

Persister cells to ofloxacin are not necessarily slow growers and do endure DNA damages, similar to their sensitive siblings.
Abstract
Bacterial persistence refers to the capacity of small subpopulations within clonal populations to tolerate antibiotics. Persisters are thought to originate from dormant cells in which antibiotic targets are less active and cannot be corrupted. Here, we report that in exponentially growing cultures, ofloxacin persisters originate from metabolically active cells: These cells are dividing before the addition of ofloxacin and do endure DNA damages during the treatment, similar to their nonpersister siblings. We observed that growth rate, DNA content, and SOS induction vary among persisters, as in the bulk of the population and therefore do not constitute predictive markers for persistence. Persister cells typically form long polynucleoid filaments and reach maximum SOS induction after removal of ofloxacin. Eventually, cell division resumes, giving rise to a new population. Our findings highlight the heterogeneity of persister cells and therefore the need to analyze these low-frequency phenotypic variants on a case-by-case basis at the single-cell level.
INTRODUCTION
Failure to eradicate bacterial infections is typically attributed to the selection of antibiotic resistance mutations or the acquisition of resistance determinants by horizontal gene transfer (1–3). Nevertheless, bacterial persistence, a less well-understood phenomenon, appears to play a pivotal role in the reoccurrence of infections and the evolution of antibiotic resistance (4–6). In contrast to resistance, persistence is not genetically acquired and relies on phenotypic heterogeneity (5). Persister cells capable of tolerating antibiotic treatment constitute small subpopulations generated at low frequency (10−2 to 10−6) within clonal populations (7). Upon removal of the antibiotic, persister cells are able to resume growth and give rise to a viable progeny that is genetically identical to the initial population.
In their pioneering work, Balaban and colleagues observed persister cells in a microfluidic device at the single-cell level using Escherichia coli mutants showing high-persistence frequency (hip mutants) (8). This paved the way to the definition of different types of persister cells: Type I persisters are thought to be triggered by stress such as stationary phase, starvation, or host environment, whereas type II persisters are considered slow- or nongrowing cells that are stochastically generated in exponentially growing populations. Persister cells are therefore commonly thought to be in a dormant, metabolically inactive state that protects them from antibiotic insults (8–10). This dormant physiological state would provide tolerance to different antibiotics, a hallmark of persistence. However, it is clear that the nondividing state is not sufficient for survival (10, 11) and that only a small fraction of dormant cells have the capacity to regrow after the antibiotic treatment (12–14). For instance, in Mycobacterium smegmatis, persister cells to isoniazid (INH) are generated by actively growing cells before the antibiotic treatment (15), and several reports highlight the involvement of active antibiotic efflux in individual persister cells (16). Persistence thus appears to be a complex phenomenon, with most likely multiple underlying molecular mechanisms, strongly depending on the experimental model and conditions and, particularly, on the class of antibiotic tested (4, 17). Whether persistence is a selected trait also remains under debate. While some groups have proposed that persistence constitutes a bet-hedging strategy involved in bacterial adaption to fluctuating environments (18, 19), Levin and co-workers proposed that persistence is rather the product of different kinds of errors like mutations are [PASH (persistence as stuff happens) hypothesis] (20, 21).
In the case of E. coli persistence to fluoroquinolones and in line with the dormancy hypothesis, persister cells were initially thought to consist of a subpopulation of cells experiencing spontaneous errors of DNA synthesis leading to induction of the SOS response and to growth arrest (22). Preexistence of a subpopulation of nongrowing persister cells was later refuted by the Lewis group, as they found that persister cells were formed upon exposure to ciprofloxacin, via the induction of the SOS response (23, 24). They showed that in addition to SOS-dependent DNA repair mechanisms, the TisB pore-forming toxin, belonging to the type I tisAB toxin-antitoxin system, is required for persistence. Expression of TisB leads to the decrease of the proton motive force and adenosine triphosphate levels (25). Persistence to fluoroquinolones is therefore quoted as drug-induced persistence and does not appear to rely on a stochastic switch (23). However, in stationary phase cultures, persistence to fluoroquinolones was later found to be independent of TisB and rather requires DNA repair mechanisms during the recovery phase, after antibiotic removal (26).
An important drawback of all precited studies on persistence to fluoroquinolones and of most persistence studies in general is that they describe the persistence phenomenon at the population level by the use of time-kill curves or flow cytometry techniques (26–29). However, because of the scarce and unstable nature of persister cells, population-based approaches fail to capture the specificity of these cells as their signal is diluted in the bulk of nonpersister cells. Persister cells cannot be isolated from the bulk of nonpersister cells, as no specific markers for persistence have been identified. A few studies managed to enrich the fraction of persister cells by sorting and enrichment of dormant cells (11, 14). However, persister cells were found to be scarce even among such dormant populations, as more than 99% of these cells remained antibiotic sensitive upon cell sorting (11). Therefore, observation of persister cells at the single-cell level appears to be the most appropriate approach. There are only a few reports regarding the observation of persister cells at the single-cell level. However, experiments either were conducted on high-persistent mutants (8) or were later shown to be problematic, as discussed in (30–33). As a result, the very concept of stochastic switching and the fate of the resulting persister cells have never been described up to this day.
Here, we developed a single-cell approach, based on microfluidics coupled with fluorescence microscopy. As a model, we analyzed persistence of E. coli wild-type cells to ofloxacin in steady-state growth conditions using fluorescent reporters to monitor the dynamics of the SOS response and to visualize the nucleoids in individual cells. On the contrary to the prevailing hypothesis, we observed that persister cells are not necessarily slow growers and that both persister and ofloxacin-sensitive cells endure comparable levels of DNA damages during ofloxacin exposure, as indicated by a similar induction of the SOS response in both cell types. Therefore, neither growth rate nor SOS induction can be used as a marker to predict the fate of a particular cell to become persister. Our analyses revealed persister-specific traits during the recovery phase, after antibiotic removal. First, the SOS induction was prolonged during the early recovery phase, reaching its maximum peak a few hours after ofloxacin removal. Persister cells recovery was further characterized by the formation of long polynucleoïd bacterial filaments, and cell division ultimately resumed at multiple locations in the filament after nucleoid segregation, giving rise to a viable progeny.
RESULTS
Setting up an experimental framework for tracking bacterial persister cells at the single-cell level
The microfluidic experimental setup comprised three different phases: (i) After inoculation of liquid cultures in the microfluidics device, cells were perfused with MOPS-glucose medium for 5 to 7 hours. (ii) Cells were then perfused with MOPS-glucose medium supplemented with ofloxacin (5 μg/ml) [60-fold the minimal inhibitory concentration (MIC) for the wild-type strain used in this work, 0.08 ± 0.01 μg/ml] for 5 to 7 hours. (iii) Cells were reperfused with MOPS-glucose medium for 24 hours, allowing for persister cells recovery and expansion. Figure 1A shows the time-kill curve of the batch culture performed in the same conditions. At 5 hours of treatment, only the persister cells are surviving (second part of the curve, with a low killing rate). For the live imaging experiments, pictures were taken every 15 min over the course of the three phases, enabling us to track back the history of cells identified as persisters and monitor their behavior during growth. In this way, we monitored cellular parameters, such as cell area and generation time on a large number of cells, at every step of the experiment. We used a psulA::gfp reporter to monitor the induction of the SOS response as well as an HU-GFP reporter strain to visualize nucleoids and quantify cellular DNA content. This setup provides a robust analysis tool to obtain quantitative data and to compare the characteristics of persister cells with their many antibiotic-sensitive siblings.
Fig. 1. Single-cell imaging of persister and nonpersister cells to ofloxacin.
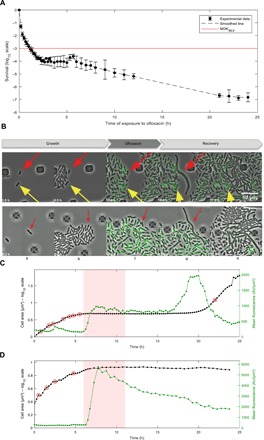
(A) Time-kill curve of MG1655 strain. Batch cultures were performed in similar conditions to microfluidic experiments. The MG1655 strain was grown in MOPS-based medium supplemented with 0.4% glucose and then challenged with ofloxacin (final concentration of 5 μg/ml). Survival was monitored at indicated times as described in the Materials and Methods. Data points are mean values of nine independent experiments, and error bars represent SDs. The red line represents the MDK99.9 (minimum duration for killing 99.9% of the population). (B) Fluorescence microscopy snapshots of the MG1655 strain containing the psulA::gfp reporter plasmid during time-lapse microscopy in the microfluidic chamber. Top panels show a persister cell that did not divide during the 7-hour growth phase in the microfluidic chamber after inoculation with an overnight culture (red arrow) and a nongrowing cell that is not a persister cell (yellow arrow). Bottom panels represent the persister p2 that was dividing after inoculation of the microfluidic chamber with an exponentially growing culture (red arrow). Time is indicated at the bottom-left corner of each picture. (C and D) Life cycle of persister p2 (C) and nonpersister cell np8 (D). The cumulated area (log10 scale) and the mean fluorescence of persister cell p2 and nonpersister cell np8 are plotted as a function of time. Red circles indicate cell division events before the addition of ofloxacin (5 μg/ml) and the first cell division event during the recovery phase (C). Red shaded area indicates ofloxacin treatment.
In a first attempt to observe persister cells in exponentially growing populations, stationary phase cells were perfused in the microfluidic plate with culture medium for 7 hours to allow the cells to grow before ofloxacin treatment. In these conditions, only type I persister cells originating from nongrowing cells and carried over from stationary phase were observed (Fig. 1B, top panels). The frequency of type I persisters was quantified in a representative experiment (Fig. 1B). Of the 2125 cells present at the time of treatment, we observed 17 cells that did not divide, but only 1 of these cells was able to survive the antibiotic treatment. We cannot exclude that among the 16 cells that did not give rise to persisters, some were not viable before the treatment. Figure 1B shows the growth dynamics of that particular type I persister cell (Fig. 1B, top panels, red arrows). One can directly observe that the persister cell did not divide during the first growth phase, while most cells within the same field were actively dividing. Upon addition of ofloxacin, all the cells stopped dividing. After reperfusion of the chamber with medium devoid of ofloxacin, only the persister cell underwent massive filamentation. The filament then divided into individual cells to form a viable progeny. The vast majority of the cells remained frozen in the initial state; they neither formed filaments nor divided after reperfusion with medium devoid of ofloxacin. In the same field, a second nondividing cell before ofloxacin treatment was observed (Fig. 1B, top panels, yellow arrows). This cell was unable to form a filament and to divide after removal of ofloxacin, indicating that the nongrowing state is not sufficient to trigger persistence, in accordance with previous observations (10, 11).
To increase the probability of observing persister cells generated by actively growing cells at treatment time, we added an additional step of growth to ensure that most of the cells would be in steady-state growth. Stationary phase cultures were diluted to an OD600nm of ~0.01 into culture flasks and grown to midlog phase (OD600nm ~ 0.3) before the inoculation of the microfluidic chamber. Growth of the persister cells that were identified after removal of ofloxacin was monitored to confirm that they were actively dividing before the treatment (Fig. 1B, bottom panels, and movie S1). Figure 1B shows one of the 23 persister cells that we observed in the course of this work (persister p2). This persister cell originated from growing ancestors and not from a dormant cell. It only ceased growth and cell division upon ofloxacin treatment. As previously observed for the type I persister cell, after removal of the antibiotic, the p2 persister cell formed a long filament followed by cell division and further growth. Persister frequency was determined on a representative subset of 47,000 cells. Among this population of 47,000 cells, we were able to detect 6 persister cells, corresponding to a frequency of ~1 in 104, a ratio close to that observed in flask cultures after 5 hours of ofloxacin exposure (Fig. 1A). Generation time before ofloxacin treatment was 63 ± 10 min and 75 ± 14 min in the microfluidic system and in liquid cultures, respectively.
The SOS response is heterogeneous both in ofloxacin-treated and untreated conditions
To follow the induction and dynamics of the SOS response at the single-cell level, we made use of a low–copy number plasmid carrying a transcriptional fusion between the sulA promoter and the gfp gene (psulA::gfp). To validate the SOS reporter, we monitored fluorescence by flow cytometry in the wild-type strain containing the psulA::gfp plasmid, as well as in mutants in which the SOS response is either constitutively induced (SOS ON, LexA51 ΔSulA) or noninducible (SOS OFF, LexA3) (fig. S1A). As a control, fluorescence of cells with a promoterless gfp plasmid was monitored in the wild-type strain untreated or treated with ofloxacin (fig. S1A). In the wild-type strain containing the psulA::gfp plasmid, fluorescence was similar to that observed in the control in absence of ofloxacin, although a small subpopulation (0.13% of the total cells) showed high fluorescence. After 5 hours of ofloxacin treatment, the mean fluorescence significantly increased (25-fold) in comparison to the untreated sample. Fluorescence intensity showed a broad distribution within the population, indicating that SOS induction is quite heterogeneous. In the SOS OFF mutant, the mean fluorescence in the absence of treatment was comparable to that of the wild-type strain. After treating cells with ofloxacin for 5 hours, the mean fluorescence increased only 3.2-fold, indicating that the SOS response is ~90% repressed upon ofloxacin treatment (as compared to the wild-type), and as observed for the wild-type strain, the distribution of fluorescence intensities broadened, indicating similar cell-to-cell heterogeneity. In the SOS ON mutant, the mean fluorescence was ~60-fold higher than that of the wild-type strain in steady-state growth conditions. Nonetheless, exposure to ofloxacin for 5 hours still led to a slight increase (1.3-fold). Together, these data indicate that the psulA::gfp transcriptional fusion accurately and specifically reports SOS induction. Moreover, it shows that spontaneous SOS induction occurs at low frequency in the wild-type strain and that the induction of the SOS response varies from cell-to-cell upon ofloxacin treatment, as previously reported by Pennington and Rosenberg (34).
The level of induction of the SOS reporter is likely to reflect the amount of DNA damages
The next question was whether the level of induction of the psulA::gfp reporter correlates to the amount of DNA damages. One of the limitations of the reporter could be that gyrase poisoning by the concentration of ofloxacin used in this work (5 μg/ml) leads to rapid transcription inhibition and cell death. Therefore, cells incurring high levels of DNA damage may show lower reporter expression. To test this hypothesis, the transcription and translation capacities of cells treated with ofloxacin (5 μg/ml) were assessed by flow cytometry using a fluorescence-based two-color system (28). This system is composed of two plasmids, one carrying the gfp gene under the control of an isopropyl-β-d-thiogalactopyranoside (IPTG)–inducible promoter and the other carrying the crimson gene under the control of a constitutive promoter (table S1). The Crimson fluorescence remained stable during the course of the experiment, both in treated and untreated cells. Upon addition of IPTG, as expected, GFP fluorescence of the untreated control cells increased with time (fig. S1B). GFP fluorescence of cells treated with ofloxacin and IPTG increased for the first 2 hours of treatment (65-fold increase as compared to 125-fold increase for the untreated control) and remained stable for the next 3 hours, indicating that transcription occurs in treated cells, although to a lesser extent than in the untreated control (~53% decrease as compared to the untreated control). Moreover, only a very small proportion of cells (~1%) were stained by propidium iodide (PI) after 5 hours in the presence of ofloxacin (fig. S1C), suggesting that during the course of the treatment, most of the ofloxacin-treated cells did not present membrane defects. A 4′,6-diamidino-2-phenylindole (DAPI) counterstain was used to make sure that PI-negative cells were not DNA-free cells (fig. S1C). This conclusion was supported as treating the cells at high temperature or with 70% isopropanol after the antibiotic treatment increased the frequency of PI-positive cells to almost 100% (fig. S1C). Together, these data indicate that ofloxacin-treated cells remain metabolically active and do not present membrane defects at least during the first 2 hours of treatment. The apparent discrepancy between the PI-staining data and the loss of viability as measured by plating the treated cells suggests that cell death might not occur during the ofloxacin treatment but rather on plates, during the recovery period.
Next, we asked whether induction of SOS may reach saturation once all available LexA is cleaved, thereby not reporting additional DNA damages. Wild-type cells containing the SOS reporter were treated with ofloxacin (1 and 5 μg/ml), and both survival and fluorescence were monitored as a function of time (fig. S1, D and E). For both concentrations, viability was gradually lost and fluorescence was gradually increasing. Note that loss of viability was more important at 1 μg/ml of ofloxacin than at 5 μg/ml, as was fluorescence. This phenomenon is known as the paradoxical effect of quinolones: High concentrations of quinolones are less bactericidal (35). Loss of viability as measured on plates was highly correlated with the increase in fluorescence of the psulA::gfp reporter (Spearman correlation coefficient = −1, P < 10−3 at 5 μg/ml of ofloxacin) in that time frame, indicating that expression of this reporter is likely to report the extent of DNA damage. In addition, the level of fluorescence was higher at 1 μg/ml than at 5 μg/ml of ofloxacin, indicating that saturation of the SOS response does not occur under our experimental conditions.
SOS induction before ofloxacin treatment does not trigger persister cell formation
Since we observed wild-type cells with high fluorescence at low frequency in the absence of ofloxacin (fig. S1A), we asked whether these cells were more likely to generate persister cells in exponentially growing populations, as previously suggested (22). The frequency of SOS spontaneous induction in cells growing in the microfluidic setup (0.08% of 47,000 analyzed cells) was similar to that observed in liquid culture, as measured by flow cytometry (0.13% of 225,000 events). Forty wild-type cells in which the SOS response was ON before ofloxacin treatment were tracked in microfluidic experiments. None of these SOS ON cells was able to give rise to a viable progeny after removal of ofloxacin (see movie S2 for a representative example). Survival and MDK99.9 (minimum duration for killing 99.9% of the population) measurements of the SOS ON mutant at the population level confirmed that constitutive SOS system does not increase persister cell frequency (Fig. 2, A and B). A slight and significant 1.9-fold decrease of the MDK99.9 was even observed as compared to the wild-type strain (Fig. 2, A and B). The ∆SulA mutant was tested as a control, and both survival and MDK99.9 were comparable to that of the wild-type strain. As expected, survival and MDK99.9 were strongly reduced for the SOS OFF mutant as compared to the wild-type strain (68.1- and 5.8-fold; Fig. 2, A and B), confirming previous published observations (22, 23, 26). Since expression of TisB was shown to lead to persistence, we tested the ∆TisAB mutant but could not observe any significant differences in survival or MDK99.9 as compared to the wild-type strain. To better understand such discrepancy with previously published data (24), we tested the survival of the ∆TisAB mutant in the experimental setup described in the original paper. Under these conditions, the previously published phenotype was reproducible (25-fold decrease in survival after 5 hours of ciprofloxacin treatment as compared to the wild-type strain), indicating that TisB might play a role in persistence to fluoroquinolones in very specific experimental conditions. Note that the MICs of the different mutants were comparable to that of the wild-type strain (SOS OFF = 0.08 ± 0.01 μg/ml, ∆SulA = 0.10 ± 0.01 μg/ml, SOS ON = 0.09 ± 0.04 μg/ml, and ∆TisAB = 0.09 ± 0.01 μg/ml). Overall, in agreement with what was previously reported for stationary phase persisters (26), these data show that while a functional SOS response contributes to persistence to ofloxacin, induction of the SOS response before the antibiotic treatment does not increase persistence frequency in steady-state growth conditions. In addition, the impact of the TisB toxin appears to be condition dependent and does not solely explain the role of the SOS response in persistence to fluoroquinolones, at least not in the conditions tested here.
Fig. 2. The SOS response is required for persistence to ofloxacin but not prior treatment.
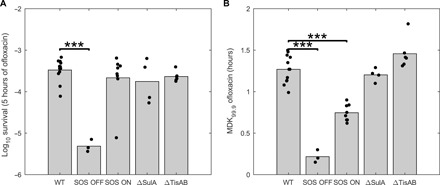
Survival fraction after 5 hours of ofloxacin treatment (5 μg/ml) (A) and MDK99.9 (time to kill 99.9% of the population) (B) are represented for the wild-type strain and isogenic LexA3 (SOS OFF), LexA51 ΔSulA (SOS ON), ΔSulA and ΔTisAB mutants. Bar graphs represent mean values of at least three independent experiments. Independent replicate values are plotted as dots. Student’s t-tests were performed between data for the wild-type (WT) and mutant strains (***P < 0.001).
Persister and sensitive cells similarly respond to DNA damage during ofloxacin treatment
It is generally assumed that persister cells are slow growers or nongrowers, making them “refractory” to antibiotics. The rationale is that antibiotics target physiological processes (in the case of ofloxacin, DNA replication via gyrase poisoning) that are less active in slowly growing cells (36). Using the psulA::gfp reporter plasmid, we monitored the fluorescence intensity as a proxy for DNA damage levels. Before treatment (after 6 hours of growth), the mean fluorescence of the population was low, and the fluorescence distribution showed some degree of heterogeneity [quartile-based coefficient of variation (QCV) = 22%; Table 1 and Fig. 3A]. No significant difference was observed between fluorescence of persister cells, a subset of 14 randomly chosen cells, and that of the population (Table 1 and Fig. 3A; see Materials and Methods for how the random subset of cells was defined). After 5 hours of ofloxacin treatment, as expected, mean fluorescence had increased in all cell types: 12.6-fold increase in the bulk of the population, 14.9-fold increase in persister cells, and 9.6-fold increase in the subset of sensitive cells (Table 1 and Fig. 3A). In addition, dispersion of the data increased as well (QCV = 79, 105, and 91% for the population, persisters, and sensitive cells, respectively), indicating that the ofloxacin treatment generated cell-to-cell heterogeneity within the population (Table 1 and Fig. 3A). As no significant difference was observed between SOS induction in persister cells, in the subset of sensitive cells and in the bulk of the population (Table 1 and Fig. 3B), we conclude that, during ofloxacin treatment, the levels of DNA damage are similar in persister and ofloxacin-sensitive cells.
Table 1. Induction of the SOS response at the population level, in persister cells and in sensitive cells throughout microfluidic time-lapse experiments.
Mean fluorescence and SDs for the wild-type strain containing the psulA::gfp reporter plasmid during time-lapse microscopy in the microfluidic chamber for the bulk of the population, the 23 persister cells and a subset of 14 random nonpersister cells (see Materials and Methods for how the random subset of cells was defined) at indicated times. In the case of persister cells early recovery, the maximum value of fluorescence was used and therefore corresponds to different times of recovery. The indicated time corresponds to the mean time at which persister cells reached maximum fluorescence together with the corresponding SD. The fourth column represents the mean fluorescence fold increase as compared to the untreated condition. The fifth column represents the number of cells that were analyzed. The sixth column represents the QCV, i.e., the interquartile range divided by the median, and is a robust measurement of heterogeneity within a sample (see Materials and Methods). AU, arbitrary units.
| Time |
Mean fluorescence (AU/μm2 × 103) |
Fold increase | Sample size | QCV (%) | |
| Population | 6 hours of growth | 0.29 ± 0.16 | 14,946 | 22.3 | |
| 5 hours of ofloxacin | 3.69 ± 2.49 | 12.6 | 20,022 | 79.2 | |
| 3.5 hours of recovery | 3.54 ± 3.40 | 12.1 | 18,027 | 90.5 | |
| 8.5 hours of recovery | 3.03 ± 3.71 | 10.4 | 16,448 | 101.3 | |
| Persister cells | 6 hours of growth | 0.30 ± 0.09 | 23 | 36.0 | |
| 5 hours of ofloxacin | 4.39 ± 2.69 | 14.9 | 23 | 104.5 | |
| 3.6 ± 2.4 hours of recovery | 7.28 ± 3.95 | 24.7 | 23 | 85.9 | |
| 8.5 hours of recovery | 2.21 ± 1.26 | 7.5 | 23 | 75.9 | |
| Sensitive cells | 6 hours of growth | 0.35 ± 0.16 | 14 | 44.2 | |
| 5 hours of ofloxacin | 3.42 ± 3.11 | 9.6 | 14 | 91.3 | |
| 3.5 hours of recovery | 3.22 ± 3.37 | 9.1 | 14 | 88.5 | |
| 8.5 hours of recovery | 2.97 ± 2.98 | 8.4 | 14 | 83.7 |
Fig. 3. Persister cells are characterized by a maximal induction of the SOS response during the early recovery phase.
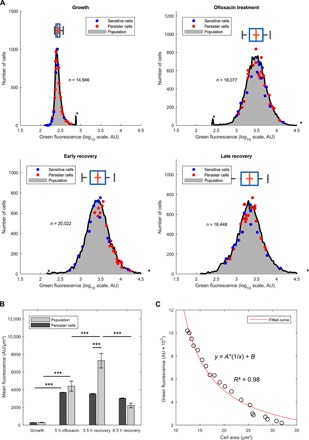
(A) Distribution of mean fluorescence is represented at the population level for the wild-type strain containing the psulA::gfp reporter plasmid during time-lapse microscopy in the microfluidic chamber before treatment, during treatment (5 hours of ofloxacin treatment), and after antibiotic removal (3.5 and 8.5 hours of recovery). Red dots indicate mean fluorescence of individual persister cells (n = 23) at the corresponding time points. Blue dots indicate mean fluorescence of a random subset of nonpersister cells (n = 14) at the corresponding time points. (B) Mean fluorescence (i.e., fluorescence per square millimeter) of the population is compared with that of persister cells in the same experiment as in (A). Bars represent mean values and error bars represent SEM. Student’s t-tests were used to identify significant differences (***P < 0.001). Dataset sizes are given in (A) for the population and are of n = 23 for persister cells. (C) Fluorescence dilution in persister cells solely accounts for the loss in fluorescence after SOS maximal induction peak. As an example, mean fluorescence of persister p17 is represented as a function of cell area after SOS maximal induction (circles). An inverse model (y = a.x−1) regression line was used to fit the experimental data points (red line). Determination coefficient is indicated next to the regression line.
Persister cells early recovery is characterized by maximal SOS induction and filamentation
Upon removal of ofloxacin, persister cells mean fluorescence rapidly increased (24.7-fold after 3.6 ± 2.4 hours of recovery; Fig. 3, A and B, Table 1, and fig. S2). Such fluorescence peak in early recovery phase is highly significant (P < 10−3; 1.7-fold increase as compared to fluorescence at treatment time) and specific to persister cells, as shown by the absence of a significant increase at the population level and in the sensitive cells (Fig. 3, A and B). Concomitantly (1.4 ± 1.0 hours after ofloxacin removal), persister cells started to elongate and generated long filaments, indicative of cell division inhibition (final filament area of 18.3 ± 8.2 μm2; fig. S2 and movies S1, S3, S4, and S6). We observed that both the kinetics and increase in fluorescence after ofloxacin removal varied among persister cells (fig. S2). In 13 of the 23 persister cells, the fluorescence reached a plateau during ofloxacin treatment, and a second fluorescence peak was observed during the early recovery phase (Fig. 1C and fig. S2A), while in the 10 others, fluorescence gradually increased during the ofloxacin treatment and subsequent recovery phase (fig. S2B). Nevertheless, in all cases, maximum fluorescence was reached during early recovery phase (3.6 ± 2.4 hours of recovery) as opposed to the subset of sensitive cells for which maximum fluorescence was reached during antibiotic treatment (4.75 ± 4.66 hours of treatment; Fig. 1D and fig. S2C). With time, filamentation of persister cells led to a strictly proportional decrease in fluorescence, indicating that the SOS response was shut down in persister cells after reaching their maximum fluorescence peak (3.3-fold drop at 8.5 hours of recovery, P < 10−3; Fig. 3 and Table 1). If the SOS response was still induced in persister cells after reaching their maximum fluorescence peak, then one would expect the data to not follow a strict inverse relationship. We found the inverse regression model to tightly fit the data, with all 23 persister cells having an R2 coefficient higher than 0.8 and 78% of the 23 persister cells having an R2 coefficient higher than 0.9 (Fig. 3C illustrates this relationship for persister p17 as a representative example).
Subsequently (6.9 ± 1.5 hours of recovery), persister cells eventually underwent cell divisions at multiple locations within the filament and gave rise to viable daughter cells (see below). Comparable phenotypes were observed in the case of type I persister cells (Fig. 1B).
Persister filaments are formed in the absence of the SOS response but with reduced size
To test whether filamentation is SOS and SulA dependent, we performed microfluidic experiments using the SOS OFF and ΔSulA mutants. In both cases, we observed filamentation during the recovery phase at a frequency similar to that of the wild-type strain (Fig. 4A and movie S5). However, for the SOS OFF mutant, the frequency of productive filament was low (see below), confirming the defect in persistence observed in flask experiments (Fig. 2). Moreover, SOS OFF filaments did not show any induction of the psulA::gfp fusion, ruling out any activation of the SOS system in these cells (movie S5). For the ΔSulA mutant, we observed and analyzed 18 persister cells. These persister cells were undergoing filamentation as observed for the wild-type strain. However, the maximal cell area of the ΔSulA filaments was significantly lower than that of wild-type filaments (12.67 ± 7.06 μm2 and 18.3 ± 8.2 μm2, respectively; Fig. 4C). Thus, although SulA-independent filamentation occurs in the ΔSulA and SOS mutant persister cells, we cannot exclude that SulA contributes to the filamentation observed in the wild-type persister cells.
Fig. 4. Characterization of persister filaments.
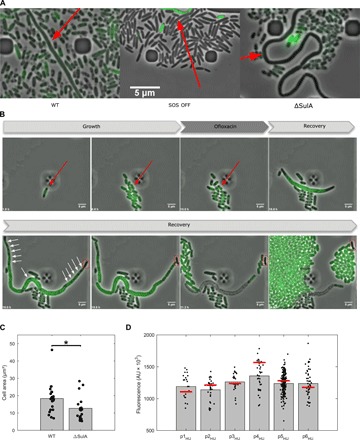
(A) Persister cells filamentation does not solely rely on the SOS response. Fluorescence microscopy snapshots of persister filaments of the wild-type strain (left), the SOS OFF mutant (middle), and the ΔSulA mutant (right) during recovery in microfluidic time-lapse experiments are shown. The different strains contain the psulA::gfp reporter plasmid. Persister filaments are indicated by a red arrow. (B) Persister cells form polynucleated filaments during recovery. Fluorescent microscopy snapshots of the MG1655 strain containing the hupA::gfp reporter during time-lapse experiments in the microfluidic chamber are shown. The red arrow indicates a persister cell during the growth and treatment phases. White arrows indicate DNA-free regions separating well-segregated nucleoids within the filament. A DNA-free pole of the persister filament is boxed in red. Time at which pictures were taken is indicated at the bottom-left corner of each image. (C) Persister filament maximal area is slightly decreased in a ΔSulA mutant during recovery. The maximal area (μm²) of persister filaments was monitored for the MG1655 wild-type strain and the isogenic ΔSulA mutant. Bar graphs represent mean values, and values for all persister cells are plotted as dots. A Student’s t-test was performed between data for the wild-type strain and the ΔSulA mutant (*P < 0.05). (D) Persister cells do not contain reduced levels of DNA before the ofloxacin treatment. Fluorescence of the MG1655 wild-type strain containing the HU-GFP reporter plasmid is represented for persister (bars) and nonpersister cells (black dots) within a same microcolony immediately before ofloxacin treatment during time-lapse microscopy in the microfluidic chamber. The number of cells within every microcolony is n = 25 (p1HU), n = 31 (p2HU), n = 24 (p3HU), n = 38 (p4HU), n = 188 (p5HU), and n = 52 (p6HU).
Persister cells generate polynucleoid filaments during the recovery phase
Next, we asked whether persister filaments were multinucleated as suggested by the large number of cell division events occurring simultaneously or in a short time frame (movie S1). Using a wild-type strain carrying an HU-GFP fusion, we studied the dynamics of DNA replication and nucleoid segregation during the entire persistence cycle (Fig. 4B and movie S6). HU-GFP “stains” the chromosome uniformly and colocalizes with DAPI staining as shown previously (37). Note that the microfluidics growth conditions used with this strain were different from those used above. Since the strain is unable to grow in MOPS-glucose medium, the experiment was performed in LB medium. Similar phenotypes were observed under these conditions, and six additional persister cells were analyzed.
During the growth phase before the ofloxacin treatment, fluorescence was diffuse within the cells and occupied a large volume of the cytoplasmic space (Fig. 4B, panels 1 and 2). Quantification of the level of fluorescence immediately before the treatment for 358 nonpersister cells and 6 persister cells revealed that both cell types contained similar cellular DNA amounts [1240 ± 181 AU (arbitrary unit) and 1263 ± 160 AU, respectively; Fig. 4D]. Upon ofloxacin treatment, fluorescence coalesced at mid-cell, indicating nucleoid compaction (Fig. 4B, panel 3), a phenomenon reported to rely on an active SOS response (38). After removal of ofloxacin, the persister cell elongated, and fluorescence expanded within the filament to occupy most of the cytoplasmic space (except near the poles of the filament), suggesting that DNA replication had resumed (Fig. 4B, panel 4). With time, fluorescence in the distal regions of the filament separated into large foci, indicating that chromosome segregation was occurring (Fig. 4B, panel 5). Cell division then occurred at multiple positions within the distal regions, giving rise to daughter cells that contain DNA and are able to divide. Two DNA-free cells originating from the poles of the filament were cast off (Fig. 4B, panels 6 to 8).
Cell division is a late and limiting step of persister cells recovery, which requires a functional SOS response
In Fig. 4B (panels 5 and 6), fluorescence in a large central portion of the filament did not coalesce into foci, indicating that nucleoid segregation did not occur. This portion of the filament lysed over the course of several hours as evidenced by the loss of contrast and fluorescence (Fig. 4B, panels 6 to 8). Thus, cell division only occurs following nucleoid segregation. We next asked whether persister cells had reached their maximal growth rate during cell filamentation or only after the first cell division event. We therefore measured the elongation rate of the 23 wild-type persister filaments and found that exponential growth takes place within persister filaments, before the first cell division, at a rate comparable to the mean population growth rate in normal growth conditions (doubling time values of 90 ± 42 min and 76 ± 9 min, respectively). As previously observed for the SOS induction, the doubling time of persister filaments was highly heterogeneous, ranging from 189 min for the slowest recovery to 44 min for the fastest recovery. Elongation continued for several hours (5.5 ± 1.5 hours) before cell division resumed. Although all persisters that eventually resumed robust growth had initially undergone extensive filamentation, only 21% of the wild-type filaments that appeared during recovery were able to resume cell division and to generate a viable progeny. This abortive recovery was even more notable in the SOS OFF mutant, in which filamentation occurred at a similar frequency than in the wild-type strain (0.053 and 0.074%, respectively), with only 5% of the filaments able to generate a viable progeny. The higher failure rate in this mutant underlines the importance of SOS-dependent DNA repair for cell division to resume.
Persister cells are not slow growers before exposure to ofloxacin
As mentioned above, it is commonly thought that persister cells arise from subpopulations of nongrowing or slow-growing cells. To determine whether the persister cells that we observed belonged to this definition, we quantified the growth rate of persister cells, of the random subset of sensitive cells previously described, and of the bulk of the population. For this purpose, we defined a metric called “single-cell instantaneous growth rate,” or SIGR. As an example, Fig. 5 shows the tracking of persister p22 during the growth phase, before antibiotic treatment. Cell area is measured at every time point during growth, giving rise to a typical saw-tooth profile (Fig. 5, A and B) reflecting cell elongation and cell division cycles. The drop in cell area due to cell division events was compensated by adding the area of the cell of interest to that of its sister cell generated by the same division events and that of the sister cells generated at the previous division events (Fig. 5A). This calculation yielded a linear increase in cell area as a function of time. The SIGR is defined as the slope (μm2/hour) of the regression line used to fit the data during the last 1.5 hours of the growth phase (Fig. 5C) and corresponds to the rate at which a cell elongates immediately before the ofloxacin treatment.
Fig. 5. Slow growth is not a general rule for persister cells.
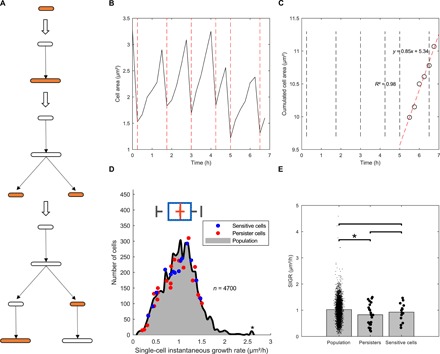
(A to C) Schemes illustrating the methodology used to measure SIGR. Briefly, elongation was monitored at the single-cell level during the growth phase (A). Cell area was plotted against time for persister p22 as an example, yielding a characteristic saw-tooth profile, with positive slopes corresponding to cell elongation and negative slopes corresponding to cell division (B). The drop in cell area due to cell division events is compensated as described in the text, giving rise to a linear increase in cell area as a function of time (shown for persister p22 as an example). SIGR is defined as the slope of that line for the last six time points (90 min) before the antibiotic treatment (C). (D) Distribution of SIGR for 4700 nonpersister cells. Red and blue dots indicate the SIGR of 23 persister cells and the 14 randomly chosen ofloxacin-sensitive cells, respectively. (E) Comparison of the SIGR values for the population, the persister cells, and the random subset of sensitive cells. Single-cell data are plotted as dots. Student’s t-tests were performed between data (*P < 0.05).
A first important observation is that the SIGR of both persister and susceptible cells is heterogeneous and follows a normal distribution (Fig. 5D). If we consider the mean SIGR for persisters, then a slight but significant decrease was observed in persister cells as compared to the population (0.83 ± 0.41 μm2/hour versus 1.02 ± 0.38 μm2/hour, respectively; P = 0.03; Fig. 5E). However, it is well known that trivially small effects will be found very significant with very large sample sizes (here, n = 4700 for the population), as a result of increased statistical power (1 − β) (39). The risk is then to perform a highly sensitive test detecting very small size effects and therefore being meaningless. To circumvent such drawback in significance testing, we tested whether we could find a significant difference between persister cells and the subset of randomly chosen nonpersister cells, but this was not the case (0.83 ± 0.41 μm2/hour versus 0.93 ± 0.35 μm2/hour, respectively; P = 0.48; Fig. 5E). To conclude, our data strongly suggest that persister cells are not slow growers.
DISCUSSION
In this work, we set up a unique experimental framework for tracking and monitoring persister cells during steady-state growth using microfluidics coupled to time-lapse microscopy. To our knowledge, it is the first time that persister cells originating from a wild-type E. coli strain in steady-state growth were observed at the single-cell level from the initial growth phase before the antibiotic treatment to the recovery phase after antibiotic removal. The SIGR and the induction of the SOS response were monitored in 23 persister cells, in a subset of 14 randomly chosen ofloxacin-sensitive cells, and in the bulk of the population. Our data indicate that these two parameters are highly heterogeneous both in persisters and in sensitive cells. Analysis of the SIGR before the ofloxacin treatment revealed that persister cells do not constitute a slowly growing subpopulation of cells. Our results thus rule out the “slow growth” hypothesis in steady-state growth conditions in E. coli wild-type cells. In addition, we observed that persister cells endure similar levels of DNA damages as compared to sensitive cells during ofloxacin treatment, as judged by comparable SOS induction and morphological changes such as DNA compaction.
What makes persister cells distinguishable from their sensitive siblings is, by definition, the ability to recover from the antibiotic treatment. The persister cells that we observed here present unique characteristics at the time of recovery: Upon ofloxacin removal, the SOS response continuously increased in the 23 persister cells, reaching a maximal peak at 3.6 ± 2.4 hours of recovery. Again, the behavior of persister cells was heterogeneous. In about half of the persister cells, the SOS response gradually increased during the treatment and the recovery phase, while in the other half, a plateau was reached during the treatment, and the SOS response sharply increased in the early recovery phase. Concomitantly, with the increase of the SOS induction, persister cells were forming long filaments. We speculate that filamentation leads to a decrease of the intracellular concentration of ofloxacin that might allow partial resumption of DNA replication and/or chromosome decatenation, as observed in cells expressing the HU-GFP fusion. However, some ofloxacin-poisoned topoisomerase complexes may remain and block replication and transcription machineries, provoking replication forks to collapse and generating double-stranded DNA breaks. This would account for the continuous induction of the SOS response during early recovery. We observed that with time, cell division resumed at multiple locations within the filaments, giving birth to viable cells but only in the regions where nucleoid segregation occurred as judged by the HU-GFP fluorescence. This observation suggests a tight coupling between replication/segregation and cell division. Cell filamentation was also observed in a ΔSulA mutant, although filaments were of smaller size as compared to the wild-type filaments. It is possible that in the wild-type strain, both SulA-dependent and independent mechanisms (such as SlmA or DamX) might contribute to cell division inhibition (40, 41). Mutants incapable of inducing the SOS response were found to generate as many filaments as the wild-type strain but with a much higher chance of abortive recovery. Some SOS-dependent repair functions such as RecA-mediated homologous recombination and RuvAB-dependent recombination are most probably required to repair DNA lesions at the time of recovery as it has been suggested previously (23, 26, 42).
While this work paves the way for persister cells observation and characterization, a number of questions remain open. Whether persister cells are in a dormant, metabolically inactive state has been addressed by several laboratories using different models (e.g., stationary-phase or exponentially growing cells, artificial systems to induce persistence such as overexpression of toxins or pretreatment with bacteriostatic antibiotics or any chemical that stop bacterial growth) and still remains under investigation [see e.g., (8, 10, 11, 28, 42–46)]. In M. smegmatis, the question of dormancy before treatment was addressed at the single-cell level by different groups. While the McKinney group showed that single-cell growth rate before INH (a mycolic acid synthesis inhibitor) treatment does not correlate with persistence (15), the Fortune group showed that there is a correlation: Fast-growing cells are more sensitive to different antibiotics, notably INH and peptidoglycan synthesis inhibitors (47). However, slow-growing cells were as sensitive to rifampicin, a transcription inhibitor (47). From these different works, it appears quite clear that dormancy alone is not sufficient to make cells persistent. Independent laboratories showed both in vitro (10, 11) and in vivo (48) that, while dormant cells generate the highest fractions of persister cells, they are very rare, thereby limiting their overall impact. It was also proposed that persister cells to fluoroquinolones are induced by the antibiotic via the induction of the SOS response rather than being stochastically generated and preexisting the treatment (23). There is no doubt that the SOS system contributes to persistence to ofloxacin, as SOS mutants show a drastic reduction in persister frequency (23, 26, 49). As mentioned above, SOS-dependent repair functions are most likely important for persistence. However, our data are indicating that TisB is not essential for persistence in the conditions that we tested. Since the level of SOS induction in persister cells during the ofloxacin treatment is comparable to that of the population, it indicates that SOS is not sufficient for persistence and that yet unknown physiological parameters are involved. In addition to the primary antibiotic-target interactions, several recent reports are showing that global downstream changes in gene expression occur upon exposure to antibiotic, leading notably to the rewiring of the entire metabolism and, as for fluoroquinolones, the induction of stress responses. These changes in cell physiology play an important role in the antibiotic-mediated lethality and likely persistence (50–54). Therefore, it is essential to better characterize the physiology of persisters at the single-cell level to be able to determine whether or not persister cells are in a particular state before the antibiotic treatment. The complexity and the plasticity of the bacterial physiology as well as the great heterogeneity of persister cells found in this work emphasize the need to study persisters on a case-by-case basis at the single-cell level and reinforce the idea that attempting to reduce persistence to a unique and defined trait may lead to a great oversimplification of the persistence phenomenon (17, 20).
As stated above, the differences between persister and sensitive cells highlighted here occur at the time of recovery, when persisters are elongating and undergoing a second SOS induction. While filamentation appears to be a hallmark of persister cells, it appears not to be sufficient, as only 20% of the filaments are able to give rise to viable cells. The kinetics and maximum of SOS induction during the recovery phase are variable from one persister to the other, indicating that the extent and the timing of induction of SOS do not represent a limiting step for persister cells recovery. However, we monitored the SOS response using a single reporter fusion (psulA::gfp). Fluctuation of the expression level of other SOS functions notably those involved in DNA double-stranded break repair, also occurs and influences the efficiency of DNA repair and thereby the generation of viable cells at the time of recovery. We cannot exclude cell-to-cell heterogeneity of other functions such as porins or active efflux pumps that would decrease the intracellular ofloxacin concentration in persister cells and thereby contribute to persistence (16, 55). As a follow-up of this initial analysis, it will be important to dissect the recovery phase process by expanding the number and nature of fluorescent reporters and by determining the molecular mechanism involved in cell filamentation.
MATERIALS AND METHODS
Bacterial strains and plasmids
Strains and plasmids used in this study are described in table S1. Strains are MG1655 derivatives (56). P1 transduction was used to introduce the lexA3 malB::Tn9 allele (SMR820 strain, provided by S. Rosenberg) in MG1655, the ΔsulA::kan allele [obtained from the Keio collection (57)] in MG1655, and the lexA51 malB::Tn9 allele (SMR4568 strain, provided by S. Rosenberg) in MG1655. The tisAB locus was deleted using the mini-λ system (58) and the pKD3 plasmid as template DNA for amplification of the chloramphenicol resistance cassette [ΔtisAB, 5′ GTGCCGTCAGCGCCTTAACCCCGCGTGAGCACACTGTGTTGTGTAGGCTGGAGCTGCTTC (forward); 5′ GGTTTCCCGCTCCCCTTTGGTGCGACTTGAATCTGAATTACATATGAATATCCTCCTTA (reverse)] as described by Datsenko and Wanner (59). The kanamycin and chloramphenicol resistance cassettes associated with sulA and tisAB deletions were removed by using the helper pCP20 thermosensitive plasmid encoding the FLP recombinase as described by Datsenko and Wanner (59). The fluorescence-based two-color system comprising two compatible plasmids (pET-GFP and pC17-Crimson) (28) was transformed in DJ624 (60), a derivative of MG1655 carrying a lacIq allele to allow for enhanced repression of the IPTG-inducible promoter upstream of the gfp gene.
Media and growth conditions
Experiments were performed in MOPS-based medium supplemented with 0.4% glucose prepared as described (61). Strain constructions by P1 transduction and microfluidic experiments with the hupA::gfp strain were performed in LB medium.
Persistence assays
Persistence assays were essentially performed as described in (61) with increased sampling frequency. Ofloxacin was used at 5 μg/ml, corresponding to 60-fold the MIC for the MG1655 wild-type strain. The frequency of persistence was calculated as the ratio of the number of colony-forming units per milliliter at a given time to the number of colony-forming units per milliliter at the treatment time.
MIC determination
MIC measurements were performed using the agar dilution method as described in (62). Briefly, overnight cultures were spotted on LB plates containing increasing concentrations of ofloxacin. The MIC value was defined as the lowest concentration of antibiotic inhibiting growth.
Transcription and translation activity
The DJ624 strain containing the pET-GFP and pC17-Crimson plasmids (28) was grown overnight in MOPS-based medium supplemented with 0.4% glucose and diluted to an OD600nm of ~0.01 in the same medium. Cultures were grown for 5 hours to mid-log phase (OD600nm ~0.3) and then treated with ofloxacin (5 μg/ml) and/or 1 mM IPTG. Samples were analyzed by flow cytometry at given time points as described below. Red fluorescence was used as a control since the Crimson protein is constitutively produced. Green fluorescence was monitored to assess transcription and translation activity.
Analysis of SOS induction at the population level by flow cytometry
Overnight cultures grown in MOPS-based medium supplemented with 0.4% glucose were diluted to an OD600nm of ~0.01 in the same medium. Cultures were grown for 5 hours to mid-log phase (OD600nm ~0.3) and diluted in phosphate-buffered saline (PBS) buffer before injection into an Attune NXT flow cytometer. In each experiment, 75,000 events were analyzed with a blue laser (488 nm) and a 530/30 emission filter for GFP and with a yellow laser (561 nm) and a 620/15 emission filter for Crimson (red fluorescence). Subsequent analyses were performed using FlowJo 10.4 and MATLAB (R2017b, MathWorks), assisted by custom-made scripts.
Viability testing by flow cytometry
The MG1655 strain carrying the psulA::gfp reporter plasmid was grown to mid-exponential phase (OD600nm ~ 0.3) in MOPS-based medium supplemented with 0.4% glucose and subsequently treated with ofloxacin (final concentration of 5 μg/ml). One-milliliter samples were withdrawn before antibiotic treatment and after 5 hours of ofloxacin exposure and were either left at room temperature for 1 hour, exposed to strong heat shock (72°C) for 1 hour, or treated with 70% isopropyl alcohol (final concentration) for 1 hour at room temperature. Samples were washed in PBS and then stained with PI (final concentration of 10 μg/ml) and DAPI (final concentration of 10 μg/ml) and incubated at room temperature in the dark for 20 min. Cells were washed in PBS, resuspended to a final OD600nm ~ 0.01, and analyzed by flow cytometry. Seventy-five thousand events were analyzed with a blue laser (488 nm) and a 530/30 emission filter for GFP, with a yellow laser (561 nm) and a 620/15 emission filter for PI and with a violet laser (405 nm) and a 440/50 emission filter for DAPI. Analyses were further performed using FlowJo 10.4. High fluorescent gates were designed on a nonfluorescent control, i.e., a culture of exponentially growing MG1655 wild-type cells carrying a promoterless pUA66(gfp) plasmid (with no staining). High fluorescent gates were set to 400, 400, and 600 AU for GFP, PI, and DAPI, respectively. The fraction of highly fluorescent cells was defined as the number of highly fluorescent cells divided by the total number of cells.
Real-time cellular imaging in microfluidic culture device
The commercially available microfluidic culture device CellASIC ONIX B04A-03 Microfluidic Plate (Millipore, USA) and the CellASIC ONIX Microfluidic System were used. The microfluidic B04A plate is made of four independent units, each containing five inlet wells, a cell inlet, a cell outlet, and a large outlet well. Each row of wells connects the corresponding culture chamber. The plates were preprimed with a PBS solution. The culture chamber dimensions are 2.0 mm by 1.2 mm in area with trap heights of 0.7, 0.9, 1.1, 1.3, 2.3, and 4.5 μm, respectively. The microfabricated chamber gently holds cells against the glass imaging surface to maintain a single focal plane during perfusion-based imaging experiments. Before each microfluidic experiment, a preculture step was performed in 10 ml of MOPS-based medium supplemented with 0.4% glucose in flasks at 37°C with orbital shaking at 180 rpm. Flasks were inoculated with an isolated colony grown on LB solid medium overnight. At mid-log phase, cells were harvested by centrifugation and washed two times in PBS. Cells were then suspended in MOPS-based medium supplemented with 0.4% glucose and diluted to a final OD600nm of around 0.03. A 100-μl aliquot of the final solution was injected in the inoculation well of the microfluidic plate. Cells were injected in the culture chamber in which they were trapped, and they grew in a single focal plane. In this chamber, cells were under continuous perfusion of culture medium due to the five inlets. Detailed protocols for the cell loading and cell culture can be found in the user guide provided by the supplier. Image acquisition was performed at 15-min intervals. Temperature was set at 37°C during the time of the experiment, and the microfluidic chamber was placed on a 100× oil immersion objective on an inverted microscope (Axio Observer, Zeiss, Germany). Illumination was provided by an HXP (mercury short-arc) lamp (120 V). For fluorescence measurements, 38 high-efficiency filter set was used (Zeiss, Germany). Images were collected with a Hamamatsu ORCA-Flash 4.0 digital camera. Image analysis was performed with the freeware Fiji (63), using the MicrobeJ plugin (64), and data analyses were performed in MATLAB (R2017b, MathWorks), assisted by custom-made scripts.
Selection of a random subset of 14 nonpersister cells
Numbers were assigned to all cells at treatment time during microfluidic assays using the MicrobeJ plugin in ImageJ. Cells were chosen by running the “randi” function in MATLAB, thereby creating sequences of pseudorandom numbers among the set of previously identified cells. In this process, two constraints were applied: First, the 14 nonpersister cells were equally distributed among seven independent microfluidic experiments, and second, two different nonpersister cells could not be chosen from the same microcolony.
Significance tests
Student’s t test was used for two-group comparisons. Comparison between persister cells fluorescence at different time points was performed using paired two-tailed t test. Comparison of the population at different time points was performed using unpaired two-tailed t test. Comparison of persister cells with the population was performed using unpaired two-tailed t test. Analyses were performed using MATLAB (R2017b, MathWorks) assisted by custom-made scripts.
Correlation tests
Spearman rank order coefficient was determined as the relationship between the variables (log10 survival and log10 mean fluorescence) was shown to be nonlinear (sigmoidal).
Measuring cell-to-cell heterogeneity
QCV was used, as it is considered far more robust to outliers than the CV (coefficient of variation) and is a valid metric for skewed distributions on the contrary to the CV. The QCV is defined as the interquartile range (a robust measure of spread) divided by the median (a robust measure of central tendency).
Supplementary Material
Acknowledgments
We thank I. Matic, M. Maurizi, and N. Fraikin for helpful discussions; N. Mine for technical support; and R. Hallez for constructing the TisAB mutant. We are grateful to M. Maurizi for critical and careful reading of this manuscript and suggestions for improving the presentation. We thank T. Tenson, S. Rosenberg, and I. Matic for donating strains. Funding: This work was supported by the Fonds National de la Recherche Scientifique (FNRS; T.0147.15F PDR and J.0061.16F CDR), the Fonds d’Encouragement à la Recherche ULB (FER-ULB), the Interuniversity Attraction Poles Program initiated by the Belgian Federal Science Policy Office (MicroDev), the Fonds Jean Brachet, and the Fondation Van Buuren. Author contributions: F.G. and L.V.M. designed the research. F.G. performed the research. F.G. and L.V.M. analyzed data. F.G. and L.V.M. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav9462/DC1
Table S1. Bacterial strains and plasmids used in this work.
Fig. S1. The level of induction of the SOS reporter is likely to reflect the extent of DNA damage.
Fig. S2. Growth and fluorescence profiles of 23 persister and 14 nonpersister cells before, during, and after ofloxacin treatment.
Movie S1. Single-cell imaging of 10 representative ofloxacin persister cells in an active growth state before the antibiotic treatment.
Movie S2. Spontaneous induction of the SOS response does not lead to persistence.
Movie S3. Single-cell imaging and monitoring of a slow-growing ofloxacin persister cell.
Movie S4. Single-cell imaging and monitoring of a fast-growing ofloxacin persister cell.
Movie S5. Persister cells elongation does not rely on the SOS response.
Movie S6. DNA localization and dynamics in ofloxacin persister cells.
Reference (65)
REFERENCES AND NOTES
- 1.Blair J. M. A., Webber M. A., Baylay A. J., Ogbolu D. O., Piddock L. J. V., Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Davies J., Davies D., Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry J. A., Wright G. D., Forces shaping the antibiotic resistome. Bioessays 36, 1179–1184 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Helaine S., Kugelberg E., Bacterial persisters: Formation, eradication, and experimental systems. Trends Microbiol. 22, 417–424 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Levin-Reisman I., Ronin I., Gefen O., Braniss I., Shoresh N., Balaban N. Q., Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Vogwill T., Comfort A. C., Furió V., MacLean R. C., Persistence and resistance as complementary bacterial adaptations to antibiotics. J. Evol. Biol. 29, 1223–1233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brauner A., Fridman O., Gefen O., Balaban N. Q., Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Balaban N. Q., Merrin J., Chait R., Kowalik L., Leibler S., Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Jõers A., Kaldalu N., Tenson T., The frequency of persisters in Escherichia coli reflects the kinetics of awakening from dormancy. J. Bacteriol. 192, 3379–3384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roostalu J., Jõers A., Luidalepp H., Kaldalu N., Tenson T., Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol. 8, 68 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orman M. A., Brynildsen M. P., Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 57, 3230–3239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keren I., Shah D., Spoering A., Kaldalu N., Lewis K., Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keren I., Minami S., Rubin E., Lewis K., Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2, e00100–e00111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah D., Zhang Z., Khodursky A. B., Kaldalu N., Kurg K., Lewis K., Persisters: A distinct physiological state of E. coli. BMC Microbiol. 6, 53 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakamoto Y., Dhar N., Chait R., Schneider K., Signorino-Gelo F., Leibler S., McKinney J. D., Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Pu Y., Zhao Z., Li Y., Zou J., Ma Q., Zhao Y., Ke Y., Zhu Y., Chen H., Baker M. A. B., Ge H., Sun Y., Xie X. S., Bai F., Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 62, 284–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaldalu N., Hauryliuk V., Tenson T., Persisters—As elusive as ever. Appl. Microbiol. Biotechnol. 100, 6545–6553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van den Bergh B., Michiels J. E., Wenseleers T., Windels E. M., Boer P. V., Kestemont D., De Meester L., Verstrepen K. J., Verstraeten N., Fauvart M., Michiels J., Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 1, 16020 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Kussell E., Kishony R., Balaban N. Q., Leibler S., Bacterial persistence: a model of survival in changing environments. Genetics 169, 1807–1814 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin B. R., Concepción-Acevedo J., Udekwu K. I., Persistence: A copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr. Opin. Microbiol. 21, 18–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson P. J. T., Levin B. R., Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLOS Genet. 9, e1003123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debbia E. A., Roveta S., Schito A. M., Gualco L., Marchese A., Antibiotic persistence: The role of spontaneous DNA repair response. Microb. Drug Resist. 7, 335–342 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Dörr T., Lewis K., Vulić M., SOS response induces persistence to fluoroquinolones in Escherichia coli. PLOS Genet. 5, e1000760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dörr T., Vulić M., Lewis K., Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLOS Biol. 8, e1000317 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unoson C., Wagner E. G. H., A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol. Microbiol. 70, 258–270 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Völzing K. G., Brynildsen M. P., Stationary-phase persisters to ofloxacin sustain DNA damage and require repair systems only during recovery. mBio 6, e00731–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verstraeten N., Knapen W. J., Kint C. I., Liebens V., van den Bergh B., Dewachter L., Michiels J. E., Fu Q., David C. C., Fierro A. C., Marchal K., Beirlant J., Versées W., Hofkens J., Jansen M., Fauvart M., Michiels J., Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 59, 9–21 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Jõers A., Tenson T., Growth resumption from stationary phase reveals memory in Escherichia coli cultures. Sci. Rep. 6, 24055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fridman O., Goldberg A., Ronin I., Shoresh N., Balaban N. Q., Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513, 418–421 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Goormaghtigh F., Fraikin N., Putrinš M., Hallaert T., Hauryliuk V., Garcia-Pino A., Sjödin A., Kasvandik S., Udekwu K., Tenson T., Kaldalu N., Van Melderen L., Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli Type II Persister Cells. MBio 9, e00640-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goormaghtigh F., Fraikin N., Putrinš M., Hauryliuk V., Garcia-Pino A., Udekwu K., Tenson T., Kaldalu N., Van Melderen L., Reply to holden and errington, “Type II toxin-antitoxin systems and persister cells”. MBio 9, e01838-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harms A., Fino C., Sørensen M. A., Semsey S., Gerdes K., Prophages and growth dynamics confound experimental results with antibiotic-tolerant persister cells. MBio 8, e01964-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holden D. W., Errington J., Type II toxin-antitoxin systems and persister cells. MBio 9, e01574-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pennington J. M., Rosenberg S. M., Spontaneous DNA breakage in single living Escherichia coli cells. Nat. Genet. 39, 797–802 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eagle H., Musselman A. D., The rate of bactericidal action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. J. Exp. Med. 88, 99–131 (1948). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhar N., McKinney J. D., Microbial phenotypic heterogeneity and antibiotic tolerance. Curr. Opin. Microbiol. 10, 30–38 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Wery M., Woldringh C. L., Rouviere-Yaniv J., HU-GFP and DAPI co-localize on the Escherichia coli nucleoid. Biochimie 83, 193–200 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Odsbu I., Skarstad K., DNA compaction in the early part of the SOS response is dependent on RecN and RecA. Microbiology 160, 872–882 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Johnson D. H., The insignificance of statistical significance testing. K. Wildl. Manage. 63, 763–772 (1999). [Google Scholar]
- 40.Wu L. J., Errington J., Nucleoid occlusion and bacterial cell division. Nat. Rev. Microbiol. 10, 8–12 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Khandige S., Asferg C. A., Rasmussen K. J., Larsen M. J., Overgaard M., Andersen T. E., Møller-Jensen J., DamX controls reversible cell morphology switching in uropathogenic Escherichia coli. MBio 7, e00642-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mok W. W. K., Brynildsen M. P., Timing of DNA damage responses impacts persistence to fluoroquinolones. Proc. Natl. Acad. Sci. U.S.A. 115, E6301–E6309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Y., Vulic M., Keren I., Lewis K., Role of oxidative stress in persister tolerance. Antimicrob. Agents Chemother. 56, 4922–4926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helaine S., Cheverton A. M., Watson K. G., Faure L. M., Matthews S. A., Holden D. W., Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vázquez-Laslop N., Lee H., Neyfakh A. A., Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 188, 3494–3497 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kudrin P., Varik V., Oliveira S. R. A., Beljantseva J., del Peso Santos T., Dzhygyr I., Rejman D., Cava F., Tenson T., Hauryliuk V., Subinhibitory concentrations of bacteriostatic antibiotics induce relA-dependent and relA-independent tolerance to β-lactams. Antimicrob. Agents Chemother. 61, e02173-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aldridge B. B., Fernandez-Suarez M., Heller D., Ambravaneswaran V., Irimia D., Toner M., Fortune S. M., Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335, 100–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Claudi B., Spröte P., Chirkova A., Personnic N., Zankl J., Schürmann N., Schmidt A., Bumann D., Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158, 722–733 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Theodore A., Lewis K., Vulić M., Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway. Genetics 195, 1265–1276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohanski M. A., Dwyer D. J., Collins J. J., How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 8, 423–435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blazquez J., Rodríguez-Beltrán J., Matic I., Antibiotic-induced genetic variation: How it arises and how it can be prevented. Annu. Rev. Microbiol. 72, 209–230 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Dwyer D. J., Belenky P. A., Yang J. H., MacDonald I. C., Martell J. D., Takahashi N., Chan C. T. Y., Lobritz M. A., Braff D., Schwarz E. G., Ye J. D., Pati M., Vercruysse M., Ralifo P. S., Allison K. R., Khalil A. S., Ting A. Y., Walker G. C., Collins J. J., Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A. 111, E2100–E2109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lobritz M. A., Belenky P., Porter C. B. M., Gutierrez A., Yang J. H., Schwarz E. G., Dwyer D. J., Khalil A. S., Collins J. J., Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. U.S.A. 112, 8173–8180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belenky P., Ye J. D., Porter C. B. M., Cohen N. R., Lobritz M. A., Ferrante T., Jain S., Korry B. J., Schwarz E. G., Walker G. C., Collins J. J., Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep. 13, 968–980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sánchez-Romero M. A., Casadesús J., Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. U.S.A. 111, 355–360 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blattner F. R., Plunkett G. III, Bloch C. A., Perna N. T., Burland V., Riley M., Collado-Vides J., Glasner J. D., Rode C. K., Mayhew G. F., Gregor J., Davis N. W., Kirkpatrick H. A., Goeden M. A., Rose D. J., Mau B., Shao Y., The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H., Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharan S. K., Thomason L. C., Kuznetsov S. G., Court D. L., Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 4, 206–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vanderpool C. K., Gottesman S., The novel transcription factor SgrR coordinates the response to glucose-phosphate stress. J. Bacteriol. 189, 2238–2248 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goormaghtigh F., Van Melderen L., Optimized method for measuring persistence in Escherichia coli with improved reproducibility. Methods Mol. Biol. 1333, 43–52 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Wiegand I., Hilpert K., Hancock R. E. W., Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ducret A., Quardokus E. M., Brun Y. V., MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat. Microbiol. 1, 16077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marceau A. H., Bahng S., Massoni S. C., George N. P., Sandler S. J., Marians K. J., Keck J. L., Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J 30, 4236–4247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav9462/DC1
Table S1. Bacterial strains and plasmids used in this work.
Fig. S1. The level of induction of the SOS reporter is likely to reflect the extent of DNA damage.
Fig. S2. Growth and fluorescence profiles of 23 persister and 14 nonpersister cells before, during, and after ofloxacin treatment.
Movie S1. Single-cell imaging of 10 representative ofloxacin persister cells in an active growth state before the antibiotic treatment.
Movie S2. Spontaneous induction of the SOS response does not lead to persistence.
Movie S3. Single-cell imaging and monitoring of a slow-growing ofloxacin persister cell.
Movie S4. Single-cell imaging and monitoring of a fast-growing ofloxacin persister cell.
Movie S5. Persister cells elongation does not rely on the SOS response.
Movie S6. DNA localization and dynamics in ofloxacin persister cells.
Reference (65)