treated with entinostat for 48 h. The expression of BRCA1, BRCA2 and RAD51 was down-regulated dose-dependently following entinostat treatment in TC-71, SK-ES-1, CHLA-258 and COG-E-352 cells (Figure 3B). In addition, entinostat enhanced histone H3 acetylation, which is a clinical pharmacodynamic biomarker for predicting response to HDAC inhibitors, and concurrently increased p-H2AXSer139 expression, which is a DSBs marker, dose-dependently, suggesting that entinostat induced DNA damage (Figure 3B). Meanwhile, entinostat treatment decreased Ku70 expression in TC-71, SK-ES-1, CHLA-258 and COG-E-352 cells, and reduced Ku80 expression in all tested cell lines except TC-71 cell line (Supplementary Figure S2). The expression of DNA-PKcs was down-regulated in TC-71, CHLA-258 and COG-E-352 cells following treatment with entinostat for 48 h, but was slightly up-regulated in SK-ES-1 cells (Supplementary Figure S2). The expression of p-DNA-PKcsThr2609 was down-regulated by entinostat in TC-71, SK-ES-1 and COG-E-352 cells, while up-regulated in CHLA-258 cells (Supplementary Figure S2). These data suggest that the regulation of entinostat on the expression of DNA repair proteins on the NHEJ pathway is cell-line dependent.
To explore the correlation between EWS-FLI1 and HDAC1 and HDAC3, we examined the expression of HDAC1 and HDAC3 after inhibiting the function of EWS-FLI1 by using specific siRNAs and a pharmacological inhibitor. Depletion of EWS-FLI1 expression by two different siRNAs (siFLI1-#1 and siFLI1-#3) dramatically decreased the protein levels of cyclin D1 (positive control) and HDAC3, but not HDAC1, in A673 and SK-ES-1 cells when compared with siControl treatment (Figure 3C–D). To further verify these results, we used YK-4–279, a reported small molecule inhibitor that blocks the interactions between EWS-FLI1 and RNA helicase A (RHA) [28], as well as DDX5 [29], to abrogate the transcriptional function of EWS-FLI1. Consistently, our data show that the protein expression of cyclin D1 and HDAC3 was significantly reduced in a dose-dependent manner upon the treatment of YK-4-279 for 12 h in A673 and SK-ES-1 cells, while the expression of HDAC1 was barely changed (Figure 3C–D). Meanwhile, YK-4-279 treatment minimally affected the protein expression of EWS-FLI1 and RHA at the indicated concentrations (Supplementary Figure S3). Taken together, our data strongly suggest that HDAC3, but not HDAC1, is a downstream mediator of EWS-FLI1 regulation.
HDAC3 is a transcriptional target of EWS-FLI1
To evaluate whether EWS-FLI1 binds to the HDAC3 promoter, we first analyzed the HDAC3 promoter sequence and identified eleven GGAA elements in the 2.1-kb promoter region (Figure 4A), which are core EWS-FLI1-binding motifs [30]. We then performed ChIP-PCR experiments in both A673 and TC-71 cells to assess the potential interaction between EWS-FLI1 and the HDAC3 promoter in vivo. As shown in Figure 4B, except for site 1, the specific PCR fragments corresponding to the EWS-FLI1 binding sites 2, 3 and 4 in the HDAC3 promoter were detected in the anti-FLI1 immunoprecipitation using A673 and TC-71 cell extracts. The sequences of these PCR fragments were confirmed by Sanger sequencing (data not shown). No PCR fragment was detected in the control IgG immunoprecipitation or when the DNA template was absent. These data demonstrate that EWS-FLI1 binds to the HDAC3 promoter in ES cells.
Figure 4. EWS-FLI1 binds to and activates the HDAC3 promoter.
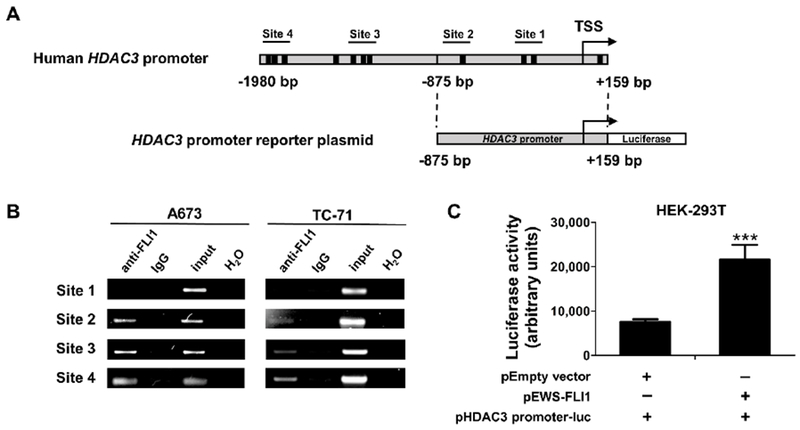
A, Schematic representation of human HDAC3 promoter and the luciferase reporter plasmid containing human HDAC3 promoter (−875 to +159 bp from the transcription start site). The black boxes represent the GGAA sites, which are core EWS-FLI1 binding motifs. The transcription start site (TSS) is denoted by an arrow. The putative EWS-FLI1 binding sites with PCR primers used to amplify each fragment are numbered. B, ChIP-PCR demonstrates that EWS-FLI1 binds to the HDAC3 promoter. ChIP experiments were performed in A673 and TC-71 cells using anti-FLI1 and normal rabbit IgG antibodies. Four sets of primers were designed to cover the potential EWS-FLI1 binding sites within the HDAC3 promoter and then were used to amplify immunoprecipitated DNA by PCR. Input is a portion of the sheared chromatin prior to immunoprecipitation. H2O represents a no DNA template control. C, HDAC3 promoter luciferase assay showing EWS-FLI1 activates the HDAC3 promoter in HEK-293T cells. HEK-293T cells were co-transfected with EWS-FLI1 expression plasmid (pEWS-FLI1) plus the HDAC3 promoter luciferase construct (pHDAC3 promoter-luc) or empty vector (pEmpty vector) plus pHDAC3 promoter-luc at a 1:1 ratio. Total plasmid DNA amount in each well was 100 ng. Luciferase activity was measured 48 hours post transfection. Data were presented as mean ± SD of three independent experiments (n = 4). ***p < 0.001 by Student’s t test.
To further evaluate the functional outcome of the interaction between EWS-FLI1 and the HDAC3 promoter, we transfected HEK-293T cells with the EWS-FLI1 expression plasmid and the HDAC3 promoter (from −875 to +159 bp relative to the transcription start site) reporter construct at a 1:1 ratio. Our data showed that exogenous expression of EWS-FLI1 significantly increases the HDAC3 promoter activity by 2.8-fold when compared with the empty vector control (Figure 4C). These experiments were repeated three times with different combination ratios (2:1 and 3:1) between the EWS-FLI1 expression plasmid and the HDAC3 promoter reporter, resulting in at least a 2.5-fold increase in promoter activity (data not shown). These data demonstrate that EWS-FLI1 can influence, either directly or indirectly, the transcription of HDAC3.
HDAC3 is critical for cell viability and genomic stability maintenance in ES cells
To delineate the functions of HDAC1 and HDAC3, which are two major and distinct isoforms in Class I HDACs, we evaluated the effects of HDAC1 and HDAC3 on cell viability and the HR repair pathway. We found that RNAi silencing of HDAC3, but not HDAC1, significantly reduced cell viability in A673 cells when compared with CHLA-258 cells, due to the much higher transfection efficiency in A673 cells as indicated by the treatment with siPLK1 (served as a positive control) (Figure 5A–B). Significantly, knockdown of HDAC3, and to a much lesser extent, HDAC1, led to the depletion of BRCA1 and RAD51 in A673 and CHLA-258 cells (Figure 5C–D). These data suggest that HDAC3, but not HDAC1, is critical for cell viability and genomic stability maintenance in ES cells.
Figure 5. HDAC3, but not HDAC1, is critical for ES cell survival and genomic stability maintenance.
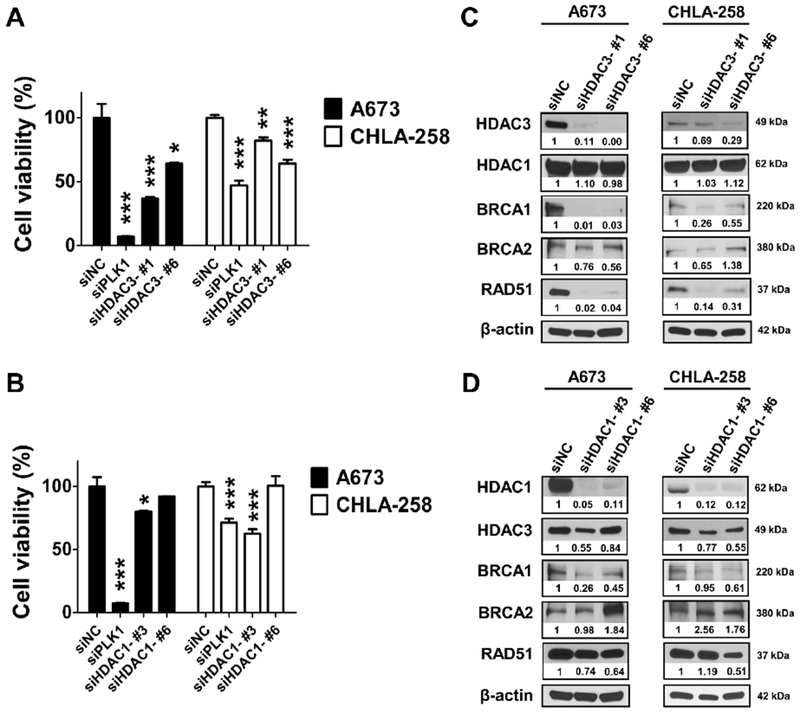
A, Cell viability of A673 and CHLA-258 cells was detected after being treated with siNC, siPLK1, siHDAC3-#1 and siHDAC3-#6, respectively, for 72 hours. Data, mean ± SD. B, Cell viability of A673 and CHLA-258 cells was detected after being treated with siNC, siPLK1, siHDAC1-#3 and siHDAC1-#6, respectively, for 72 hours. Data, mean ± SD. * p < 0.05, ** p < 0.01, ***p < 0.001 vs. siNC by Student’s t test. C, Representative Western blots showing the protein expression changes of HDAC1, HDAC3, BRCA1, BRCA2 and RAD51 72 hours post siHDAC3 transfection in A673 and CHLA-258 cells. β-actin served as a loading control. The relative intensities of protein bands are shown under the immunoblots after normalization for the levels of β-actin. D, Representative Western blots showing the protein expression changes of HDAC1, HDAC3, BRCA1, BRCA2 and RAD51 72 hours post siHDAC1 transfection in A673 and CHLA-258 cells. β-actin served as a loading control. The relative intensities of protein bands are shown under the immunoblots after normalization for the levels of β-actin. siNC served as a negative control and siPLK1 targeting polo-like kinase 1 served as a positive control.
Support for in vivo preclinical proof of principle studies
Pharmacokinetic studies were conducted to establish the relationships between systemic exposure and dose, route of administration and dose frequency for entinostat. We demonstrated that the regimens employed in the in vivo preclinical proof of principle studies achieved systemic exposures of entinostat, exceeding the IC50 values determined in vitro. Resultant mean (± SD) plasma drug concentration-time profiles for days one and five for entinostat administered orally at 24.5 mg/kg following daily consecutive doses are shown in the Supplementary Figure S4A. The derived entinostat non-parametric pharmacokinetic parameters are summarized in the Supplementary Table S6. As shown in Supplementary Figure S4B, systemic exposure of entinostat substantially decreased in a nearly linear manner upon repeat dose administration, suggesting increased drug metabolism during absorption in mice [31]. To verify that this observation wasn’t a result of decomposition of the dosing solution, the stability of the entinostat dosing solution stored at 4 °C for a period of 5 days was evaluated and found not to have changed (data not shown). Despite the possible autoinduction, mean entinostat Cmax values observed on days 1 through day 5 ranged from 12,400 to 1,920 ng/ml (32.94 to 5.10 μΜ), exceeding in vitro IC50 values ranging from 418 to 1,077 ng/ml (1.11 to 2.86 μΜ) determined in ES cell lines.
Entinostat significantly reduced tumor burden and increased survival in murine xenograft models of ES
It is noteworthy that the doubling time of CHLA-258 cells (89 hours) is approximately four-fold slower than TC-71 cells (21 hours) [32]. Therefore, to examine the antitumor efficacy of entinostat in vivo,we selected CHLA-258 and TC-71 cell lines to generate ES xenograft mouse models, which represent slow-growing xenografts and fast-growing xenografts, respectively. In the CHLA-258 study, mice were randomized to receive treatment with either vehicle or entinostat (25 mg/kg) on day 17th when tumors averaged 165 mm3. After 36 days of treatment, entinostat strikingly inhibited the growth of ES xenografts, as demonstrated by the tumor volume in entinostat group decreased by an average of 97.4% (p < 0.001) relative to the vehicle control group (Figure 6A). In addition, the entinostat treatment significantly reduced tumor weight by 99.4% (p < 0.001) when compared with the vehicle treatment (Figure 6B). A potential limitation of this efficacy study is that the drug administration was terminated on day 36th due to IACUC limits on allowed tumor burden (4,000 mm3) in the control animals, therefore the overall median survival benefit could not be established. We can only speculate that additional treatments of entinostat might have led to complete tumor eradication in these tumor-bearing mice.
Figure 6. Entinostat significantly inhibited the growth of CHLA-258 xenografts in mice.
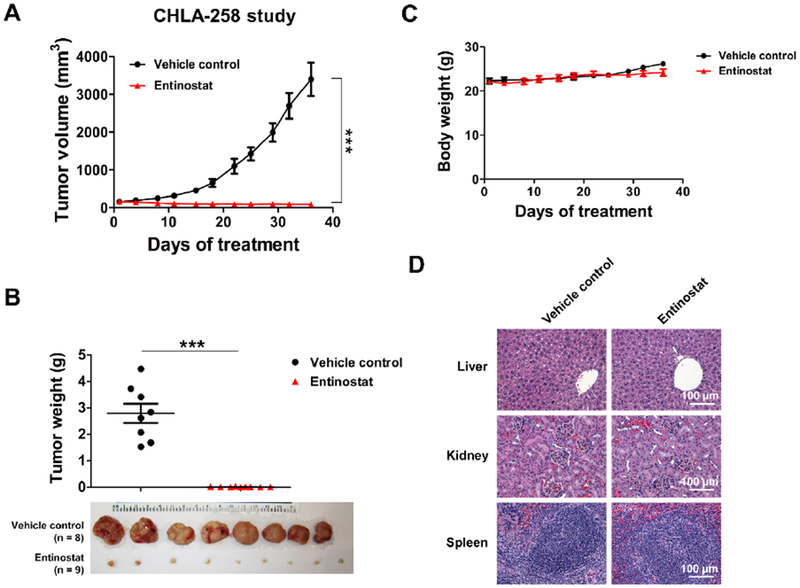
A, Tumor growth of CHLA-258 xenografts in nude mice treated with vehicle or entinostat (25 mg/kg, p.o., qd) for 36 days. N = 8 – 9 mice per treatment group. Data, mean ± SEM. *** p < 0.001 by Student’s t test. B, Dot plot of tumor weights of each treatment group in CHLA-258 xenograft mouse study. Image of tumors following sacrifice in CHLA-258 xenograft mouse study was shown. Data, mean ± SEM. *** p < 0.001 by Student’s t test. C, Mice body weights throughout the treatment period in the CHLA-258 xenograft mouse study. Data, mean ± SEM. D, Representative H&E staining photographs of livers, kidneys and spleens from each treatment group in the CHLA-258 xenograft mouse study. Bar = 100 μm.
Next, we performed a second preclinical proof of principle study in the ES xenograft mouse model by subcutaneous inoculation of TC-71 cells. In the TC-71 model, drug treatment began on day 14th when tumors averaged 877 mm3. Our results show that 25 mg/kg of entinostat significantly delayed the progression of ES xenografts by extending the median survival to 15.5 days (p < 0.001), as compared to the vehicle treatment (8.5 days) (Supplementary Figure S5A). We then estimated the tumor growth rate for each treatment group by fitting a linear mixed model with random intercept and time slope to the log-transformed tumor size data. Whereas the estimated growth rate for the vehicle control group was 1.20 (20% increase per day), rate for the entinostat group was 1.12 (p < 0.001) (Supplementary Figure S5B).
In both in vivo efficacy studies, the treatment of entinostat was well tolerated in the mice without causing toxicity-related deaths, significant weight loss, or other discernible adverse effects (Figure 6C & Supplementary Figure S5C). No gross pathologic changes were observed in the livers, kidneys or spleens of animals in the two pilot studies (Figure 6D & Supplementary Figure S5D).
Discussion
The present study has demonstrated a novel result showing entinostat, a selective HDAC1 and HDAC3 inhibitor, exerts potent anti-tumor activity against ES in preclinical models, through suppression of EWS-FLI1/HDAC3/HSP90 signaling. In vitro,entinostat induces substantial apoptosis of ES cells through inducing cell cycle arrest, increasing intracellular ROS levels, damaging DNA and impairing HR DNA repair. In vivo,entinostat markedly reduces tumor burden and prolongs survival. More importantly, for the first time, we demonstrated that HDAC3 is likely a transcriptional target of EWS-FLI1, and it is essential for cell survival and the maintenance of genomic stability in ES cells. Our studies demonstrate the feasibility and therapeutic efficacy of targeting the downstream mediators of EWS-FLI1 that contribute to the oncogenesis of ES, which serves as an alternative and promising therapeutic option for the treatment of these malignancies. Such a strategy could be applied to a variety of cancers driven by dominant oncogenes, especially those in which the oncogenic events have not been successfully exploited therapeutically.
In addition, epigenetic dysregulation is increasingly recognized as an important mechanism in the pathogenesis of ES. Recent next-generation sequencing studies have identified that EWS-FLI1 drives widespread epigenetic reprogramming and chromatin remodeling in ES [33, 34]. As such, epigenetic therapy has been considered as an attractive treatment strategy in ES. Pattenden et al. [35] previously identified that a cluster of HDAC inhibitors including entinostat significantly decreased EWS-FLI1-dependent chromatin accessibility by decreasing EWS-FLI1 levels, which explained our predictions in in silico screen that entinostat can reverse the ES disease signature and EWS-FLI1-mediated transcriptional signature. Besides entinostat, other HDAC inhibitors (e.g., romidepsin, vorinostat, sodium butyrate) have also shown potent anti-ES activity [22, 36, 37], which provides convincing evidence of the epigenome as a therapeutic target in ES.
Due to the intrinsic defects in HR repair [38] and the synthetic lethality conferred by EWS-FLI1 [39], ES is extraordinarily sensitive to the agents that induce DNA damage and/or prevent its repair [40]. Consistent with these findings, our studies demonstrated that entinostat preferentially and potently inhibits the proliferation of ES cells with different subtypes of EWS-FLI1 fusion both in vitro and in vivo. Furthermore, we extended this observation to the ES cells with the second most common genomic alteration, EWS-ERG fusion. The EWS-FLI1 or EWS-ERG-dependency in the ES cells sensitivity to entinostat further supports our in silico prediction that entinostat can reverse the EWS-FLI1 transcriptional program, suggesting entinostat targets the downstream effectors of EWS-FLI1 or EWS-ERG to mitigate its oncogenic functions.
Given the EWS-FLI1 fusion gene encodes for a chimeric transcription factor, the relationship between EWS-FLI1 and HDAC1/3 was examined. Genetic or pharmacological inhibition of EWS-FLI1 led to the decreased expression of HDAC3, but not HDAC1, suggesting that EWS-FLI1 is a potential regulator of HDAC3 expression. ChIP-PCR and promoter-luciferase reporter assays further demonstrate that EWS-FLI1 either directly or indirectly binds to the HDAC3 promoter and induces its expression. Moreover, knockdown of HDAC3, but not HDAC1, caused significant cell death and a dramatic reduction in the expression of BRCA1 and RAD51 in ES cells, which is most likely because HDAC3 is an HSP90 deacetylase and inhibition of HDAC3 induces hyperacetylation and inhibition of nuclear HSP90 [41]. In line with these findings, our studies demonstrated that entinostat caused hyperacetylation of HSP90 in ES cells, which leads to the depletion of the validated HSP90 client proteins including EWS-FLI1, BRCA1, BRCA2 and RAD51 [24, 26]. Taken together, our results revealed an underappreciated pro-oncogenic role for the EWS-FLI1/HDAC3/HSP90 signaling axis, which is critical for maintaining ES cell viability and genomic stability, as well as driving ES progression (Figure 7). Undoubtedly, these discoveries refine our knowledge of ES biology and open new opportunities for the translational studies of HDAC inhibitors in the treatment of ES.
Figure 7. Scheme shows the mechanisms through which entinostat kills ES cells.
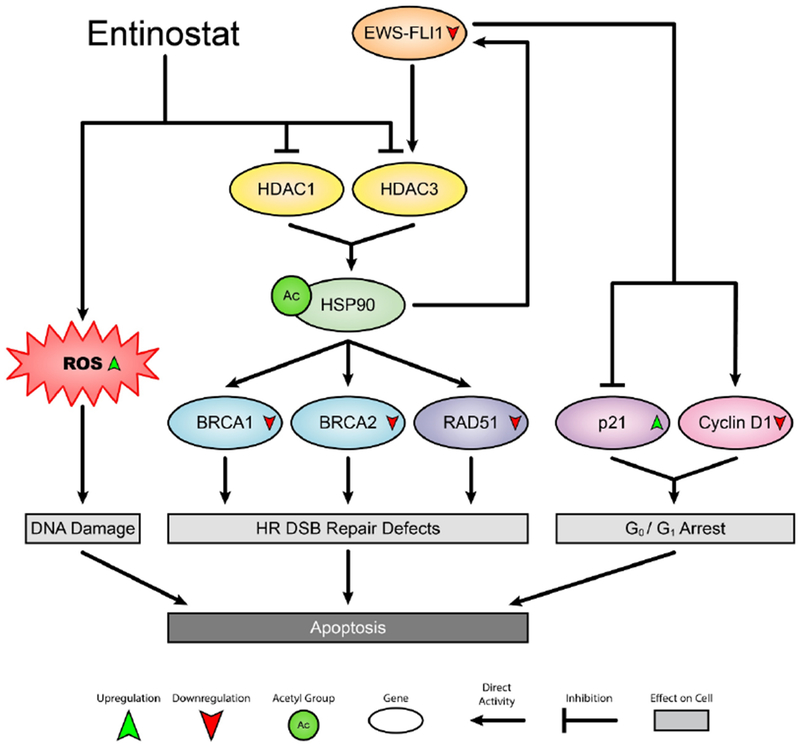
HR, homologous recombination; DSB, double-strand break; ROS, reactive oxygen species.
Mounting evidence has indicated that HDAC isoenzymes have distinct and redundant functions as regulators in the control of cell proliferation, differentiation and tumorigenesis [42]. It is unclear what functional contributions of individual HDAC isoenzyme are in the pathogenesis of ES. HDAC3 is upregulated in many different malignancies including leukemia, lymphoma, renal cancer, gastric cancer, colorectal cancer and liver cancer, and its aberrant overexpression promotes the proliferation of cancer cells and predicts poor survival in cancer patients [43]. However, little is known regarding the correlation between HDAC3 and Ewing sarcomagenesis, and the mechanisms by which HDAC3 drives the epigenetic and transcriptional reprogramming during ES progression remain enigmatic. It is worth deciphering the roles of HDAC3 and other HDACs in the pathogenesis of ES, which will advance our understanding of the epigenetic dysregulation in ES and open new avenues for targeted therapies in ES. More importantly, insights gained from the functions and potential drug targets specific to selected HDACs will be essential to guide the selection of HDAC inhibitors and stratify patients in the clinical trials for ES.
In our study, HSP90 acetylation led to the down-regulation of EWS-FLI1, which subsequently modulated the expression of EWS-FLI1 transcriptional targets including p21Waf1/Cip1 and cyclin D1 and triggered G0/G1 arrest. Interestingly, we only observed the accumulation of TC-71 cells, but not CHLA-258 cells, in the G0/G1 phase following entinostat treatment, suggesting that other mechanisms may counteract the effects of cyclin D1 and p21Waf1/Cip1 on the cell cycle control in CHLA-258 cells. This phenotypic difference might also explain the superior efficacy of entinostat in CHLA-258 xenograft-bearing mice and beg for discovery of companion biomarkers to select patients who would most likely obtain the greatest benefit from this therapy. Meanwhile, we found that entinostat increased the ROS levels in ES cells, which might be attributed to the reduced activities of the intracellular antioxidants (e.g.,thioredoxin) upon treatment with entinostat [44]. The increased oxidative stress caused by entinostat induced DNA DSBs, as evidenced by the increased p-H2AXSer139 levels, and entinostat inactivated the HR repair pathway by down-regulating the expression levels of BRCA1, BRCA2 and RAD51, thus leading to HR repair defects. In addition, in the presence of HR deficiency, entinostat regulates the NHEJ pathway in a cell line-dependent manner, suggesting that entinostat promotes genomic instability. Collectively, our studies demonstrated that the cell cycle arrest, the elevated ROS and the impaired HR DSB repair machinery contribute to the entinostat-mediated apoptosis in ES cells (Figure 7), further supporting the idea that therapeutic targeting of cell cycle regulators, redox homeostasis and DNA repair machinery in combination of entinostat may represent a novel and more effective strategy for the treatment of ES.
It is noteworthy that entinostat has a long half-life (between 30 – 80 hours) in humans [45, 46], which allows continuous drug exposure in cancer patients with either once-weekly or biweekly oral dosing. The half-life of entinostat in rats dosed orally was also previously reported to be 9.8 hours [47]. However, our pharmacokinetics data showed that the half-life of oral entinostat in the mice plasma (between 32 – 58 minutes) is substantially shorter than that observed in rats and cancer patients. This striking pharmacokinetic discrepancy between humans, rats and mice suggests a species-related difference in the pharmacokinetics of entinostat. Possible explanations could include 1) the significant difference in the metabolic capacity of the species per m2 of body surface area and 2) the significant higher binding of entinostat to human plasma proteins [48], which results in a slower distribution and elimination of entinostat in humans when compared to rats and mice. In addition, the enhanced metabolism of entinostat observed in our studies may be associated with the activation of CYP450 isoforms [49]. Although the autoinduction of entinostat metabolism was observed in mice after continuous oral administration, we and others [21] have demonstrated that entinostat exerts potent anti-ES activity in vivo,suggesting that plasma entinostat concentrations were maintained above a minimum value to achieve the desired anticancer effect. Together, our results along with previously published data [47] suggest that the preclinical pharmacokinetic studies of entinostat in laboratory animals may not predict its pharmacokinetic properties in humans due to the significant interspecies differences in genetics, physiology and metabolism. Special concerns should be taken when translating mouse studies of entinostat alone or in combination with other chemotherapies or biologic agents into the clinic, especially in pediatric patients. Of note, a Phase 1 clinical trial of entinostat (NCT02780804) has been initiated in pediatric patients with recurrent or refractory solid tumors, where the maximum tolerated dose and the pharmacokinetics profile of entinostat in the patients aged from 12 months to 21 years will be addressed.
In conclusion, our studies demonstrated that this oral histone deacetylase inhibitor should be considered as a promising therapeutic for the treatment of children and young adults diagnosed with relapsed or refractory ES, given its exquisite selectivity, outstanding in vitro and in vivo anticancer potency, long half-life and excellent safety and tolerability profiles in cancer patients. More importantly, our mechanistic studies point out the dependency of ES cells on the EWS-FLI1/HDAC3/HSP90 signaling for survival and genomic stability maintenance, suggesting that combinatorial use of entinostat and HSP90 inhibitors, PARP inhibitors or DNA damaging agents may be particularly effective at treating ES. Therefore, our preclinical proof of principle studies provide support for the targeted clinical development of the entinostat-based therapies for the treatment of patients with ES.
Supplementary Material
Key messages.
Entinostat potently inhibits ES both in vitro and in vivo.
EWS-FLI1 and EWS-ERG confer sensitivity to entinostat treatment.
Entinostat suppresses the EWS-FLI1/HDAC3/HSP90 signaling.
HDAC3 is a direct transcriptional target of EWS-FLI1.
HDAC3 is essential for ES cell viability and genomic stability maintenance.
Acknowledgments
The authors gratefully acknowledge Dr. Jeff Hirst for technical help in flow cytometry assays and animal studies, Ms. Tara Meyer for technical assistance with H&E and immunohistochemical staining, Dr. Rashna Madan for reviewing the pathology slides, Dr. Richard C. Hastings for technical help in flow cytometry assays and data analysis, Mr. Mitch Braun and Ms. Carolyn Vivian for assistance with animal studies. We also acknowledge the support of the University of Kansas Cancer Center’s Biospecimen Repository Core Facility and Lead Development and Optimization Shared Resource (P30 CA168524) and the University of Kansas Medical Center’s Flow Cytometry Core Laboratory (P30 GM103326).
Funding information
This work was supported in part by an MCA Partners Advisory Board grant from Children’s Mercy Hospital and The University of Kansas Cancer Center (to KMC and AKG), the Braden’s Hope for Childhood Cancer Foundation (to GS & AKG), the NIH/NIGMS P20 GM130423–01 (to AKG), and the Kansas Bioscience Authority Eminent Scholar Program (to AKG). AKG is the Chancellors Distinguished Chair in Biomedical Sciences Endowed Professor.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Balamuth NJ, Womer RB (2010) Ewing’s sarcoma. Lancet Oncol 11: 184–192. DOI 10.1016/S1470-2045(09)70286-4 [DOI] [PubMed] [Google Scholar]
- 2. Jain S, Kapoor G (2010) Chemotherapy in Ewing’s sarcoma. Indian J Orthop 44: 369–377. DOI 10.4103/0019-5413.69305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perkins SM, Shinohara ET, DeWees T, Frangoul H (2014) Outcome for children with metastatic solid tumors over the last four decades. PLoS One 9: e100396 DOI 10.1371/journal.pone.0100396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, Marcus K, Sailer S, Healey JH, Dormans JP, et al. (2012) Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol 30: 4148–4154. DOI 10.1200/JCO.2011.41.5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamilton SN, Carlson R, Hasan H, Rassekh SR, Goddard K (2017) Long-term Outcomes and Complications in Pediatric Ewing Sarcoma. Am J Clin Oncol 40: 423–428. DOI 10.1097/COC.0000000000000176 [DOI] [PubMed] [Google Scholar]
- 6.May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, Zucman J, Thomas G, Denny CT (1993) Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci U S A 90: 5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arvand A, Denny CT (2001) Biology of EWS/ETS fusions in Ewing’s family tumors. Oncogene 20: 5747–5754. DOI 10.1038/sj.onc.1204598 [DOI] [PubMed] [Google Scholar]
- 8.DuBois SG, Krailo MD, Lessnick SL, Smith R, Chen Z, Marina N, Grier HE, Stegmaier K, Children’s Oncology G (2009) Phase II study of intermediate-dose cytarabine in patients with relapsed or refractory Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer 52: 324–327. DOI 10.1002/pbc.21822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baruchel S, Pappo A, Krailo M, Baker KS, Wu B, Villaluna D, Lee-Scott M, Adamson PC, Blaney SM (2012) A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children’s Oncology Group. Eur J Cancer 48: 579–585. DOI 10.1016/j.ejca.2011.09.027 [DOI] [PubMed] [Google Scholar]
- 10.Wagner LM, Fouladi M, Ahmed A, Krailo MD, Weigel B, DuBois SG, Doyle LA, Chen H, Blaney SM (2015) Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer 62: 440–444. DOI 10.1002/pbc.25334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michelagnoli M, Whelan J, Forsyth S, Otis Trial Management Group SI (2015) A phase II study to determine the efficacy and safety of oral treosulfan in patients with advanced pre-treated Ewing sarcoma ISRCTN11631773. Pediatr Blood Cancer 62: 158–159. DOI 10.1002/pbc.25156 [DOI] [PubMed] [Google Scholar]
- 12.Grohar PJ, Glod J, Peer CJ, Sissung TM, Arnaldez FI, Long L, Figg WD, Whitcomb P, Helman LJ, Widemann BC (2017) A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS-FLI1 fusion transcript. Cancer Chemother Pharmacol 80: 645–652. DOI 10.1007/s00280-017-3382-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsafou K, Tiwari PB, Forman-Kay JD, Metallo SJ, Toretsky JA (2018) Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J Mol Biol 430: 2321–2341. DOI 10.1016/j.jmb.2018.04.008 [DOI] [PubMed] [Google Scholar]
- 14.Pessetto ZY, Chen B, Alturkmani H, Hyter S, Flynn CA, Baltezor M, Ma Y, Rosenthal HG, Neville KA, Weir SJ, et al. (2017) In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget 8: 4079–4095. DOI 10.18632/oncotarget.13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knipstein J, Gore L (2011) Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin Investig Drugs 20: 1455–1467. DOI 10.1517/13543784.2011.613822 [DOI] [PubMed] [Google Scholar]
- 16.Hess-Stumpp H, Bracker TU, Henderson D, Politz O (2007) MS-275, a potent orally available inhibitor of histone deacetylases--the development of an anticancer agent. Int J Biochem Cell Biol 39: 1388–1405. DOI 10.1016/j.biocel.2007.02.009 [DOI] [PubMed] [Google Scholar]
- 17.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O (1999) A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A 96: 4592–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al. (2011) Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 1: 598–607. DOI 10.1158/2159-8290.CD-11-0214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB (2013) Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol 31: 2128–2135. DOI 10.1200/JCO.2012.43.7251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pili R, Quinn DI, Hammers HJ, Monk P, George S, Dorff TB, Olencki T, Shen L, Orillion A, Lamonica D, et al. (2017) Immunomodulation by Entinostat in Renal Cell Carcinoma Patients Receiving High-Dose Interleukin 2: A Multicenter, Single-Arm, Phase I/II Trial (NCI-CTEP#7870). Clin Cancer Res 23: 7199–7208. DOI 10.1158/1078-0432.CCR-17-1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaboin J, Wild J, Hamidi H, Khanna C, Kim CJ, Robey R, Bates SE, Thiele CJ (2002) MS-27–275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res 62: 6108–6115 [PubMed] [Google Scholar]
- 22.Sonnemann J, Dreyer L, Hartwig M, Palani CD, Hong le TT, Klier U, Broker B, Volker U, Beck JF (2007) Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing’s sarcoma cells. J Cancer Res Clin Oncol 133: 847–858. DOI 10.1007/s00432-007-0227-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hedrick E, Crose L, Linardic CM, Safe S (2015) Histone Deacetylase Inhibitors Inhibit Rhabdomyosarcoma by Reactive Oxygen Species-Dependent Targeting of Specificity Protein Transcription Factors. Mol Cancer Ther 14: 2143–2153. DOI 10.1158/1535-7163.MCT-15-0148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambati SR, Lopes EC, Kosugi K, Mony U, Zehir A, Shah SK, Taldone T, Moreira AL, Meyers PA, Chiosis G, et al. (2014) Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol Oncol 8: 323–336. DOI 10.1016/j.molonc.2013.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gierisch ME, Pfistner F, Lopez-Garcia LA, Harder L, Schafer BW, Niggli FK (2016) Proteasomal Degradation of the EWS-FLI1 Fusion Protein Is Regulated by a Single Lysine Residue. J Biol Chem 291: 26922–26933. DOI 10.1074/jbc.M116.752063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stecklein SR, Kumaraswamy E, Behbod F, Wang W, Chaguturu V, Harlan-Williams LM, Jensen RA (2012) BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc Natl Acad Sci U S A 109: 13650–13655. DOI 10.1073/pnas.1203326109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dote H, Burgan WE, Camphausen K, Tofilon PJ (2006) Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res 66: 9211–9220. DOI 10.1158/0008-5472.CAN-06-2181 [DOI] [PubMed] [Google Scholar]
- 28.Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, et al. (2009) A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med 15: 750–756. DOI 10.1038/nm.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, Yu S, Liu X, Paulsen MT, Ljungman ME, et al. (2015) Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc Natl Acad Sci U S A 112: E1307–1316. DOI 10.1073/pnas.1500536112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mao X, Miesfeldt S, Yang H, Leiden JM, Thompson CB (1994) The FLI-1 and chimeric EWS-FLI-1 oncoproteins display similar DNA binding specificities. J Biol Chem 269: 18216–18222 [PubMed] [Google Scholar]
- 31.Graham MJ, Lake BG (2008) Induction of drug metabolism: species differences and toxicological relevance. Toxicology 254: 184–191. DOI 10.1016/j.tox.2008.09.002 [DOI] [PubMed] [Google Scholar]
- 32.May WA, Grigoryan RS, Keshelava N, Cabral DJ, Christensen LL, Jenabi J, Ji L, Triche TJ, Lawlor ER, Reynolds CP (2013) Characterization and drug resistance patterns of Ewing’s sarcoma family tumor cell lines. PLoS One 8: e80060 DOI 10.1371/journal.pone.0080060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schonegger A, Datlinger P, Kubicek S, Bock C, Kovar H (2015) Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep 10: 1082–1095. DOI 10.1016/j.celrep.2015.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, et al. (2014) EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 26: 668–681. DOI 10.1016/j.ccell.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pattenden SG, Simon JM, Wali A, Jayakody CN, Troutman J, McFadden AW, Wooten J, Wood CC, Frye SV, Janzen WP, et al. (2016) High-throughput small molecule screen identifies inhibitors of aberrant chromatin accessibility. Proc Natl Acad Sci U S A 113: 3018–3023. DOI 10.1073/pnas.1521827113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakimura R, Tanaka K, Nakatani F, Matsunobu T, Li X, Hanada M, Okada T, Nakamura T, Matsumoto Y, Iwamoto Y (2005) Antitumor effects of histone deacetylase inhibitor on Ewing’s family tumors. Int J Cancer 116: 784–792. DOI 10.1002/ijc.21069 [DOI] [PubMed] [Google Scholar]
- 37.Souza BK, da Costa Lopez PL, Menegotto PR, Vieira IA, Kersting N, Abujamra AL, Brunetto AT, Brunetto AL, Gregianin L, de Farias CB, et al. (2018) Targeting Histone Deacetylase Activity to Arrest Cell Growth and Promote Neural Differentiation in Ewing Sarcoma. Mol Neurobiol 55: 7242–7258. DOI 10.1007/s12035-018-0874-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, Iniguez AB, Bernard X, Masamsetti VP, Roston S, et al. (2018) EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 555: 387–391. DOI 10.1038/nature25748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iniguez AB, Stolte B, Wang EJ, Conway AS, Alexe G, Dharia NV, Kwiatkowski N, Zhang T, Abraham BJ, Mora J, et al. (2018) EWS/FLI Confers Tumor Cell Synthetic Lethality to CDK12 Inhibition in Ewing Sarcoma. Cancer Cell 33: 202–216 e206 DOI 10.1016/j.ccell.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith MA, Reynolds CP, Kang MH, Kolb EA, Gorlick R, Carol H, Lock RB, Keir ST, Maris JM, Billups CA, et al. (2015) Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res 21: 819–832. DOI 10.1158/1078-0432.CCR-14-2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM, et al. (2014) Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 5: 5637–5650. DOI 10.18632/oncotarget.2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42. DOI 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barneda-Zahonero B, Parra M (2012) Histone deacetylases and cancer. Mol Oncol 6: 579–589. DOI 10.1016/j.molonc.2012.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marks PA (2006) Thioredoxin in cancer--role of histone deacetylase inhibitors. Semin Cancer Biol 16: 436–443. DOI 10.1016/j.semcancer.2006.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, et al. (2005) Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol 23: 3912–3922. DOI 10.1200/JCO.2005.02.188 [DOI] [PubMed] [Google Scholar]
- 46.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, et al. (2007) Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 109: 2781–2790. DOI 10.1182/blood-2006-05-021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X, Zhang Q, Chen M, Hu L (2014) Pharmacokinetic interaction of entinostat and lapatinib following single and co-oral administration in rats. Xenobiotica 44: 1009–1013. DOI 10.3109/00498254.2014.919431 [DOI] [PubMed] [Google Scholar]
- 48.Acharya MR, Sparreboom A, Sausville EA, Conley BA, Doroshow JH, Venitz J, Figg WD (2006) Interspecies differences in plasma protein binding of MS-275, a novel histone deacetylase inhibitor. Cancer Chemother Pharmacol 57: 275–281. DOI 10.1007/s00280-005-0058-8 [DOI] [PubMed] [Google Scholar]
- 49.Wu Q, Zhang Q, Wen C, Hu L, Wang X, Lin G (2015) The effect of MS-275 on CYP450 isoforms activity in rats by cocktail method. Int J Clin Exp Pathol 8: 9360–9367 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.