

Abstract
To identify genes associated with carotenoid accumulation in petals of Chinese cabbage, the composition and content of carotenoids were analyzed, and comparative transcriptome sequencing was performed between the yellow flower line, 92S105, and the orange flower line, 94C9. High-performance liquid chromatography (HPLC) revealed that petals of 92S105 were high in violaxanthin as well as lutein, whereas petals of 94C9 showed considerable levels of lutein and β-carotene. Transcriptome analysis showed that 3534 and 3833 genes were up- and down-regulated in 94C9, respectively. Among these differentially expressed genes (DEGs), many related to carotenoid accumulation were identified, including 12 carotenoid biosynthesis pathway genes, 4 transcription factor genes, and 1028 specifically expressed genes. β-carotene hydroxylase 1 (BrBCH1), BrBCH2, zeaxanthin epoxidase (BrZEP), and MYB transcription factor gene (BrGAMYB) were down-regulated in petals of 94C9 when compared with petals of 92S105, which caused β-carotene accumulation and may lead to orange petal color in 94C9. Expression levels of 20 DEGs were verified by qPCR and the results were highly consistent with those of transcriptome sequencing. Moreover, Gene Ontology (GO) enrichment analysis revealed that membrane, binding, and metabolic processes were the most significantly enriched GO terms in cellular component, molecular function, and biological process ontologies, respectively. In conclusion, our study analyzed the differences in composition and content of carotenoids between 92S105 and 94C9 and identified potential candidate genes related to carotenoid accumulation in petals, thereby creating a solid foundation for future studies on the mechanism regulating carotenoid accumulation in petals of Chinese cabbage.
Electronic supplementary material
The online version of this article (10.1007/s13205-019-1813-6) contains supplementary material, which is available to authorized users.
Keywords: Chinese cabbage, Carotenoid accumulation, HPLC, Transcriptome sequencing, Differentially expressed genes
Introduction
Carotenoids are synthesized in chloroplasts and are involved in photosynthesis and photoprotection in green tissues (Grotewold 2006; Walter and Strack 2011). In flowers and fruits, carotenoids that are synthesized in chromoplasts can confer distinct yellow, orange, or red colors (DellaPenna and Pogson 2006; Grotewold 2006) that act as important signals to attract pollinators and seed dispersers (Kevan and Baker 1983; Bartley and Scolnik 1995). Carotenoids can also provide precursors for the biosynthesis of the phytohormone abscisic acid (ABA) and strigolactones (Nambara and Marion-Poll 2005; Dun et al. 2009; Walter and Strack 2011).
Carotenoid accumulation in plants was regulated by the major factors including carotenoid biosynthesis and degradation (Galpaz et al. 2006; Tanaka and Ohmiya 2008). Most of the genes from carotenoid biosynthesis pathway have been identified (Fraser and Bramley 2004; Tanaka et al. 2008; Zhu et al. 2010). The transcriptional levels of these genes may affect carotenoid accumulation. For example, expression levels of phytoene desaturase (PDS) and BCH increased in parallel with carotenoid accumulation in mature petals of Sandersonia (Nielsen et al. 2003). In Ipomoea, Yamamizo et al. (2010) found that the transcriptional levels of most carotenoid biosynthesis genes were lower in white than in yellow petals. BCH played an important role in regulating carotenoid content in petals. For example, in several plants, the increase in the expression level of BCH was accompanied by the increase in carotenoid content (Zhu et al. 2003; Yamagishi et al. 2010; Yamamizo et al. 2010). In contrast, in Oncidium petals, down-regulated expression of BCH did not affect carotenoid content; however, it caused accumulation of β-carotene, which is a substrate of BCH, and resulted in orange petals (Chiou et al. 2010). In addition, in chrysanthemum, Kishimoto and Ohmiya (2006) reported no significant difference in transcriptional levels of carotenoid biosynthesis genes between white and yellow petals. Subsequently, Ohmiya et al. (2006, 2009) found that the expression level of carotenoid cleavage dioxygenase 4 (CCD4) affected the accumulation of carotenoid in chrysanthemum petals.
Technological advancement and rapid cost reduction has allowed next-generation sequencing (NGS) to become an effective tool for revealing potential molecular mechanisms of biological processes and has been widely applied to fine mapping of genes (Liu et al. 2015; Zhang et al. 2016; Chen et al. 2017) and expression analysis of whole genes (Huang et al. 2015; Zhou et al. 2017; Alghamdi et al. 2018; Wang et al. 2018; Xu et al. 2018). RNA sequencing (RNA-Seq) has served as a rapid and exact method for obtaining large amounts of gene expression data used to identify critical genes associated with various important agronomic traits in many horticultural plants, such as almonds (Hosseinpour et al. 2018), cucumber (Zhang et al. 2014), radish (Feng et al. 2017), and Brassica rapa (Huang et al. 2015; Zhou et al. 2017; Li et al. 2019).
In this study, carotenoid profiles of yellow and orange petals were analyzed using high-performance liquid chromatography (HPLC). RNA-Seq was used for obtaining data of global gene expression in yellow and orange petals. Key differentially expressed genes (DEGs) associated with carotenoid accumulation were identified using DEG comprehensive analysis and HPLC analysis. The results are expected to provide a preliminary understanding of the regulation of carotenoid accumulation in Chinese cabbage petals.
Materials and methods
Plant materials
Yellow-flowered 92S105 and orange-flowered 94C9 lines of Chinese cabbage used in this study (Fig. 1) were provided by the Chinese cabbage research group at Northwest A&F University (Yangling, China). The yellow- and orange-flowered plants were cultured in the same experimental field at Northwest A&F University. At full-bloom stage, yellow and orange petals of fully open flowers from three different plants were chosen for HPLC analysis and transcriptome profiling. All samples were immediately frozen in liquid nitrogen, and stored at − 80 °C.
Fig. 1.

Phenotypic characterization of flowers from Chinese cabbage yellow-flowered line 92S105 (a) and orange-flowered line 94C9 (b)
Carotenoid analysis
Carotenoid extraction from fresh petals and detection were performed based on the methods previously described by Cao et al. (2012). Carotenoid detection and quantification were conducted using a Shimadzu HPLC (LC-2010AHT, Shimadzu Corporation, Kyoto, Japan). Carotenoids were separated using an YMC C30 column (YMC, Kyoto, Japan; 250 × 4.6; 5 μm) and identified based on the typical retention time obtained from the standards of violaxanthin (Sigma-Aldrich, Saint Louis, America), α-carotene and β-carotene (Wako, Osaka, Japan), and lutein (Solarbio, Beijing, China). Individual identified carotenoids were quantified based on previous methods (Morris et al. 2004). All means and standard errors were calculated using data from three biological replicates.
RNA isolation, cDNA library construction, and RNA-seq
Total RNA was isolated from yellow and orange petals using Trizol Reagent (Invitrogen, Carlsbad, USA) in accordance with the manufacturer’s instructions. RNA quality and purity were assessed using 1.0% agarose gels and a NanoDrop 8000 spectrophotometer (Thermo Scientific, Waltham, USA), and RNA integrity was evaluated with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA).
Sequencing libraries were generated following the manufacturer’s instructions (Illumina, San Diego, USA). Eukaryotic mRNA was enriched from total RNA by Oligo(dT) beads, while prokaryotic mRNA was purified by removing rRNA with a Ribo-ZeroTMMagnetic Kit (Epicentre, Madison, USA), and then the obtained mRNA was broken into short fragments using fragmentation buffer. These short fragments were used to synthesize first-strand cDNA with random primers and second-strand cDNA synthesis was conducted with DNA polymerase I, RNase H, dNTP, and buffer. cDNA fragments were purified using a QiaQuick PCR extraction kit and end reparation and addition of poly(A) were performed, and then fragments were ligated to Illumina sequencing adapters. Suitably sized ligation products were selected for amplification by PCR. Finally, cDNA libraries were sequenced using Illumina HiSeq™2500 by Sagene Biotech Co. Ltd (Guangzhou, China). The obtained raw data from constructed cDNA libraries was deposited in NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra/) under the accession number: BioProject PRJNA525538.
DEG analysis and GO and KEGG enrichment analysis of DEGs
Raw reads from RNA-seq were filtered to obtain high-quality clean reads; they were then aligned to the B. rapa reference genome obtained from BRAD (http://brassicadb.org/brad) using TopHat2 software (Kim et al. 2013). Individual gene expression level was calculated based on the fragments-per-kilobases-per-million-mapped reads (FPKM) method. To identify DEGs between yellow and orange petals, a corrected p value < 0.05 and the absolute value of log2 (fold change) ≥ 2 were selected as thresholds for evaluating the significance of the differences in gene expression. Fold change was the ratio of FPKM values for gene expression between the two petal colors.
To identify possible biological functions of DEGs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed. Gene Ontology enrichment analysis was used to obtain the GO terms significantly enriched in DEGs. Kyoto Encyclopedia of Genes and Genomes enrichment analysis provided the metabolic or signal transduction pathways that were significantly enriched in DEGs. For GO and KEGG enrichment analyses, all DEGs were mapped to GO and KEGG terms in the database, and significantly enriched GO and KEGG terms in DEGs were then searched by comparing to the genome background with an adjusted p value < 0.05 as threshold.
Quantitative real-time PCR (qPCR) validation
Twenty selected DEGs were validated using qPCR. The first-strand cDNA synthesis was performed with PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara, Dalian, China) following manufacturer instructions. Specific primers of the selected DEGs were designed by Primer Premier 5.0 software (http://www.premierbiosoft.com/primerdesign/) (Table S1) and synthesized by Sangon Biotech Co., Ltd (Shanghai, China). Chinese cabbage elongation-factor-1-α (EF-1-α) gene was used as internal reference (Qi et al. 2010). The qPCR analyses were performed in triplicate on an iCycler iQ5 real-time PCR detection system (Bio-Rad, Hercules, USA) with SYBR® Premix Ex Taq™ II (Takara, Dalian, China) following manufacturer instructions. Each reaction (20 µL volume) consisted of 2 μL of cDNA template (100 ng/µL), 0.5 μL of each forward and reverse primers (10 µmol), 10 μL 2 × SYBR Green PCR Master Mix and 7 µL ddH2O. The following PCR program was used: 95 °C for 30 s, 40 cycles of 95 °C for 10 s, 58 °C for 30 s, and 72 °C for 30 s. Relative gene expression levels were calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001).
Results
Carotenoid accumulation in yellow and orange petals
To study whether accumulation of different carotenoids resulted in the differences in petal color, the analysis of carotenoid profiles in yellow and orange petals was performed using HPLC. By comparing retention times of sample carotenoid peaks with those of standard compounds, four carotenoids were identified, including violaxanthin, lutein, α-carotene, and β-carotene. HPLC analysis showed that petals of 92S105 contained mainly violaxanthin and lutein; whereas petals of 94C9 contained mostly lutein and β-carotene. Violaxanthin content in petals of 92S105 was significantly higher than that in petals of 94C9, but larger amounts of lutein and β-carotene were accumulated in petals of 94C9 than in petals of 92S105 (Fig. 2). These results indicated that the difference in petal color between 92S105 and 94C9 lines was most likely due to differences in the composition and content of carotenoids.
Fig. 2.
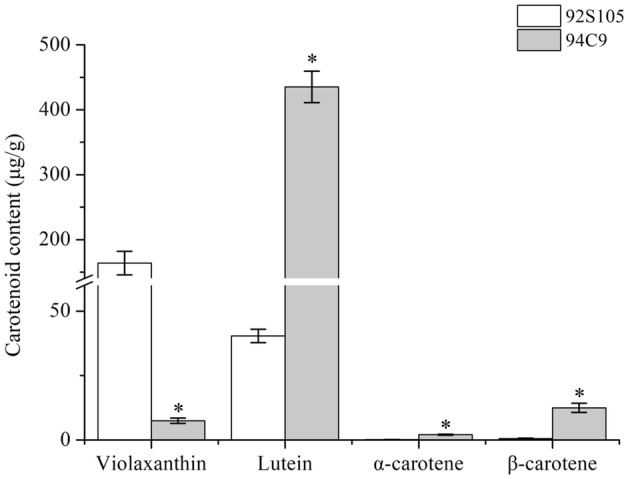
Carotenoid composition and content in yellow and orange petals of Chinese cabbage flowers. Bars represent mean ± SE of triplicate assays, and asterisks represent significant difference between 92S105 and 94C9 (t test, p < 0.05)
Transcriptome sequencing and mapping of sequence reads
Transcriptome sequencing of the two petal cDNA libraries separately constructed from 92S105 and 94C9 was performed using Illumina HiSeq™ 2500 to obtain the differentially expressed genes presumably regulating petal color in Chinese cabbage. The cDNA libraries constructed from the yellow and the orange petals were named S105 and C9, respectively. After adaptor sequences and low-quality reads were removed, a total of 113 million clean reads were obtained including 58 million from S105 and 55 million from C9 and the Q20 percentage was above 96% (Table 1), indicating that the sequencing results could be used for further analysis.
Table 1.
Summary of clean transcriptome sequencing reads and reads mapping to the Brassica rapa reference genome
| Sample | Clean reads | High-quality clean reads (%) | Q20 (%) | GC content (%) | Total mapped reads (%) | Multiple mapped reads (%) | Uniquely mapped reads (%) |
|---|---|---|---|---|---|---|---|
| S105 | 58,193,087 | 99.27 | 97.20 | 46.70 | 73.66 | 0.83 | 72.83 |
| C9 | 55,485,687 | 99.15 | 97.05 | 46.84 | 75.18 | 0.84 | 74.34 |
The clean reads obtained from S105 and C9 were mapped to the B. rapa reference genome. The results revealed that 72.83% and 74.34% reads were uniquely matched to the reference genome in S105 and C9, respectively, while 0.84% reads were mapped to multiple locations of the reference genome in the two samples (Table 1).
Identification and analysis of DEGs
To determine the exact differences in gene expression between S105 and C9, gene expression levels were normalized by FPKM. The transcript abundance level of a gene was used to directly represent the relative expression level. The FPKM value of each gene was calculated. Analysis of the FPKM interval distribution of all genes showed that the most abundant FPKM interval was 0–1 (22,679, 53.55% of all genes in S105; and 21,058, 49.72% of all genes in C9), followed by 3–15 (7394, 17.46% of all genes in S105; and 7970, 18.82% of all genes in C9) (Table 2).
Table 2.
The FPKM distribution of the total genes obtained from S105 and C9
| FPKM interval | S105 (%) | C9 (%) |
|---|---|---|
| 0–1 | 22,679 (53.55) | 21,058 (49.72) |
| 1–3 | 4899 (11.57) | 4821 (11.38) |
| 3–15 | 7394 (17.46) | 7970 (18.82) |
| 15–60 | 4395 (10.38) | 5163 (12.19) |
| > 60 | 2986 (7.05) | 3341 (7.89) |
Based on the restrictive threshold, a set of 7367 genes exhibiting significant differences in expression between S105 and C9 was identified. Among these DEGs, 3534 up-regulated and 3833 down-regulated genes were found by the comparison between S105 and C9 (Fig. 3, Table S2). These findings revealed that the percentage of up-regulated genes was almost equal to that of down-regulated genes. Moreover, specifically expressed genes (SEGs) were defined as genes that were not expressed in one sample but had read numbers > 11 in the other sample (Tao et al. 2012). We identified 632 specifically expressed genes (SEGs) in S105 and 396 SEGs in C9 (including 178 novel genes); Among SEGs, a total of 426 genes (including 10 novel genes), including 269 genes (including 5 novel genes) in S105 and 157 (including 5 novel genes) in C9, were divided into 21 functional categories and one other category (the number of genes in functional categories ≤ 2). A high proportion of SEGs was associated with nucleotide-binding catalytic activity and heterocyclic compound binding; additionally, 158 SEGs (including 2 novel genes) in S105 and 96 SEGs (including 3 novel genes) in C9 were identified, which encode unknown proteins (Table 3, Table S3).
Fig. 3.
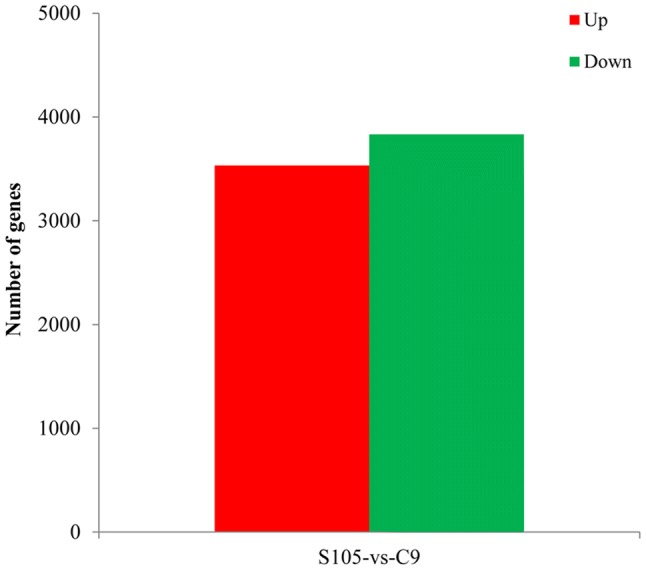
Differentially expressed genes between S105 and C9. The results were summarized for the number of up-regulated and down-regulated genes
Table 3.
Functional categories for specifically expressed genes from S105 and C9
| Gene functional categories | S105 | C9 |
|---|---|---|
| Heterocyclic compound binding | 11 | 12 |
| Catalytic activity | 21 | 16 |
| Ion binding | 12 | 4 |
| Enzyme regulator activity | 9 | 1 |
| Hydrolase activity | 4 | 1 |
| Transmembrane transporter activity | 7 | 2 |
| Small molecule binding | 6 | 1 |
| Transferase activity | 7 | 3 |
| Nucleic acid binding | 5 | 8 |
| Protein binding | 6 | 5 |
| Nucleotide binding | 33 | 17 |
| Organic cyclic compound binding | 8 | 7 |
| Hydrolase activity | 10 | 3 |
| Cation binding | 8 | 3 |
| Enzyme inhibitor activity | 6 | 1 |
| Transporter activity | 5 | 3 |
| Molecular function regulator | 3 | 0 |
| Oxidoreductase activity | 4 | 2 |
| Hydrolase activity | 4 | 2 |
| Metal ion binding | 9 | 6 |
| Transcription factor activity | 2 | 9 |
| Other categories | 89 | 51 |
| No categories | 205 | 143 |
| Unknown genes | 158 | 96 |
| Total | 632 | 396 |
Other categories represent all the categories in which the number of genes is not more than 2
Differentially expressed transcription factor genes related to carotenoid accumulation
Previous studies revealed that accumulated carotenoids are responsible for three colors (yellow, orange, and red) in flower petals (DellaPenna and Pogson 2006; Grotewold 2006). Based on reported transcription factor genes associated with carotenoid accumulation in tomato and maize (Lee et al. 2012; Jin et al. 2018), 14 homologous genes in Chinese cabbage were obtained using blastp tool in BRAD (Table S4). Screening of differentially expressed transcription factor genes (DETFG) related to carotenoid accumulation was performed; thus, four DETFGs were identified, including genes encoding two ANAC074 transcription factors (Bra011037 and Bra024194), one Dof-type zinc finger domain-containing protein (Bra002504), and one GAMYB transcription factor (Bra005597). Our comparison between S105 and C9 revealed that, in addition to Bra005597, all other DETFGs were up-regulated (Table 4).
Table 4.
Identification of DEGs related to carotenoid accumulation in Chinese cabbage
| Gene source | Gene ID | Annotation | Log2 fold change | P value |
|---|---|---|---|---|
| Carotenoid biosynthesis pathway gene | Bra032770 | Phytoene dehydrogenase | 5.1063 | 0.0013 |
| Bra019145 | Carotene beta-ring hydroxylase 1 | − 3.0485 | 3.6170E–29 | |
| Bra003121 | Carotene beta-ring hydroxylase 2 | − 2.1296 | 1.0266E–11 | |
| Bra012127 | Zeaxanthin epoxidase | − 4.5784 | 2.7199E–16 | |
| Bra018616 | Violaxanthin de-epoxidase | − 2.8937 | 1.0904 E–22 | |
| Bra027336 | Nine-cis-epoxycarotenoid dioxygenase 3 | − 2.8193 | 2.8569E–12 | |
| Bra021558 | Nine-cis-epoxycarotenoid dioxygenase 3 | − 2.4328 | 1.1478E–08 | |
| Bra001552 | Nine-cis-epoxycarotenoid dioxygenase 3 | − 2.3323 | 2.5049E–15 | |
| Bra020970 | Nine-cis-epoxycarotenoid dioxygenase 4 | − 5.9653 | 5.8468E–11 | |
| Bra013386 | Abscisic acid 8′-hydroxylase | − 4.5881 | 9.0919 E–13 | |
| Bra021965 | Abscisic acid 8′-hydroxylase | − 5.3545 | 3.9087 E–56 | |
| Bra027602 | Abscisic acid 8′-hydroxylase | − 5.4120 | 5.5361 E–51 | |
| Transcription factor gene | Bra011037 | NAC domain-containing protein 74 | 2.6317 | 5.7602E–19 |
| Bra024194 | NAC domain-containing protein 74 | 2.0641 | 1.6701E–12 | |
| Bra002504 | Dof-type zinc finger domain-containing protein | 2.2070 | 2.3106E–08 | |
| Bra005597 | GAMYB protein-like | − 10.8578 | 6.5519E–55 |
Functional enrichment analysis of DEGs
To gain a better insight into the function of the identified DEGs, GO enrichment analysis was conducted. All DEGs in the two petal colors were mapped to different functional GO terms and the significantly enriched GO terms were selected based on the threshold, corrected p values < 0.05. As a result, a total of 48 GO terms were significantly enriched in 3 GO ontologies, including 16 GO terms in the cellular component group, 13 GO terms in the molecular function group, and 19 GO terms in the biological process group. In the cellular component ontology, the two most significantly enriched GO terms were membrane and membrane part. The binding was the most significantly enriched in the molecular function ontology. As for the biological process ontology, the metabolic process category was the most significantly enriched (Fig. 4).
Fig. 4.

GO functional enrichment analysis of S105 vs. C9. X and Y axis represent enriched GO terms and the number of DEGs, respectively
For further identification of metabolic pathways enriched by detected DEGs, 4810 DEGs were assigned to all KEGG pathways. Among these pathways, the most significantly enriched was plant–pathogen interaction (Table S5), with most DEGs in this pathway exhibiting down-regulation in C9; furthermore, most of these down-regulated DEGs were calcium (Ca2+)-binding genes (Table S6). Finally, there were 12 DEGs involved in the carotenoid biosynthesis pathway, 11 of which were down-regulated in C9 (Table 4).
qPCR validation of gene expression
To verify our RNA-seq results, 20 genes were tested by qPCR. These genes were classified into four categories, including eight carotenoid biosynthesis pathway genes (Bra032770, Bra019145, Bra003121, Bra012127, Bra027336, Bra021558, Bra001552, and Bra020970); four transcription factor genes associated with carotenoid accumulation (Bra011037, Bra024194, Bra002504, and Bra005597); four specifically expressed genes encoding unknown proteins (Bra001015, Bra010007, Bra017584, and Bra019906); and four Ca2+ binding genes from plant–pathogen interaction (Bra003712, Bra004165, Bra009420, and Bra011605). The results of qPCR showed that the relative expression levels of some of the selected genes were different from the data determined by RNA-seq; however, the expression trends of all the selected genes were consistent with those obtained from RNA-seq (Fig. 5), indicating that our analysis of RNA-seq was accurate and reliable.
Fig. 5.

qPCR validation of 20 DEGs related to carotenoid accumulation in S105 and C9. Bars represent mean ± SE of triplicate tests, and asterisks indicate significant differences between S105 and C9 (t test, p < 0.05)
Discussion
In flowers, accumulated carotenoids are responsible for petal colors in many plants, such as Oncidium (Chiou et al. 2010), Osmanthus fragrans (Han et al. 2014), and B. napus (Zhang et al. 2015). In B. napus, Zhang et al. (2015) reported that the main carotenoid in yellow flowers was violaxanthin and a similar result was found in yellow flowers of Oncidium (Chiou et al. 2010). Studies revealed that orange petals of Oncidium and Osmanthus fragrans accumulated significantly more β-carotene than the yellow petals (Chiou et al. 2010; Han et al. 2014). In this study, HPLC analysis revealed that violaxanthin and lutein were the most abundant carotenoids in yellow petals, whereas lutein and β-carotene were more abundant in orange petals. These results were consistent with those from previous investigations (Chiou et al. 2010; Han et al. 2014; Zhang et al. 2015) indicating that the characteristic orange color of petals on flowers of B. rapa line 94C9 was most likely due to the accumulation of β-carotene to a greater extent than accumulation of the same pigment in yellow petals on flowers of B. rapa, line 92S105.
RNA-seq has become a powerful tool for the elucidation of gene expression patterns; as such, RNA-seq can contribute to the identification of potential candidate genes associated with target traits. In this study, a comparative RNA-seq analysis was performed between yellow and orange petals of B. rapa lines 92S105 and 94C9, respectively. Thus, we obtained 112 million high-quality reads of which approximately 74% were uniquely mapped to B. rapa reference genome. We identified 3534 up-regulated and 3833 down-regulated genes in samples of the two petal colors. Among them, 632 and 396 genes were specifically expressed in S105 and C9, respectively. Although functional annotation analysis revealed that annotated SEGs were not associated with carotenoid metabolism, we speculated that some SEGs encoding unknown proteins may be involved in carotenoid accumulation in Chinese cabbage petals.
Generally, carotenoid content and composition correlate with the transcriptional level of carotenogenic genes. Down-regulated expression of β-carotene hydroxylase (OgBCH) and zeaxanthin epoxidase (OgZEP) in Oncidium petals led to the accumulation of β-carotene, whereby floral tissues appeared orange (Chiou et al. 2010). In this study, 12 genes from the carotenoid biosynthesis pathway showed different expression patterns in the two lines under study. Among these DEGs, BrBCH and BrZEP, which encode proteins that control the conversion of β-carotene to zeaxanthin and that of zeaxanthin to violaxanthin, respectively, showed down-regulation in C9, compared with S105 (Table 4, Fig. 5); such was presumably the cause of the accumulation of β-carotene that might have indirectly contributed to the accumulation of lutein in 94C9. Moreover, while CCD4 was reportedly involved mainly in carotenoid degradation in petals (Ohmiya 2009; Zhang et al. 2015), the expression of CCD4 in S105 and C9 did not differ in this study.
Although the main genes in the carotenoid metabolic pathway have been identified for many crops, the genes regulating the expression of genes directly involved in the pathway remain largely unknown. Up to present, only a few transcription factors that affect carotenoid accumulation have been identified in tomato. Thus, for example, transcription factor LE15G11 showed a negative correlation with lutein and β-carotene (Lee et al. 2012). In this study, homologous genes of LE15G11 in Chinese cabbage, Bra011037 and Bra024194, were up-regulated in C9, indicating that these two genes are not likely to be the cause of the accumulation of lutein and β-carotene. In maize, transcription factors ZmPBF and ZmGAMYB independently activated the expression of ZmBCH2 (Jin et al. 2018); in our study, Bra002504 and Bra005597, which are homologous of the two maize genes (ZmPBF and ZmGAMYB) in Chinese cabbage, respectively, exhibited up- and down-regulation in C9. Based on the observed expression of BrBCH in S105 and C9, it seems that Bra005597 might play a key role in the formation of orange petals in Chinese cabbage.
In plants, ABA is synthesized mainly through the carotenoid biosynthesis pathway. Previous investigations revealed that ABA enhanced plant resistance to pathogens by promoting callosum deposition on the cell wall (Mauch-Mani and Mauch 2005; García-Andrade et al. 2011) and stomatal closure (Melotto et al. 2006; Cao et al. 2011). As part of the plant–pathogen interaction pathway, calmodulin (CaM)/calmodulin-like (CML) proteins and calcium-dependent protein kinases (CDPKs) have been shown to be involved in cell wall reinforcement and stomatal closure. Moreover, the expression of many CaM/CML and CDPK genes was induced by ABA in plant tissues (Breviario et al. 1995; Yoon et al. 1999; White and Broadley 2003; Zhou et al. 2008). Therefore, ABA might be involved in the plant–pathogen interaction through the regulation of CaM/CML and CDPK gene expression. In the present study, two genes involved in the carotenoid biosynthesis pathway, BrZEP and BrNCED, which play an important role in ABA biosynthesis, were down-regulated in C9 (Table 4). In the most significantly enriched plant–pathogen interaction pathway, most DEGs were CaM/CML and CDPK genes involved in cell wall reinforcement and stomatal closure. Among them, nearly all genes were down-regulated in C9 (Table S6). Altogether, these findings indirectly indicated that carotenoid biosynthesis might be the significantly enriched pathway between S105 and C9. In addition, according to previous studies, ABA was primarily associated with seed dormancy and responses to abiotic and biotic stress (Audenaert et al. 2002; Fujii et al. 2007; Fan et al. 2009; Wang et al. 2013; Ma et al. 2017; Vishwakarma et al. 2017). However, up to present, there is no report that ABA might be directly involved in petal color formation.
Conclusion
Our study provides a transcriptome-based analysis of differentially expressed genes associated with carotenoid accumulation in petals of Chinese cabbage. The main carotenoids found in yellow petals were violaxanthin and lutein, whereas lutein and β-carotene were more abundant in orange petals. Our transcriptome comparison between yellow-petal line 92S105 and orange-petal line 94C9, suggested four genes, including BrBCH1, BrBCH2, BrZEP, and BrGAMYB, that might be considered as possible candidate genes associated with carotenoid accumulation in petals and their expression patterns were validated. These findings would facilitate better understanding of the regulatory mechanism of carotenoid accumulation in Chinese cabbage petals.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2017YFD0101802), the National Science and Technology Support Program of China (2014BAD01B0802), and the Natural Science Basic Research Plan in Shaanxi Province of China (2019JQ-228).
Author contributions
LZ and NZ conceived and designed the experiments. NZ performed HPLC and sequenced dada analysis and qPCR validation of gene expression, and wrote the paper. YX and YS prepared the RNA-seq samples. RL and XM participated in the HPLC analysis. SN provided helpful advice on data analysis and revised the paper. LZ provided the B. rapa materials, revised the paper and supervised the research.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Alghamdi SS, Khan MA, Ammar MH, Sun QW, Huang LH, Migdadi HM, El-Harty EH, Al-Faifi SA. Characterization of drought stress-responsive root transcriptome of faba bean (Vicia faba L.) using RNA sequencing. 3 Biotech. 2018;8:502–520. doi: 10.1007/s13205-018-1518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert K, De Meyer GB, Höfte MM. Abscisic acid determines basal susceptibility of tomato to Botrytis cinerea and suppresses salicylic acid-dependent signaling mechanisms. Plant Physiol. 2002;128:491–501. doi: 10.1104/pp.010605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley GE, Scolnik PA. Plant carotenoids: pigments for photoprotection, visual attraction, and human health. Plant Cell. 1995;7:1027–1038. doi: 10.1105/tpc.7.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breviario D, Morello L, Giani S. Molecular cloning of two novel rice cDNA sequences encoding putative calcium-dependent protein kinase. Plant Mol Bio. 1995;127:953–967. doi: 10.1007/BF00037023. [DOI] [PubMed] [Google Scholar]
- Cao FY, Yoshioka K, Desveaux D. The roles of ABA in plant–pathogen interactions. J Plant Res. 2011;124:489–499. doi: 10.1007/s10265-011-0409-y. [DOI] [PubMed] [Google Scholar]
- Cao HB, Zhang JC, Xu JD, Ye JL, Yun Z, Qiang X, Xu J, Deng XX. Comprehending crystalline β-carotene accumulation by comparing engineered cell models and the natural carotenoid-rich system of citrus. J Exp Bot. 2012;63:4403–4417. doi: 10.1093/jxb/ers115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FF, Fu BB, Pan YP, Zhang CW, Wen HF, Weng YQ, Chen P, Li YH. Fine mapping identifies CsGCN5 encoding a histone acetyltransferase as putative candidate gene for tendril-less1 mutation (td-1) in cucumber. Theor Appl Genet. 2017;130:1549–1558. doi: 10.1007/s00122-017-2909-1. [DOI] [PubMed] [Google Scholar]
- Chiou CY, Pan HA, Chuang YN, Yeh KW. Differential expression of carotenoid-related genes determines diversified carotenoid coloration in floral tissues of Oncidium cultivars. Planta. 2010;232:93–948. doi: 10.1007/s00425-010-1222-x. [DOI] [PubMed] [Google Scholar]
- DellaPenna D, Pogson BJ. Vitamin synthesis in plants: tocopherols and carotenoids. Annu Rev Plant Biol. 2006;57:711–738. doi: 10.1146/annurev.arplant.56.032604.144301. [DOI] [PubMed] [Google Scholar]
- Dun EA, Brewer PB, Beveridge CA. Strigolactones: discovery of the elusive shoot branching hormone. Trends Plant Sci. 2009;14:364–372. doi: 10.1016/j.tplants.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Fan J, Hill L, Crooks C, Doerner P, Lamb C. Abscisic acid has a key role in modulating diverse plant–pathogen interactions. Plant Physiol. 2009;150:1750–1761. doi: 10.1104/pp.109.137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HY, Xu L, Wang Y, Tang MJ, Zhu XW, Zhang W, Sun XC, Nie SS, Muleke EM, Liu LY. Identifcation of critical genes associated with lignin biosynthesis in radish (Raphanus sativus L.) by de novo transcriptome sequencing. Mol Genet Genomics. 2017;292:1151–1163. doi: 10.1007/s00438-017-1338-9. [DOI] [PubMed] [Google Scholar]
- Fraser PD, Bramley PM. The biosynthesis and nutritional uses of carotenoids. Prog Lipid Res. 2004;43:228–265. doi: 10.1016/j.plipres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Fujii H, Verslues PE, Zhu JK. Identification of two protein kinases required for abscisic acid regulation of seed germination, root growth, and gene expression in Arabidopsis. Plant Cell. 2007;19:485–494. doi: 10.1105/tpc.106.048538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galpaz N, Ronen G, Khalf Z, Zamir D, Hirschberg J. A chromoplast-specific carotenoid biosynthesis pathway is revealed by cloning of the tomato white-flower locus. Plant Cell. 2006;18:1947–1960. doi: 10.1105/tpc.105.039966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Andrade J, Ramírez V, Flors V, Vera P. Arabidopsis ocp3 mutant reveals a mechanism linking ABA and JA to pathogen-induced callose deposition. Plant J. 2011;67:783–794. doi: 10.1111/j.1365-313X.2011.04633.x. [DOI] [PubMed] [Google Scholar]
- Grotewold E. The genetics and biochemistry of floral pigments. Annu Rev Plant Biol. 2006;57:761–780. doi: 10.1146/annurev.arplant.57.032905.105248. [DOI] [PubMed] [Google Scholar]
- Han YJ, Wang XH, Chen WC, Dong MF, Yuan WJ, Liu X, Shang FD. Differential expression of carotenoid-related genes determines diversified carotenoid coloration in flower petal of Osmanthus fragrans. Tree Genet Genomes. 2014;10:329–338. doi: 10.1007/s11295-013-0687-8. [DOI] [Google Scholar]
- Hosseinpour B, Sepahvand S, Aliabad KK, Bakhtiarizadeh M, Imani A, Assareh R, Salami SA. Transcriptome profiling of fully open flowers in a frost-tolerant almond genotype in response to freezing stress. Mol Genet Genomics. 2018;293:151–163. doi: 10.1007/s00438-017-1371-8. [DOI] [PubMed] [Google Scholar]
- Huang SN, Liu ZY, Yao RP, Li DY, Feng H. Comparative transcriptome analysis of the petal degeneration mutant pdm in Chinese cabbage (Brassica campestris ssp. pekinensis) using RNA-Seq. Mol Genet Genomics. 2015;290:1833–1847. doi: 10.1007/s00438-015-1041-7. [DOI] [PubMed] [Google Scholar]
- Jin X, Bai C, Bassie L, Nogareda C, Romagosa I, Twyman RM, Christou P, Zhu CF. ZmPBF and ZmGAMYB transcription factors independently transactivate the promoter of the maize (Zea mays) β-carotene hydroxylase 2 gene. New Phytol. 2018 doi: 10.1111/nph.15614. [DOI] [PubMed] [Google Scholar]
- Kevan PG, Baker HG. Insects as flower visitors and pollinators. Annu Rev Entomol. 1983;28:407–453. doi: 10.1146/annurev.en.28.010183.002203. [DOI] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto S, Ohmiya A. Regulation of carotenoid biosynthesis in petals and leaves of chrysanthemum (Chrysanthemum morifolium Ramat.) Physiol Plant. 2006;128:437–447. doi: 10.1111/j.1399-3054.2006.00761.x. [DOI] [Google Scholar]
- Lee JM, Joung JG, McQuinn R, Chung MY, Fei Z, Tieman D, Klee H, Giovannoni J. Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J. 2012;70:191–204. doi: 10.1111/j.1365-313X.2011.04863.x. [DOI] [PubMed] [Google Scholar]
- Li QQ, Yang SQ, Ren J, Ye XL, Jiang X, Liu ZY. Genome-wide identification and functional analysis of the cyclic nucleotide-gated channel gene family in Chinese cabbage. 3 Biotech. 2019;9:114–127. doi: 10.1007/s13205-019-1647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HQ, Meng HW, Pan YP, Liang XJ, Jiao JQ, Li YH, Chen SX, Cheng ZH. Fine genetic mapping of the white immature fruit color gene w to a 33.0-kb region in cucumber (Cucumis sativus L.) Theor Appl Genet. 2015;128:2375–2385. doi: 10.1007/s00122-015-2592-z. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma L, Hu L, Fan J, Amombo E, Khaldun ABM, Zheng Y, Chen L. Cotton GhERF38 gene is involved in plant response to salt/drought and ABA. Ecotoxicology. 2017;26:841–854. doi: 10.1007/s10646-017-1815-2. [DOI] [PubMed] [Google Scholar]
- Mauch-Mani B, Mauch F. The role of abscisic acid in plant–pathogen interactions. Curr Opin Plant Biol. 2005;8:409–414. doi: 10.1016/j.pbi.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Melotto M, Underwood W, Koczan J, Nomura K, He SY. Plant stomata function in innate immunity against bacterial invasion. Cell. 2006;126:969–980. doi: 10.1016/j.cell.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Morris WL, Ducreux L, Grifths DW, Stewart D, Davies HV, Taylor MA. Carotenogenesis during tuber development and storage in potato. J Exp Bot. 2004;55:975–982. doi: 10.1093/jxb/erh121. [DOI] [PubMed] [Google Scholar]
- Nambara E, Marion-Poll A. Abscisic acid biosynthesis and catabolism. Annu Rev Plant Biol. 2005;56:165–185. doi: 10.1146/annurev.arplant.56.032604.144046. [DOI] [PubMed] [Google Scholar]
- Nielsen KM, Lewis DH, Morgan ER. Characterization of carotenoid pigments and their biosynthesis in two yellow flowered lines of Sandersonia aurantiaca (Hook) Euphytica. 2003;130:25–34. doi: 10.1023/A:1022328828688. [DOI] [Google Scholar]
- Ohmiya A. Carotenoid cleavage dioxygenases and their apocarotenoid products in plants. Plant Biotech. 2009;26:351–358. doi: 10.5511/plantbiotechnology.26.351. [DOI] [Google Scholar]
- Ohmiya A, Kishimoto S, Aida R, Yoshioka S, Sumitomo K. Carotenoid cleavage dioxygenase (CmCCD4a) contributes to white color formation in chrysanthemum petals. Plant Physiol. 2006;142:1193–1201. doi: 10.1104/pp.106.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmiya A, Sumitomo K, Aida R. Yellow Jimba: suppression of carotenoid cleavage dioxygenase (CmCCD4a) expression turns white chrysanthemum petals yellow. J Jpn Soc Hort Sci. 2009;78:450–455. doi: 10.2503/jjshs1.78.450. [DOI] [Google Scholar]
- Qi JN, Yu SC, Zhang FL, Shen XQ, Zhao XY, Yu YJ, Zhang DS. Reference gene selection for real-time quantitative polymerase chain reaction of mRNA transcript levels in Chinese cabbage (Brassica rapa L. ssp. pekinensis) Plant Mol Biol Rep. 2010;28:597–604. doi: 10.1007/s11105-010-0185-1. [DOI] [Google Scholar]
- Tanaka Y, Ohmiya A. Seeing is believing: engineering anthocyanin and carotenoid biosynthetic pathways. Curr Opin Biotechnol. 2008;19:190–197. doi: 10.1016/j.copbio.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Sasaki N, Ohmiya A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 2008;54:733–749. doi: 10.1111/j.1365-313X.2008.03447.x. [DOI] [PubMed] [Google Scholar]
- Tao X, Gu YH, Wang HY, Zheng W, Li X, Zhao CW, Zhang YZ. Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam.] PLoS One. 2012;7:e36234. doi: 10.1371/journal.pone.0036234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishwakarma K, Upadhyay N, Kumar N, Yadav G, Singh J, Mishra RK, Kumar V, Verma R, Upadhyay RG, Pandey M, Sharma S. Abscisic acid signaling and abiotic stress tolerance in plants: a review on current knowledge and future prospects. Front Plant Sci. 2017;8:161–172. doi: 10.3389/fpls.2017.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MH, Strack D. Carotenoids and their cleavage products: biosynthesis and functions. Nat Prod Rep. 2011;28:663–692. doi: 10.1039/c0np00036a. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tao X, Tang XM, Xiao L, Sun JL, Yan XF, Li D, Deng HY, Ma XR. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genomics. 2013;14:841–854. doi: 10.1186/1471-2164-14-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Yang YY, Chi DF. Transcriptome analysis of abscisic acid induced 20E regulation in suspension Ajuga lobata cells. 3 Biotech. 2018;8:320–339. doi: 10.1007/s13205-018-1352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PJ, Broadley PJ. Calcium in plant. Ann Bot. 2003;92:487–511. doi: 10.1093/aob/mcg164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JM, Yan ZM, Xu ZG, Wang YH, Xie ZQ. Transcriptome analysis and physiological responses of the potato plantlets in vitro under red, blue, and white light conditions. 3 Biotech. 2018;8:394–404. doi: 10.1007/s13205-018-1410-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi M, Kishimoto S, Nakayama M. Carotenoid composition and changes in expression of carotenoid biosynthetic genes in sepals of Asiatic hybrid lily. Plant Breed. 2010;129:100–107. doi: 10.1111/j.1439-0523.2009.01656.x. [DOI] [Google Scholar]
- Yamamizo C, Kishimoto S, Ohmiya A. Carotenoid composition and carotenogenic gene expression during Ipomoea petal development. J Exp Bot. 2010;61:709–719. doi: 10.1093/jxb/erp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon GM, Cho HS, Ha HJ, Liu JR, Lee HSP. Characterization of NtCDPK1, a calcium-dependent protein kinase gene in Nicotiana tabacum, and the activity of its encoded protein. Plant Mol Bio. 1999;39:991–1001. doi: 10.1023/A:1006170512542. [DOI] [PubMed] [Google Scholar]
- Zhang N, Zhang HJ, Zhao B, Sun QQ, Cao YY, Li R, Wu XX, Weeda S, Li L, Ren SX, Reiter RJ, Guo YD. The RNA-seq approach to discriminate gene expression profiles in response to melatonin on cucumber lateral root formation. J Pineal Res. 2014;56:39–50. doi: 10.1111/jpi.12095. [DOI] [PubMed] [Google Scholar]
- Zhang B, Liu C, Wang YQ, Yao X, Wang F, Wu JS, King GJ, Liu KD. Disruption of a carotenoid cleavage dioxygenase 4 gene converts flower colour from white to yellow in Brassica species. New Phytol. 2015;206:1513–1526. doi: 10.1111/nph.13335. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Wu JQ, Dai ZH, Qin ML, Hao LY, Ren YJ, Li QF, Zhang LG. Allelism analysis of Br Rfp locus in different restorer lines and map-based cloning of a fertility restorer gene, Br Rfp1, for pol CMS in Chinese cabbage (Brassica rapa L.) Theor Appl Genet. 2016;130:539–547. doi: 10.1007/s00122-016-2833-9. [DOI] [PubMed] [Google Scholar]
- Zhou JX, Hu XW, Zhang HW, Huang RF. Regulatory role of ABA in plant response to biotic stresses. J Agric Biotechnol. 2008;16:169–174. [Google Scholar]
- Zhou X, Liu ZY, Ji RQ, Feng H. Comparative transcript profling of fertile and sterile flower buds from multiple-allele-inherited male sterility in Chinese cabbage (Brassica campestris ssp. pekinensis) Mol Genet Genomics. 2017;292:967–990. doi: 10.1007/s00438-017-1324-2. [DOI] [PubMed] [Google Scholar]
- Zhu C, Yamamura S, Nishihara M, Koiwa H, Sandmann G. cDNA for the synthesis of cyclic carotenoids in petals of Gentiana lutea and their regulation during flower development. Biochim Biophys Acta. 2003;1625:305–308. doi: 10.1016/S0167-4781(03)00017-4. [DOI] [PubMed] [Google Scholar]
- Zhu C, Bai C, Sanahuja G, Yuan D, Farré G, Naqvi S, Shi L, Capell T, Christou P. The regulation of carotenoid pigmentation in flowers. Arch Biochem Biophys. 2010;504:132–141. doi: 10.1016/j.abb.2010.07.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.