

Blocking of sortilin ligand binding prevents neuropathic pain by inhibiting BDNF-induced spinal KCC2 down-regulation.
Abstract
Neuropathic pain is a major incurable clinical problem resulting from peripheral nerve trauma or disease. A central mechanism is the reduced expression of the potassium chloride cotransporter 2 (KCC2) in dorsal horn neurons induced by brain-derived neurotrophic factor (BDNF), causing neuronal disinhibition within spinal nociceptive pathways. Here, we demonstrate how neurotensin receptor 2 (NTSR2) signaling impairs BDNF-induced spinal KCC2 down-regulation, showing how these two pathways converge to control the abnormal sensory response following peripheral nerve injury. We establish how sortilin regulates this convergence by scavenging neurotensin from binding to NTSR2, thus modulating its inhibitory effect on BDNF-mediated mechanical allodynia. Using sortilin-deficient mice or receptor inhibition by antibodies or a small-molecule antagonist, we lastly demonstrate that we are able to fully block BDNF-induced pain and alleviate injury-induced neuropathic pain, validating sortilin as a clinically relevant target.
INTRODUCTION
Neuropathic pain is a debilitating clinical pain syndrome arising from nerve injury. In contrast to the beneficial role of acute pain, neuropathic pain persists after the initial injury has healed. The condition is notoriously resistant to treatment, and with a prevalence of 7 to 10% in the general population, neuropathic pain constitutes a major socioeconomic problem (1). Extensive and converging lines of research have made it clear that neuropathic pain is a manifestation of pathological alterations in the central nervous system (CNS), including loss of inhibition (disinhibition) in the spinal dorsal horn (SDH). Disinhibition thus appears to be a fundamental feature of neuropathic pain, resulting from reduced spinal expression of the potassium chloride cotransporter 2 (KCC2) following injury to peripheral nerves (2) or to the spinal cord (3). The consequent disruption of anion homeostasis in superficial SDH neurons, one of the main spinal nociceptive output pathways, results in increased excitability of the spinal pain circuitry and imbalanced pain signaling relayed to the brain (4).
Brain-derived neurotrophic factor (BDNF) signaling via tropomyosin receptor kinase B (TrkB) receptors appears to be a key culprit responsible for spinal KCC2 down-regulation (5). We previously described how sortilin, a type 1 receptor expressed by neurons and glial cells of the peripheral nervous system (PNS) and CNS, associates with Trk receptors to facilitate their anterograde trafficking and enhances signaling by neurotrophins (6). Sortilin is also known as neurotensin receptor 3 (NTSR3) and is expressed in several neurotensin-responsive tissues and cells (7, 8). The involvement of neurotensin signaling pathways in neuropathic pain is well documented, and the activation of NTSR1 and NTSR2 has been demonstrated to have analgesic effects in experimental pain models (9–12). Notably, previous studies demonstrate how sortilin, capable of binding to the neuropeptide with high affinity (0.1 to 0.3 nM) but devoid of apparent consensus signaling motifs, may modulate neurotensin signaling (7, 11, 13, 14). Modulation of either the BDNF/TrkB or neurotensin pathways provides an appealing approach to circumventing injury-induced neuropathic pain. It is, however, unknown whether these are independent pathways or whether they ultimately converge. Our study establishes sortilin as a regulatory link that integrates neurotensinergic inhibitory pathways and spinal BDNF/TrkB signaling, controlling spinal KCC2 levels. Furthermore, we demonstrate that small-molecule inhibition of sortilin represents a highly efficient means to alleviate neuropathic pain.
RESULTS
Sort1−/− mice are protected against neuropathic pain and spinal KCC2 down-regulation
We previously reported that the neuronal composition of dorsal root ganglia (DRG) and the sciatic nerve of the PNS is unaffected by sortilin deficiency; Sort1−/− mice display normal responses to acute mechanical (von Frey filaments) and thermal (Hargreaves test) stimuli (6). To explore the sensory phenotype following peripheral nerve injury, we tested here the development of mechanical allodynia, a pathological condition that is reliant on reduced expression of spinal KCC2. We took advantage of the spared nerve injury (SNI) model, which is a well-described paradigm for induction of neuropathic pain in rodents (15). Whereas wild-type (WT) mice developed considerable mechanical allodynia on the injured (ipsilateral) side, reaching a stable plateau after 2 days and remaining so for several weeks, Sort1−/− mice were fully protected throughout the 2-week test period (Fig. 1A). This difference was accompanied by substantial reduction in KCC2 expression in the SDH of WT mice (55.0 ± 1.4%, P = 7.9 × 10−5) but not in the SDH of Sort1−/− mice, as determined by Western blot quantification (Fig. 1, B and C). A further analysis by quantitative immunohistochemistry (IHC) confirmed that peripheral nerve injury caused the down-regulation of KCC2 in the affected segment of superficial lumbar SDH [identified by a reduction in isolectin B4 (IB4) binding] in WT mice but not in Sort1−/− mice (Fig. 1, D to G).
Fig. 1. KCC2 down-regulation is prevented in sortilin-deficient mice.
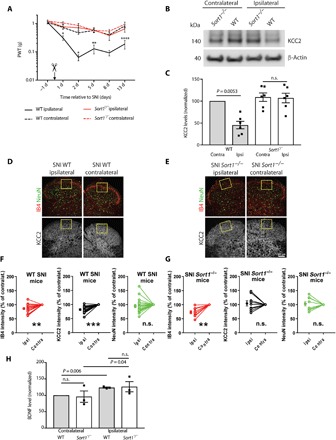
(A) Paw withdrawal threshold (PWT) to tactile stimuli of ipsilateral versus contralateral sides of WT and Sort1−/− mice before and after SNI (day 0). *P < 0.02, **P < 0.009, and ****P < 0.0001; n.s., not significant; n = 7 to 8, two-way repeated measures (RM) analysis of variance (ANOVA) with post hoc Tukey’s test [F(3,26) = 31.52, P < 0.0001], means ± SEM. (B) Representative Western blot of KCC2 in L3-L5 SDH 6 days after SNI. (C) KCC2 levels in L3-L5 SDH quantified by Western blot and normalized to WT contralateral 6 days after SNI. n = 6, one-way RM ANOVA with post hoc Tukey’s test [F(1.997,9.985) = 15.06, P = 0.001], means ± SEM. (D and E) IHC analysis showing IB4, NeuN, and KCC2 expression in the ipsilateral and contralateral SDH of WT and Sort1−/− mice. Scale bar, 100 μm. (F and G) Comparisons of average pixel intensity are shown across SNI animals of WT versus Sort1−/− mice in the region of interest (ROI). Nerve injury resulted in decreased IB4 intensity in the ROI in WT mice (contralateral versus ipsilateral: paired t test, t = 3.749; df = 18, P = 0.0015; n = 19) as in Sort1−/− mice (contralateral versus ipsilateral: paired t test, t = 4; df = 8, P = 0.004; n = 9). Nerve injury caused the down-regulation of KCC2 expression in the dorsal horn of WT mice but not in Sort1−/− mice [contralateral versus ipsilateral: (WT mice) paired t test, t = 6.24; df = 18, P < 0.0001; n = 19; and (Sort1−/− mice) t = 0.2093; df = 8, P = 0.839; n = 9]. No loss of neurons, measured as the difference in the average NeuN immunostaining intensities, was observed between ipsilateral and contralateral sides in both WT and Sort1−/− mice [contralateral versus ipsilateral: (WT mice) paired t test, t = 1.206; df = 18, P = 0.2436; n = 19; and (Sort1−/− mice) t = 0.3838; df = 8, P = 0.7111; n = 9]. **P < 0.01 and ***P < 0.0001; intensity units (i.u.) are shown as means ± SEM. (H) BDNF levels 6 days after SNI in L3-L5 SDH relative to WT contralateral [n = 3, pooled samples from eight mice for each run, paired t test within genotype (WT: t = 13.42, df = 2; Sort1−/−: t = 4.62, df = 2) and unpaired t test between genotypes (means ± SEM)].
Peripheral nerve injury stimulates release of signaling molecules from primary afferents into the SDH, initiating a neuroinflammatory response that ultimately leads to KCC2 down-regulation (16). As primary afferents (6) and microglia (13) both express sortilin, we considered whether the impaired KCC2 down-regulation in Sort1−/− mice is a consequence of impaired neuroinflammatory response visualized by compromised microglia reactivity. However, microglia reactivity (Iba1+) was also observed in the ipsilateral SDH of Sort1−/− mice after SNI comparable to WT mice (fig. S1).
As KCC2 down-regulation depends on BDNF (5), we next measured BDNF levels in the SDH by enzyme-linked immunosorbent assay and found a similar increase in WT and Sort1−/− mice following SNI (32.5 ± 8.9% in WT mice and 30.3 ± 15.6% in Sort1−/− mice; Fig. 1H). Measurements of the spinal neurotransmitters γ-aminobutyric acid (GABA), glycine, and glutamate, as assessed by high-performance liquid chromatography (HPLC) analysis, demonstrated identical levels in WT and Sort1−/− mice (fig. S2). We lastly assessed whether the “pain circuitry” is functionally intact in Sort1−/− mice, i.e., whether KCC2 function is normal and, subsequently, whether the inhibition of KCC2 function could induce mechanical allodynia in Sort1−/− mice comparable to WT mice. To accomplish this, we examined the transporter activity in reverse mode by measuring K+-driven uptake of Cl− in superficial dorsal horn neurons of lumbar spinal cord slices but found no difference in KCC2-mediated Cl− transport between genotypes (fig. S3A). Next, we applied the highly specific KCC2 antagonist VU0463271 (17) to naïve WT and Sort1−/− mice by intrathecal injections into the lumbar spinal cord. Subsequent von Frey measurements revealed that blockade of KCC2 in Sort1−/− mice induced mechanical allodynia fully comparable to the effect observed in WT mice (fig. S3B), arguing that the observed phenotype in Sort1−/− mice upon SNI relies on altered functional properties rather than on developmental changes in the spinal pain processing circuitry.
Sort1−/− mice are resistant to BDNF-induced pain
We next tested the responsiveness of Sort1−/− mice to BDNF stimulation. Lumbar intrathecal injections of BDNF induced robust transient mechanical allodynia in WT mice. In contrast, BDNF injections did not induce allodynia in Sort1−/− mice (Fig. 2A), demonstrating that these mice are functionally unresponsive to BDNF. Identical spinal TrkB levels for WT and Sort1−/− mice demonstrated that this observation was not an effect of reduced TrkB expression (Fig. 2, B and C). In a previous study, we reported that sortilin physically associates with Trk receptors to enhance their anterograde transport, exposure on the plasma membrane, and signaling (6). Accordingly, cultured DRG sensory neurons from Sort1−/− mice demonstrated blunted extracellular signal–regulated kinase 1 and 2 (ERK1/2) signaling upon neurotrophin stimulation. In marked contrast to this observation, we observed no changes in subcellular localization of TrkB in the SDH of Sort1−/− mice (fig. S4), proposing that spinal TrkB missorting is not the underlying cause of blunted BDNF-induced allodynia in Sort1−/− mice. Other studies have demonstrated that the detection of endogenous phospho-TrkB is notoriously difficult in extracts from adult CNS and PNS (18, 19), an observation we could confirm. Hence, we examined activation of ERK1/2 in the SDH of WT and Sort1−/− mice following intrathecal administration of BDNF. Phospho-ERK1/2 was increased by 128 ± 22% in WT mice 3 hours after BDNF injection. In contrast, ERK1/2 signaling in the Sort1−/− mice did not stand out from their unstimulated controls (Fig. 2D). Together, the data suggest that BDNF signaling is blocked downstream of TrkB, suggesting that additional signals converging on the BDNF pathway are required for the induction of neuropathic pain.
Fig. 2. Spinal BDNF signaling is altered in the absence of sortilin.
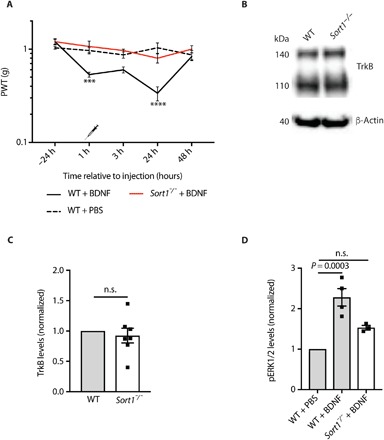
(A) PWT to tactile stimuli of WT and Sort1−/− mice before and after intrathecal BDNF or vehicle injection. ***P < 0.0009 and ****P < 0.0001, versus WT + vehicle; n = 6, two-way RM ANOVA with post hoc Dunnett’s test [F(2,15) = 10.96, P < 0.0012], means ± SEM. (B) Representative Western blot of TrkB in L3-L5 SDH. (C) TrkB levels in L3-L5 SDH in naïve WT and Sort1−/− mice quantified by Western blotting. n = 7, t test, means ± SEM. (D) pERK1/2 levels in L3-L5 SDH 3 hours after intrathecal phosphate-buffered saline (PBS) or BDNF injection at spinal L3-L5. n = 3 to 4, one-way ANOVA with post hoc Dunnett’s test [F(2,8) = 22.58, P = 0.0003), means ± SEM.
The Sort1−/− phenotype can be reversed by NTSR2 inhibition
The involvement of neurotensin pathways in neuropathic pain is well documented (9–12). Anatomically, neurotensin can be detected in several brain regions directly involved in nociceptive transmission and modulation by descending projections, e.g., in the periaqueductal gray (PAG) and various raphe nuclei in the rostral ventromedial medulla (20, 21). Furthermore, neurotensin, NTSR1 and NTSR2, are all concentrated in fibers and cell bodies in laminae I to III of the SDH in both rodents and humans (9, 10, 20, 21), and neurotensin has been reported to be up-regulated in the lumbar spinal cord of rodents with neuropathic pain (10, 22, 23). Sortilin is expressed in several neurotensin-responsive tissues and cells (7, 8, 24) where it can bind the neuropeptide with high affinity (0.1 to 0.3 nM) comparable to that of NTSR1 and NTSR2 (0.1 to 0.3 nM and 3 to 10 nM, respectively) (11). Previous studies demonstrate that sortilin, despite being devoid in apparent consensus signaling motifs, partakes in neurotensin signaling (13). Hence, we speculated that sortilin might be implicated in neurotensin signaling in the SDH by acting as a regulator of extracellular neurotensin levels and, consequently, by affecting NTSR1/NTSR2 activity. To test this hypothesis, we administered the NTSR1/NTSR2 antagonist SR142948A to WT and Sort1−/− mice subjected to SNI. Administration of SR142948A induced ipsilateral mechanical allodynia in Sort1−/− mice at a level equivalent to that of WT mice, whereas the contralateral side was unaffected by the antagonist (Fig. 3A). Upon return to Sort1−/− baseline levels, re-injections with the NTSR1/NTSR2 antagonist again induced robust allodynia on the ipsilateral side. In contrast to Sort1−/− mice, the pain threshold in WT mice was unaffected by the antagonist. Conclusively, these data demonstrate how NTSR1/NTSR2 signaling inhibits allodynia in Sort1−/− mice, as blocking of these receptors render Sort1−/− mice susceptible to SNI-induced pain. Our data also suggest that endogenous NTSR1/NTSR2 signaling is insufficient to provide analgesic effects following SNI in WT mice, i.e., in the presence of sortilin.
Fig. 3. Sortilin deletion unmasks NTSR2 signaling.
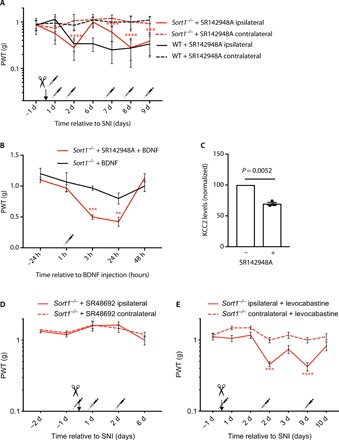
(A) PWT to tactile stimuli of WT and Sort1−/− mice before and after SNI and intrathecal SR142948A injection. ***P < 0.0004 and ****P < 0.0001, versus the contralateral side of Sort1−/− mice; n = 6 to 8, two-way RM ANOVA with post hoc Dunnett’s test [F(3,24) = 35.53, P < 0.0001], means ± SEM, with syringes representing double injections per day. (B) PWT to tactile stimuli of Sort1−/− mice before and after intrathecal BDNF ± SR142948A injection. **P < 0.009 and ***P < 0.0008; n = 6, two-way RM ANOVA with post hoc Sidak’s test [F(1,10) = 10.49, P = 0.0089], means ± SEM, with the syringe representing double intraperitoneal SR142947A injections on days −2, −1, and 0 and one intrathecal BDNF injection on day 0, 1 hour before testing. (C) KCC2 levels in ipsilateral L3-L5 SDH of Sort1−/− mice with and without intrathecal SR142948A injection 6 days after SNI. n = 3, paired t test, means ± SEM. (D) PWT to tactile stimuli of Sort1−/− mice before and after SNI and intraperitoneal SR48692. n = 6, two-way RM ANOVA with post hoc Dunnett’s test, means ± SEM, with syringes representing double injections per day. (E) PWT to tactile stimuli of Sort1−/− mice before and after SNI and intraperitoneal levocabastine. ***P < 0.0003 and ****P < 0.0001; n = 7, two-way RM ANOVA with post hoc Dunnett’s test [F(3,24) = 39.06, P < 0.0001], means ± SEM, with syringes representing single injections.
BDNF/TrkB signaling is a principal driver of spinal disinhibition in WT mice (5, 25, 26), but we found BDNF unable to induce allodynia in Sort1−/− mice (Fig. 2A). As SR142948A was able to sensitize Sort1−/− mice following SNI, we asked whether this compound could also sensitize these mice to BDNF. To test this, BDNF was administered by lumbar intrathecal injections to SR142948A-treated Sort1−/− mice, and we found that BDNF then induced transient allodynia (Fig. 3B). Western blotting (WB) analysis of the SDH substantiated that KCC2 levels, which were otherwise unaffected ipsilaterally following SNI in Sort1−/− mice (Fig. 1C), were significantly reduced following SR142948A treatment (Fig. 3C). The contralateral side was unaffected by SR142948A treatment (data not shown). Last, to clarify the involvement of NTSR1 versus NTSR2, we injected selective antagonists against either receptor (SR48692 and levocabastine, respectively) and found that only the inhibition of NTSR2 could induce allodynia in Sort1−/− mice (Fig. 3, D and E). Injection of SR48692 and levocabastine in WT mice had no effect (data not shown).
The sortilin extracellular domain is sufficient to prevent allodynia
Sortilin carries several sorting signals in the cytoplasmic tail and has been demonstrated to engage in endocytosis and Golgi-endosome transport, as well as in anterograde transport along neurites (7). However, we did not find any changes in either neurotensin mRNA or protein levels nor in NTSR2 protein levels between uninjured WT and Sort1−/− lumbar SDH or following SNI (fig. S5), arguing that the function of sortilin observed here is accounted for neither by extracellular clearance nor by lysosomal degradation of this ligand. A number of studies have demonstrated that sortilin may be cleaved off the plasma membrane, giving rise to a soluble protein encompassing the entire extracellular domain (ECD) of sortilin, capable of ligand binding—an observation that may explain the presence of soluble sortilin in plasma and cerebrospinal fluid (CSF) from humans (27–30). Hence, we speculated that sortilin may act outside the cells. To test this, polyclonal anti-sortilin antibodies were injected intrathecally into WT mice, and their response to von Frey filaments following SNI was monitored. We found that administration of control (pre-immune) immunoglobulin G (IgG) at the time of SNI had no effect, whereas anti-sortilin antibodies could completely prevent mechanical allodynia (Fig. 4A). The effect of a single dose lasted approximately 2 days, with normal levels of mechanical allodynia appearing around day 3. We further attempted to reverse induced allodynia by intrathecal antibody injections 6 days after SNI; injection of control antibodies at day 6 after injury had no effect, but anti-sortilin antibodies were able to partially reverse mechanical allodynia. Again, the effect lasted for 2 to 3 days, after which pre-injection levels were observed (Fig. 4, B to D).
Fig. 4. The extracellular domain of sortilin is sufficient for BDNF/TrkB signaling.
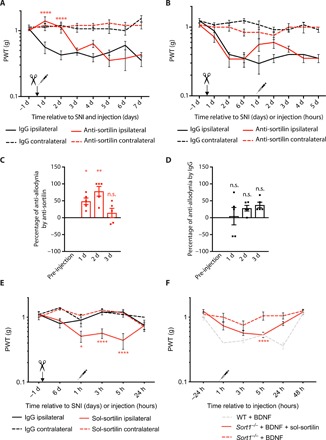
(A) PWT to tactile stimuli of WT mice before and after SNI and intrathecal anti-sortilin antibodies or immunoglobulin G (IgG) injection at day 0. ****P < 0.0001 for ipsilateral anti-sortilin versus ipsilateral IgG; n = 6, two-way RM ANOVA with post hoc Dunnett’s test [F(3,20) = 48.09, P < 0.0001], means ± SEM. (B) PWT to tactile stimuli of WT mice before and after SNI (day 0) and intrathecal anti-sortilin antibodies or IgG injection 6 days after SNI, means ± SEM. (C) Percentage of anti-allodynia by anti-sortilin 1, 2, and 3 hours versus pre-injection baseline (average of days 2 and 6 of the same mouse). *P < 0.03 and **P < 0.001; n = 5, one-way RM ANOVA with post hoc Dunnett’s test [F(3,12) = 9.691, P = 0.0016], means ± SEM. (D) Percentage of anti-allodynia by IgG 1, 2, and 3 hours versus average of days 2 and 6 of the same mouse. n = 5, one-way RM ANOVA with post hoc Dunnett’s test, means ± SEM. (E) PWT to tactile stimuli of Sort1−/− mice before and after SNI (day 0) and intrathecal sol-sortilin or IgG injection (0 hours). *P < 0.02 and ****P < 0.0001, versus the ipsilateral side of intrathecal IgG; n = 4 to 7, two-way RM ANOVA with post hoc Dunnett’s test [F(3,18) = 15.17, P < 0.0001], means ± SEM. (F) PWT to tactile stimuli of WT and Sort1−/− mice before and after intrathecal BDNF ± sol-sortilin injection. ****P < 0.0001 versus Sort1−/− + BDNF injection; n = 6 to 12, two-way RM ANOVA with post hoc Dunnett’s test [F(2,23) = 10.85, P = 0,0005], means ± SEM.
To substantiate whether the sortilin ECD is indeed sufficient for mechanical allodynia development, we introduced the entire ECD as soluble protein (sol-sortilin) into the lumbar spinal cord of Sort1−/− mice. Intrathecal injections of sol-sortilin 6 days after SNI resulted in transient mechanical allodynia in the otherwise pain-resistant Sort1−/− mice (Fig. 4E). These results prompted us to investigate whether rescue by sol-sortilin could also restore BDNF-mediated sensitization. We found that, while intrathecal BDNF failed to induce transient mechanical allodynia in Sort1−/− mice, co-injecting sol-sortilin with BDNF caused sensitization in Sort1−/− mice (Fig. 4F). Collectively, these data are compatible with a model in which sortilin scavenges neurotensin from binding to NTSR2, modulating its inhibition on the BDNF-mediated down-regulation of KCC2 and the induction of mechanical allodynia (fig. S6).
Inhibition of ligand binding to sortilin prevents injury-induced tactile allodynia
The binding of a 324-Da small-molecule ligand (AF38469) to the neurotensin binding site in sortilin has been thoroughly characterized by modeling of the crystal structures obtained for sortilin in complex with neurotensin or a small-molecule ligand. It was demonstrated that the four C-terminal amino acids of neurotensin bind to a pocket in the tunnel of the sortilin Vps10p domain and that AF38469 inhibits this binding by fitting into the binding pocket (14, 31, 32). We therefore tested AF38469 in the SNI model by lumbar intrathecal injection in WT and Sort1−/− mice. Injection of AF38469 at the time of SNI delayed the onset of mechanical allodynia in WT mice for 1 day, with a normal post-SNI mechanical threshold appearing at day 2 (Fig. 5A). No effect was observed on the WT contralateral side or in Sort1−/− mice. Control injections with an inactive form of AF38469 (the stereoisomer AF38540), which is unable to bind sortilin, had no analgesic effect. Injection of AF38469 2 days following SNI transiently induced a full rescue (Fig. 5, B to D). Moreover, AF38469 was able to impair BDNF-induced allodynia in WT mice (Fig. 5E), demonstrating that specific inhibition of sortilin ligand binding effectively impairs BDNF-dependent mechanical allodynia.
Fig. 5. Pharmacological inhibition of sortilin prevents mechanical allodynia.
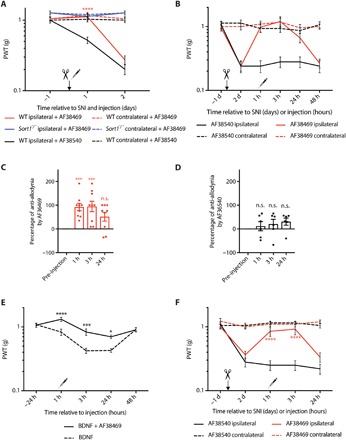
(A) PWT to tactile stimuli of WT and Sort1−/− mice before and after SNI and intrathecal AF38469 (active) or AF38540 (inactive) injection. ****P < 0.0001 versus the ipsilateral side of WT + AF38540; n = 13 to 17, two-way RM ANOVA with post hoc Dunnett’s test [F(5,66) = 17.75, P < 0.0001], means ± SEM. (B) PWT to tactile stimuli of WT mice after intrathecal AF38469 (active) or AF38540 (inactive) injection 2 days after SNI. (C) Percentage of anti-allodynia by AF38469 1, 2, and 3 hours versus pre-injection baseline (day 2 of the same mouse). ***P < 0.0007; n = 9, one-way RM ANOVA with post hoc Dunnett’s test [F(3,24) = 8.597, P = 0.0005], means ± SEM. (D) Percentage of anti-allodynia by AF38540 1, 2, and 3 hours versus pre-injection baseline (day 2 of the same mouse). n = 6, one-way RM ANOVA with post hoc Dunnett’s test, means ± SEM. (E) PWT to tactile stimuli of WT mice before and after intrathecal BDNF ± AF38469 (active) injection. *P < 0.03, ***P < 0.0004, and ****P < 0.0001; n = 6 to 7, two-way RM ANOVA with post hoc Sidak’s test [F(1,11) = 19, P = 0.0011], means ± SEM. (F) PWT to tactile stimuli of hum-sortilin-KI mice after intrathecal AF38469 (active) or AF38540 (inactive) injection 2 days after SNI. ****P < 0.0001 versus ipsilateral side with AF38540; n = 8, two-way RM ANOVA with post hoc Dunnett’s test [F(3,28) = 45.67, P < 0.0001, means ± SEM.
Last, to test the translational perspective of AF38469 or similar molecules for the treatment of neuropathic pain in humans, we generated a humanized mouse model in which murine sortilin is replaced by human sortilin (hum-sortilin-KI) under the control of the endogenous mouse Sort1 promoter (fig. S7). Our experiments demonstrated that hum-sortilin-KI mice behaved identically to WT mice in terms of baseline mechanical sensitivity and the development of mechanical allodynia following SNI. Furthermore, application of AF38469 2 days following SNI fully suppressed mechanical allodynia (Fig. 5F), demonstrating that AF38469 targets human sortilin in vivo and substantiates sortilin as a target for neuropathic pain treatment.
DISCUSSION
It has previously been demonstrated how NTSR1 and NTSR2 agonists have analgesic effects following peripheral nerve injury (9–11), but it has not been studied how this effect relates to the BDNF/TrkB-mediated induction of neuropathic pain. Here, we show that inhibition of sortilin prevents the BDNF-induced KCC2 down-regulation in the SDH and impairs neuropathic pain in an NTSR2-dependent manner. Neurotensin acts as an endogenous analgesic and is detected in brain regions such as PAG and various raphe nuclei involved in descending inhibitory control. In addition, neurotensin exerts local inhibitory action at the spinal cord level via expression of neurotensin and NTSR1/NTSR2 in fibers and cell bodies in laminae I to III (20, 21). Our data suggest that NTSR2 signaling is able to inhibit the BDNF-mediated KCC2 down-regulation in SDH neurons and that sortilin functions as an endogenous regulator of NTSR2 signaling (fig. S6).
How can sortilin exert this control? We have previously described how sortilin regulates Trk receptor signaling by facilitating Trk anterograde transport (6). This finding arose primarily from studies of the PNS and hippocampus, but our data here argue that sortilin does not affect spinal TrkB cellular localization. Rather, our results show that the effect is mediated by the sortilin ECD, whether membrane bound as full-length sortilin or shedded as soluble sortilin, and can be narrowed down to the neurotensin binding site. In contrast to NTSR1 and NTSR2, sortilin is not considered to be a signaling receptor upon ligand binding. Rather, we reason that sortilin is able to regulate availability of extracellular neurotensin for NTSR2 activation. We have previously shown how sortilin binds progranulin, thereby regulating progranulin CSF levels by 2.5- to 5-fold (33). As for progranulin, full-length sortilin has been demonstrated to bind and internalize neurotensin (34, 35). Considering that the Kd values for binding of neurotensin to NTSR2 and sortilin have been determined to be 3 to 10 nM and 0.1 to 0.3 nM (11), respectively, sortilin may therefore indeed be able to compete with NTSR2 for ligands in the SDH and thus modulate the analgesic activity of NTSR2.
The presence of sol-sortilin has been demonstrated in the CSF (30), proposing that sortilin does not necessarily need to be expressed by NTSR2+ neurons or proximal cells to modulate NTSR2 activity. Sol-sortilin (approximately 15 ng/ml) was detected in human CSF (30), and both neurons and microglia have been reported to be able to shed sortilin ECD (13, 36), providing a cellular source of sol-sortilin to the CSF. Notably, it has further been reported how neurotensin increases surface expression and shedding of sortilin (13), suggesting a negative feedback loop by which increased neurotensin levels promote subsequent clearance.
The effect of intrathecal administration of sol-sortilin, anti-sortilin antibodies, and the small-molecule ligand AF38469 indicates that the sortilin effect is at the level of the lumbar spinal cord. This is in accordance with the presence of NTSR2 in the SDH, particularly in the substantia gelatinosa, which is highly innervated by Aδ and C afferent fibers (10). A minor diffusion to the brain stem or cerebrum following lumbar intrathecal injections may nevertheless occur, as demonstrated by an earlier study (37). Neurotensin has potent antinociceptive effects when injected into the PAG, and the PAG endogenous neurotensin level has been reported to increase in response to peripheral nerve injury in rodents (23). It is reasonable to speculate that if intrathecally injected sortilin inhibitors diffuse to the brainstem, then the increased neurotensin activity in the PAG induced by injury could also promote the observed analgesic effect via activation of descending inhibitory pathways. Thus, sortilin inhibition may potentially both enforce local lumbar neurotensinergic inhibition and activate PAG/raphe nuclei–mediated descending pathways.
Our finding that the effect of sortilin inhibition depends on NTSR2 does not rule out that there may be other endogenous ligands than neurotensin; the related neuropeptide Neuromedin N (NN), e.g., was recently identified as an NTSR2 agonist (38). NN and neurotensin are processed from the same precursor and are thus under identical transcriptional regulation. NN may thus act as an endogenous analgesic transmitter in the same pathways as neurotensin. Importantly, the four C-terminal amino acids (Pro-Tyr-Ile-Leu), which are essential for neurotensin binding to sortilin, are identical in NN, predicting equivalent binding affinity to sortilin (14, 38). Current knowledge of NN is, however, very limited, and further studies on the involvement of NN in pain pathways are warranted.
NTSR2 agonists have been proposed for the treatment of neuropathic pain (10). Considering the profound involvement of neurotensin signaling in various CNS functions, including endocrine and behavioral systems (39), it may turn out to be very difficult to titrate for selective analgesic effects. In contrast to NTSR2 agonists leading to global NTSR2 activation, regulation of neurotensin availability (or other NTSR2/sortilin ligands) by blocking the binding site on sortilin would be expected to preserve neurotensin signaling dynamics, including that of descending inhibitory pathways, by only strengthening NTSR2 activity in areas of neurotensin release. A similar strategy might underlie, e.g., the more refined and dynamic intervention of serotonin activity by selective serotonin reuptake inhibitor antidepressants relative to systemic application of serotonin receptor agonists.
In conclusion, our experiments have unraveled that blocking ligand binding to spinal sortilin impairs development of neuropathic pain following peripheral nerve injury. This occurs by strengthening NTSR2 inhibitory signaling, which, as demonstrated here, impairs the BDNF/TrkB-mediated down-regulation of spinal KCC2. Targeting sortilin might therefore provide a novel strategy to overcome spinal disinhibition.
MATERIALS AND METHODS
Mice
C57Bl/6J mice were purchased from Taconic Biosciences (Denmark). Sort1−/− mice have been described previously (6). C57BL/6J mice expressing the human SORT1 gene under the control of the endogenous mouse Sort1 promoter (hum-sortilin-KI) were generated by TaconicArtemis (Germany). The coding region in mouse exon 1 was replaced with a human SORT1 complementary DNA (cDNA) engineered to improve the expression of the human SORT1 cDNA in mouse cells and to prevent downstream transcription of the remaining mouse Sort1 gene. The human SORT1 cDNA was engineered by inserting the human SORT1 intron 7 between exons 7 and 8 and introducing the mouse Sort1 3′ untranslated region (3′UTR) downstream of the translation termination codon in exon 20 (fig. S7A). An additional polyadenylation signal (human growth hormone polyadenylation signal) was inserted downstream of the 3′UTR. The mouse genomic sequence downstream of exon 1 was left intact to preserve potential regulatory elements. Mouse genomic fragments were obtained from the C57BL/6J RPCIB-731 bacterial artificial chromosome library. For generation of heterozygous-targeted C57Bl/6J embryonic stem (ES) cells, the targeting vector was introduced by homologous recombination. ES cells were grown on a mitotically inactivated feeder layer composed of mouse embryonic fibroblasts in ES cell culture medium containing leukemia inhibitory factor and fetal bovine serum. Cells were electroporated with the linearized DNA targeting vector according to standard procedures. Resistant ES cell colonies with a distinct morphology were isolated and validated by Southern blotting and polymerase chain reaction (PCR). Homologous recombinant ES cell clones were expanded and frozen in liquid nitrogen. Hum-sortilin-KI mice were generated by breeding with C57BL/6J transgenic Flp_deleter mice. Mice were genotyped by PCR detecting the constitutive humanized allele, and protein expression was confirmed by WB (fig. S7B).
Behavioral studies
C57BL/6J, C57BL/6JSort1−/−, and hum-sortilin-KI mice (males and females, aged 8 to 10 weeks) were subjected to the SNI model, and mechanical allodynia was assessed by von Frey testing, as described previously (40). Briefly, the common peroneal and tibial branches of the sciatic nerve were ligated and cut distally to the ligation, just distal to the branching of the sural nerve, which was left untouched. Testing was performed by the same female researcher throughout the experiments to avoid person-to-person variation and gender bias (41). von Frey experiments on WT and Sort1−/− mice subjected to SNI were repeated by two independent researchers, blinded to genotype and treatment types. Isoflurane gas (IsoFlo vet, Abbott) was applied with a Univentor 1200 anesthesia unit (Univentor). Analgesia, Lidocaine SAD (10 mg/ml; Amgros I/S) applied on wound; temgesic, buprenorphine (0.3 mg/ml; RB Pharmaceuticals); and antibiotics, pentrexyl (250 mg/ml; Bristol-Myers Squibb). Temgesic and pentrexyl were mixed and diluted 1:10 in isotonic saline (9 mg/ml; Fresenius Kabi) and were injected subcutaneously at 0.1 ml. Mice were housed with littermates at all times with water and chow ad libitum with a 12-hour light/dark cycle (tests were performed during the light phase). All experiments were approved by the Danish Animal Experiment Inspectorate (permission no. 2006/561-1206 and 2017-15-0201-01192).
Intrathecal injections (anti-sortilin, BDNF, sol-sortilin, small-molecule ligand, and VU0463271)
Mice were single injected intrathecally around lumbar vertebral segments L4/L5 or L5/L6 in a 30° to 45° angle using a Hamilton syringe (30 gauge) eliciting a Straub tail response (37).
Antibodies
Anti-sortilin (8 μl) (2 μg per mouse; AF2934, R&D Systems) and normal goat IgG (8 μl) (2 μg per mouse; AB-108-C, R&D Systems), both diluted in sterile phosphate-buffered saline (PBS; Life Technologies), were used. Intrathecal injections were performed immediately after SNI (Fig. 4A). Human BDNF (7.5 μl) (1.5 μg per mouse; B-250, Alomone Labs) diluted in sterile PBS and sol-sortilin (8 μl; 10 μg per mouse) diluted in sterile PBS were prepared, as previously described (14).
Small-molecule ligands
AF38469 (7 μl) (14, 31, 32) binding to the neurotensin binding site in sortilin and the inactive stereoisomer AF38540 [both 20 μM, dissolved in dimethyl sulfoxide (DMSO) and diluted in sterile PBS] were provided by H. Lundbeck A/S (Denmark). For injection of a combination of BDNF and AF39469, concentrations similar to individual injections were mixed before the injection (Fig. 5E). Intrathecal injections were performed immediately after SNI (Fig. 5A).
KCC2 antagonist
VU0463271 (8 μl) (20 μM, 4719, R&D Systems) was dissolved in DMSO and diluted in sterile PBS.
Intraperitoneal injections (SR142948A, SR48692, and levocabastine)
Mice were injected intraperitoneally with the following neurotensin receptor antagonists: NTSR1/NTSR2 antagonist SR142948A (1 mg/kg; SML00015, Sigma-Aldrich) at injection intervals of twice a day for three consecutive days on day 0 before and after SNI and on days 1, 2, 7, 8, and 9, with two injections approximately 6 hours apart (Fig. 3A), and at injection intervals of twice a day on days 4 to 6 after SNI with tissue collection on day 6 after SNI (Fig. 3C); selective NTSR1 antagonist SR48692 (3 mg/kg; SML0278, Sigma-Aldrich) at injection intervals of twice a day for three consecutive days on day 0 before and after SNI and on days 1 and 2, with two injections approximately 6 hours apart (Fig. 3D); and selective NTSR2 antagonist levocabastine (2.5 mg/kg; L3042, Sigma-Aldrich) at injection intervals of single injections on day 0 before SNI and on days 2 and 9 approximately 2 hours before testing (Fig. 3E). All substances were dissolved according to the manufacturer’s protocol and diluted in sterile PBS. Injection volumes were 200 μl per injection twice a day (alternating sides). Mice were injected intraperitoneally with SR142948A at injection intervals of twice a day on days −2 to 0, approximately 6 hours apart, and intrathecally with BDNF once a day on day 0, 1 hour before testing (Fig. 3B).
Western blotting
Spinal cords were extruded with ice-cold PBS (pH 7.4), 6 days after injury, and the dorsal horn of the lumbar enlargement corresponding to L3 to L5 was isolated. Following BDNF injections, spinal cords were extruded with ice-cold PBS (pH 7.4) at times 0 (no injection), 3, 6, and 24 hours, and the entire lower lumbar enlargement was used for WB. The tissue was lysed in standard Tris-NaCI-EDTA lysis buffer supplemented with protease and phosphatase inhibitors. Following SDS–polyacrylamide gel electrophoresis (PAGE) and blotting, the proteins were probed against NTSR-3 (1:1000; 61200, BD Biosciences), KCC2 (1:1000; 07-432, Millipore), TrkB (1:1000; AF1494, R&D Systems), ERK1/2 (1:1000; 4695, Cell Signaling Technology), pERK1/2 (1:1000; 9101, Cell Signaling Technology), Akt (1:500; 9272, Cell Signaling Technology), pAkt (1:300, 9275, Cell Signaling Technology), NKCC1 (1:1000; Lytle, C., Developmental Studies Hybridoma Bank, NICHD, The University of Iowa), pNKCC1 (1:1000; donated by B. Forbush, Yale University, New Haven, CT), and β-actin (1:5000; Sigma-Aldrich), visualized with horseradish peroxidase–conjugated secondary antibody (Dako) and ECL substrate (Amersham) by LAS 4000 (Fujifilm Life Science) and quantified using Multi Gauge version 3.2 (Fujifilm Life Science).
Enzyme-linked immunosorbent assay
BDNF Emax ImmunoAssay System (Promega) was applied with Nunc-Immuno MaxiSorp plates (Nunc) according to the manufacturer’s protocol on tissue processed as for WB.
Quantitative PCR
Spinal cords were isolated as for WB and stored in the RNAlater Stabilization Solution at 4°C, and cDNA was obtained according to the manufacturer’s protocol (both Invitrogen). Quantitative PCR was performed with primers for Gfap (Mm01253033_m1), Hprt (Mm03024075_m1), Nts (Mm00481140_m1), Ntsr1 (Mm00444459_m1), and Ntsr2 (Mm01270334_m1) according to the manufacturer’s protocol on the 7500 Fast Real-Time PCR System (Thermo Fisher Scientific).
Immunohistochemistry
Spinal cords were extruded and processed for cryosectioning. Cryosections (10 μm; Leica CM1900 cryostat with CryoJane tape transfer system, Leica) were incubated against astrocyte or microglia markers (glial fibrillary acidic protein: 1:1000, Z0334, Dako or Iba1: 1:500, 019-19741, Wako, respectively) and, subsequently, with Alexa Fluor 488–conjugated secondary antibody (A-11059, Invitrogen). Astrocytes and microglia were quantified with ImageJ (NIH). The contralateral uninjured SDH served as a control in each section.
Quantitative IHC was performed as follows: 35-μm sections cut on a sledge freezing microtome Leica SM2000 R (Leica Microsystems) were permeabilized in PBS (pH, 7.4) with 0.2% Triton X-100 (PBS-T) for 10 min, washed twice in PBS, and incubated for 12 hours at 4°C in primary antibodies diluted in PBS-T containing 10% normal goat serum [rabbit anti-KCC2 (1:1000; 07-432, Upstate, EMD Millipore)]. Chicken anti-NeuN (1:1000; ABN91, EMD Millipore) was used as a control marker in the superficial dorsal horn. After washing in PBS, the tissue was incubated for 2 hours at room temperature in a solution containing a mixture of the Cy3 goat anti-rabbit purified secondary antibody (1:500; 111-165-144, Jackson ImmunoResearch Laboratories Inc.), Alexa Fluor 647 goat anti-chicken purified secondary antibody (1:500; A-21449, Thermo Fisher Scientific), and Isolectin B4 [Alexa Fluor 488–conjugated IB4 (1:500; I21411, Invitrogen), used to identify lesions due to the loss of nonpeptidergic terminals in SNI mice in the L4 and L5 lumbar segments of superficial dorsal horns] diluted in PBS-T (pH 7.4) containing 10% normal goat serum. Sections were mounted on superfrost gelatin-subbed slides (Fisherbrand), allowed to dry overnight at 4°C, and coverslipped using a fluorescence mounting medium (S3023, Dako). During image acquisition, the experimenter was blind to the genotype of the mouse spinal cord. Images were obtained with the confocal LSM 880 Zeiss Laser Scanning Microscope equipped with a Zeiss 20× plan apochromatic air objective (numerical aperture, 0.8). Laser power was adequately chosen to avoid saturation and limit photobleaching. All the acquisitions were performed with the same settings [laser, power, and photomultiplier tube (PMT) settings; image size; pixel size; and scanning time]. Acquisitions were 12-bit images of size 2048 × 2048 pixels with a pixel dwell time of 1.02 μs and a pixel resolution of 0.3 μm. In spinal cord sections, KCC2, IB4, and NeuN average pixel intensities in superficial dorsal horn were measured using 64-bit Fiji-ImageJ (version 1.52i). Loss of IB4 staining was used to define the region of interest (ROI) analyzed in ipsilateral side of SNI mice. Corresponding contralateral regions were used as an internal control in each ipsilateral side of SNI mouse.
Synaptosomal preparation from spinal cord
Spinal cord tissue was isolated as for WB, and the SDH of spinal L3 to L5 were isolated. The procedure for fractionation of spinal cords from mice into synaptosomes, synaptic vesicle fractions, synaptic plasma membrane, and postsynaptic densities was performed, as previously described (6). All procedures were performed at 4°C. Briefly, spinal cords were isolated from eight WT and eight Sort1−/− mice (8 to 12 weeks of age), pooled, and homogenized following addition of 10 volumes of ice-cold 0.32 M sucrose, 4 mM Hepes, and protease inhibitor (pH 7.4; cOmplete, Roche), using a glass Teflon homogenizer. WT and Sort1−/− samples were processed in parallel for direct fraction comparison. After centrifugation of the homogenate for 10 min at 1000g, P1 and S1 corresponded to pellet and supernatant, respectively. S1 was further centrifuged for 15 min at 10,000g to obtain supernatant S2 and pellet P2, a crude synaptosomal preparation. Solubilized P2 was centrifuged for 15 min at 10,000g, and the resulting pellet was lysed by hypoosmotic shock in H2O, rapidly adjusted to 4 mM Hepes (pH 7.4), and stirred for 30 min. The lysate was centrifuged at 25,000g for 20 min, generating the synaptosomal membrane fraction P3 and a supernatant S3 enriched in presynaptic vesicles. S3 was further centrifuged at 165,000g for 2 hours, generating pelleted synaptic vesicle preparation enriched in synaptic vesicles. Solubilized P3 was applied to a discontinuous sucrose gradient containing 0.8, 1.0, and 1.2 M sucrose and centrifuged at 150,000g for 2 hours. The fraction between the 1.0 and 1.2 M sucrose layers was recovered and diluted to 0.32 M sucrose, after which it was centrifuged again at 150,000g for 30 min, resulting in the pelleted synaptosomal plasma membrane (SPM) fraction. The SPM fraction was resuspended with 0.4% Triton X-100 in 50 mM Hepes (pH 7.4) and 2 mM EDTA containing protease inhibitors and was centrifuged at 35,000g for 20 min to obtain the pelleted postsynaptic density fraction I (PSDI) containing postsynaptic densities. Purity was increased by resolubilizing the PSDI fraction, followed by centrifugation at 200,000g for 20 min yielding PSDII. The total protein concentration in the fractions was determined using a bicinchoninic acid kit (Sigma-Aldrich). Equal amounts of proteins of each fraction were separated by reducing SDS-PAGE and analyzed by WB for the presence of TrkB (1:1000; AF1494, R&D Systems), PSD95 (1:1000; P-246, Sigma-Aldrich), and synaptophysin (1:1000; MAB5258, Chemicon).
Imaging of reverse Cl− transport in SDH lamina I neurons
Parasagittal lumbar spinal slices (thickness, 350 μm) of WT and Sort1−/− mice were prepared and kept in bubbled artificial CSF (ACSF) at 34°C. Slices were then incubated in ACSF containing 5 mM of the Cl− indicator MQAE (N-6-methoxyquinolinium acetoethylester, Molecular Probes) for 30 to 40 min and transferred to a perfusion chamber (2 ml/min). Extracellular MQAE was washed out for 10 min in the presence of 1 μM tetrodotoxin, 10 μM CNQX (6-cyano-7-nitroquinoxaline-2,3-dione), 40 μM AP5, 1 μM strychnine, and 10 μM gabazine to minimize KCC2-independent Cl− transport. Fluorescence lifetime imaging of MQAE was conducted, as previously described (42). Instrument response function of the detection path was acquired using an 80-nm gold nanoparticle suspension to generate second-harmonic signal. Recorded cells were visually identified by merging transmitted light and MQAE fluorescence. Lifetime images were acquired every 10 s for a period of 7 min. After a control period of 50 s, the perfusion solution was switched to ACSF (controlled perfusion rate at 2 ml/min) containing 15 mM KCl (osmolarity adjusted using mannitol) to reverse KCC2-mediated Cl− transport. Lifetime in each cell was averaged over the whole-cell body area and extracted for each time point using a custom-made MATLAB software. Lifetime changes for each slice were then expressed as the mean of changes occurring in each cell. Briefly, based on the work of Digman et al. (43), we converted the photon timing histograms of each acquired lifetime image to phasor plots. Then, for every time point, ROIs corresponding to cell bodies were selected and added to a new phasor. Lifetime of all the cells was averaged for each slice at each time point to generate the lifetime time course.
Mass spectrometry analysis
Tissue processing for liquid chromatography–parallel-reaction monitoring–mass spectrometry
Spinal cord tissue was isolated as for WB and immediately placed on dry ice. Hot MilliQ water (300 μl) was added to frozen spinal cords (WT, 3.9 mg ±0.3; KO, 3.8 mg ± 0.3) and immediately homogenized using a blender, followed by heating for 10 min at 95°C in a water bath. The tissue was further homogenized using a Dounce tool with 10 strokes of each piston. Supernatant was recovered by centrifugation at 16,000g for 15 min at 4°C. The pellet was resuspended in 200 μl of cold 0.25% acetic acid/50% acetonitrile/water, and once again, the supernatant was recovered by centrifugation. The two supernatants were pooled, and isotope-labeled neurotensin peptide (pyro-QLYENK[+8]PRRPYIL[+7]) was added. Supernatant was applied to a 10 K cutoff filter (Millipore) and centrifuged at 14,000g for 30 min at 4°C. The filter was subsequently washed with 50 μl of 0.5 M NaCl, followed by another round of centrifugation. The peptide fraction passing through the filter was lyophilized using a SpeedVac concentrator, and the final volume was adjusted to 50 μl (pH 8) by adding 2.5 μl of 2 M tris-HCl (pH 8). Then, the peptide fraction was digested for 12 hours using 0.5 μg of trypsin (sequence grade, Sigma-Aldrich) at 37°C. Digested samples were prepared for mass spectrometry (MS), as previously described (44). LC–parallel reaction monitoring (PRM)–MS analysis was performed on a TripleTOF 6600 instrument (SCIEX) equipped with an in-line Eksigent ekspert nanoLC 400 system and a NanoSpray III source (AB SCIEX) and operated under Analyst TF 1.7.0 control. Samples were analyzed as previously described (45) using a targeted PRM method against light and heavy tryptic neurotensin peptides.
Post-acquisition extracted ion chromatogram
PRM-MS files were converted to Mascot generic format (MGF) using the AB SCIEX MS Data Converter beta 1.3 (AB SCIEX) and “ProteinPilot MGF” parameters. The peak lists were used to match the mouse neurotensin sequence in the Swiss-Prot database (2017_09) using Mascot 2.5.1 (Matrix Science, London, UK), and search results were imported to MS Data Miner version 1.3.0 (46). Trypsin was selected as the digestion enzyme, allowing two missed cleavages. The data were searched with a mass tolerance of the precursor and product ions of 10 parts per million and 0.2 Da using ESI-QUAD-TOF (electrospray ionization quadrupole time-of-flight) as the instrument setting. The significance threshold (P) was set to 0.05, and the ion score expected cutoff was set to 20. Mascot DAT files were extracted and used to build a spectral library in Skyline (MacCoss Lab, University of Washington). A Skyline PRM analysis was performed using TOF settings. The method monitored intact neurotensin peptide and the two neurotensin peptides derived by trypsin digestion. Only the N-terminal tryptic peptide was observed in the analysis represented by seven product ions (y2, y3, y4, y5, y6, b2, and b3) for both the light and heavy peptide [pyro-QLYENKPR (+2 m/z), pyro-QLYENK[+8]PR (+2 m/z)]. The light-to-heavy ratio between WT and Sort1−/− groups was used for statistical analysis.
HPLC analysis
Spinal cords were extruded as for WB, weighed, and snap-frozen on dry ice. Spinal cords were sonicated before further analysis. Electrochemical detection of amino acids was performed according to the Neuroactive Amino Acids Method and Maintenance Guide (ESA Magellan Biosciences). Briefly, samples were separated by isocratic elution (UltiMate 3000-series HPLC system with electrochemical detection, Dionex, Thermo Fisher Scientific). HPLC separation was performed on an HR-80 catecholamine column (Thermo Fisher Scientific) maintained at 35°C and with a flow rate 1.0 ml/min. Dual electrochemical detectors were set to +150 and +550 mV, respectively.
The mobile phase for isocratic separation consisted of 100 mM disodium hydrogen phosphate anhydrous, 22% methanol, and 3.5% acetonitrile adjusted to pH 6.75 with phosphoric acid. For the derivatization procedure, the o-Phthalaldehyde (OPA) stock solution was prepared by using 27 mg of OPA dissolved in 1 ml of MeOH and then adding 5 μl of 2-mercaptoethanol and 9 ml of OPA diluent. Fresh working OPA solution was prepared daily by using 2 ml of OPA stock solution and then adding 6 ml of OPA diluent. Spinal cord tissue was homogenized in 0.3 N perchloric acid by sonication on ice and then diluted 10 times with water. Lysates were cleared by centrifugation for 5 min at maximum gravity at 4°C. 20 μl was mixed with 50 μl of OPA working solution. After derivatization for 2 min, 10 μl was injected for analysis. Amino acid signals were quantified as area under the curve (Chromeleon software, Thermo Fisher Scientific). For absolute quantification, amino acid stock solutions were used to prepare dilution ranges that were analyzed in parallel. Tissue concentrations were obtained by normalizing the concentration of each sample to the weight of tissue used to prepare the lysate.
Statistical analysis
Statistical significance was determined as specified in figure legends using GraphPad Prism 7.0d software. Differences were considered significant at P < 0.05. All data are reported as means ± SEM.
Supplementary Material
Acknowledgments
We thank K. B. Riskjær and M. S. Terkildsen for important participation in the earliest phase of this project. Funding: This study was funded by The Lundbeck Foundation grants R77-A6802 (to C.B.V. and L.T.P.), R118-A11413 (to M.R.), R90-2011-7723 (to M.R., L.T.P., M.U., H.L., O.M.A., C.B.V., and A.N.), and R248-2017-431 and DANDRITE-R248-2016-2518 (to A.N.); The Danish Council for Independent Research | Medical Sciences DFF grants 4183-00494 (to C.B.V.) and 7016-00261 (to A.N.); The Carlsberg Foundation grant 2010_01-0784 (to C.B.V.); the EU FP7 grant 603191 PAINCAGE (to M.R., L.T.P., C.B.V., and A.N.); The Danish National Research Foundation grant DNRF133 (to A.N.); Dagmar Marshall’s Fund (to M.R., L.T.P., and C.B.V.); Simon Fougner Hartmanns Familiefond (to C.B.V.); Aase and Ejnar Danielsens Fond (to C.B.V.); and The Canadian Institutes of Health Research (to Y.D.K.). Author contributions: M.R., C.B.V., and A.N. conceived and designed the project. M.R., O.J.B., Y.D.K., C.B.V., and A.N. supervised the experiments. M.R., L.T.P., M.U., E.T.P., T.H.H., H.L., A.C., L.-E.L., and N.P.G. performed the experiments. L.C.B.R. and I.J.M. generated hum-sortilin-KI mice. M.R., L.T.P., E.T.P., A.C., L.-E.L., O.M.A., K.L.-H., J.J.E., Y.D.K., O.J.B., C.B.V., and A.N. analyzed the data and contributed to the interpretation of results. M.R., C.B.V., and A.N. wrote the manuscript. All authors read and discussed the manuscript. Competing interests: The authors declared that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav9946/DC1
Fig. S1. Injury-induced microglia reactivity in WT and Sort1−/− lumbar spinal cord.
Fig. S2. Neurochemical markers remain intact in sortilin-deficient mice.
Fig. S3. Direct KCC2 inhibition induces pain hypersensitivity.
Fig. S4. Subcellular localization of TrkB is unchanged in SDH of sortilin-deficient mice.
Fig. S5. mRNA and protein levels of neurotensin and NTSR2 are unchanged in sortilin-deficient mice.
Fig. S6. Sortilin allows direct manipulation of the pain response to neuronal injury.
Fig. S7. Hum-sortilin-KI mouse model.
REFERENCES AND NOTES
- 1.van Hecke O., Austin S. K., Khan R. A., Smith B. H., Torrance N., Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 155, 654–662 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Coull J. A. M., Boudreau D., Bachand K., Prescott S. A., Nault F., Sík A., De Koninck P., De Koninck Y., Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Lu Y., Zheng J., Xiong L., Zimmermann M., Yang J., Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. J. Physiol. 586, 5701–5715 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doyon N., Vinay L., Prescott S. A., De Koninck Y., Chloride regulation: A dynamic equilibrium crucial for synaptic inhibition. Neuron 89, 1157–1172 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Coull J. A. M., Beggs S., Boudreau D., Boivin D., Tsuda M., Inoue K., Gravel C., Salter M. W., De Koninck Y., BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Vaegter C. B., Jansen P., Fjorback A. W., Glerup S., Skeldal S., Kjolby M., Richner M., Erdmann B., Nyengaard J. R., Tessarollo L., Lewin G. R., Willnow T. E., Chao M. V., Nykjaer A., Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 14, 54–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nykjaer A., Willnow T. E., Sortilin: A receptor to regulate neuronal viability and function. Trends Neurosci. 35, 261–270 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Sarret P., Krzywkowski P., Segal L., Nielsen M. S., Petersen C. M., Mazella J., Stroh T., Beaudet A., Distribution of NTS3 receptor/sortilin mRNA and protein in the rat central nervous system. J. Comp. Neurol. 461, 483–505 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Sarret P., Esdaile M. J., Perron A., Martinez J., Stroh T., Beaudet A., Potent spinal analgesia elicited through stimulation of NTS2 neurotensin receptors. J. Neurosci. 25, 8188–8196 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tétreault P., Beaudet N., Perron A., Belleville K., René A., Cavelier F., Martinez J., Stroh T., Jacobi A. M., Rose S. D., Behlke M. A., Sarret P., Spinal NTS2 receptor activation reverses signs of neuropathic pain. FASEB J. 27, 3741–3752 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Kleczkowska P., Lipkowski A. W., Neurotensin and neurotensin receptors: Characteristic, structure–activity relationship and pain modulation—A review. Eur. J. Pharmacol. 716, 54–60 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Guillemette A., Dansereau M. A., Beaudet N., Richelson E., Sarret P., Intrathecal administration of NTS1 agonists reverses nociceptive behaviors in a rat model of neuropathic pain. Eur. J. Pain 16, 473–484 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Patel A. B., Tsilioni I., Leeman S. E., Theoharides T. C., Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. U.S.A. 113, E7049–E7058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quistgaard E. M., Madsen P., Grøftehauge M. K., Nissen P., Petersen C. M., Thirup S. S., Ligands bind to Sortilin in the tunnel of a ten-bladed β-propeller domain. Nat. Struct. Mol. Biol. 16, 96–98 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Decosterd I., Woolf C. J., Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Beggs S., Trang T., Salter M. W., P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 15, 1068–1073 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delpire E., Baranczak A., Waterson A. G., Kim K., Kett N., Morrison R. D., Scott Daniels J., David Weaver C., Lindsley C. W., Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: Development of a highly selective and more potent in vitro probe. Bioorg. Med. Chem. Lett. 22, 4532–4535 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knusel B., Rabin S. J., Hefti F., Kaplan D. R., Regulated neurotrophin receptor responsiveness during neuronal migrationand early differentiation. J. Neurosci. 14, 1542–1554 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Lieto A., Rantamäki T., Vesa L., Yanpallewar S., Antila H., Lindholm J., Rios M., Tessarollo L., Castrén E., The responsiveness of TrkB to BDNF and antidepressant drugs is differentially regulated during mouse development. PLOS ONE 7, e32869 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jennes L., Stumpf W. E., Kalivas P. W., Neurotensin: Topographical distribution in rat brain by immunohistochemistry. J. Comp. Neurol. 210, 211–224 (1982). [DOI] [PubMed] [Google Scholar]
- 21.Wang J., Zhang H., Feng Y.-P., Meng H., Wu L.-P., Wang W., Li H., Zhang T., Zhang J.-S., Li Y.-Q., Morphological evidence for a neurotensinergic periaqueductal gray-rostral ventromedial medulla-spinal dorsal horn descending pathway in rat. Front. Neuroanat. 8, 112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vachon P., Massé R., Gibbs B. F., Substance P and neurotensin are up-regulated in the lumbar spinal cord of animals with neuropathic pain. Can. J. Vet. Res. 68, 86–92 (2004). [PMC free article] [PubMed] [Google Scholar]
- 23.Williams F. G., Beitz A. J., Chronic pain increases brainstem proneurotensin/neuromedin-N mRNA expression: A hybridization-histochemical and immunohistochemical study using three different rat models for chronic nociception. Brain Res. Rev. 611, 87–102 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Hermey G., The Vps10p-domain receptor family. Cell. Mol. Life Sci. 66, 2677–2689 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beggs S., Salter M. W., Microglia-neuronal signalling in neuropathic pain hypersensitivity 2.0. Curr. Opin. Neurobiol. 20, 474–480 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X., Ratnam J., Zou B., England P. M., Basbaum A. I., TrkB signaling is required for both the induction and maintenance of tissue and nerve injury-induced persistent pain. J. Neurosci. 29, 5508–5515 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarro V., Vincent J.-P., Mazella J., Shedding of the luminal domain of the neurotensin receptor-3/sortilin in the HT29 cell line. Biochem. Biophys. Res. Commun. 298, 760–764 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Hermey G., Sjøgaard S. S., Petersen C. M., Nykjær A., Gliemann J., Tumour necrosis factor α-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem. J. 395, 285–293 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buttenschøn H. N., Demontis D., Kaas M., Elfving B., Mølgaard S., Gustafsen C., Kaerlev L., Petersen C. M., Børglum A. D., Mors O., Glerup S., Increased serum levels of sortilin are associated with depression and correlated with BDNF and VEGF. Transl. Psychiatry 5, e677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mølgaard S., Demontis D., Nicholson A. M., Finch N. A., Petersen R. C., Petersen C. M., Rademakers R., Nykjaer A., Glerup S., Soluble sortilin is present in excess and positively correlates with progranulin in CSF of aging individuals. Exp. Gerontol. 84, 96–100 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Schrøder T. J., Christensen S., Lindberg S., Langgård M., David L., Maltas P. J., Eskildsen J., Jacobsen J., Tagmose L., Simonsen K. B., Biilmann Rønn L. C., de Jong I. E. M., Malik I. J., Karlsson J.-J., Bundgaard C., Egebjerg J., Stavenhagen J. B., Strandbygård D., Thirup S., Andersen J. L., Uppalanchi S., Pervaram S., Kasturi S. P., Eradi P., Sakumudi D. R., Watson S. P., The identification of AF38469: An orally bioavailable inhibitor of the VPS10P family sorting receptor Sortilin. Bioorg. Med. Chem. Lett. 24, 177–180 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Andersen J. L., Schrøder T. J., Christensen S., Strandbygård D., Pallesen L. T., García-Alai M. M., Lindberg S., Langgård M., Eskildsen J. C., David L., Tagmose L., Simonsen K. B., Maltas P. J., Rønn L. C. B., de Jong I. E. M., Malik I. J., Egebjerg J., Karlsson J.-J., Uppalanchi S., Sakumudi D. R., Eradi P., Watson S. P., Thirup S., Identification of the first small-molecule ligand of the neuronal receptor sortilin and structure determination of the receptor-ligand complex. Acta Crystallogr. D Biol. Crystallogr. 70, 451–460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu F., Padukkavidana T., Vægter C. B., Brady O. A., Zheng Y., Mackenzie I. R., Feldman H. H., Nykjaer A., Strittmatter S. M., Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro V., Martin S., Sarret P., Nielsen M. S., Petersen C. M., Vincent J.-P., Mazella J., Pharmacological properties of the mouse neurotensin receptor 3. Maintenance of cell surface receptor during internalization of neurotensin. FEBS Lett. 495, 100–105 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Morinville A., Martin S., Lavallée M., Vincent J.-P., Beaudet A., Mazella J., Internalization and trafficking of neurotensin via NTS3 receptors in HT29 cells. Int. J. Biochem. Cell Biol. 36, 2153–2168 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Evans S. F., Irmady K., Ostrow K., Kim T., Nykjaer A., Saftig P., Blobel C., Hempstead B. L., Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J. Biol. Chem. 286, 29556–29567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hylden J. L. K., Wilcox G. L., Intrathecal morphine in mice: A new technique. Eur. J. Pharmacol. 67, 313–316 (1980). [DOI] [PubMed] [Google Scholar]
- 38.Tóth F., Mallareddy J. R., Tourwé D., Lipkowski A. W., Bujalska-Zadrozny M., Benyhe S., Ballet S., Tóth G., Kleczkowska P., Synthesis and binding characteristics of [3H]neuromedin N, a NTS2 receptor ligand. Neuropeptides 57, 15–20 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Boules M., Li Z., Smith K., Fredrickson P., Richelson E., Diverse roles of neurotensin agonists in the central nervous system. Front. Endocrinol. 4, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richner M., Bjerrum O. J., Nykjaer A., Vaegter C. B., The spared nerve injury (SNI) model of induced mechanical allodynia in mice. J. Vis. Exp. 18, 3092 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorge R. E., Martin L. J., Isbester K. A., Sotocinal S. G., Rosen S., Tuttle A. H., Wieskopf J. S., Acland E. L., Dokova A., Kadoura B., Leger P., Mapplebeck J. C. S., McPhail M., Delaney A., Wigerblad G., Schumann A. P., Quinn T., Frasnelli J., Svensson C. I., Sternberg W. F., Mogil J. S., Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat. Methods 11, 629–632 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Ferrini F., Trang T., Mattioli T.-A. M., Laffray S., del’Guidice T., Lorenzo L.-E., Castonguay A., Doyon N., Zhang W., Godin A. G., Mohr D., Beggs S., Vandal K., Beaulieu J.-M., Cahill C. M., Salter M. W., de Koninck Y., Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nat. Neurosci. 16, 183–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Digman M. A., Caiolfa V. R., Zamai M., Gratton E., The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 94, L14–L16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poulsen E. T., Dyrlund T. F., Runager K., Scavenius C., Krogager T. P., Højrup P., Thøgersen I. B., Sanggaard K. W., Vorum H., Hjortdal J., Enghild J. J., Proteomics of Fuchs’ endothelial corneal dystrophy support that the extracellular matrix of Descemet’s membrane is disordered. J. Proteome Res. 13, 4659–4667 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malmos K. G., Bjerring M., Jessen C. M., Nielsen E. H. T., Poulsen E. T., Christiansen G., Vosegaard T., Skrydstrup T., Enghild J. J., Pedersen J. S., Otzen D. E., How glycosaminoglycans promote fibrillation of salmon calcitonin. J. Biol. Chem. 291, 16849–16862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dyrlund T. F., Poulsen E. T., Scavenius C., Sanggaard K. W., Enghild J. J., MS Data Miner: A web-based software tool to analyze, compare, and share mass spectrometry protein identifications. Proteomics 12, 2792–2796 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav9946/DC1
Fig. S1. Injury-induced microglia reactivity in WT and Sort1−/− lumbar spinal cord.
Fig. S2. Neurochemical markers remain intact in sortilin-deficient mice.
Fig. S3. Direct KCC2 inhibition induces pain hypersensitivity.
Fig. S4. Subcellular localization of TrkB is unchanged in SDH of sortilin-deficient mice.
Fig. S5. mRNA and protein levels of neurotensin and NTSR2 are unchanged in sortilin-deficient mice.
Fig. S6. Sortilin allows direct manipulation of the pain response to neuronal injury.
Fig. S7. Hum-sortilin-KI mouse model.