

Abstract
Mucin 4 (MUC4) is a high molecular weight glycoprotein that is differentially overexpressed in pancreatic cancer (PC), functionally contributes to disease progression, and correlates with poor survival. Further, due to its aberrant glycosylation and extensive splicing, MUC4 is a potential target for cancer immunotherapy. Our previous studies have demonstrated the utility of amphiphilic polyanhydride nanoparticles as a useful platform for the development of protein-based prophylactic and therapeutic vaccines. In the present study, we encapsulated purified recombinant human MUC4-beta (MUC4β) protein in polyanhydride (20:80 CPTEG:CPH) nanoparticles (MUC4β-nanovaccine) and evaluated its ability to activate dendritic cells and induce adaptive immunity. Immature dendritic cells when pulsed with MUC4β-nanovaccine exhibited significant increase in the surface expressions of MHC I and MHC II and costimulatory molecules (CD80 and CD86), as well as, secretion of pro-inflammatory cytokines (IFN-γ, IL-6, and IL-12) as compared to cells exposed to MUC4β alone or MUC4β mixed with blank nanoparticles (MUC4β+NP). Following immunization, as compared to the other formulations, MUC4β-nanovaccine elicited higher IgG2b to IgG1 ratio of anti-MUC4β-antibodies suggesting a predominantly Th1-like class switching. Thus, our findings demonstrate MUC4β-nanovaccine as a novel platform for PC immunotherapy.
Keywords: MUC4, pancreatic cancer, cancer vaccine, immunotherapy, nanoparticle
INTRODUCTION
Pancreatic cancer (PC) has a dismal prognosis with an overall survival rate of 8%, due to the limited efficacy of existing treatment modalities including surgery, chemotherapy, and radiation [1, 2]. Furthermore, PC has an elaborate immunosuppressive microenvironment comprised of high desmoplasia, immune-suppressive cells and an anti-inflammatory cytokine milieu [3]. Due to the high level of chemotherapy-induced toxicity, PC patients seldom benefit from chemotherapy. Recent studies have shown that immunotherapy-based strategies like cancer vaccines can provide therapeutic benefit by breaking the immunological tolerance to self-derived tumor associated antigens (TAAs) and overcoming immunosuppression, thereby improving the overall survival and quality of life [4, 5]. However, the development of efficacious anti-cancer vaccines is arduous due to the challenges in finding TAAs, as the majority of these antigens behave as “self”, and therefore, are immunologically ignored by the host immune system [4].
Mucins are high molecular-weight glycoproteins that are overexpressed on various epithelial surfaces for protection and lubrication. Several mucins are aberrantly overexpressed in pancreatic cancer where they play tumor-promoting role. Due to their aberrant expression and glycosylation, functional involvement in the pathogenesis and correlation with poor prognosis, mucin family members have emerged as ideal TAAs for PC and are currently being exploited for cancer immunotherapy [6]. Mucin1 (MUC1) is one of the well-studied targets for cancer vaccine development [7]. MUC1 peptide and glycopeptide-based vaccine studies have shown their potential in eliciting anti-tumor responses in various malignancies [8–13]. However, the limited immunogenic epitopes provided by peptide-based MUC1 vaccines have achieved suboptimal clinical success in PC patients [10, 14, 15]. Unlike MUC1, Mucin4 (MUC4) is undetectable in normal pancreatic tissue and its expression progressively increases with PC progression [16]. MUC4 is putatively cleaved at a Gly-Asp-Pro-His (GDPH) site in an autocatalytic manner, generating two subunits: a large N-terminal subunit called MUC4α that contains the characteristic tandem repeat domain, and a smaller membrane-tethered subunit termed MUC4β [17–19]. The MUC4β region is considered functionally important as it has 3 EGF-like domains that interact with HER-2 and promote cancer cell proliferation [6, 19, 32]. In a previous study, it was shown that the mice immunized with MUC4 glycopeptides conjugated to tetanus toxoid induced strong immune responses and predominantly produced IgG1 antibodies [20]. However, such “cherry-picked” immuno-dominant peptides limit the epitopes that can be employed to elicit immune responses in an unbiased manner, and thus are of limited translational value. While the large size of MUC4 can potentially provide a large epitope repertoire for eliciting potent immune responses, the production and purification of intact megadalton MUC4 protein is challenging. To circumvent these problems, this study investigated the utility of recombinant MUC4β subunit for tumor vaccine development.
One of the major challenges of vaccine delivery vehicles is to ensure protein stability and release over a sustained period [21, 22]. Amphiphilic polyanhydride nanoparticles (NPs), composed of 1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane (CPTEG) and 1,6-bis(p-carboxyphenoxy) hexane (CPH) have been shown to stabilize the structure and activity of encapsulated proteins while providing sustained release via a surface erosion mechanism [23, 24]. Furthermore, these NPs have been shown to be readily internalized by antigen presenting cells (APCs), such as dendritic cells (DCs) and macrophages, leading to the upregulation of cell surface activation markers including major histocompatibility complexes class I and II (MHC I and MHC II), co-stimulatory molecules (CD80, CD86, CD40), secretion of inflammatory cytokines and generation of humoral responses [25–28].
In the present study, we encapsulated endotoxin-free recombinant human MUC4β in 20:80 CPTEG:CPH NPs (MUC4β-nanovaccine). The relationships between antigen release kinetics, the ability of MUC4β-nanovaccine to activate APCs, and the nature of immune responses elicited were investigated. These studies demonstrated that the MUC4β-nanovaccine activated DCs, and induced a Th1 type of immune response. It was further observed that MUC4β-nanovaccine-immunized mice produced more IgG2b anti-MUC4β antibodies than IgG1 antibodies, suggesting that MUC4β-nanovaccine induces sufficient IFN-γ to promote antibody isotype switching consistent with a Th1-like immune response. Therefore, the recombinant human MUC4β-based polyanhydride nanovaccine has the potential to be an effective immunotherapeutic modality against PC and other MUC4-overexpressing malignancies.
RESULTS
Encapsulation of MUC4β into polyanhydride NPs provides sustained antigen release
The 20:80 CPTEG:CPH NPs loaded with 3% wt/wt MUC4β (endotoxin level < 0.01EU/mg) were synthesized via solid-oil-oil double emulsion. Scanning electron microscopy showed the NPs to be relatively spherical with a geometric mean diameter of 147 nm (with a geometric standard deviation of 1.3) (Figure 1A). The release kinetics of MUC4β from 20:80 NPs showed a burst of approximately 20% at early time points followed by slow and sustained release of protein (Figure 1B). The data showed that after four days, the 20:80 CPTEG:CPH particles released 23% of the encapsulated protein in a near-zero order release profile, which was consistent with previous work on protein release kinetics from CPTEG:CPH polyanhydride formulations [23, 24, 28–30]. Finally, the encapsulation efficiency of the MUC4β was determined to be 32 ± 1%.
Figure 1. Encapsulation and release kinetics of MUC4β-nanovaccine.

Endotoxin-free recombinant MUC4β protein was isolated from E. coli RosettaTM 2 (DE3) strain bacteria and purified by affinity chromatography. 20:80 CPTEG:CPH NPs encapsulating 3% MUC4β were synthesized via a solid/oil/oil double emulsion flash nanoprecipitation process. A. Scanning electron microscope images of 20:80 CPTEG:CPH NP-encapsulated recombinant : MUC4β. B. Antigen release kinetics were characterized by incubating the NPs in PBS and measuring MUC4β released at regular intervals with a microBCA assay. 3% MUC4β-loaded 20:80 CPTEG:CPH NPs exhibited an initial burst (20%) release of protein followed by sustained release. The encapsulation efficiency of protein was determined to be ~32%.
MUC4β-nanovaccine enhances the surface expression of MHC and co-stimulatory molecules on DCs
While the functional role of MUC4 in PC pathobiology has been studied extensively [16, 31–38], the utility of MUC4 as a candidate for vaccine development remains to be explored systematically. To examine the antigenicity of MUC4β and characterize the potential of MUC4β-nanovaccine in activating mature DCs, flow cytometry was used to measure the expression of cell surface markers such as MHC II and MHC I, co-stimulatory molecules CD80 and CD86, CD40, and C-type lectin CD205 (DEC-205, a DC maturation marker). After 9 days of IL-4 and GM-CSF treatment, more than 60% of the bone marrow-derived cells in culture differentiated into CD11c+ DCs. MUC4β-nanovaccine resulted in significantly higher activation of DCs as compared to other treatments as shown by the increased levels of costimulatory marker CD86 on the CD11c+ population (Figure 2A). There was no significant difference in the proportion of CD11c+CD86+ cells (activated DCs) across various treatments with the exception of cells treated with LPS as a positive control (Supplementary Figure 1). However, an increase in mean fluorescence intensity (MFI) indicated that the MUC4β-nanovaccine upregulated DC activation markers on the CD11c+CD86+ population as compared to the unstimulated control and other treatment groups. On the CD11c+CD86+ gated cells, there was a significant upregulation of MHC I, CD80, and CD40 following LPS treatment. As compared to the unstimulated control and other treatment groups (MUC4β alone, NP alone, and MUC4β+NP), treatment with MUC4β-nanovaccine significantly enhanced the expression of MHC I, MHC II, and CD40 (Figure 2B-2E). As compared to unstimulated control, MUC4β-nanovaccine resulted in a 1.9-fold increase in MHC I and a 1.83-fold increase in MHC II on CD11c+CD86+cells (Figure 2B-2C). Similarly, there was a significant increase in the expression of CD80 on CD11c+CD86+ cells following treatment with MUC4β-nanovaccine (2.25-fold) and MUC4β+NP (1.76-fold) as compared to untreated control, while treatment with MUC4β alone or blank NPs had no effect (Figure 2D). Further, as compared to the unstimulated control, MUC4β-nanovaccine treatment significantly enhanced the surface expression of CD40 (1.72-fold; p < 0.001) on CD11c+CD86+cells, while there was no change following treatment with MUC4β alone, blank NP or MUC4β+NP (Figure 2E). Lastly, the expression of CD205 was significantly upregulated on MUC4β-nanovaccine-stimulated DCs (2.6-fold; p < 0.001) when compared to the unstimulated control group (Figure 2F). These results suggest that the MUC4β-nanovaccine upregulates MHC I, MHC II, and costimulatory molecules and promotes antigen presentation by DCs i in vitro.
Figure 2. MUC4β-nanovaccine activates DCs and induces expression of MHC II and co-stimulatory molecules.
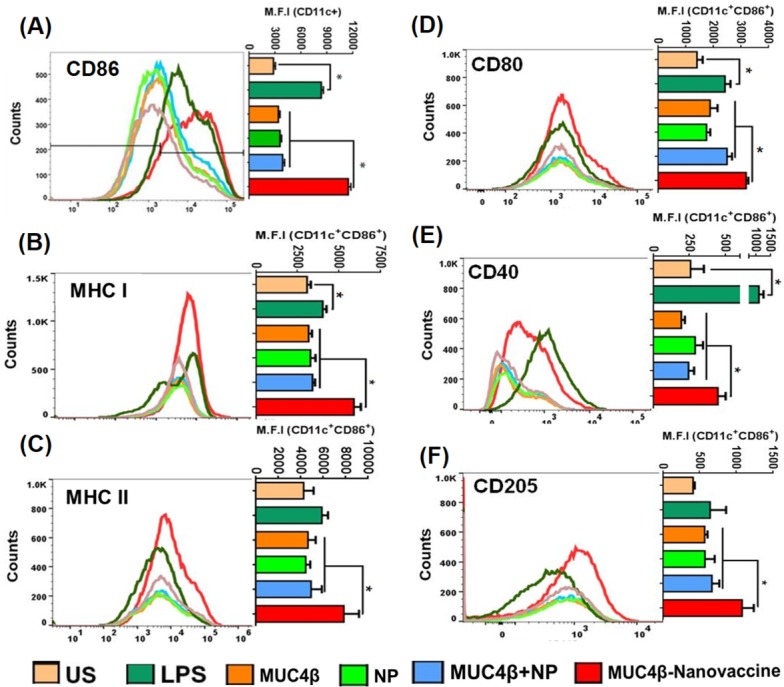
Flow cytometry analysis of in vitro antigen-pulsed DCs shows that MUC4β-nanovaccine activates DCs robustly as compared to other treatment groups. A. MUC4β-nanovaccine significantly upregulates CD86 expression in the CD11c+ population as analyzed by comparing the Mean Fluorescence Intensity (MFI) among all the treatment groups. LPS served as a positive control in the experiment, and unstimulated (US) DCs as negative control. B-F. Analysis of costimulatory and activation markers in CD11c+CD86+ cells. MUC4β-nanovaccine treatment upregulated the expression levels of MHC I (B), MHC II (C), CD80 (D), CD40 (E), and CD205 (F) after 48 h of stimulation using MUC4β protein (3 μg/mL), NP (100 μg/mL), or MUC4β+NP and MUC4β-nanovaccine (100 μg/mL). The fluorophore-conjugated antibodies were used at a 1:300 dilution except anti-CD40 antibody, which was used at a 1:200 dilution. The representative histograms for each surface marker have been presented with adjacent bar graphs showing the post-treatment MFI change. Each histogram plot represents a single experiment and the MFI plots summarize all experiments (n = 3). One-way ANOVA was used for data comparison for each group and pair wise comparison was performed using Student's paired t-test. The statistical significance was set at p < 0.001.
MUC4β-nanovaccine induces pro-inflammatory DC cytokine secretion
DCs direct immune responses not only by interacting with lymphocytes and presenting antigen [39, 40], but also by secreting an array of cytokines that modulate these responses. The supernatant of DCs cultured with MUC4β-nanovaccine contained significantly higher levels of IL-12p40, IL-6, and IFN-γ as compared to supernatants from untreated DCs or DCs stimulated with MUC4β alone or MUC4β+NP (Figure 3). The amounts of IL-6 and IL-12/ IL-23p40 in culture supernatants treated with MUC4β-nanovaccine were 2-fold and 1.5-fold higher, respectively, as compared to the unstimulated control (Figure 3A & 3B). The level of IFN-γ after MUC4β-nanovaccine treatment was found to be 1.25-fold higher than unstimulated control groups (Figure 3C). DCs treated with MUC4β alone or MUC4β+NP expressed low levels of cytokines, which were not significantly different than the levels expressed by unstimulated DCs. Similar to enhanced surface expression of MHC II and co-stimulatory molecules, DCs secreted higher levels of cytokines when treated with encapsulated MUC4β in 20:80 CPTEG:CPH NPs.
Figure 3. MUC4β-nanovaccine induces Th1 DC cytokine secretion.

Cytokine analysis demonstrated that MUC4β-nanovaccine-activated DCs secrete Th1 cytokines. Free MUC4β mixed with empty NPs (MUC4β+NP)-pulsed DCs and MUC4β -pulsed DCs produced Th1 cytokines at levels compared to the unstimulated (US) DCs. Only encapsulation of MUC4β could modulate and reprogram DCs to secrete higher levels of Th1 cytokines IL-6 (A), IL-12/IL-23p40 (B), and IFN-γ (C). For each treatment group, the sample size was n = 4. Statistical significance was set at p < 0.05. ANOVA analysis of the data was p < 0.05. Statistical comparison between MUC4β, MUC4β+NP and US with MUC4β-nanovaccine is denoted by #, * & + respectively.
Immunization with MUC4β-nanovaccine elicits robust anti-MUC4 humoral responses
Polyanhydride nanovaccines have been shown to induce the formation of germinal center B-cells that result in sustained serum antibody responses after a single dose [41]. The presence of high levels of antigen-specific IgG2b over IgG1 indicates isotype-switching mediated by IFN-γ, whereas a low IgG2b:IgG1 ratio is indicative of Th2-like T-cell-response [42]. To investigate if immunization with MUC4β-nanovaccine induced robust humoral immune responses, animals were immunized subcutaneously twice (days 0 and 14) following the prime-boost regimen with MUC4β-nanovaccine or various control formulations (MUC4β emulsified in Freuend's adjuvant or MUC4β free protein mixed with empty NPs). Five days after the booster, the anti-MUC4β titer was 16,000 in the sera of mice that were immunized with the three formulations containing MUC4β. However, the reactivity was comparable and higher in mice administered with MUC4β emulsified in Freund's adjuvant (FA) and MUC4β-nanovaccine as compared to the mice that received a mixture of free protein and blank NPs (Figure 4A). Importantly, mice immunized with the MUC4β-nanovaccine demonstrated a higher IgG2b:IgG1 ratio than mice immunized with MUC4β+FA (Figure 4B). These results indicate that MUC4β protein encapsulated in 20:80 CPTEG:CPH NPs predominantly elicits a Th1-type immune response.
Figure 4. MUC4β-nanovaccine generates anti-MUC4β Th1 humoral response in mice.

A. Titers of anti-MUC4β antibodies were determined by ELISA in the sera of mice immunized with various formulations, and suggest that MUC4β emulsified in Freund's adjuvant (MUC4β +FA), or mixed with empty nanoparticles (MUC4β+NP), or encapsulated in the nanoparticles (MUC4β-nanovaccine)-elicited anti-MUC4 antibodies. B. Isotyping of anti-MUC4β antibodies (at 1:1000 dilution) demonstrates that isotype switching was predominantly to Th1 IgG2b in MUC4β-nanovaccine-immunized mice, whereas in MUC4β-immunized mice it was predominantly the Th2 IgG1 isotype. For each treatment group sample size was n = 3. Statistical significance was set at p < 0.05. ANOVA analysis of the data was p < 0.05. Statistical comparison between MUC4β+FA and MUC4β-nanovaccine is denoted by #.
Immunization with MUC4β-nanovaccine enhances the circulating levels of inflammatory cytokines
Next, the presence of pro-inflammatory cytokines (IL-6, IL-12, and IFN-γ) in the sera of immunized mice was investigated. Sera from MUC4β-nanovaccine-immunized mice had significantly higher levels of IL-6 compared to MUC4β+FA- and MUC4β+NP-immunized mice (Figure 5A). The sera from MUC4β-nanovaccine-immunized mice had significantly higher amounts of IL-12/IL-23p40 (~2.7-fold increase) and inflammatory cytokine IFN-γ (~5.2-fold increase) when compared to PBS-treated control mice (Figure 5B, 5C). Similarly, as compared to sera from the mice immunized with MUC4β+FA, we observed a significant increase of IL-12/ IL-23p40 (~2.2-folds) and IFN-γ (~4.5-folds) cytokines in the sera of MUC4β-nanovaccine-immunized mice (Figure 5B, 5C). Significant upregulation of IFN-γ, as compared to control group as well as other treatment groups (Figure 5C), suggests that MUC4β-nanovaccine promotes Th1 mediated isotype switching of antibodies in these mice.
Figure 5. Presence of pro-inflammatory cytokines in the sera of mice immunized with MUC4β-nanovaccine.

Levels of IL6 (A), IL-12 (B) and IFN-γ (C) in the sera of mice with immunied with various formulations. The sera of MUC4β-nanovaccine-immunized mice had significantly higher levels of IL6, IL-12/IL23p40, and IFN-γ as compared to mice immunized with MUC4β+FA, and MUC4β+NP. For each treatment group, the sample size was n = 3. Statistical significance was set at p < 0.05. ANOVA analysis of the data was p < 0.05. Statistical comparison between MUC4β+FA, and MUC4β+NP with MUC4β-nanovaccine is denoted by # & * respectively.
DISCUSSION
To date vaccine development involving mucins has been based on selected peptides that have a limited repertoire of immunogenic epitopes and have predominantly focused on the tandem repeat domains which are typically O-glycosylated. Recombinant proteins could address these limitations by presenting the entire spectrum of possible epitopes present in the original antigens in an unbiased manner [3]. In this study, the β-subunit of MUC4 expressed and purified from a bacterial expression system was used to investigate its potential as an effective immunogen. The data presented show that free MUC4β alone induces relatively low expression of MHC I and MHC II complexes, co-stimulatory molecule CD80 (Figure 2B-2D), and inflammatory cytokines in DCs (Figure 3A-3C) in vitro. Immunization of mice with MUC4β in combination with Freund's adjuvant likely induces a Th-2 like response based on the induction of significantly higher levels of IgG1 antibodies (Figure 4). Th2 immune responses have been well established to promote tumor pathogenesis and aggressiveness, whereas shifting the immune response to Th1 phenotype provides anti-tumor protection [43–45]. In this study, MUC4β was encapsulated into 20:80 CPTEG:CPH nanoparticles (MUC4β-nanovaccine) in order to investigate whether the MUC4β-nanovaccine could enhanced activation of DCs and modulated Th1-mediated humoral responses.
The MUC4β-nanovaccine enhanced surface expression of MHC I and MHC II on CD86+CD11c+ DCs (Figure 2B-2C), which are implicated in presentation of antigen, and increased the surface expression of CD80 (Figure 2D), a co-stimulatory molecule required for activation T-cells. Antigen that is taken up via CD205 enters the MHC I and MHC II antigen presentation pathways, and thus an increase in CD205 expression enhances antigen presentation by DCs to both CD8+ cytotoxic T-cells and CD4+ helper T-cells and could potentially lead to long-lived immunity by specifically targeting the cancer cells. [46, 47]. MUC4β-nanovaccine significantly enhanced surface expression of CD205 on DCs (Figure 2F) that corresponded with increased expression of MHC complexes on these DCs. Thus, upregulation of these markers by MUC4β-nanovaccine, in comparison to free MUC4β-pulsed DCs, suggests that the encapsulated formulation was better in potentiating antigen presentation by DCs. Further, it was observed that only stimulation with MUC4β-nanovaccine enhanced DC secretion of the pro-inflammatory cytokines IL-6, IL-12/IL-23p40, and IFN-γ in vitro (Figure 3) as compared to free MUC4β protein or MUC4β mixed with empty NPs (MUC4β+NP), which corroborated the observed alterations in the expression of DC activation markers. These results indicate that the encapsulation of MUC4β protein in CPTEG:CPH NPs is crucial in stimulating a cytokine profile that would enhance a Th1-like immune response.
Previously, it has been shown that a single immunization of polyanhydride nanovaccines can induce high antibody titers and provide protective immunity against Yersinia pestis in mice [29, 48]. Additionally, it is important to consider the quality of the antibody response generated by the nanovaccine, which may be characterized by the specificity, avidity and isotype profile of the antibody response [49]. It is therefore noteworthy that the MUC4β-nanovaccine-immunized mice had the highest IgG2b:IgG1 ratio, which indicates induction of isotype class switching, whereas MUC4β+FA or MUC4β in combination with blank NPs preferentially induced IgG1 anti-MUC4β antibodies (Figure 4). This supports our in vitro observation that encapsulation of MUC4β into polyanhydride NPs likely plays a crucial role in activating DCs to produce cytokines (IL-6, IL-12/ IL-23p40 and IFN-γ) (Figure 3) that would polarize the immune response towards a Th1 phenotype. This was further validated with the detection of higher levels of IFN-γ in the sera of MUC4β-nanovaccine-immunized mice (Figure 5C). The data herein shows encapsulating MUC4β into 20:80 CPTEG: CPH NPs is an effective strategy to activate DCs and modulate the response towards an anti-MUC4 Th1 phenotype and potentially generate antigen-specific cytotoxic CD8+ T-cells. IgG2b antibodies may possibly provide additional immunity against MUC4-expressing tumors by inducing antibody-dependent-cellular cytotoxicity [50, 51]. The current studies provides a basis for investigating the use of the MUC4β-nanovaccine as an immunotherapeutic strategy in cancer models that overexpress MUC4 as a tumor-associated antigen.
MATERIALS AND METHODS
MUC4β purification
The cDNA sequence encoding the 733 amino acids (2199 base pairs) of human MUC4β fragment was cloned into the bacterial expression vector pET-28a (Novagen, USA), and transformed into the E. coli RosettaTM 2 (DE3) strain. Bacterial culture was grown under standard conditions and MUC4β was purified by AKTA Ni-NTA affinity chromatography. Eluted fractions (in 6M urea) containing the recombinant MUC4β protein were assessed with SDS-PAGE gels and further confirmed with immunoblotting. All the verified fractions were pooled and concentrated using Amicon ultracentrifugal filters (50 kDa molecular weight cut-off). A step-wise dialysis was performed against a decreasing concentration of urea in 1Χ PBS to allow the protein refolding. Purified fractions were passed through an endotoxin removal spin column (Pierce). Finally, the concentration of our target protein and the level of endotoxins in the samples were measured using the BCA Protein Assay Kit (Pierce Thermo Fisher) and the Pierce LAL Chromogenic Endotoxin assay kit, respectively.
Encapsulation of MUC4β in polyanhydride nanoparticles
The 20:80 CPTEG:CPH polymer was synthesized via melt polycondensation [24]. Next the nanoparticles encapsulating MUC4β were synthesized using a solid-oil-oil double emulsion technique as described previously [48]. Briefly, purified MUC4β was dialyzed to nanopure water and lyophilized. Next, 20:80 CPTEG:CPH polymer containing 3% wt. of MUC4β was dissolved at 20 mg/ mL in methylene chloride. The solution was sonicated for 30 s to ensure even distribution of the protein. The nanoparticles were precipitated into chilled pentane (−10°C; 1:250 methylene chloride: pentane) and collected via vacuum filtration. NP morphology was verified by scanning electron microscopy (FEI Quanta 250, FEI, Hillsboro, OR) and NP size subsequently analyzed with ImageJ (ImageJ 1.48v, NIH).
The release kinetics of MUC4β from 20:80 CPTEG:CPH nanoparticles were monitored as previously described [23]. Briefly, NPs were incubated in PBS at 37°C. Periodically, the samples were centrifuged, supernatant collected, and particles were resuspended in fresh buffer. The amount of protein in the collected supernatant was quantified using a microBCA assay. Further, the encapsulation efficiency was determined by comparing the total amount of protein released to the amount theoretically encapsulated. Briefly, at the end of approximately one month, the buffer was exchanged with 40 mM sodium hydroxide to quickly degrade the NPs and release any remaining protein, which was estimated using microBCA assay.
Primary dendritic cells (DCs) isolation
C57BL/6 mice were kept under specific pathogen-free conditions at the UNMC Animal Facility in accordance with UNMC Institutional Animal Care and Use Committee (IACUC) standards. Femurs and tibiae of 6-8 week old C57BL/6 mice were removed and the marrow was flushed in RPMI medium using a 1 mL syringe attached to a 25 G needle. Cells were centrifuged at 2000 rpm for 2.5 min and the cell pellet was resuspended in 10 mL of 1x RBC lysis buffer in the dark for 5 min. Ten mL RPMI medium was added to stop the lysis. Cells were centrifuged at 2000 rpm and washed 3 times in 10 mL of RPMI medium. After the last wash, cells were resuspended homogeneously in a 1 bone: 1 mL of media ratio. Resuspended cells were passed through a cell strainer to remove clumps. Cell suspensions were poured into 10 mL RPMI medium containing 10% FBS, penicillin (100 U/mL, Sigma) and streptomycin (100 mg/mL, Sigma) in a 10 cm tissue culture plate that was then kept in an incubator for 3 h. After incubation, all nonadharent cells were collected and the plate was washed with medium twice to collect any remaining nonadharent cells into a 50 mL tube. The cells were pelleted down by centrifugation and the medium aspirated. The cells were resuspended in 10 mL complete RPMI medium containing 50 μM β-merceptoethanol (β-ME), 1 mM sodium pyruvate, and 10 mM HEPES and transferred into T75 flasks for our in vitro studies.
DC maturation and pulsing
To generate bone marrow derived DC population, we added 50 ng/mL of recombinant mouse (rm) GM-CSF and 25 ng/mL rm-IL-4 reconstituted and diluted in serum-free RPMI medium to freshly isolated DC cultures on Day 0. On Day 3, 5, and 7, the non-adherent cells were collected in 50 mL centrifuge tubes, pelleted them and re-suspended in a total volume of 10 mL of fresh RPMI medium containing 10% FBS, penicillin and streptomycin antibiotics, 10 mM HEPES, 1 mM sodium pyruvate, 50 μM β- ME, 50 ng/mL of GM-CSF and 25 ng/mL IL-4 in a fresh 6-well plate. At Day 9, immature DCs were counted and seeded in a 24 well plate for activation studies. Polyanhydride NPs were suspended in complete culture medium, sonicated briefly (30 s on ice), and added to the DC cultures at Day 9 at a concentration of 100 μg/mL. DCs were pulsed in the following groups: 3 μg/mL free MUC4β protein, free MUC4β protein (3 μg/mL) mixed with blank NPs (100 μg/mL) (MUC4β+NP) and MUC4β-nanovaccine (100 μg/mL). Unstimulated DCs (US) and DCs stimulated with LPS (200 ng/mL) were used as negative and positive controls, respectively. Cultures were incubated for 48 h (37°C, 5% CO2). Activated DCs were harvested from the 24-well plate and centrifuged at 1000 x g for 5 min to collect the culture medium and the pellet was processed further for flow cytometry studies. Supernatant was collected for ELISA studies from each treatment group.
Flow cytometry of activated DCs
DCs were resuspended and washed 3 times in FACS buffer containing 1xPBS (pH 7.2) and 1% fetal bovine serum (FBS) to remove any residual culture medium. After that, the 1X105 DCs were resuspended in a 100 μL volume of conjugated antibody cocktail for detection of DC surface markers consisting of antibodies recognizing CD11c, MHC I, MHC II, CD80, CD86, and CD205 and prepared at a 1:300 dilution except anti-CD40 antibody, which was used at 1:200 dilution in FACS Buffer. Corresponding isotype controls were also prepared at similar dilutions in FACS buffer. Following incubation with antibodies for 60 min on ice (4°C) in the dark, the cells were washed thrice with FACs buffer, and fixed with 4% paraformaldehyde for 15 min at RT. The cells were washed again and analyzed using a BD LSR-II Green Flow Cytometer (BD Biosciences). FlowJo® and BD FACSDIVA software were used to analyze the data.
Cytokine analysis by ELISA
Supernatants were preserved at −80°C. IL-6, IFN-γ and IL-12/IL-23 p40 cytokines were measured by enzyme-linked immunosorbent assay (ELISA) kits from BioLegend and the manufacturer's protocol was followed for the assay. 96-well ELISA strips were coated with capture antibodies (1:200 dilution) in carbonate-bicarbonate buffer (0.5M, pH 9.6) and incubated overnight at 4°C. The next day coated strips were washed 4 times with 1xPBST (1x PBS and 0.05% Tween 20) and blocked with 3% bovine serum albumin (BSA) in PBS for 2 hours at 37°C. Strips were then washed 4 times and 100 μL of supernatants were added to coated strips and incubated for 2 hours at 37°C. Plates were washed with PBST for 4 times, followed by incubation with detection antibody (1:200) for 1 hour at 37°C. Excess secondary antibody was washed away with 4 PBST washes. Avidin (1:1000) was added to ELISA strips and incubated for 30 min at RT in dark. Excess avidin was removed with 5 PBST washes, followed by addition of TMB (3,3′,5,5′-Tetramethylbenzidine) substrate in the dark and the strips were incubated at RT for color to develop. Absorbance was measured after the reaction was stopped (~15 min) with 1N H2SO4 at 450 nm using a SpectraMax® Plus384 microplate reader (Molecular Devices LLC, Sunnyvale, California). For serum cytokines analysis, serum samples were collected post-euthanasia and stored at −80°C. Serum samples were added at 1:10 dilution to capture-antibody coated plates for cytokine analysis.
Mice immunization
Eight-week old C57BL/6 mice were immunized subcutaneously with various formulations of recombinant MUC4β protein emulsified with Freund's incomplete adjuvant (MUC4β+FA: 20 μg/mouse/dose), encapsulated MUC4β protein (MUC4β-nanovaccine: 20 μg protein+300 μg NP/mouse/dose), protein plus empty NPs (MUC4β+NP: 20 μg MUC4β+300 μg empty NP / mouse/dose), and PBS+ Freund's adjuvant control. A booster immunization was given 2 weeks after the primary immunization and blood was collected from the submandibular vein after 5 days and processed for serum preparation.
Anti-MUC4β antibodies detection in immunized mice sera by ELISA
To detect anti-MUC4β antibodies in the sera of immunized mice, we used a modified ELISA protocol. A 96-well plate was coated with 5 μg/mL of recombinant MUC4β protein in carbonate-bicarbonate coating buffer (0.5 M, pH 9.6) and incubated overnight at 4°C. The plate was washed 2 times with 1xPBST and blocked with 3% BSA in PBS for 3 hours at 37°C. Further, the plate was washed 4 times and then serial dilutions of the serum samples were incubated in a MUC4β-coated plate for 2 hours at 37°C. Plates were washed with PBST 4 times, followed by incubation with horseradish-peroxidase (HRP) conjugated goat anti-mouse IgG (total H+L) (Thermo Fisher), IgG1 (Abcam) or IgG2b (Abcam) for 1 hour at 37°C. Excess secondary antibody was washed away with 5 PBST washes followed by the addition of TMB substrate in the dark. Absorbance was measured after the reaction was stopped (at ~15 min) with 1N H2SO4 at 450 nm using a SpectraMax® Plus384 microplate reader (Molecular Devices LLC, Sunnyvale, California).
Antibodies
Alexa Fluor® 700 anti-mouse CD11c (clone N418), anti-Mouse MHC Class I (H-2Kb) eFluor® 450 (Clone: AF6-88.5.5.3), FITC conjugated anti-mouse/rat MHC Class II (I-A/I-E; clone M5/114.15.2), APC anti-mouse CD40 (clone 1C10), PE conjugated anti-mouse CD80 (clone 16-10A1) antibodies with their respective isotype controls comprising of Alexa Fluor® 700 conjugated Armenian hamster IgG (clone eBio299Arm), FITC rat IgG2b κ (clone eB149/10H5), Rat IgG2a k Isotype Control APC (clone eBR2a), PE-conjugated rat IgG2a (clone eBR2a), Rat IgG2a k Isotype Control PE-Cyanine7 (clone eBR2a), and mouse IgG2a k Isotype Control eFluor® 450 (clone eBM2a) were purchased from eBioscience™ (San Diego, CA). PE/Cy7™ rat anti-mouse CD86 (Clone GL1) was purchased from BD Pharmingen™. PE/Cy 5.5 anti-mouse CD205 (MMR, clone NLDC-145) and PerCP/ Cy5.5 Rat IgG2a, κ Isotype control were procured from BioLegend®.
Statistical analysis
Differences among group means were tested by one-way analysis of variance (ANOVA) F-test using GraphPad Prism v. 7.0 (GraphPad, La Jolla, CA). If the F-test was significant, Tukey's t-tests were performed for pairwise comparisons of group means. Significance was defined at p < 0.05 or p < 0.001 as indicated in the respective figure legends.
SUPPLEMENTARY FIGURE
Acknowledgments
This work and authors are supported, in part, by the following grants from the National Institute of Health: U01 CA213862, R01 CA195586, U01 CA200466, R01 CA133774, R01 CA183459, P50 CA127297, P30 CA036727, R21 CA223429, and R21 AA026428. The UNMC Flow Cytometry Research Facility is administrated through the Office of the Vice Chancellor for Research and supported by state funds from the Nebraska Research Initiative (NRI) and The Fred and Pamela Buffett Cancer Center's National Cancer Institute Cancer Center Support Grant.
Footnotes
CONFLICT OF INTEREST
Authors declare no conflict of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333–48. doi: 10.1038/s41575-018-0005-x. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee K, Kumar S, Ross KA, Gautam S, Poelaert B, Nasser MW, Aithal A, Bhatia R, Wannemuehler MJ, Narasimhan B, Solheim JC, Batra SK, Jain M. Emerging trends in the immunotherapy of pancreatic cancer. Cancer Lett. 2018;417:35–46. doi: 10.1016/j.canlet.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delitto D, Wallet SM, Hughes SJ. Targeting tumor tolerance: A new hope for pancreatic cancer therapy? Pharmacol Ther. 2016;166:9–29. doi: 10.1016/j.pharmthera.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Kajihara M, Takakura K, Kanai T, Ito Z, Matsumoto Y, Shimodaira S, Okamoto M, Ohkusa T, Koido S. Advances in inducing adaptive immunity using cell-based cancer vaccines: Clinical applications in pancreatic cancer. World J Gastroenterol. 2016;22:4446–58. doi: 10.3748/wjg.v22.i18.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia R, Gautam SK, Cannon A, Thompson C, Hall BR, Aithal A, Banerjee K, Jain M, Solheim JC, Kumar S, Batra SK. Cancer-associated mucins: role in immune modulation and metastasis. Cancer Metastasis Rev. 2019 doi: 10.1007/s10555-018-09775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres MP, Chakraborty S, Souchek J, Batra SK. Mucin-based targeted pancreatic cancer therapy. Curr Pharm Des. 2012;18:2472–81. doi: 10.2174/13816128112092472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glaffig M, Stergiou N, Schmitt E, Kunz H. Immunogenicity of a fully synthetic MUC1 glycopeptide antitumor vaccine enhanced by poly(I: C) as a TLR3-activating adjuvant. Chem Med Chem. 2017;12:722–7. doi: 10.1002/cmdc.201700254. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Wang Y, Yu F, Zhang Z, Yang Z, Zhang W, Wang PG, Zhao W. Potentiating the immune response of MUC1-based antitumor vaccines using a peptide-based nanovector as a promising vaccine adjuvant. Chem Commun (Camb) 2017;53:9486–9. doi: 10.1039/c7cc04386d. [DOI] [PubMed] [Google Scholar]
- 10.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H, Day R, Troetschel M, Finn OJ. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immunother. 2005;54:254–64. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheikl-Gatard T, Tosch C, Lemonnier F, Rooke R. Identification of new MUC1 epitopes using HLA-transgenic animals: implication for immunomonitoring. J Transl Med. 2017;15:154. doi: 10.1186/s12967-017-1254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stergiou N, Glaffig M, Jonuleit H, Schmitt E, Kunz H. Immunization with a synthetic human MUC1 glycopeptide vaccine against tumor-Associated MUC1 breaks tolerance in human MUC1 transgenic mice. Chem Med Chem. 2017;12:1424–8. doi: 10.1002/cmdc.201700387. [DOI] [PubMed] [Google Scholar]
- 13.Tosch C, Bastien B, Barraud L, Grellier B, Nourtier V, Gantzer M, Limacher JM, Quemeneur E, Bendjama K, Preville X. Viral based vaccine TG4010 induces broadening of specific immune response and improves outcome in advanced NSCLC. J Immunother Cancer. 2017;5:70. doi: 10.1186/s40425-017-0274-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kondo H, Hazama S, Kawaoka T, Yoshino S, Yoshida S, Tokuno K, Takashima M, Ueno T, Hinoda Y, Oka M. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Res. 2008;28:379–87. [PubMed] [Google Scholar]
- 15.Rong Y, Qin X, Jin D, Lou W, Wu L, Wang D, Wu W, Ni X, Mao Z, Kuang T, Zang YQ, Qin X. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin Exp Med. 2012;12:173–80. doi: 10.1007/s10238-011-0159-0. [DOI] [PubMed] [Google Scholar]
- 16.Singh AP, Moniaux N, Chauhan SC, Meza JL, Batra SK. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004;64:622–30. doi: 10.1158/0008-5472.can-03-2636. [DOI] [PubMed] [Google Scholar]
- 17.Moniaux N, Escande F, Batra SK, Porchet N, Laine A, Aubert JP. Alternative splicing generates a family of putative secreted and membrane-associated MUC4 mucins. Eur J Biochem. 2000;267:4536–44. doi: 10.1046/j.1432-1327.2000.01504.x. [DOI] [PubMed] [Google Scholar]
- 18.Moniaux N, Escande F, Porchet N, Aubert JP, Batra SK. Structural organization and classification of the human mucin genes. Front Biosci. 2001;6:D1192–206. doi: 10.2741/moniaux. [DOI] [PubMed] [Google Scholar]
- 19.Moniaux N, Nollet S, Porchet N, Degand P, Laine A, Aubert JP. Complete sequence of the human mucin MUC4: a putative cell membrane-associated mucin. Biochem J. 1999;338(2):325–33. [PMC free article] [PubMed] [Google Scholar]
- 20.Cai H, Palitzsch B, Hartmann S, Stergiou N, Kunz H, Schmitt E, Westerlind U. Antibody induction directed against the tumor-associated MUC4 glycoprotein. Chembiochem. 2015;16:959–67. doi: 10.1002/cbic.201402689. [DOI] [PubMed] [Google Scholar]
- 21.Bilati U, Allemann E, Doelker E. Strategic approaches for overcoming peptide and protein instability within biodegradable nano- and microparticles. Eur J Pharm Biopharm. 2005;59:375–88. doi: 10.1016/j.ejpb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Bilati U, Allemann E, Doelker E. Nanoprecipitation versus emulsion-based techniques for the encapsulation of proteins into biodegradable nanoparticles and process-related stability issues. AAPS PharmSciTech. 2005;6:E594–604. doi: 10.1208/pt060474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres MP, Determan AS, Anderson GL, Mallapragada SK, Narasimhan B. Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials. 2007;28:108–16. doi: 10.1016/j.biomaterials.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J Biomed Mater Res A. 2006;76:102–10. doi: 10.1002/jbm.a.30510. [DOI] [PubMed] [Google Scholar]
- 25.Chavez-Santoscoy AV, Roychoudhury R, Pohl NL, Wannemuehler MJ, Narasimhan B, Ramer-Tait AE. Tailoring the immune response by targeting C-type lectin receptors on alveolar macrophages using “pathogen-like” amphiphilic polyanhydride nanoparticles. Biomaterials. 2012;33:4762–72. doi: 10.1016/j.biomaterials.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 26.Haughney SL, Ross KA, Boggiatto PM, Wannemuehler MJ, Narasimhan B. Effect of nanovaccine chemistry on humoral immune response kinetics and maturation. Nanoscale. 2014;6:13770–8. doi: 10.1039/c4nr03724c. [DOI] [PubMed] [Google Scholar]
- 27.Petersen LK, Ramer-Tait AE, Broderick SR, Kong CS, Ulery BD, Rajan K, Wannemuehler MJ, Narasimhan B. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32:6815–22. doi: 10.1016/j.biomaterials.2011.05.063. [DOI] [PubMed] [Google Scholar]
- 28.Torres MP, Wilson-Welder JH, Lopac SK, Phanse Y, Carrillo-Conde B, Ramer-Tait AE, Bellaire BH, Wannemuehler MJ, Narasimhan B. Polyanhydride microparticles enhance dendritic cell antigen presentation and activation. Acta Biomater. 2011;7:2857–64. doi: 10.1016/j.actbio.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joshi VB, Geary SM, Carrillo-Conde BR, Narasimhan B, Salem AK. Characterizing the antitumor response in mice treated with antigen-loaded polyanhydride microparticles. Acta Biomater. 2013;9:5583–9. doi: 10.1016/j.actbio.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haughney SL, Petersen LK, Schoofs AD, Ramer-Tait AE, King JD, Briles DE, Wannemuehler MJ, Narasimhan B. Retention of structure, antigenicity, and biological function of pneumococcal surface protein A (PspA) released from polyanhydride nanoparticles. Acta Biomater. 2013;9:8262–71. doi: 10.1016/j.actbio.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bafna S, Kaur S, Momi N, Batra SK. Pancreatic cancer cells resistance to gemcitabine: the role of MUC4 mucin. Br J Cancer. 2009;101:1155–61. doi: 10.1038/sj.bjc.6605285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, Singh PK, Hollingsworth MA, Mehta PP, Batra SK. MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Res. 2008;68:2065–70. doi: 10.1158/0008-5472.CAN-07-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gautam SK, Kumar S, Cannon A, Hall B, Bhatia R, Nasser MW, Mahapatra S, Batra SK, Jain M. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin Ther Targets. 2017;21:657–69. doi: 10.1080/14728222.2017.1323880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaur S, Sharma N, Krishn SR, Lakshmanan I, Rachagani S, Baine MJ, Smith LM, Lele SM, Sasson AR, Guha S, Mallya K, Anderson JM, Hollingsworth MA, et al. MUC4-mediated regulation of acute phase protein lipocalin 2 through HER2/AKT/NF-kappaB signaling in pancreatic cancer. Clin Cancer Res. 2014;20:688–700. doi: 10.1158/1078-0432.CCR-13-2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakshmanan I, Seshacharyulu P, Haridas D, Rachagani S, Gupta S, Joshi S, Guda C, Yan Y, Jain M, Ganti AK, Ponnusamy MP, Batra SK. Novel HER3/MUC4 oncogenic signaling aggravates the tumorigenic phenotypes of pancreatic cancer cells. Oncotarget. 2015;6:21085–99. doi: 10.18632/oncotarget.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mimeault M, Johansson SL, Senapati S, Momi N, Chakraborty S, Batra SK. MUC4 down-regulation reverses chemoresistance of pancreatic cancer stem/progenitor cells and their progenies. Cancer Lett. 2010;295:69–84. doi: 10.1016/j.canlet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rachagani S, Macha MA, Ponnusamy MP, Haridas D, Kaur S, Jain M, Batra SK. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis. 2012;33:1953–64. doi: 10.1093/carcin/bgs225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senapati S, Gnanapragassam VS, Moniaux N, Momi N, Batra SK. Role of MUC4-NIDO domain in the MUC4-mediated metastasis of pancreatic cancer cells. Oncogene. 2012;31:3346–56. doi: 10.1038/onc.2011.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ni K, O'Neill HC. The role of dendritic cells in T cell activation. Immunol Cell Biol. 1997;75:223–30. doi: 10.1038/icb.1997.35. [DOI] [PubMed] [Google Scholar]
- 40.Behrens G, Li M, Smith CM, Belz GT, Mintern J, Carbone FR, Heath WR, Helper T. cells, dendritic cells and CTL Immunity. Immunol Cell Biol. 2004;82:84–90. doi: 10.1111/j.1440-1711.2004.01211.x. [DOI] [PubMed] [Google Scholar]
- 41.Vela Ramirez JE, Tygrett LT, Hao J, Habte HH, Cho MW, Greenspan NS, Waldschmidt TJ, Narasimhan B. Polyanhydride nanovaccines induce germinal center B cell formation and sustained serum antibody responses. J Biomed Nano technol. 2016;12:1303–11. doi: 10.1166/jbn.2016.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–50. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 43.Deligne C, Metidji A, Fridman WH, Teillaud JL. Anti-CD20 therapy induces a memory Th1 response through the IFN-gamma/IL-12 axis and prevents protumor regulatory T-cell expansion in mice. Leukemia. 2015;29:947–57. doi: 10.1038/leu.2014.275. [DOI] [PubMed] [Google Scholar]
- 44.Pradhan P, Qin H, Leleux JA, Gwak D, Sakamaki I, Kwak LW, Roy K. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35:5491–504. doi: 10.1016/j.biomaterials.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang CY, Chen JJ, Shen KY, Chang LS, Yeh YC, Chen IH, Chong P, Liu SJ, Leng CH. Recombinant lipidated HPV E7 induces a Th-1-biased immune response and protective immunity against cervical cancer in a mouse model. PLoS One. 2012;7:e40970. doi: 10.1371/journal.pone.0040970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shrimpton RE, Butler M, Morel AS, Eren E, Hue SS, Ritter MA. CD205 (DEC-205): a recognition receptor for apoptotic and necrotic self. Mol Immunol. 2009;46:1229–39. doi: 10.1016/j.molimm.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee H-W, Park CG, Steinman RM, Nussenzweig MC. Differential Antigen Processing by Dendritic Cell Subsets in Vivo. Science. 2007;315:107. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 48.Ulery BD, Kumar D, Ramer-Tait AE, Metzger DW, Wannemuehler MJ, Narasimhan B. Design of a protective single-dose intranasal nanoparticle-based vaccine platform for respiratory infectious diseases. PLoS One. 2011;6:e17642. doi: 10.1371/journal.pone.0017642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson-Welder JH, Torres MP, Kipper MJ, Mallapragada SK, Wannemuehler MJ, Narasimhan B. Vaccine adjuvants: current challenges and future approaches. J Pharm Sci. 2009;98:1278–316. doi: 10.1002/jps.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richards JO, Albers AJ, Smith TS, Tjoe JA. NK cell-mediated antibody-dependent cellular cytotoxicity is enhanced by tamoxifen in HER2/neu non-amplified, but not HER2/neu-amplified, breast cancer cells. Cancer Immunol Immunother. 2016;65:1325–35. doi: 10.1007/s00262-016-1885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol. 2015;6:368. doi: 10.3389/fimmu.2015.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.