

Abstract
The kinetics and mechanism(s) of the reactions of [K(Krypt)][LCuO2] (Krypt = 4,7,13,16,21,24-hexaoxa-1,10- diazabicyclo[8.8.8]hexacosane, L = a bis(arylcarboxamido)- pyridine ligand) with 2,2,6,6-tetramethylpiperdine-N-hydroxide (TEMPOH) and the para-substituted phenols XArOH (X = para substituent NO2, CF3, Cl, H, Me, tBu, OMe, or NMe2) at low temperatures were studied. The reaction with TEMPOH occurs rapidly (k = 35.4 ± 0.3 M−1 s−1) by second-order kinetics to yield TEMPO• and [LCuOOH]− on the basis of electron paramagnetic resonance spectroscopy, the production of H2O2 upon treatment with protic acid, and independent preparation from reaction of [NBu4][LCuOH] with H2O2 (Keq = 0.022 ± 0.007 for the reverse reaction). The reactions with XArOH also follow second-order kinetics, and analysis of the variation of the k values as a function of phenol properties (Hammett σ parameter, O−H bond dissociation free energy, pKa, E1/2) revealed a change in mechanism across the series, from proton transfer/electron transfer for X = NO2, CF3, Cl to concerted-proton/electron transfer (or hydrogen-atom transfer) for X = OMe, NMe2 (data for X = H, Me, tBu are intermediate between the extremes). Thermodynamic analysis and comparisons to previous results for LCuOH, a different copper−oxygen intermediate with the same supporting ligand, and literature for other [CuO2]+ complexes reveal significant differences in proton-coupled electron-transfer mechanisms that have implications for understanding oxidation catalysis by copper-containing enzymes and abiological catalysts.
Graphical Abstract
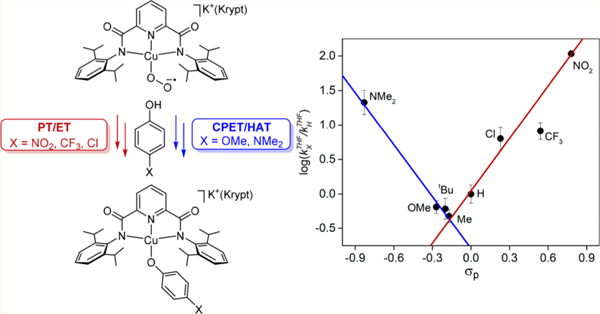
INTRODUCTION
A key to understanding enzymes1 and synthetic catalysts2 that use single copper sites to perform challenging oxidative transformations is discerning the nature of the involved copper−oxygen intermediates. Postulates of such intermediates are shown in Figure 13–5 in which supporting ligands are omitted and each species is shown in a form emphasizing either proposed formal oxidation states (blue) or overall charge for the core (black, in brackets). Synthetic complexes that incorporate many of these cores have been characterized and their reactivity studied, shedding important light on their viability in catalysis.4–10 Yet, compounds incorporating many of the indicated cores have not been unequivocally isolated to date, and much is still unknown about the fundamental chemistry of those that have been prepared, particularly with respect to their interconversions (potentially involving reactions with organic substrates) by the indicated proton and/or electron transfers (PT, vertical arrows; ET, horizontal arrows; PCET, proton-coupled electron transfer, red diagonal arrows). An overarching research goal, and one which underlies the work reported herein, is to discern the relative propensities for these reaction paths (kinetics and thermodynamics) and how these are influenced by the conditions (supporting ligand environment, secondary coordination sphere, temperature, solvent, etc.). Such research ultimately aims to develop rules for oxidase and/or oxygenase catalyst design.
Figure 1.

Intermediates proposed for O2 activation at a single copper site, emphasizing proton transfers (PT, vertical arrows), electron transfers (ET, horizontal arrows), and proton-coupled electron transfers (PCET, diagonal red arrows) that connect them. Arrows are drawn for simplicity as unidirectional, although the processes are equilibria characterized by parameters pKa (PT), E1/2 (ET), or BDFE (PCET), respectively, where BDFE = bond dissociation free energy. Gray boxes indicate the square schemes A and B emphasized in this work, and each intermediate is written in two forms: indicating possible formal oxidation states (blue) or overall charge (black). Figure inspired by and adapted from one published in ref 3a.
Deep insights into the nature of proton-coupled electron-transfer mechanisms11,12 such as those depicted in Figure 1 have been attained through examinations of the reactivity of metal− oxygen species with phenols (important substrates in biological proton-coupled electron-transfer processes) that have tunable properties because of the presence of variable para substituents.13–16 By examination of the course and kinetics of the reactions with a series of para-substituted phenols, trends can be ascertained that, in concert with thermodynamic analysis of relevant square schemes, enable stepwise (PT/ET or ET/PT) and concerted proton-coupled electron transfer (CPET) mechanisms to be distinguished. In a recent application of this approach, we examined the kinetics of the reactions of LCuOH comprising the [CuOH]2+ core supported by strongly electron-donating bis(arylcarboxamido)pyridine ligands (L2−; Figure 2) with para-substituted phenols XArOH (X = para-substituent NO2, CF3, Cl, H, Me, OMe, NMe2) using low-temperature stopped-flow methods and reference to the square scheme B (Figure 1).16 A key finding was that a CPET (or hydrogen-atom transfer, HAT) pathway was followed for all X, but with anomalies in the cases of X = NO2 and CF3 that were interpreted to indicate contributions from a PT/ET route for acidic phenol substrates.
Figure 2.

Copper complexes comprising [CuOH]2+ and [CuO2]+ cores supported by bis(arylcarboxamido)pyridine ligands. Krypt = Krypto-fix222.
At the other end of the cascade of possible copper−oxygen intermediates (Figure 1) is the initially formed 1:1 Cu/O2 adduct with the [CuO2]+ core (typically formally CuII−O2−•) that must be involved in any system that uses a single copper ion to bind and activate O2. A number of synthetic complexes this core have been characterized,4–8 including one ([K-(Krypt)][LCuO2])17 with the same supporting ligand as in LCuOH (Figure 2). We envisioned that unique insights would be attained by comparing the reactivity of these two different monocopper−oxygen cores supported by the same ligand. Moreover, such comparisons would address an important fundamental question with parallels in many other metal−oxygen systems:18,19 How do the reactivities and mechanisms traversed differ for species that contain an intact O−O bond (like [CuO2]+) versus those that do not (such as [CuOH]2+)? with Herein, we describe the results of studies aimed at answering this general question in a specific case by examining the kinetics of the reactions of [K(Krypt)][LCuO2] with the same series of phenols XArOH used in previous studies16 of LCuOH. We also studied the reactivity of [K(Krypt)][LCuO2] with a substrate having a particularly weak O−H bond, 2,2,6,6-tetramethylpi- perdine-N-hydroxide (TEMPOH). Analysis via reference to square scheme A and comparisons to the previous results for LCuOH and literature for other [CuO2]+ complexes reveal significant differences in proton-coupled electron-transfer mechanisms. These differences have implications for under-standing the reactivity of copper−oxygen species proposed in enzymatic and other catalytic systems.
RESULTS
Detailed studies of the reactivity of [LCuO2]− were enabled by the discovery (reported in detail elsewhere17b) that stable solutions (t1/2 ∼12 h at −60 °C) in predominantly tetrahydrofuran (THF) with minimal CH3CN (THF/CH3CN ratios 10−40:1) could be obtained by using 4,7,13,16,21,24- hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane (Kryptofix222, abbreviated further here as “Krypt”) to solubilize the reactant KO2. The resulting solutions of the complex [K(Krypt)]- [LCuO2] contain a characteristic UV−vis feature at 628 nm (ε ≈ 1400 M−1 cm−1) that was monitored in reactions with external substrates.
Reactivity of [K(Krypt)][LCuO2] with TEMPOH.
Addition of a solution of TEMPOH in THF (40−200 mM) to a freshly prepared solution of [K(Krypt)][LCuO2] (0.65 mM) in THF/ CH3CN (v/v 30:1) at −80 °C resulted in the rapid decay of the UV−vis feature at 628 nm to yield a final spectrum with absorptions at 485 nm (ε = 386 M−1 cm−1) and 655 nm (238 M−1 cm−1), with a shoulder at 513 nm (Figure 3). The final product spectroscopic features remained unchanged for long periods up to −20 °C, but decomposed at room temperature. Single-wavelength monitoring at 628 nm showed the reactions with excess TEMPOH to obey first-order kinetics on the basis of fits to simple exponential decays (Figure S3). A plot of the observed pseudo-first-order rate constants (kobs) versus [TEMPOH]0 was fit to a straight line with an effectively zero intercept within error, consistent with an overall second-order reaction with slope = k of 34.9 ± 1.5 M−1 s−1 (inset to Figure 3). This result was corroborated by an alternative analysis using multiwavelength global fitting of the UV−vis spectra that gave k = 35.4 ± 0.3 M−1 s−1 (Figure S4).
Figure 3.
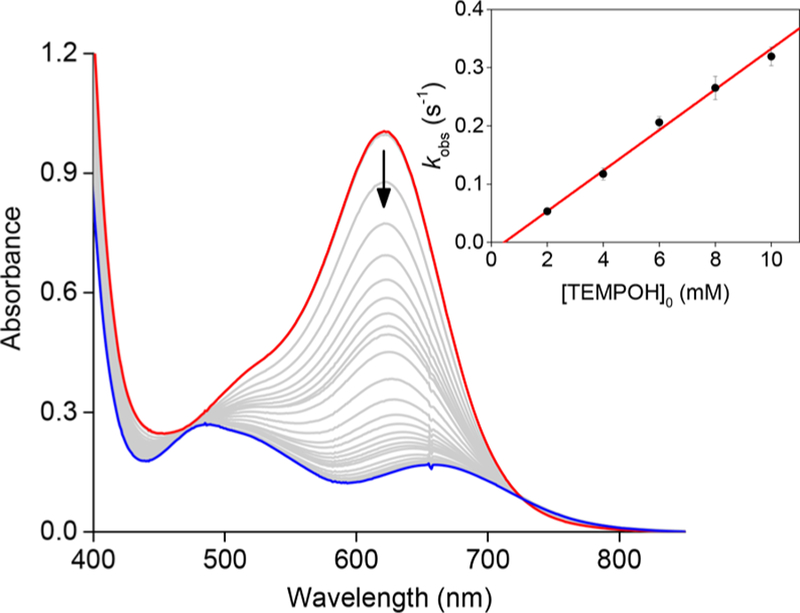
UV−vis spectra as a function of time observed upon reaction of [K(Krypt)][LCuO2] (red) with TEMPOH. Conditions: [Cu] = 0.7 mM, [TEMPOH]0 = 1.4 mM, THF:MeCN 10:1, −80 °C. Inset shows linear fit in plot of kobs versus [TEMPOH]0 (slope = 34.9 ± 1.5, intercept = −0.016 ± 0.005, R2 = 0.993).
We propose on the basis of evidence described below that the products of the reactions with TEMPOH are the TEMPO• radical and the new complex [K(Krypt)][LCuOOH] (Scheme 1). The X-band electron paramagnetic resonance (EPR) spectrum of the product mixture from the reaction of [K(Krypt)][LCuO ] and 10 equiv of TEMPOH displayed a characteristic signal for a LCu species overlaid with that for free TEMPO• radical (g = 2.005; Figure S5). Spin integration compared to either a Cu(II) or TEMPO• standard indicated two S = 1/2 systems in a 1:1 ratio; no excess TEMPO• was formed, indicating that the Cu(II) reaction product does not react further with TEMPOH. The copper hyperfine splitting constant (Az = 578 MHz) and gz value (2.185) for the Cu(II) signal were found to be comparable to those reported previously for other LCuII complexes (Table S3).
Scheme 1.

Reaction of [K(Krypt)][LCuO2] with TEMPOH
The nature of the Cu(II) product of the reactions of [K(Krypt)][LCuO2] with TEMPOH was further probed by adding triflic acid (HOTf) to the product mixture and assaying it for H2O2. In the experiment, addition of HOTf in Et2O (20 mM) to a THF solution of [K(Krypt)][LCuOOH] (made from addition of 2 equiv of TEMPOH to [K(Krypt)][LCuO2] at −60 °C) resulted in the immediate appearance of bands at 406 (2450 M−1 cm−1) and 642 nm (370 M−1 cm−1) (Figure S6). Titration of the product solution into saturated NaI in MeCN revealed 73% yield of H2O2 relative to copper,20 consistent with protonolysis of the proposed hydroperoxide species [K- (Krypt)][LCuOOH]. Further corroborating the formulation of the latter complex, addition of a solution of H2O2 in Et2O (predried with MgSO4 to remove most H2O) to a solution of [NBu4][LCuOH] in THF yielded a UV−vis spectrum closely analogous to that of the solution resulting from reaction of [K(Krypt)][LCuO2] with TEMPOH (Figure S7). The reaction with H2O2 was reversed upon addition of H2O, consistent with the equilibrium eq 1, for which Keq was estimated to be 0.022 ± 0.007 (Figure S8, Supporting Information). Taken together, the data support formation of [LCuOOH]− via both routes. Unfortunately, attempts to identify the [CuOOH]+ core more directly by resonance Raman spectroscopy on solution state samples prepared by addition of H O in ether (1 equiv) to [NBu4][LCuOH] (10 mM in THF) at −20 °C (90° backscattering geometry) were unsuccessful; discernible peaks derived from the sample were not observed using any of the available excitation wavelengths (406, 457, 515, 561, and 660 nm).
| (1) |
Finally, in an attempt to determine the E1/2 for the [CuOOH]2+/+ couple, we measured the cyclic voltammogram of the solution obtained upon reaction of [NBu4][LCuOH] (2 mM) with anhydrous H2O2 in Et2O (1 equiv) in 1,2- difluorobenzene at 0 °C (0.2 M NBu4PF6, Figure S10). An irreversible oxidation wave was observed with Ep,a = −0.177 V versus Fc+/Fc at a scan rate of 100 mV/s; no return wave was observed even at high scan rates (2000 mV/s). The data suggest that oxidation to the [CuOOH]2+ core occurs, but that this core is unstable at 0 °C. The irreversible voltammograms were batch-simulated using the Digisim 3.03b software package from BASi (Figure S11). Data collected at scan rates of 50, 100, 200, 750, 1500, and 2000 mV/s were simultaneously modeled using a two-step mechanism: initial heterogeneous electron transfer (eq S6) followed by a fast, homogeneous chemical event which quenches the oxidized copper species (eq S7). From this model, a theoretical redox potential (that is necessarily approximate) for the [CuOOH]2+/+ couple was derived with a value of E1/2 = −0.215 V (versus Fc+/Fc) (Figure S11).
Reactivity of [K(Krypt)][LCuO2] with para-Substituted Phenols.
To solutions of K(Krypt)][LCuO2] in THF/ CH3CN (18:1) were added solutions of the substituted phenols XArOH (20 equiv, X = para-substituent NO2, CF3, Cl, H, Me, tBu, OMe, or NMe2) in THF at −60 °C (Figure 4). Monitoring the reactions by UV−vis spectroscopy showed that the features due to [K(Krypt)][LCuO2] decayed, ultimately affording spectra with weak bands >400 nm associated with Cu(II) products (Figure S12). Those products were identified as salts of the phenolate complexes [LCuOArX]− on the basis of comparison to data obtained for independently synthesized and characterized complexes for several cases (X = NO2, H, OMe, NMe2; the formulations for X = CF3, tBu, Me, Cl were made by analogy). Thus, the complexes [NR4][LCuOArX] were prepared either by reaction of (a) LCu(CH3CN) with [NR4][OArX] (R = Bu, X = NO2;16 R = Et, X = H or OMe) or (b) [NBu4][LCuOH] with XArOH (X = NMe2). The products were isolated and characterized by UV−vis and EPR spectroscopy (Figures S14 and S1, respectively), CHN analysis or high-resolution mass spectrometry (X = NMe2), and X-ray crystallography (illustrative structure for X = H in Figure 4; others shown in Figure S2). Importantly, the close similarity of the UV−vis spectroscopic features for the isolated complexes and the final solutions resulting from the reactions of [K(Krypt)][LCuO2] with XArOH indicated that the final products of the latter were the respective complexes [LCuOArX]−.
Figure 4.

Reactions of [K(Krypt)][LCuO2] with phenols XArOH, with a representation of the X-ray crystal structure for the case X = H, showing all nonhydrogen atoms as 50% thermal ellipsoids (hydrogen atoms and NEt4 counterion omitted). Selected interatomic distances (Å) and angles (deg): Cu1−O1: 1.8767(13), Cu1−N2: 1.9296(14), Cu1−N1: 2.0064(15), Cu1−N3: 2.0220(15), O1−Cu1−N2: 172.98(6), O1−Cu1−N1: 96.94(6), O1−Cu1−N3: 102.46(6) N1− Cu1−N3: 160.44(6).
The UV−vis spectra as a function of time for the reactions of [K(Krypt)][LCuO2] with XArOH were analyzed by multi-wavelength global analysis (Figures S12 and S13). In all cases, the initial exponential decays of [LCuO2]− were fit to a simple second-order model, yielding second-order rate constants (k) listed in Table 1. For the cases X = NMe2 and OMe, data acquired over longer time periods indicated a more elaborate rate law (see below), but the k values in Table 1 were found to be reasonable for comparative purposes. To evaluate the data using thermodynamic parameters reported in dimethyl sulfoxide (DMSO),21,22 these rate constants were converted to values in that solvent (kDMSO) according to a previously described protocol16 specific for HAT/CPET reactions.23 Plots of log(kDMSO) versus phenol σ values (Hammett), pKa’s, and E1/2 values (for the XArOH0/+ couples) are shown in Figure 5. In each plot, the data point for X = NMe2 is clearly an outlier and a distinct break in continuity is apparent by the indicated linear fits, which occurs approximately at X = Me, with maximum rates at the extremes X = NO2 and NMe2. Such a break in these plots points to a change in mechanism across the series. We hypothesize that two mechanisms are operative, one being rate-determining PT (line through point for X = NO2, rate increases as pKa decreases) and the other being rate-determining CPET/HAT (line through point for X = NMe2, which has lowest E1/2 and highest pKa).
Table 1.
Second-Order Rate Constants (k) for the Reactions between [K(Krypt)][LCuO2] and XArOH at −60 °C
| X | k (M−1 s−1) | log(k) | log(kDMSO) |
|---|---|---|---|
| NMe2 | 30(13) | 1.4(2) | 0.1(2) |
| OMe | 0.7(1) | −0.16(9) | −1.45(9) |
| tBu | 0.8(3) | −0.2(2) | −1.4(2) |
| Me | 0.5(1) | −0.3(1) | −1.6(1) |
| H | 1.2(3) | 0.0(1) | −1.3(1) |
| Cl | 8(3) | 0.8(2) | −0.7(2) |
| CF3 | 10(3) | 0.9(1) | −0.7(1) |
| NO2 | 114(6) | 2.06(2) | 0.29(2) |
Figure 5.
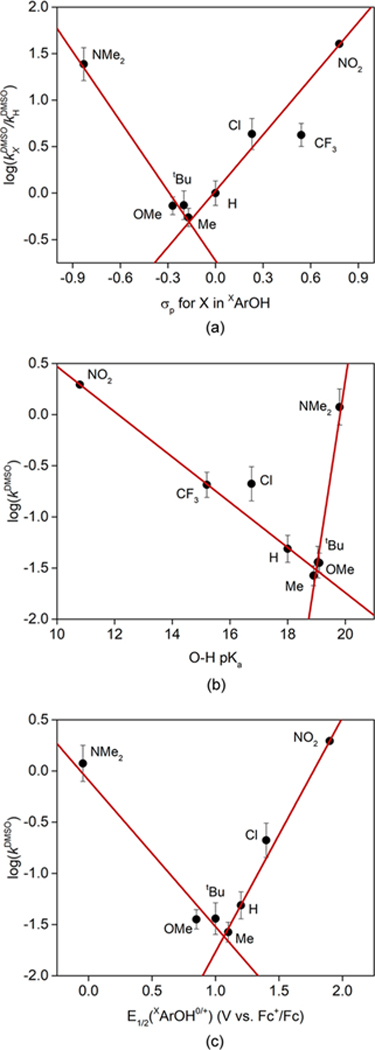
(a) Hammett plot of log(kXDMSO/kHDMSO) versus XArOH σp, with X labeled. Separate linear fits are shown to data for X = NMe2, OMe, tBu, and Me (slope ρ = −2.5, R2 = 0.97) and for X = Me, H, Cl, CF3, and NO2 (slope ρ = +2.0, R2 = 0.96). (b) Plot of log(kDMSO) versus XArOH pKa. Separate linear fits are shown to data for X = NMe2, OMe, tBu, and Me (slope = +1.84, R2 = 0.924) and for X = Me, H, Cl, CF3, and NO2 (slope = −0.22, R2 = 0.998). (c) Plot of log(kDMSO) versus XArOH0/+ E1/2. Separate linear fits are shown to data for X = NMe2, OMe, tBu, and Me (slope = −1.43, R2 = 0.92) and for X = Me, H, Cl, and NO2 (slope = +2.29, R2 = 0.996).
Further evaluation of the kinetic UV−vis spectroscopic data over longer reaction times revealed additional evidence in support of the above mechanistic hypotheses. For the reactions with XArOH (X = NMe2, OMe, and to a lesser extent tBu, and Me), distinct losses of isosbestic points in the UV−vis traces over long time periods are apparent (Figure 6, Figure S16) and an intermediate spectrum (purple, “Int.”) slowly converts to that of the final [LCu(OArX)]− product (blue). The two different time courses are illustrated for select wavelengths in the inset to Figure 6. Additional kinetics runs with XArOH (X = OMe) were performed using variable concentrations of substrate (5−25 equiv based on Cu), followed over 1 h at −60 °C by UV−vis spectroscopy. In all cases, multiwavelength global analysis of the full kinetic traces were fit to a two-phase reaction mechanism (eq 2), with average rate constants of k1 = 0.44(9) M−1 s−1 and k2 = 0.12(2) M−1 s−1 (Figure S17).24 Multiwavelength global analysis was also used to fit the reaction of [LCuO2]− and NMe2ArOH (20 equiv) to a two-phase reaction mechanism, with k1 = 13 M−1 s−1 and k2 = 1.1 M−1 s−1 (Figures S16 and S17, Scheme S1). Furthermore, included in the model was the appearance of the transient phenoxyl radical, NMe2ArO•, with a fitted spectrum closely matching that reported previously (λmax = 495 nm).25 The appearance of this radical is consistent with a CPET/HAT mechanism for X = NMe2. The spectrum of the initially formed intermediate in both reactions (X = OMe and NMe2; “Int.” in eq 2) is indicative of LCu(II) species, and is not consistent with formally Cu(III) complexes that are characterized by intense ligand to metal charge transfer (LMCT) absorptions.26–28 We hypothesize that the intermediate is [LCuOOH]− derived from CPET/HAT, which is favorable for substrates with relatively weak O−H bonds (especially X = NMe2, BDFE = 79 kcal/mol; OMe, BDFE = 83 kcal/mol; Figure 7).21 The intermediate is then consumed in a protonolysis event by excess phenol (k2). In support of this hypothesis, reaction of the independently synthesized [NBu4][LCuOOH] with NMe2ArOH (20 equiv) resulted in a UV−vis spectrum matching that of [LCuOArNMe2]− and a calculated second-order rate eq 4, eq 3 may be simplified to eq 5, which relates ΔG to the constant of k = 0.54 M−1 s−1 (Figure S18). We surmise that because these phenols are weak acids (X = NMe, pK = 19.8; OMe, pKa = 19.1), the protonolysis is relatively slow, thus enabling observation of the intermediate.
Figure 6.

UV−vis spectral traces for the reaction of [K(Krypt)]- [LCuO2] (red) with XArOH (X = OMe, 20 equiv) at −60 °C, 19:1 THF:CH3CN, over ∼30 min. Inset shows the decay trace over time following the signal at 628 (black squares) and 775 nm (red triangles). “Int.” is defined as the initially formed Cu(II) intermediate and is highlighted in the UV−vis spectrum in purple.
Figure 7.

Proposed PT/ET (red) and CPET (blue) mechanisms for the reactions of [K(Krypt)][LCuO2] with XArOH. The data are ambiguous for X = H, Me, tBu, suggesting that in these cases both pathways may be operative (not shown).
For substrates that are more acidic (especially X =NO2, pKa = 10.8; CF3, pKa = 15.2)21,22 the reactions exhibit clean isosbestic points and no intermediate is observed. We propose that the rate-limiting step in these cases is an initial PT event that yields a highly reactive and unstable (formally Cu(III)) species LCuOOH. This species could decay quickly via ET from the phenoxide anion and subsequent protonolysis, or by some other decay pathway (such as Cu−O bond homolysis)29,30 followed simply by phenoxide coordination (Figure 7). Because of the larger E1/2 values for the XArO•/XArO− couple (X = NO2 and CF3; E1/2 = 0.314 and 0.079 V versus Fc+/Fc)21,22 compared to the approximated [CuOOH]2+/+ couple (E1/2 =−0.215 V versus Fc+/Fc), pathway (b) is unlikely from a thermodynamic standpoint. Furthermore, reactions with [LCuO2]− and only 1 equiv of XArOH (X = NO2, CF3; Figure S20) yield the same phenoxide product in the same amount (calculated by extinction of final UV−vis spectrum) as those performed with an excess of phenol substrate, favoring pathway (a) where excess XArOH is not needed (Figure 7). Only ClArO− (E1/2 = −0.232 V versus Fc+/Fc)21 could undergo a thermodynamically favorable ET to [LCuOOH] by pathway (b). Formulation of the mechanisms for X = H, Me, tBu are less clear because of their position in the plots (Figure 5) at the intersection of the linear fits, and we suspect that contributions from both pathways may occur in these intermediate cases. Further aspects of these mechanistic hypotheses are described below (see Discussion section).
Thermodynamic Analysis.
To obtain a deeper under-standing of the reactivity of [K(Krypt)][LCuO2] with the phenols XArOH, we attempted to evaluate the thermodynamics of PCET via the square scheme A (Figure 1). Complicating the analysis is the instability of all the species in the scheme (except [K(Krypt)][LCuO2]). Nonetheless, we were able to obtain a rough estimate of key pKa and redox potential values, enabling estimation of the O−H BDE for the [CuOOH]+ core. As noted above, cyclic voltammograms measured on solutions of [LCuOOH]− prepared by reaction of [LCuOH]− with H2O2 contained an irreversible wave ascribed to oxidation of the [CuOOH]+ core with Ep,a = −0.177 V versus Fc+/Fc. By simulating the half-wave at varying scan rates, a value for E1/2 for the [CuOOH]2+/+ couple was estimated at −0.215 V versus Fc+/Fc. To estimate the pKa value of the [CuOOH]2+ complex, we make the crude assumption that at the mechanistic crossover point in the plots shown in Figure 5 (about at X = Me) the thermodynamic driving forces for PT and CPET are approximately the same (ΔGPT ∼ ΔGCPET).31 The driving force for the CPET reaction (ΔGCPET) is estimated by the difference between the BDFE of MeArOH in DMSO and the BDE of [LCuOO−H]− in THF (eq 3). The [LCuOO−H]− BDE is represented by eq 4, where pKa is the acidity of LCuIIIOO−H, E° is approximated by E1/2 of [LCuOOH]− (−0.215 V), and a value of 66 kcal mol−1 is used for the constant CH (in THF).32 With use of the reported value of 87.1 kcal mol−1 for the BDFE of MeArOH,21 and the incorporation of eq 4, eq 3 may be simplified to eq 5, which relates ΔGCPET to the unknown pKa of LCuIIIOOH. The driving force for proton transfer (ΔGPT) is estimated from the difference in pKa of MeArOH and LCuIIIOOH, the latter being the unknown variable (eq 6, where R is the ideal gas constant and T is the temperature). With use of the known pKa of MeArOH (19.1, DMSO),21 eq 6 simplifies to eq 7. Working under the assumption that ΔGPT ∼ ΔGCPET, eq 5 and eq 7 may be set to be equal to one another, enabling calculation of the pKa of LCuIIIOOH to be ∼19 (THF). Inserting this value back into eq 4 enables calculation of the BDE of [LCuIIOOH]− as ∼87 kcal mol−1. We hypothesize that this approximated value represents an upper estimate; on the basis of the proposed CPET mechanism followed in the reaction of [LCuO2]− and OMeAr- OH (BDFE = 83 kcal/mol),21 it is likely that the true BDE of [LCuOO−H]− falls in the range 83−87 kcal/mol.
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
DISCUSSION
Comparisons to other previously reported complexes with [CuO2]+ cores for which reactivity with TEMPOH (or derivatives) and phenols have been studied are useful for placing our findings in perspective (Figure 8).4,14,33–39 All are postulated to exhibit end-on η1 binding of superoxide, except ArLCuO2; however, this η2 complex was proposed to exist in equilibrium with an end-on isomer.39 The complex [K(Krypt)]- [LCuO2] differs from the complexes shown in Figure 8 with respect to overall charge and supporting ligand geometry and donor set. These structural differences are accompanied by disparities in their reactivity with TEMPOH (or derivatives) and phenols, as discussed below. We also discuss reactivity variations between [K(Krypt)][LCuO2] and LCuOH, which contains a different monocopper−oxygen core supported by the same ligand.
Figure 8.
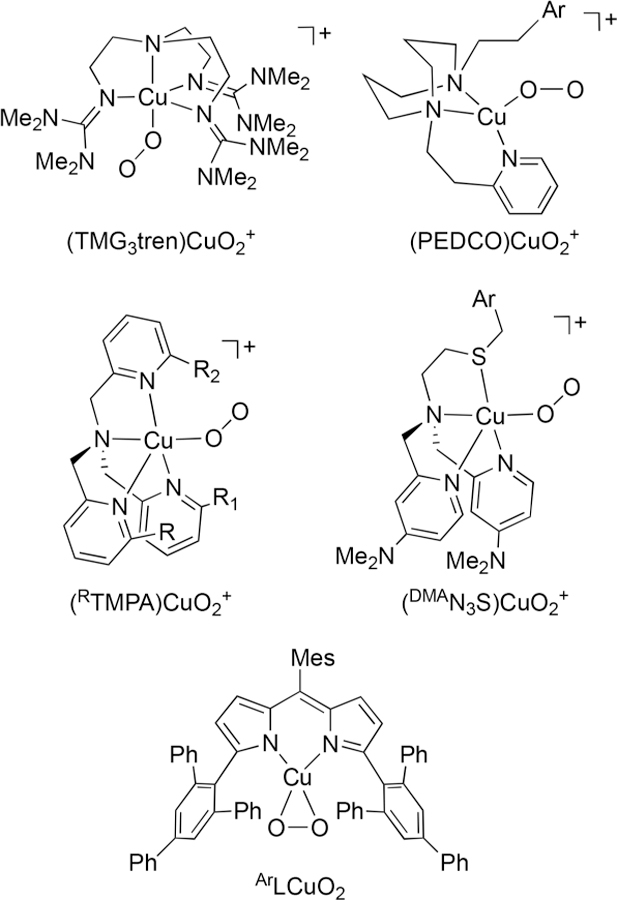
Complexes with [CuO2]+ cores for which reactivity studies with TEMPOH (or derivatives) and phenols have been reported. DMATMPA: R = NMe2, R1 = R2 = H; DMMTMPA: R = OMe, R1 = R2 = H; PVTMPA: R = NH(CO)tBu, R1 = R2 = H; NEOTMPA: R = R1 = NH(neopentyl), R2 = H). Ar = aryl, Mes = mesityl. Note: The complex ArLCuO2 is proposed to equilibrate with an end-on (η1) isomer (ref 39).
Reaction with TEMPOH.
Reaction of [K(Krypt)][LCuO2] with TEMPOH (Scheme 1) yields TEMPO• and a Cu(II) product that we formulate as [LCuOOH]− on the basis of EPR spectroscopy, the production of H2O2 upon treatment with protic acid, independent preparation from reaction of [NBu4]- [LCuOH] with H2O2 (eq 1, Keq = 0.022 ± 0.007), and analogy to previously reported and more stable complexes [LCuOOR]− (R = cumyl, tBu).28 The rapid CPET/HAT reaction followed the second-order rate law: rate = k[complex][TEMPOH], with k = 34.9 ± 1.5 M−1 s−1 at −80 °C. Similar conversions were reported previously for [CuO2]+ complexes supported by TMG3tren,33 NEOTMPA,38 PEDCO,14 and ArL− 39 (Figure 8). It would thus appear that, for the substrate TEMPOH having a quite weak O−H bond (BDFE ∼67.5 kcal/mol),40 a common reaction course involving apparent HAT is followed for the [CuO2]+ core irrespective of the overall charge and supporting ligand in the complexes. Nonetheless, in the one other report of reaction kinetics having been evaluated ((PEDCO)CuO2 +),14 the second-order rate constant (k = 2.4 ± 0.05 M−1 s−1 at −85°C) is about an order of magnitude smaller than that we observed for [K(Krypt)][LCuO2]. The complex LCuOH also abstracts an H atom from TEMPOH at −80 °C, but at a rate so fast that we have not been able to monitor it with much precision or accuracy. This greater PCET reactivity for LCuOH is reflected in the phenol reaction kinetics as well.
Reactions with Phenols.
The reactions of [K(Krypt)]- [LCuO2] with phenols XArOH (X = para-substituent NO2, CF3, Cl, H, Me, tBu, OMe, or NMe2) yields the complexes [LCuOArX]−. These products were identified by spectroscopic comparison to independently synthesized samples, several of which were characterized by X-ray crystallography (X = NO2, H, OMe, NMe2; Figure 4 and Figure S2); similar formulations for X = CF3, tBu, Me, Cl were made by analogy. Kinetics studies of the reactions monitored by UV−vis spectroscopy showed that the decay of the features due to [K(Krypt)][LCuO2] followed second-order kinetics (k and k DMSO values listed in Table 1). The initial formation of an intermediate that slowly transformed to the final [LCuOArX]− product was indicated for X = NMe2 and OMe (also tBu and Me, although the data were less clear). We postulate that, for these cases, the intermediate is the same product seen in the TEMPOH reactions, [LCuOOH]−, and that the mechanism traversed differs from those for the phenols with other X substituents.
The plots of log(kDMSO) versus phenol σ values (Hammett), pKa’s, and E1/2 values (for the XArOH0/+ couples) shown in Figure 5 corroborate a change in mechanism across the series of X groups.23 For the series X = NO2, CF3, Cl, H, Me the rate constant increases with increasing electron-withdrawing properties (ρ = +2.0, Figure 5a), increasing acidity (lower pKa, Figure 5b), and increasing redox potential for the phenol substrates (Figure 5c). These results are consistent with a mechanism involving rate-determining proton transfer (PT) from the phenol to [K(Krypt)][LCuO2] to yield the formally Cu(III) complex LCuOOH (Figure 7, top). Supporting the feasibility of such a complex, related species LCuOOR (R = tBu, cumyl) have been identified by spectroscopy.28 The failure to observe LCuOOH as an intermediate suggests that it reacts rapidly, with one possible pathway involving reduction by the phenolate ion to yield [LCuOOH]− that then undergoes fast protonolysis by the excess (acidic) phenol (Figure 7, path (b)). A similar PT process but an alternative subsequent decay pathway was proposed to explain the results of reactions of (PEDCO)CuO + with a series of para-substituted phenols XArOH (X = OPh, Me, H, F, Cl)14 and ArLCuO2 with XArOH (X = H, NO2) and 2- amino-4-tert-butylphenol.39 These reactions also yielded phenolate complexes, and mechanisms were proposed involving initial PT, but followed by Cu−O homolysis and loss of HO2 radical that then disproportionates to yield H2O2 and O2 (Figure 7, path (a)). We cannot unequivocally distinguish between paths (a) and (b) with the available evidence, although the observation of the same yield of final product in reactions using only 1 equiv of substrate phenol argues against path (b). Importantly, both paths involve ET, such that the overall reaction is a PT/ET process.
The lines through the data for X = NMe2, OMe, tBu, and Me in Figure 5 paint a different picture. The rate constant decreases with increasing electron-withdrawing properties (ρ = −2.5, Figure 5a), increasing acidity (lower pKa, Figure 5b), and increasing redox potential for the phenol substrates (Figure 5c). We interpret these results, as well as the observation in these cases of an intermediate formulated as [LCuOOH]−, to indicate that a CPET/HAT process is followed, akin to that seen in the reaction with TEMPOH (Figure 7, blue). This contrasting reactivity is analogous to that reported for ArLCuO2, which performs HAT with 2-hydroxy-2-azaadamantane but PT/ET with some phenols.39 Yet, to our knowledge, the observation of a changeover in reaction pathway for one type of substrate that we observe is unique in copper−oxygen chemistry.
Deeper insights may be drawn by comparing the data for the reactions of XArOH with [K(Krypt)][LCuO2] to those we reported previously for LCuOH,16 which features a different [CuOH]2+ core but with the same supporting ligand (Figure 9). Overall, the rate constants for the reactions with LCuOH measured at −80 °C are significantly larger (log kDMSO range from ca. 0 to +5) relative to those for [K(Krypt)][LCuO2] (log kDMSO range from ca. −1.6 to +0.4) measured at −60 °C.41 Clearly, at parity of the supporting ligand, the [CuOH]2+ core is more reactive than the [CuO2]+ unit. Even more striking are the essentially opposite slopes to the linear fits (blue versus red, Figure 9). In the log(kDMSO) versus O−H BDFE plot (a), for [CuOH]2+ (blue) the rate constant increases as the bond strength decreases, as expected for a predominate CPET/HAT process. The data for the [CuO2]+ complex is much more scattered, with a fit to the data for X = Me, H, Cl, CF3, and NO2, indicating a poor correlation (R2 = 0.779) and an opposite trend (faster rate for substrates with stronger O−H bonds). Notably, similar rate constants are observed for X = NO2 and NMe2 that have drastically different O−H BDFE values (93 versus 79 kcal/ mol, respectively). These data argue against a predominant CPET/HAT process and support a mechanism change across the series. Similarly, the Hammett plot (b) and the log(kDMSO) versus E1/2 plot (d) show divergent slopes for the [CuOH]2+ (blue) and [CuO2]+(red) cores, with the data for X = NMe2 for the [CuO2]+ case being a clear outlier. The log(kDMSO) versus pKa plot (c) is particularly revealing. For the case of the [CuOH]2+ (blue) complex, the rate decreases as the acidity increases, but with X = NO2 being an outlier. In contrast, for the [CuO2]+ (red) complex, the rate increases as the acidity increases, but with X = NMe2 being an outlier. We rationalize these opposite behaviors by (a) a predominantly CPET/HAT pathway for the [CuOH]2+ core, with a PT/ET contributor for the most acidic (X = NO2) phenol (indicated by arrow), and (b) a predominantly PT/ET pathway for the [CuO2]+ core that shifts to CPET/HAT for the least acidic (X = NMe2) phenol (indicated by arrow). The observation of anomalously high rates for the substrate with X = NMe2 that has the weakest O−H bond, most electron-donating substituent, and lowest reduction potential also is consistent with a CPET/HAT mechanism for its reaction with the [CuO2]+ unit.
Figure 9.

(a) log(kDMSO) versus XArOH BDFE overlay plot for the reaction of [CuO2]+ (red circles, −60 °C) and [CuOH]2+ (blue squares, −80 °C) cores with XArOH (X = NMe2, OMe, Me, H, Cl, CF3, NO2). The red line depicts an arbitrary linear correlation for the data points for X = Me, H, Cl, CF3, and NO2 (R2 = 0.779, slope = 0.29). The blue line is a fit to the data for all X (R2 = 0.932, slope = −0.35). (b) log(kDMSO) versus XArOH σp overlay plot for the [CuO2]+ (red circles, −60 °C) and [CuOH]2+ (blue squares, −80 °C) cores. The red line depicts a fit to the data for X = Me, H, Cl, CF3, and NO2 (R2 = 0.959, slope = 2.0). The blue line depicts a fit to the data for all X (R2 = 0.806, slope = −3.2). (c) log(kDMSO) versus XArOH pKa overlay plot for the [CuO2]+ (red circles, −60 °C) and [CuOH]2+ (blue squares, −80 °C) cores. The red and blue lines are fits to the data for all X except NMe2 (red, R2 = 0.992, slope = −0.22) or except NO2 (blue, R2 = 0.811, slope = 1.1). (d) log(kDMSO) versus XArOH0/+ E1/2 overlay plot for the [CuO2]+ (red circles, −60 °C) and [CuOH]2+ (blue squares, −80 °C) cores (E1/2 values referenced to the Fc+/Fc redox couple). The red line is a fit for X = Me, H, Cl, and NO2 (R2 = 0.996, slope = 2.3). The blue line is a fit to the data for all X (R2 = 0.798, slope = −2.5).
A key question concerns why the two cores exhibit such different behaviors. Comparison of the thermodynamics as reflected in the square schemes A and B (Figures 1 and 10) is revealing. The measured/estimated pKa, E1/2, and BDE values for each core supported by the same ligand L2− are shown in Figure 10.42 It is notable that the O−H BDE value for the [CuOOH]+ core is at least ∼3 kcal/mol lower than that for the [CuOH2]2+ unit, consistent with the generally faster rates for PCET by [CuOH]2+ than for [CuO2]+. Another notable difference is the basicity of these latter cores; the ∼7 pKa unit difference between the pKa values for the [CuOOH]2+ and [CuOH2]3+ cores is significant. Also significant is the difference in the E1/2 values for the [CuOOH]2+/+ and [CuOH2]2+/3+ couples (∼0.5 V higher for the latter). Both of these large differences (pKa and E1/2) are consistent with the one unit differences in charge between the species being compared; the higher charge for the [CuOH2]3+ core results in a lower pKa and higher E1/2 relative to those of the [CuOOH]2+ unit.
Figure 10.

Square schemes A and B (from Figure 1), but with measured or estimated thermodynamic parameters from this work (A) or reported previously (B)42 indicated.
The aforementioned differences in acidity and redox potential translate into key differences in thermodynamics that we postulate underlie the differences in observed reactivity. As described previously, for [CuOH]2+ (B),16 the difference between the free energy for CPET/HAT (defined as the difference in the BDE values for the phenols and the [CuOH2]2+ O−H bonds) and the free energy for PT (estimated from the difference in pKa values for the phenols in DMSO and that of the [CuOH2]3+ unit in THF) is large for most of the phenols, thus rendering PT unlikely and favoring CPET/HAT (Figure 11a). The exceptions are for the most acidic phenols (X = CF3 or NO2), where PT becomes thermodynamically feasible (dashed box). In contrast (Figure 11b), for [LCuO2]− the overall thermodynamic driving forces are smaller, the pKa for the [CuOOH]2+ core is ∼7 units higher than that for [CuOH2]3+, and the enhanced basicity of the [CuO2]+ core thus renders PT much more favorable such that only for the least acidic phenols is CPET/HAT preferred. Put another way, as graphically shown in Figure 11, the dashed box (where PT/ET and CPET/HAT paths have similar driving forces) is positioned differently along the phenol Hammett σ axis for the two cores, predominantly because of significant differences in the PT thermodynamics (more favorable for [LCuO2]−).
Figure 11.
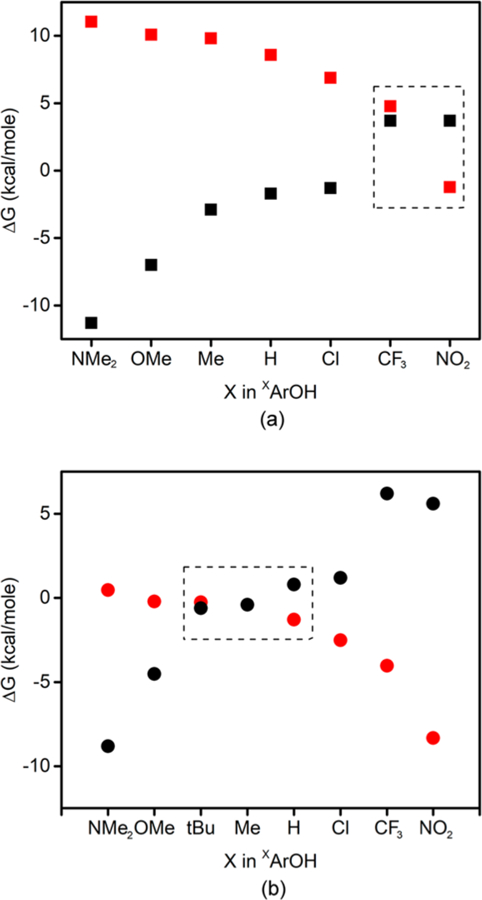
Plots of ΔG for the corresponding PT (red) or CPET (black) reactions with LCuOH (top, squares, adapted from ref 17) and [LCuO2]− (bottom, circles). The dashed regions highlight where there is thermodynamic potential for both PT and CPET mechanisms to occur.
Notably relevant to our work are the results of reactions of the [CuO2]+ complexes shown in Figure 8 with the weak O−H bonds of 2,2,6,6-tetramethylpiperdine-N-hydroxide (TEMPOH) or phenols.4,14,33–39 All of the complexes react with TEMPOH to give TEMPO• and compounds with the [CuOOH]+ core, consistent with hydrogen-atom abstraction (HAT) from the O−H bond having a BDFE ∼67.5 kcal/mol. Differences arise in reactions with phenols. The [CuO2]+ omplexes supported by TMG3tren,33 DMATMPA,34 DMMTMPA,35 and PVTMPA37 yield products derived from phenoxyl radicals and [CuOOH]+ cores upon reaction with various substituted phenols, again consistent with HAT pathways. Such a pathway for [(DMMTMPA)CuO2]+ was further supported by a detailed mechanistic study and thermodynamic analysis, which notably led to a PT/ET pathway being ruled out.35 In the context of our findings, we hypothesize that for all of these cationic complexes supported by TMG3tren and RTMPA derivatives, HAT is favored because the basicity of the [CuO2]+ core is insufficient to make initial PT feasible. Consistent with this notion, even a strong acid like trifluoroaceticcid does not protonate [(TMG3tren)CuO2]+; instead, an adduct forms that is then susceptible to reduction by octamethyl- or decamethylferrocene.43
On the other hand, reactions of [(PEDCO)CuO2]+ with a series of para-substituted phenols XArOH (X = OPh, Me, H, F, Cl) yielded copper(II)−phenolate complexes [(PEDCO)- CuOArX]+, and a pathway was proposed involving proton transfer to the superoxide (PT) followed by loss of HO2 radical that then disproportionates to yield H2O2 and O2 (essentially, a PT/ET pathway).14 Consistent with initial PT, a plot of log k versus pKa for the phenols showed an increase in rate as the acidity increased (slope of linear fit = −0.18, similar to the value of −0.22 that we observed; Figure 9c), although in the study of [(PEDCO)CuO2]+ the span in pKa’s measured was small (2 units) and there was significant deviation in the data. In studies of reactions of ArLCuO2 with phenol and tert-butyl-substituted derivatives, PT/ET was implicated for the former and HAT was found for the latter.39 The basis for the apparently similar tendency of the [CuO2]+ cores supported by PEDCO and L2− to react with phenols via initial PT, and for both cores supported by ArL− and L2− to follow either PT/ET or HAT depending on the nature of the phenol, is unclear. It would be useful to have more extensive thermodynamic data for the systems supported by PEDCO and ArL− in needed efforts to better understand how supporting ligands influence the mechanisms of reactions of [CuO2]+ cores.
Mechanistic dichotomies between PT/ET, CPET, or ET/PT within the same system have been observed previously in high valent metal−oxo species. For example, mechanistic changes between HAT and ET/PT were seen for a MnIV−oxo complex with changing temperature,44 and for an FeIV−oxo complex on addition of scandium.45 A changeover between PCET and PT/ ET has also been observed by tuning the ligand basicity in FeIVoxo species.46 The degree to which proton and electron transfer are coupled in CPET processes has recently been highlighted for an array of FeIV-oxo compounds through the introduction of the asynchronicity factor, η,47 and has been implemented experimentally to describe C−H bond activation by a terminal CoIII-oxo complex.48 In the latter system, reaction rates were found to be inversely related to the substrate pKa (as observed in our work for acidic phenols). However, experimental evidence did not support a stepwise process (PT/ET), but instead supported basic asynchronous CPET behavior, where a more asynchronous process resulted in higher rates. This example has important ramifications for other metal-oxo systems, where the relative reactivity with R−H substrates has been linked to the basicity of the oxo ligand,49 as well as to our work on systems supported by L2− that we report herein. Indeed, through an analysis akin to that reported previously involving determination of η and assessment of a and correlation plot (Figure S21),47,48 we confirmed that the reaction of LCuOH with XArOH via CPET is exergonic for phenols with X = NMe2, OMe, Me, H, Cl, but endergonic for X = NO2. Moreover, none of the phenols react through a true synchronous CPET mechanism; instead, asynchronicity in CPET favoring ET (X = NMe2) or favoring PT (X = OMe, Me, H, Cl) is proposed, while a stepwise PT/ET mechanism is proposed for X = NO2 (CPET endergonic). Unfortunately, because of our current inability to measure all thermodynamic parameters for [LCuO2]− (Figure 10 (A)), we were unable to perform a corresponding analysis of asynchronicity for this complex.
CONCLUSIONS
Studies of the reactions with the O−H bonds in TEMPOH and the series of phenols XArOH (X = para-substituent NO2, CF3, Cl, H, Me, tBu, OMe, or NMe2) have shown [K(Krypt)]- [LCuO2] to be capable of divergent proton-coupled electron-transfer mechanisms spanning PT/ET and CPET/HAT processes, depending on the nature of the substrate. Proton transfer (PT) is notably favorable with respect to kinetics and thermodynamics, such that CPET (or HAT) prevails for substrates that have relatively low acidities, low redox potentials, and weak O−H bonds. We surmise that the dominance of PT/ET paths is due in large part to the anionic nature of the complex, a unique attribute arising from the dianionic supporting ligand L2− relative to other known species with [CuO2]+ cores. The reactivity is notably unlike that of LCuOH, a particularly interesting comparison to draw because the complex contains a different [CuOH]2+ core supported by the same ligand L2−. Reactions of LCuOH with XArOH are significantly faster, and CPET/HAT paths are favored, such that PT/ET is apparent only for the substrate NO2ArOH with the highest acidity, highest redox potential, and strongest O−H bond. The significantly higher oxidizing power, lower basicity, and stronger O−H bond in the product [CuOH2]2+ core all contribute to the divergent reactivity of LCuOH relative to [LCuO2]−. An implication of these findings is that at parity of supporting ligand donors, one would expect similarly divergent reactivity of [CuO2]+ and [CuOH]2+ cores in enzymes or other catalysts. In addition, one would anticipate that for the [CuO2]+ unit, or others in the upper left quadrant of the cascade in Figure 1 (with intact O−O bonds), to exhibit reactivity competitive with the [CuOH]2+ core, or others in the lower right quadrant of Figure 1 (lacking O−O bonds), perturbations of the supporting ligand and/or local environment would be needed.
Supplementary Material
ACKNOWLEDGMENTS
Support for this research was provided by the NIH (Grant R37GM47365 to W.B.T.) and Washington University in St. Louis. We thank Dr. Timothy Zerk for help with CV simulations.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b00466.
Experimental details, including procedures, syntheses, and spectroscopic and kinetic data (PDF) X-ray crystallographic data (CIF)
The authors declare no competing financial interest.
X- ray data has been deposited in the CSD (1880915, 1880916, and 1880917) and may be accessed at https://www.ccdc.cam.ac.uk/.
REFERENCES
- (1).(a) Selected reviews: Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vu VV; Ngo ST Copper active site in polysaccharide monooxygenases. Coord. Chem. Rev 2018, 368, 134–157. [Google Scholar]; (c) Ciano L; Davies GJ; Tolman WB; Walton PH Bracing copper for the catalytic oxidation of C−H bonds. Nature Catal 2018, 1, 571–577. [Google Scholar]; (d) Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tandrup T; Frandsen KEH; Johansen KS; Berrin JG; Lo Leggio L Recent insights into lytic polysaccharide monooxygenases (LPMOs). Biochem. Soc. Trans 2018, 46, 1431–1447. [DOI] [PubMed] [Google Scholar]
- (2).(a) Selected lead references: Trammell R; See YY; Herrmann AT; Xie N; Diaz DE; Siegler MA; Baran PS; Garcia-Bosch I Decoding the Mechanism of Intramolecular Cu-Directed Hydroxlation of sp3 C-H Bonds. J. Org. Chem 2017, 82, 7887–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]; Snyder BER; Bols ML; Schoonheydt RA; Sels BF; Solomon EI Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Chem. Rev 2018, 118, 2718–2768. [DOI] [PubMed] [Google Scholar]; (c) Pegis ML; Wise CF; Martin DJ; Mayer JM Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts. Chem. Rev 2018, 118, 2340–2391. [DOI] [PubMed] [Google Scholar]; (d) Gandeepan P; Müller T; Zell D; Cera G; Warratz S; Ackermann L 3d Transition Metals for C−H Activation. Chem. Rev 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- (3).(a) Hedegård ED; Ryde U Targeting the reactive intermediate in polysaccharide monooxygenases. JBIC, J. Biol. Inorg. Chem 2017, 22, 1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hedegård ED; Ryde U Molecular mechanism of lytic polysaccharide monooxygenases. Chem. Sci 2018, 9, 3866–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Caldararu O; Oksanen E; Ryde U; Hedegård ED Mechanism of hydrogen peroxide formation by lytic polysaccharide monooxygenase. Chem. Sci 2019, 10, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Liu JJ; Diaz DE; Quist DA; Karlin KD Copper(I)- Dioxygen Adducts and Copper Enzyme Mechanisms. Isr. J. Chem 2016, 56, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper−Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mirica LM; Ottenwaelder X; Stack TDP Structure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev 2004, 104, 1013–1045. [DOI] [PubMed] [Google Scholar]
- (7).Lewis EA; Tolman WB Reactivity of Copper-Dioxygen Systems. Chem. Rev 2004, 104, 1047–1076. [DOI] [PubMed] [Google Scholar]
- (8).(a) Itoh S Mononuclear copper active-oxygen complexes. Curr. Opin. Chem. Biol 2006, 10, 115–122. [DOI] [PubMed] [Google Scholar]; (b) Itoh S Developing mononuclear copper-active-oxygen complexes relevant to reactive intermediates of biological oxidation reactions. Acc. Chem. Res 2015, 48, 2066–2074. [DOI] [PubMed] [Google Scholar]
- (9).Lee JY; Karlin KD Elaboration of copper-oxygen mediated C-H activation chemistry in consideration of future fuel and feedstock generation. Curr. Opin. Chem. Biol 2015, 25, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fukuzumi S; Karlin KD Kinetics and thermodynamics of formation and electron-transfer reactions of Cu−O2 and Cu2−O2 complexes. Coord. Chem. Rev 2013, 257, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mayer JM Proton-coupled electron transfer: a reaction chemist’s view. Annu. Rev. Phys. Chem 2004, 55, 363–390. [DOI] [PubMed] [Google Scholar]
- (12).Huynh MHV; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2007, 107, 5004–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Selected examples not involving copper: Yiu DTY; Lee MFW; Lam WWY; Lau T-C Kinetics and Mechanisms of the Oxidation of Phenols by a trans-Dioxoruthenium(VI) Complex. Inorg. Chem 2003, 42, 1225–1232. [DOI] [PubMed] [Google Scholar]; (b) Xie J; Ma L; Lam WW; Lau KC; Lau TC Hydrogen atom transfer reactions of ferrate(vi) with phenols and hydroquinone. Correlation of rate constants with bond strengths and application of the Marcus cross relation. Dalton Trans 2016, 45, 70–73. [DOI] [PubMed] [Google Scholar]; (c) Lansky DE; Goldberg DP Hydrogen Atom Abstraction by a High-Valent Manganese(V)−Oxo Corrolazine. Inorg Chem 2006, 45, 5119–5125. [DOI] [PubMed] [Google Scholar]; (d) Zaragoza JPT; Siegler MA; Goldberg DP A Reactive Manganese(IV)-Hydroxide Complex: A Missing Intermediate in Hydrogen Atom Transfer by High-Valent Metal-Oxo Porphyrinoid Compounds. J. Am. Chem. Soc 2018, 140, 4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lymar SV; Ertem MZ; Polyansky DE Solventdependent transition from concerted electron−proton to proton transfer in photoinduced reactions between phenols and polypyridine Ru complexes with proton-accepting sites. Dalton Trans 2018, 47, 15917–15928. [DOI] [PubMed] [Google Scholar]
- (14).Tano T; Okubo Y; Kunishita A; Kubo M; Sugimoto H; Fujieda N; Ogura T; Itoh S Redox Properties of a Mononuclear Copper(II)-Superoxide Complex. Inorg. Chem 2013, 52, 10431–10437. [DOI] [PubMed] [Google Scholar]
- (15).Osako T; Ohkubo K; Taki M; Tachi Y; Fukuzumi S; Itoh S Oxidation mechanism of phenols by dicopper-dioxygen (Cu2/O2) complexes. J. Am. Chem. Soc 2003, 125, 11027–11033. [DOI] [PubMed] [Google Scholar]
- (16).Dhar D; Yee GM; Markle TF; Mayer JM; Tolman WB Reactivity of the copper(III)-hydroxide unit with phenols. Chem. Sci 2017, 8, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Donoghue PJ; Gupta AK; Boyce DW; Cramer CJ; Tolman WB An Anionic, Tetragonal Copper(II) Superoxide Complex. J. Am. Chem. Soc 2010, 132, 15869–15871. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bailey WD; Gagnon NL; Elwell CE; Cramblitt AC; Bouchey CJ; Tolman WB Revisiting the Nucleophilic Reactivity of an Anionic Copper-Superoxide Complex. Inorg. Chem 2019, DOI: 10.1021/acs.inorgchem.9b00090, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Que L Jr.; Tolman WB Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- (19).Ray K; Heims F; Schwalbe M; Nam W High-valent metal-oxo intermediates in energy demanding processes: from dioxygen reduction to water splitting. Curr. Opin. Chem. Biol 2015, 25, 159–171. [DOI] [PubMed] [Google Scholar]
- (20).Kim S; Saracini C; Siegler MA; Drichko N; Karlin KD Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent. Inorg. Chem 2012, 51, 12603–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bordwell FG; Cheng J Substituent effects on the stabilities of phenoxyl radicals and the acidities of phenoxyl radical cations. J. Am. Chem. Soc 1991, 113, 1736–1743. [Google Scholar]
- (23). We recognize that this protocol is inappropriate for reactions with a significant ET/PT contribution because of significant solvent effects on driving force and solvent reorganization energies associated with generation of charged intermediates (ref 13e). However, the trends, crossover points, and slopes of fits to plots of uncorrected rate data (using k values, Figures S15) are closely similar to those generated using kDMSO values (Figures 5 and 9a) and all conclusions drawn from these plots are the same. We have chosen to use the kDMSO data in our analysis because of our desire to draw comparisons to previously published plots for LCuOH (ref 16) that use such corrected data (Figure 9).
- (24). In the two-phase reaction, k1 (0.44(9) M−1 s−1) for OMeArOH is statistically equal (3σ) to the second-order rate constant, k (0.7(1)M−1 s−1), derived from the single exponential decay phase. For continuity with other XArOH substrates, all arguments invoking rate data use k from the single exponential decay phase.
- (25).Eyer P; Lengfelder E Radical Formation During Autoxidation of 4-Dimethylaminophenol and Some Properties of the Reaction Products. Biochem. Pharmacol 1984, 33, 1005–1013. [DOI] [PubMed] [Google Scholar]
- (26).Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C−H Bond Activation by a Monocopper(III)−Hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dhar D; Yee GM; Spaeth AD; Boyce DW; Zhang H; Dereli B; Cramer CJ; Tolman WB Perturbing the Copper(III)− Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 2016, 138, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Neisen BD; Gagnon NL; Dhar D; Spaeth AD; Tolman WB Formally Copper(III)−Alkylperoxo Complexes as Models of Possible Intermediates in Monooxygenase Enzymes. J. Am. Chem. Soc 2017, 139, 10220–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lin T-Y; Wu C-H Activation of hydrogen peroxide in copper(II)/amino acid/H2O2 systems: effects of pH and copper speciation. J. Catal 2005, 232, 117–126. [Google Scholar]
- (30).Kim S; Ginsbach JW; Lee JY; Peterson RL; Liu JJ; Siegler MA; Sarjeant AA; Solomon EI; Karlin KD Amine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu-OOH Homolytic Cleavage Followed by Site-Specific Fenton Chemistry. J. Am. Chem. Soc 2015, 137, 2867–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Inherent in this assumption is the postulate that at the crossing point in the plots the rates of the PT/ET and CPET paths are approximately the same, and that the two paths have similar linear free energy relationships. We recognize that neither of these notions are necessarily strictly correct, but suggest that the deviances from these relationships are sufficiently small to enable the rough estimation of the thermodynamic parameters as described in the text.
- (32).(a) Tilset M The Thermodynamics of Organometallic Systems Involving Electron-Transfer Paths. In Electron Transfer in Chemistry; Balzani V, Ed.; Wiley-VCH: Weinheim, Germany, 2001; pp 677–713. [Google Scholar]; (b) Cappellani EP; Drouin SD; Jia G; Maltby PA; Morris RH; Schweitzer CT Effect of the Ligand and Metal on the pKa Values of the Dihydrogen Ligand in the Series of Complexes [M(H2)H(L)2]+, M= Fe, Ru, Os, Containing Isosteric Ditertiaryphosphine Ligands, L. J. Am. Chem. Soc 1994, 116, 3375–3388. [Google Scholar]
- (33).(a) Wurtele C; Gaoutchenova E; Harms K; Holthausen MC; Sundermeyer J; Schindler S Crystallographic characterization of a synthetic 1:1 end-on copper dioxygen adduct complex. Angew. Chem., Int. Ed 2006, 45, 3867–3869. [DOI] [PubMed] [Google Scholar]; (b) Maiti D; Lee D-H; Gaoutchenova K; Würtele C; Holthausen MC; Narducci Sarjeant AA; Sundermeyer J; Schindler S; Karlin KD Reactions of a Copper(II) Superoxo Complex Lead to C-H and O-H Substrate Oxygenation: Modeling Copper-Monooxygenase C-H Hydroxylation. Angew. Chem., Int. Ed 2008, 47, 82–85. [DOI] [PubMed] [Google Scholar]
- (34).Maiti D; Fry HC; Woertink JS; Vance MA; Solomon EI; Karlin KDA 1:1 Copper−Dioxygen Adduct is an End-on Bound Superoxo Copper(II) Complex which Undergoes Oxygenation Reactions with Phenols. J. Am. Chem. Soc 2007, 129, 264–265. [DOI] [PubMed] [Google Scholar]
- (35).Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic insights into the oxidation of substituted phenols via hydrogen atom abstraction by a cupric-superoxo complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kim S; Lee JY; Cowley RE; Ginsbach JW; Siegler MA; Solomon EI; Karlin KD A N3S(thioether)-Ligated CuII-Superoxo with Enhanced Reactivity. J. Am. Chem. Soc 2015, 137, 2796–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bhadra M; Lee JYC; Cowley RE; Kim S; Siegler MA; Solomon EI; Karlin KD Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc 2018, 140, 9042–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fujii T; Yamaguchi S; Hirota S; Masuda H H-atom abstraction reaction for organic substrates via mononuclear copper(II)- superoxo species as a model for DβM and PHM. Dalton Trans 2008, 164–170. [DOI] [PubMed]
- (39).Iovan DA; Wrobel AT; McClelland AA; Scharf AB; Edouard GA; Betley TA Reactivity of a stable copper-dioxygen complex. Chem. Commun 2017, 53, 10306–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Luo YR Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press LLC: Boca Raton, FL, 2003. [Google Scholar]
- (41). The same conclusion may be drawn by comparing uncorrected k values, where for LCuOH the log k range is from ca. 1.5 to + 6.3 (ref 16) relative to those for [K(Krypt)][LCuO2] (log k range from ca. −0.3 to +2.0).
- (42).Dhar D; Tolman WB Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 2015, 137, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Peterson RL; Ginsbach JW; Cowley RE; Qayyum MF; Himes RA; Siegler MA; Moore CD; Hedman B; Hodgson KO; Fukuzumi S; Solomon EI; Karlin KD Stepwise Protonation and Electron-Transfer Reduction of a Primary Copper-Dioxygen Adduct. J. Am. Chem. Soc 2013, 135, 16454–16467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jung J; Kim S; Lee Y-M; Nam W; Fukuzumi S Switchover of the Mechanism between Electron Transfer and Hydrogen-Atom Transfer for a Protonated Manganese(IV)-Oxo Complex by Changing Only the Reaction Temperature. Angew. Chem., Int. Ed 2016, 55, 7450–7454. [DOI] [PubMed] [Google Scholar]
- (45).Morimoto Y; Park J; Suenobu T; Lee Y-M; Nam W; Fukuzumi S Mechanistic Borderline of One-Step Hydrogen Atom Transfer versus Stepwise Sc3+-Coupled Electron Transfer from Benzyl Alcohol Derivatives to a Non-Heme Iron(IV)-Oxo Complex. Inorg. Chem 2012, 51, 10025–10036. [DOI] [PubMed] [Google Scholar]
- (46).Usharani D; Lacy DC; Borovik AS; Shaik S Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer During Activation of X−H Bonds (X = C, N, O) by Nonheme Iron−Oxo Complexes of Variable Basicity. J. Am. Chem. Soc 2013, 135, 17090–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bím D; Maldonado-Domínguez M; Rulísěk L; Srnec M Beyond the classical thermodynamic contributions to hydrogen atom abstraction reactivity. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E10287–E10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Goetz MK; Anderson JS Experimental Evidence for pK- Driven Asynchronicity in C-H Activation by a Terminal Co(III)-Oxo Complex. J. Am. Chem. Soc 2019, 141, 4051–4062. [DOI] [PubMed] [Google Scholar]
- (49).(a) Parsell TH; Yang MY; Borovik AS C-H Bond Cleavage with Reductants: Re-Investigating the Reactivity of Monomeric MnIII/ IV-Oxo Complexes and the Role of Oxo Ligand Basicity. J. Am. Chem. Soc 2009, 131, 2762–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zdilla MJ; Dexheimer JL; Abu-Omar MM Hydrogen Atom Transfer Reactions of Imido Manganese(V) Corrole: One Reaction with Two Mechanistic Pathways. J. Am. Chem. Soc 2007, 129, 11505–11511. [DOI] [PubMed] [Google Scholar]; (c) Fulton JR; Sklenak S; Bouwkamp MW; Bergman RG A Comprehensive Investigation of the Chemistry and Basicity of a Parent Amidor-uthenium Complex. J. Am. Chem. Soc 2002, 124, 4722–4737. [DOI] [PubMed] [Google Scholar]; (d) Roth JP; Mayer JM Hydrogen Transfer Reactivity of a Ferric Bi-Imidazoline Complex That Models the Activity of Lipoxygenase Enzymes. Inorg. Chem 1999, 38, 2760–2761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.